Regulation of cGAS-STING pathway - Implications for systemic lupus erythematosus
-
Audrey M. Hagiwara

Abstract
Type I interferon (IFN-I) is implicated in the pathogenesis of systemic lupus erythematosus (SLE) and the closely associated monogenic autoinflammatory disorders termed the “interferonopathies.” Recently, the cytosolic DNA sensor cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS) and its downstream signaling adaptor stimulator of interferon genes (STING) have been identified as having important, if not central, roles in driving IFN-I expression in response to self-DNA. This review highlights the many ways in which this pathway is regulated in order to prevent self-DNA recognition and underlines the importance of maintaining tight control in order to prevent autoimmune disease. We will discuss the murine and human studies that have implicated the cGAS-STING pathway as being an important contributor to breakdown in tolerance in SLE and highlight the potential therapeutic application of this knowledge for the treatment of SLE.
Introduction
Systemic lupus erythematosus (SLE) is a systemic autoimmune disease that has multi-organ involvement and presents with clinically varying phenotypic expression. It is more prevalent in females, with a female-to-male incidence ratio of approximately 9:1 during the fourth decade of life and may be seen more frequently in non-white populations.[1] Manifestations of SLE include leukopenia, mucocutaneous lesions, vasculitis, arthritis, nephritis, and central nervous system (CNS) disease, among many others, with periods of flares and remissions. There is also a wide spectrum of disease manifestations from relatively mild symptoms to organ-threatening disease.[2] The pathogenesis for this disease is complex and still relatively unknown, with several immunopathogenic pathways recently identified as possible contributory factors. The understanding of some of these pathways has resulted in potential new therapeutic targets, such as B cell activating factor (BAFF) or B lymphocyte stimulator (BLyS) and IFN-alpha (IFNα), among others. This has resulted in the approval of belimumab in 2011, which was the first agent specifically approved for SLE treatment since hydroxychloroquine in 1958.[3, 4]
A more recent potential target has emerged in the literature, involving the cyclic guanosine monophosphate (GMP)-adenosine monophosphate (AMP) synthase (cGAS) and stimulator of interferon genes (STING) pathway, which induces type I interferon (IFN-I) in SLE.[5] The involvement of the cGAS-STING pathway in SLE has largely stemmed from the discovery of monogenic autoinflammatory or autoimmune disorders that were characterized by defective regulation of the cGAS-STING pathway and overproduction of type I IFN, leading to them being described as “type I interferonopathies.”[6, 7] The potential that cGAS-STING may also be dysregulated in SLE, another type I interferonopathy, was rapidly realized. cGAS is a cytosolic double-stranded DNA (dsDNA) sensor discovered in 2013 that is indispensable for host anti-viral immunity.[8] Once activated, it synthesizes the second messenger cyclic GMP-AMP (cGAMP) that activates STING, an endoplasmic reticulum (ER) resident adaptor protein critical for induction of IFN-beta (IFNβ) downstream of immunostimulatory cytosolic DNA.[9, 10, 11] Here, we discuss new insights into how the cGAS-STING signaling pathway is regulated, the studies supporting a role for cGAS-STING in the pathogenesis of SLE, and the potential therapeutic implications of targeting this pathway in developing treatments for SLE.
Type I IFNs and their role in SLE
IFN-I is a well-studied factor implicated in the pathogenesis of SLE, first reported as increased in active SLE serum as far back as the 1970s.[12, 13] The IFN-I family is a major subgroup of cytokine proteins that are expressed in response to detection of viral, bacterial, or self-RNA and DNA by a group of pattern recognition receptors described as nucleic acid sensors. Subtypes include IFNα, of which there are 13 identified isoforms, as well as IFNβ, IFN-epsilon (IFNε), IFN-kappa (IFNκ), and IFN-omega (IFNω).[14] IFN-I bind to type 1 interferon receptors – which are a complex of two proteins, IFNAR1 and IFNAR2 – initiating a signal transduction cascade through activation of the Janus kinase/Signal transducer and activator of transcription (JAK/STAT) pathway, ultimately inducing the expression of IFN stimulated genes (ISGs).[15, 16, 17, 18, 19] Studies have shown that serum IFN-I levels and ISGs are elevated in peripheral blood mononuclear cells (PBMCs) of patients with SLE and positively correlate with disease activity.[13, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33] Elevated levels of IFN-I activity have also been positively correlated with anti-double-stranded DNA (anti-dsDNA) antibody positivity and hypocomplementemia in patients with SLE.[27] Furthermore, skin or kidney samples from patients with cutaneous lupus or lupus nephritis have demonstrated elevated ISG activity.[34, 35, 36] In addition, treatment of patients with viral infections such as hepatitis C or even malignancy with IFNα has resulted in induction of SLE symptoms in recipients.[37, 38] These findings have suggested IFN-I as a significant cause for the pathologic manifestations seen in SLE.[39]
RNA/DNA sensors not only recognize viral and bacterial RNA/DNA but can also recognize self or endogenous nucleic acids. Tight regulation of these sensors is therefore required to prevent their activation, type I IFN production, and development of SLE-like disease. The methods to prevent inapproapriate recognition include intracellular and extracellular RNA- and DNA-degrading enzymes, in addition to sequestration or compartmentalization of nucleic acids to prevent detection (such as in the nucleus or mitochondria). However, when cells are damaged or stressed, nucleic acids are released extracellularly (either via apoptosis or necrosis or via release in exosomes) or into the cytosol from subcellular compartments such as the mitochondria or nucleus, providing the potential for activation of nucleic acid sensors.[40, 41] Sources of cytosolic DNA in SLE include mitochondrial DNA,[42] endogenous retroviral elements,[43] genomic DNA, and oxidized DNA damage products.[44] Whereas the involvement of endosomal toll-like receptor (TLR)7 and TLR9 in nucleic acid sensing in SLE has been known for nearly 20 years,[45, 46, 47, 48] the role that the cytosolic DNA sensor cGAS might play in SLE pathogenesis or type I IFN production has only recently been uncovered.[49] Importantly, while TLR7 and TLR9 expression is largely restricted to subsets of immune cells (myeloid and B cells predominantly), cGAS and STING are broadly expressed in immune and non-immune cells, thus increasing the potential that defective cGAS signaling in either may be important in IFN-driven autoimmune disease.[50] Interestingly, both cGAS and STING are also ISGs, which is a positive feature in viral infection but less so when considering the potential amplifying effect their enhanced expression might have on IFN expression in the context of SLE.[49]
Regulation of cGAS-STING Signaling
Much of our understanding regarding the importance of regulating cGAS-STING signaling has come from the discoveries of monogenic IFN-driven diseases such as Aicardi-Goutier syndrome (AGS), which is caused by mutations in TREX1, a DNase that prevents cytosolic DNA detection via DNA degradation, and STING-associated vasculopathy with onset in infancy (SAVI), which is driven by activating mutations in STING.[6, 7] More recently an additional monogenic SLE-like disease was discovered where mutations in the COPA gene result in inappropriate trafficking and regulation of STING and enhanced IFN production.[51, 52]
Activation of the cGAS-STING pathway (outlined in Figure 1) is initiated by binding of dsDNA to cGAS, triggering a conformational change in the active site which then catalyzes the formation of cGAMP from adenosine triphosphate (ATP) and guanosine triphosphate (GTP).[8, 53, 54] cGAMP acts as a second messenger and binds to STING, embedded in the ER membrane, triggering a conformational change that promotes dimerization of STING.[55, 56] The STING dimer then translocates from the ER to the Golgi, where it recruits TANK-binding kinase 1 (TBK1) to form STING-TBK1 aggregates – the STING signalsome – which binds and recruits the transcription factor IRF3.[55, 57] Recruitment of IRF3 is facilitated by phosphorylation of serine 366 on STING by TBK1, which in turn phosphorylates IRF3 once it binds.[58, 59] Clustering of STING at the Golgi also promotes palmitoylation and full activation of STING.[60] STING can also cluster with I kappa B (IκB) Kinase (IKK) and drive activation of NFκB following the phosphorylation and degradation of IκB.[57, 58, 61, 62] IRF3 and NFκB translocate into the nucleus and function together to induce the expression of IFNβ and pro-inflammatory cytokines.[54, 63]
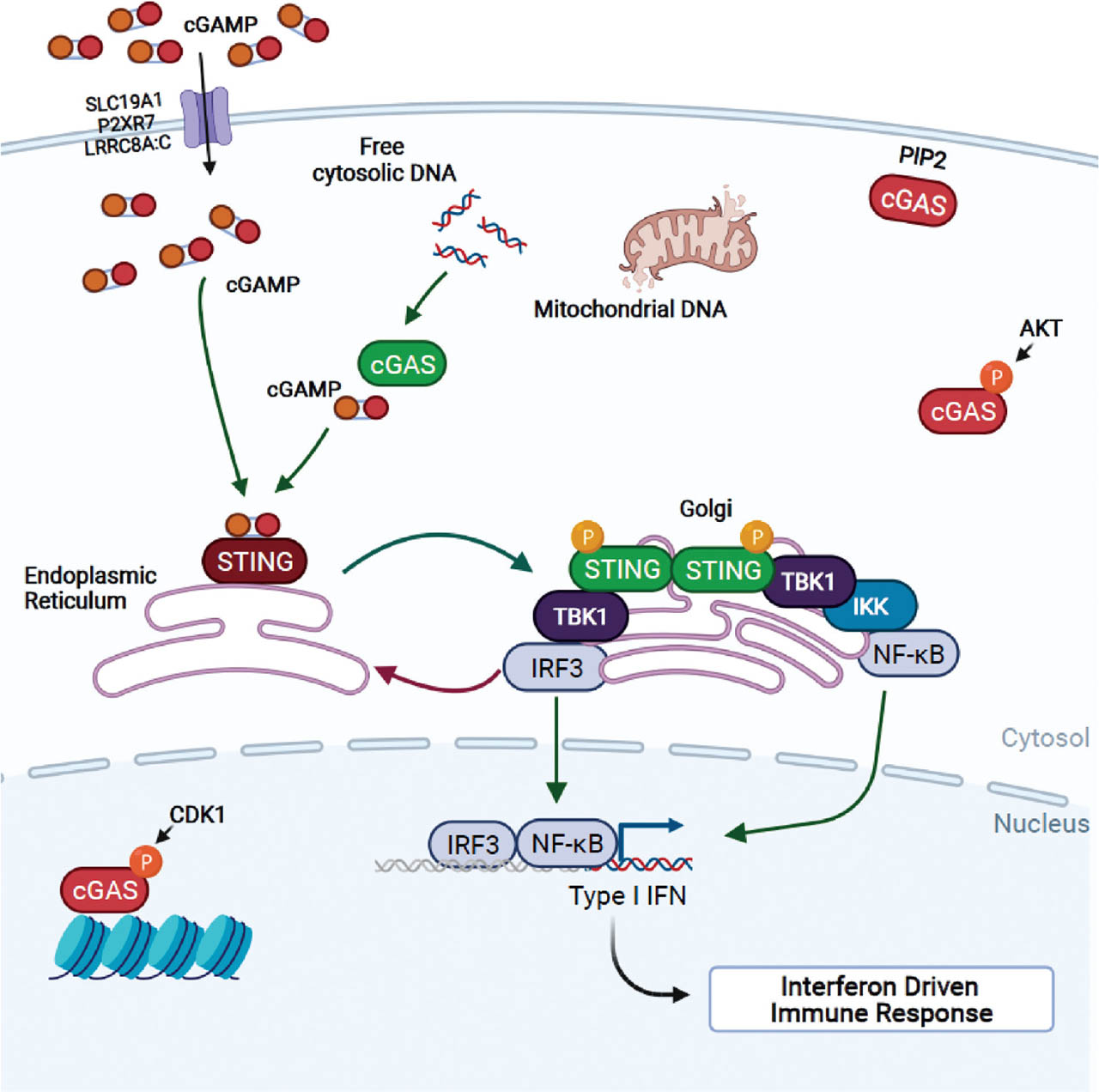
Overview of the cGAS-STING pathway. cGAS activity can be negatively regulated via phosphorylation of Serine 291 by CDK1 and AKT, triggering sequestration of cGAS to histone. cGAS binding to PIP2 also restricts its ability to interact with and sense free cytosolic DNA. Untethered cGAS may freely bind cytosolic viral, mitochondrial, bacterial, or damaged nuclear DNA and form homodimers to generate cGAMP. cGAMP generated this way or transported into the cytosol via transporter proteins SLC19A1, P2RX7, or LRRC8A:C bind to and activate STING, which then homodimerizes and translocates to the Golgi. cGAMP-bound STING homodimers recruit TBK1, which drives STING phosphorylation at Serine 366 to initiate IRF3 and IKK/NFκB phosphorylation, nuclear translocation, and transcription of the interferon-driven immune response. AMP, adenosine monophosphate; CDK1, cyclin-dependent kinase 1; cGAMP, 2′3′-cyclic guanosine monophosphate (GMP)-adenosine monophosphate (AMP); cGAS, cyclic GMP-AMP synthase; IFN, interferon; IRF3, IFN response factor 3; IKK, IκB kinase; LRRC8A:C, leucine-rich repeat-containing protein 8A:C; NFκB, nuclear factor kappa-light-chain-enhancer of activated B cells; P2RX7, P2X purinoceptor 7; PIP2, phosphatidylinositol; STING, stimulator of IFN genes; TBK1, tank-binding kinase 1. Figure created with Biorender.com.
Regulation of cGAS-STING signaling is achieved on a number of levels as shown in Figure 1 – access to cGAMP, localization of the various components, and posttranslational modifications that alter the stability and function of either cGAS or STING (reviewed by Wan[64]). For example, localization of both cGAS and STING is closely coupled with function. cGAS was first thought to be solely a cytosolic protein, thus preventing inappropriate access to self-DNA.[65] However, later studies found that cGAS can be found in the nucleus, with one study showing that nuclear tethering was essential for preventing autoreactivity,[66] and additional studies showing that chromatin tethering was via interactions with the histone H2A-H2B dimer leading to cGAS inactivation in order to block recognition of DNA during mitosis.[67, 68] cGAS also interacts with the plasma membrane inner leaflet via binding to phosphoinositol 4,5-bisphosphate (PIP2), an interaction that sequesters cGAS and prevents self-DNA detection and activation.[65] Alterations that result in mislocalization of STING also impact its activity. COPA syndrome is a recently identified inherited disorder that results from point mutations in the COPA gene. COPA is an integral part of the transport machinery that facilitates retrograde transport of proteins from the Golgi back to the ER. COPA mutations are associated with enhanced STING signaling from the Golgi as a result of failure to return STING signaling complexes to the ER.[52, 69, 70, 71, 72] Interaction with the calcium sensor stromal interaction molecule 1 (STIM1) also acts to inhibit STING by retaining it in the ER. Not surprisingly, deletion of Stim1 results in increased IFN and ISG expression following viral infection.[73, 74]
cGAS-STING activity can also be regulated through availability of the second messenger cGAMP. Recent evidence is emerging that in addition to being synthesized by cGAS, cGAMP can be transferred between cells or taken up from the extracellular milieu by cGAMP importers to directly activate STING. Regarding between cell transfer, two independent studies showed how cGAS activation by DNA virus or retrovirus drove encapsulation of cGAMP into viral particles that could then trigger an antiviral state in a STING-dependent manner in nearby target cells.[75, 76] cGAMP is also released by dead and dying cells into the extracellular milieu and thereby facilitates its role as a damage-associated molecular patterns (DAMP) or alarmin. Due to its negative charge, cGAMP must be actively imported into cells to have an effect on STING. While there is some evidence that it is internalized via clathrin-mediated endocytosis, recently the reduced folate carrier SLC19A1, which is highly expressed on myeloid cells, has been shown by two independent groups to act as a cGAMP importer on monocytes and macrophages.[77, 78] Interestingly, an association between polymorphisms in SLC19A1 with increased risk of developing SLE has been demonstrated, making SLC19A1 an interesting target for further evaluation in SLE.[79] In addition to SLC19A1, two other cGAMP importers have recently been described – P2RX7 which is found on tumor-associated macrophages and facilitates cGAMP uptake in an ATP-dependent manner,[80] and LRRC8A:C which is found on endothelial cells and facilitates cGAMP uptake.[81] cGAMP is also targeted by extracellular hydrolase ENPP1, suggesting an additional way in which cGAMP levels and activity may be dysregulated in SLE.[82, 83] Thus, understanding how cGAMP and the proteins that regulate its stability and uptake into cells will be important in understanding its contribution to cGAS-STING driven expression of IFNβ in SLE.
Posttranslational modifications also regulate the activity and stability of both cGAS and STING. B lymphocyte kinase (BLK) for example phosphorylates tyrosine 215 of cGAS in human skin fibroblast cells to promote cytosolic retention,[84] whereas AKT and CDK1 phosphorylate murine cGAS on serine 291 to inhibit its enzymatic activity; CDK1-induced phosphorylation promotes chromatin tethering during mitosis in order to prevent recognition of self-DNA.[85, 86, 87] Whether any alterations of cGAS phosphorylation occur in SLE has yet to be examined. A number of E3 ligases have also been identified as promoting cGAS activation, whether by monoubiquitination (TRIM14, TRIM56) or K27-linked polyubiquitination (RNF185).[88, 89, 90] Glutamylation and acetylation are other means of regulating cGAS activity in a dynamic way (reviewed by Wu[91]), although whether these forms of post-translational modifications are altered in SLE or in specific immune cell subsets is currently unknown.
Evidence for Role for cGAS-STING Pathway in SLE from Murine Studies
Multiple studies using murine models with lupus-like features have implicated the cGAS-STING pathway in the pathogenesis of lupus as outlined in Table 1.[92] Much of the initial work in this respect came from studies using the Trex1−/− mice.[93] TREX1 is an exonuclease that digests DNA in the cytoplasm.[94, 95, 96] Discussed above, loss of function mutations in TREX1 in humans is associated with the autoimmune and inflammatory disease AGS, an IFN-I mediated infantile disease characterized by encephalopathy and SLE,[97, 98, 99, 100, 101, 102, 103, 104, 105] whereas Trex1 deficiency in mice manifests as a noninfectious autoimmune inflammatory myocarditis, driven by self-DNA recognition via the cGAS-STING sensing pathway.[106, 107, 108, 109] Using Trex1-deficient mice, Gao et al. demonstrated that complete deletion of cGas increased their survival rates and led to the elimination of inflammatory pathology within organ tissues as well as an overall decrease in ISGs.[108] In addition, deletion of cGas in the Trex1-deficient mice significantly reduced production of autoantibodies and the number of activated T cells.[108] These findings were also reported in a separate study performed by Gray et al., who observed an increase in survival, a decrease in inflammatory destruction of tissue, and a decrease in autoreactivity and IFN-I production in Trex1/cGas double knockout mice.[110] These findings strongly suggest that cGAS activation plays an important role in the development of the pathology seen in SLE. The lethal auto-immune and myocarditis phenotype of the Trex1−/− mice is also rescued by deletion of STING, confirming the role for this pathway in this model.[106, 107, 111]
Mouse Disease Models implicating cGAS-STING in autoimmune disease indicating molecular cause of disease and phenotype
| Model | Molecular cause of disease | Notable phenotypes | References |
|---|---|---|---|
| Trex1−/− | Lacking cytosolic DNA exonuclease activity |
|
[93, 99, 107, 109, 110] |
| 129/B6.Fcgr2b−/− | Lack of inhibitory signal to autoreactive B cells |
|
[113, 115] |
| Stim1−/− | Spontaneous anterograde transport and activation of STING |
|
[73, 74] |
| COPAE241K/E241K | Ineffective retrograde transport and deactivation of STING |
|
[51, 69, 71] |
| hSTING-R284M hSTING-N154S |
Constitutively active STING |
|
[116] |
| Ogg1−/− | Raised 8-OHdG Activation of STING |
|
[126] |
| UVB exposure (500 mJ/cm2) | DNA damage and activation of neutrophils |
|
[124, 125] |
8-OHdG, 8-Oxo-2′-deoxyguanosine; IFN, interferon; ISG, IFN-stimulated gene; STING, stimulator of interferon genes; STIM1, stromal interaction molecule 1; UVB, ultraviolet B light.
Additional evidence supporting a central role for cGAS-STING pathway in SLE has come from work using mice deficient in Fcgr2b, a SLE-susceptibility gene, deletion of which is known to cause lupus-like disease in mice.[112, 113, 114] In a study conducted by Thim-Uam et al., researchers utilized Fcgr2b-deficient mice on the 129 background backcrossed to C57BL6 mice (129/B6.Fcgr2b−/−), which carries an auto-immune susceptibility locus containing interferon-inducible Ifi200 family genes, including Ifi202, which is thought to utilize STING. These mice develop overt autoreactivity and fatal lupus-like disease. STING expression was found to be increased in 129/B6.Fcgr2b−/− mice and disruption of STING signaling by crossing these mice to Stinggt/gt mice rescued the autoimmune phenotype.[115] In addition, when compared with the sera from the Fcgr2b−/− mice, sera from the Fcgr2b−/−. Stinggt/gt mice contained lower antinuclear antibody (ANA) and anti-dsDNA antibody levels, indicating the significant role STING plays in developing autoimmunity. Evaluation of kidney tissue from the Fcgr2b-deficient mice exhibited upregulation of ISG expression in the presence of STING and demonstrated diffuse proliferative glomerulonephritis in comparison to the Fcgr2b−/−. Stinggt/gt mice. Thim-Uam et al. also conducted an adoptive transfer of STING-activated bone marrow dendritic cells from Fcgr2b-deficient samples into wildtype and double-deficient mice.[115] In the wildtype mice, this adoptive transfer resulted in the development of autoimmunity, including production of anti-dsDNA and expansion of T and B cells. In the Fcgr2b−/−. Stinggt/gt mice, the adoptive transfer resulted in increased levels of anti-dsDNA. In both groups, the adoptive transfer resulted in increased immune complex deposition and inflammatory cell infiltration in kidney tissues. In this murine model, STING was implicated in the immunopathogenesis and development of autoimmune manifestations observed in SLE. This is supported by analysis of mice bearing a constitutive active mutation in STING, which resulted in enhanced ISG expression, immune infiltration into tissues, and an SLE-like disease.[116] Although the majority of murine studies substantiate the role of the cGAS-STING pathway in augmenting IFN-I responses and producing the inflammatory pathologies seen in SLE, studies have conversely described a suppressive role for STING in both the MRLlpr and pristane models.[117, 118] Crossing STING−/− with the MRLlpr mice resulted in enhanced splenomegaly, lymphadenopathy, autoantibody production, and increased proteinuria in comparison to wildtype mice. Similar findings were also observed in STING−/− animals using the pristane model of IFN-driven SLE. To explain these findings the authors showed that macrophages from STING−/− mice were hyperresponsive to TLR7 and TLR9 ligands, and that expression of negative regulators of these pathways (A20, SOCS1, and SOCS3) were decreased in both mice and macrophages from the STING-deficient mice.[118] Similar opposing roles for STING and TLR-driven pathways have also been shown in models of infection.[119] Thus, it will be important to understand the relative contribution of STING and TLRs to IFN induction in human SLE in order to support targeting it therapeutically.
Recently, the role of cGAS-STING signaling in regulating UVB-driven IFN-I responses in the skin and subsequent systemic disease has been elucidated in both murine and human studies. A large majority of SLE patients exhibit some form of sensitivity to UVB, which consequently can manifest in localized skin disease or provoke systemic SLE flares.[120] Critically, exposure to UVB drives oxidative stress, oxidized DNA damage, and accumulation of 8-OH-dG lesions in the skin.[121, 122] Oxidized DNA, including 8-OH-dG, is also abundant in UV-exposed skin lesion of lupus patients. Critically, it is insensitive to TREX1 degradation and immune sensing of UV-damaged DNA is dependent on both cGAS and STING is sensed by cGAS.[123] Subsequent studies by Skopelja-Gardner et al. demonstrated that exposure of healthy human skin to a single dose of UVB resulted in an increased IFN-I signature, thereby indicating UVB exposure as an important source of IFN-I activation in skin.[124] In mice, the investigators showed that a single dose of UVB exposure resulted in increased ISG expression in the skin in keeping with analysis of human skin.[124] Furthermore, increased ISG expression in both peripheral blood cells and kidney tissue following UVB exposure to skin indicated that UVB exposure can induce a systemic inflammatory response. When exposing cGas-deficient mice to UVB, researchers observed a near-complete elimination of early cutaneous ISG expression. In addition, peripheral blood from cGas-deficient mice contained lower levels of ISG in comparison to wildtype mice following UVB exposure, implicating cGAS as a consequential factor contributing to a systemic immune response. Interestingly, Skopelja-Gardner et al. also observed that the skin of cGas-deficient mice, in comparison to the skin of wildtype mice, had significantly reduced neutrophil and inflammatory monocyte infiltration as well as decreased chemokine expression following UVB exposure.[124] A follow-up study demonstrated that skin exposure to UVB triggered migration of neutrophils to the kidney from the skin, demonstrating a direct link between skin inflammation by UV light and kidney injury.[125] The reduction of inflammatory cell infiltration in the setting of cGas deficiency indicates the importance of the cGAS-STING pathway in augmenting the innate immune cell response. Such findings from this murine model demonstrate how UVB exposure, a common cause of flares in SLE, can incite both local and systemic IFN-I responses, predominantly induced by the cGAS-STING pathway. Interestingly, the DNA repair enzyme Ogg1 catalyzes the repair of 8-OH-dG DNA lesions that form following UVB exposure in the skin. Deletion of Ogg1 in mice resulted in accelerated skin inflammation in the pristane model of IFN-inducible SLE, with further analysis implicating the cGAS-STING in driving enhanced IFNβ expression detected in the mice. Furthermore, OGG1 expression in lesional areas from SLE with cutaneous lupus erythematosus (CLE) was reduced compared with non-lesional areas, suggesting that OGG1 protects against cutaneous involvement in SLE by reducing 8-OH-dG-driven IFN responses.[126]
Evidence for Role for cGAS-STING Pathway in SLE Patients
The first indication that the cGAS-STING pathway may be clinically important came from the discovery of an extremely rare autoinflammatory disease driven by autosomal dominant mutations in TMEM173, the gene encoding STING, presenting with clinical and histological evidence of systemic inflammation, vasculopathy, and pulmonary inflammation.[127] Termed SAVI, patients typically presented with systemic inflammation, interstitial lung disease (ILD) on computed tomography of the chest, and severe cutaneous vasculopathy.[128] Patients also demonstrated a marked increase in ISG expression in their peripheral blood, indicating that TMEM173 mutations led to a gain of function in STING, resulting in increased interferon production, marking SAVI as an interferonopathy.[127, 129] Compared with healthy controls, skin biopsies of SAVI patients with active lesions revealed upregulated inflammatory markers in their vascular endothelial cells. Such results implicate the cGAS-STING pathway in augmenting the inflammatory process, thereby contributing to the clinical and histologic signs of inflammation. More recently, hereditary mutations in the calcium sensor STIM1, which acts to restrain STING, have been reported to result in a syndrome of immunodeficiency (recurrent bacterial and viral infections), autoimmune hemolytic anemia and hepatosplenomegaly, resulting from impaired lymphocyte activation in the patients.[130] As discussed above, mutations in COPα, which gives rise to COPA syndrome, manifests as either diffuse alveolar hemorrhage or ILD. As discussed above, multiple groups have not only shown a role for type I IFN in driving the pathology of COPA but have also shown that failure to relocate STING from the Golgi post activation results in overactivation of STING-induced IFN induction.[51, 52, 69, 70, 71, 72, 131, 132, 133, 134] Recent work has also shown that C9orf72, a protein associated with the development of amyotrophic lateral sclerosis (ALS), regulates STING stability and that the absence of C9orf72 results in STING-induced inflammatory disease.[135] Thus, enhanced exposure to cytosolic DNA through incomplete DNA degradation in the case of AGS or a reduction in activity of DNA repair pathways targeting oxidized lesions, or lack of regulation of STING may contribute to enhanced signaling through the cGAS-STING pathway in SLE. Research into how precisely this pathway is dysregulated in SLE using cells and tissues from patients is ongoing and is ultimately required to address this.
In SLE patients, the role of the cGAS-STING pathway was first demonstrated by An et al.[5] When cGAS expression was measured in the PBMCs from SLE patients and healthy patients, cGAS expression in the SLE patients was significantly higher than in the healthy controls, with approximately half of the SLE patients measured demonstrating a modest increase in cGAS expression. Additionally, when comparing cGAS expression in PBMCs from SLE patients to an inflammatory disease control group consisting of rheumatoid arthritis (RA) patients, cGAS expression was found to be lower in the RA samples than in the SLE samples. After demonstrating an increase in cGAS expression in the SLE samples, an investigation into the downstream effects and levels of cGAMP was examined using mass spectrometry. Importantly, 15% of SLE patients had detectable cGAMP, while no cGAMP was detectable in the samples from the RA patients and the healthy control group, indicating more enzymatic activity of cGAS in the SLE group.[5] These results suggest a potential role for cGAS in driving IFNβ expression in SLE.
In support of a role for cGAS in SLE, Kato et al. measured serum IFN-I activity and ISG activity in Japanese patients with SLE, systemic sclerosis (SSc), primary Sjogren syndrome (SjS), and healthy controls.[136] In comparison to the other samples, sera from the SLE patients were shown to have significantly higher IFN-I bioactivity and ISG activity. In addition, Kato et al. demonstrated that STING-knockout reporter cells had reduced ISG-inducing activity when stimulated with SLE sera, indicating that the cGAS-STING pathway plays a role in activation of ISG. Furthermore, a correlation was found between STING-dependent ISG-inducing activity and IFN-I independent ISG-inducing activity of the sera from the SLE patients, suggesting that the activation of the cGAS-STING pathway may contribute to IFN-I levels in SLE.
The effect of the cGAS-STING pathway on IFN-I production in SLE patients was again demonstrated by Murayama et al[137]. Using PBMCs from SLE patients and healthy patients, the frequency of IFN-α producing cells was measured after the samples were stimulated with cGAMP. The frequency of IFN-α producing cells in the SLE patients was significantly higher when compared with the healthy controls, indicating that cGAMP activation of the cGAS-STING pathway increased IFN-α production. Additionally, STING expression in monocytes from SLE patients was increased in comparison to the healthy patients.[137] Furthermore, when cGAMP stimulated monocytes from healthy controls were treated with IFN-α, there was an increase in the production of IFN-α and STING expression, as well as an increase in the presence of TBK1, indicating that elevated levels of IFN-α seen in SLE may augment activation of the cGAS-STING pathway. Murayama et al. also found a positive correlation between the frequency of IFN-α producing monocytes with SLE disease activity and serum IFN-α levels.
Although studies are limited thus far in patients, there is growing appreciation of the role of the cGAS-STING pathway in the activation and production of IFN-I and its role in human disease (as outlined in Table 2), which is fundamental to the systemic inflammation observed in SLE. However, more extensive studies are needed to understand whether defects in this pathway are common to all SLE patients or whether regulation of these pathways is only altered in a specific subset or subgroup.
cGAS-STING in human autoimmune/autoinflammatory disorders highlighting disease name, gene involved, symptoms and molecular cause of disease
| Disease | Affected allele(s) | Molecular cause of disease | Notable symptoms | References |
|---|---|---|---|---|
| Aicardi-Goutières syndrome | TREX1 | Lacking cytosolic DNA exonuclease activity |
|
[98, 100, 102, 103, 104, 145] |
| SLE | DNASE1 FCGR2B TMEM173 TREX1 |
Multiple |
|
[97, 113, 117] |
| CRAC Channelopathies | STIM1 | Spontaneous anterograde transport and activation of STING |
|
[130] |
| SAVI | TMEM173 | Constitutive activation of STING |
|
[127, 128, 129] |
| COPA Syndrome | COPA | Ineffective retrograde transport and deactivation of STING |
|
[51, 69, 71] |
COPA, coatomer subunit alpha; CRAC, calcium release activated channel; IFN, interferon; ISG, IFN-stimulated gene; ILD, interstitial lung disease; STING, stimulator of interferon genes; STIM1, stromal interaction molecule 1; SAVI, STING-associated vasculopathy with onset in infancy; SLE, systemic lupus erythematosus.
Therapeutic implications of cGAS-STING and Future Directions
While there is relatively limited research on the cGAS-STING pathway (and even fewer studies using human subjects), contemporary findings have demonstrated compelling evidence supporting this pathway as a key mediator in the production of IFN-I and the development of systemic inflammation in SLE. Also, while treatment for SLE has generally involved broad-based immunosuppression, there is a growing interest in more targeted therapies, resulting in the approval of belimumab, a monoclonal antibody to BAFF or BlyS, and most recently, anifrolumab in August 2021. Additionally, some commonly used therapies for SLE, primarily antimalarials such as hydroxychloroquine, have been shown to attenuate the cGAS-STING pathway to some degree.[138, 139] More recently, therapeutic agents targeting the cGAS-STING pathway have been described. Lama et al. identified human-cGAS small-molecule inhibitors effective in human macrophages using high-throughput screening and targeted medicinal chemistry optimization, highlighting the potential therapeutic development of cGAS antagonists.[140] Other small molecules targeting the active site have also been described and tested in murine studies (reviewed by McWhirter and Jefferies[49], Decout et al[139]). Understanding the localization of cGAS activity may also provide another prospective therapeutic target in the cGAS-STING pathway. As described previously, cGAS activity has been shown to be dependent on subcellular localization, such as plasma tethering, which allows for interaction with cytosolic DNA and subsequent activation.[65, 141, 142] cGAS is also typically thought to be compartmentalized to the cytosol but has been shown to exist within the nucleus and is closely tethered to intact nuclear chromatin.[66, 143] Thus, targeting localization of cGAS may be another opportunity for developing potential therapeutic agents.
STING antagonists have targeted STING palmitoylation – a necessary step to fully activate STING – inhibiting type I IFN responses and signs of systemic inflammation in the Trex1−/− mice.[60, 144] Furthermore, Haag et al. identified a potent small-molecule inhibitor of STING identified as H-151, which binds to STING at the transmembrane cysteine residue at position 91. As H-151 was shown to inhibit both murine and human STING activation, its clinical considerations are significant and may potentially be used in the development of cGAS-STING targeting agents.[144] The challenge currently is translating the the utility of these targeting strategies in animal models to human diseases, to provide more precise knowledge of the roles of the cGAS-STING pathway in IFN-driven disease and SLE. Thus, continued research investigating the relationship between SLE and the cGAS-STING pathway is essential for the development of this new class of promising immunomodulating agents, thereby expanding the therapeutic treatments currently available for SLE patients.
Conflict of Interest
There is no conflict of interest declared.
References
[1] Gergianaki I, Bortoluzzi A, Bertsias G. Update on the Epidemiology, Risk Factors, and Disease Outcomes of Systemic Lupus Erythematosus. Best Pract Res Clin Rheumatol, 2018;32(2): 188–205.10.1016/j.berh.2018.09.004Search in Google Scholar PubMed
[2] Cervera R, Khamashta MA, Font JO, et al. Systemic Lupus Erythematosus: Clinical and Immunologic Patterns of Disease Expression in a Cohort of 1,000 Patients. The European Working Party on Systemic Lupus Erythematosus. Medicine (Baltimore), 1993;72(2):113–124.10.1097/00005792-199303000-00005Search in Google Scholar
[3] Wallace DJ. Advances in Drug Therapy for Systemic Lupus Erythematosus. BMC Med, 2010;8(1):77.10.1186/1741-7015-8-77Search in Google Scholar PubMed PubMed Central
[4] Bag-Ozbek, A. and J.S. Hui-Yuen, Emerging B-Cell Therapies in Systemic Lupus Erythematosus. Ther Clin Risk Manag, 2021. 17: p. 39–54.10.2147/TCRM.S252592Search in Google Scholar PubMed PubMed Central
[5] An J, Durcan L, Karr RM, et al. Expression of Cyclic GMP-AMP Synthase in Patients with Systemic Lupus Erythematosus. Arthritis Rheumatol, 2017;69(4):800–807.10.1002/art.40002Search in Google Scholar PubMed
[6] Lee-Kirsch MA. The Type I Interferonopathies. Annu Rev Med, 2017;68: 297–315.10.1146/annurev-med-050715-104506Search in Google Scholar PubMed
[7] Rodero MP, Crow YJ. Type I Interferon-Mediated Monogenic Autoinflammation: The Type I Interferonopathies, A Conceptual Overview. J Exp Med, 2016;213(12): 2527–2538.10.1084/jem.20161596Search in Google Scholar PubMed PubMed Central
[8] Sun L, Wu J, Du F, et al. Cyclic GMP-AMP Synthase is a Cytosolic DNA Sensor that Activates the Type I Interferon Pathway. Science, 2013;339(6121):786–91.10.1126/science.1232458Search in Google Scholar PubMed PubMed Central
[9] Li XD, Wu J, Gao D, et al. Pivotal Roles of cGAS-cGAMP Signaling in Antiviral Defense and Immune Adjuvant Effects. Science, 2013;341(6152): 1390–1394.10.1126/science.1244040Search in Google Scholar PubMed PubMed Central
[10] Ablasser A, Goldeck M, Cavlar T, et al. cGAS Produces a 2′-5′-Linked Cyclic Dinucleotide Second Messenger that Activates STING. Nature, 2013;498(7454): 380–384.10.1038/nature12306Search in Google Scholar PubMed PubMed Central
[11] Li X, Shu C, Chaton CT, et al. Cyclic GMP-AMP Synthase is Activated by Double-stranded DNA-induced Oligomerization. Immunity, 2013;39(6):1019–1031.10.1016/j.immuni.2013.10.019Search in Google Scholar PubMed PubMed Central
[12] Hooks JJ, Jordan GW, Cupps T, et al. Multiple Interferons in the Circulation of Patients with Systemic Lupus Erythematosus and Vasculitis. Arthritis Rheum, 1982;25(4):396–400.10.1002/art.1780250406Search in Google Scholar PubMed
[13] Hooks JJ, Moutsopoulos HM, Geis SA, et al. Immune Interferon in the Circulation of Patients with Autoimmune Disease. N Engl J Med, 1979;301(1):5–8.10.1056/NEJM197907053010102Search in Google Scholar PubMed
[14] Uze G, Schreiber G, Piehler J, et al. The Receptor of the Type I Interferon Family. Curr Top Microbiol Immunol, 2007;316: 71–95.10.1007/978-3-540-71329-6_5Search in Google Scholar
[15] de Padilla CML, Niewold TB. The Type I Interferons: Basic Concepts and Clinical Relevance in Immune-Mediated Inflammatory Diseases. Gene, 2016;576(1):14–21.10.1016/j.gene.2015.09.058Search in Google Scholar PubMed PubMed Central
[16] Aaronson DS, Horvath CM. A Road Map for those Who Don’t Know JAK-STAT. Science, 2002;296(5573):1653–1655.10.1126/science.1071545Search in Google Scholar PubMed
[17] Mowen K, David M. Role of the STAT1-SH2 Domain and STAT2 in the Activation and Nuclear Translocation of STAT1. J Biol Chem, 1998;273(46):30073–30076.10.1074/jbc.273.46.30073Search in Google Scholar PubMed
[18] Takeuchi O, Akira S. Pattern Recognition Receptors and Inflammation. Cell, 2010;140(6):805–820.10.1016/j.cell.2010.01.022Search in Google Scholar PubMed
[19] Jefferies CA. Regulating IRFs in IFN Driven Disease. Front Immunol 2019;10:325.10.3389/fimmu.2019.00325Search in Google Scholar PubMed PubMed Central
[20] Ytterberg SR, Schnitzer TJ. Serum Interferon Levels in Patients with Systemic Lupus Erythematosus. Arthritis Rheum, 1982;25(4): 401–406.10.1002/art.1780250407Search in Google Scholar PubMed
[21] Bengtsson AA, Sturfelt G, Truedsson L, et al. Activation of Type I Interferon System in Systemic Lupus Erythematosus Correlates with Disease Activity but not with Antiretroviral Antibodies. Lupus, 2000;9(9):664–671.10.1191/096120300674499064Search in Google Scholar PubMed
[22] Baechler EC, Batliwalla FM, Karypis G, et al. Interferon-inducible Gene Expression Signature in Peripheral Blood Cells of Patients with Severe Lupus. Proc Natl Acad Sci U S A, 2003;100(5):2610–2615.10.1073/pnas.0337679100Search in Google Scholar PubMed PubMed Central
[23] Bennett L. Palucka AK, Arce E, et al. Interferon and Granulopoiesis Signatures in Systemic Lupus Erythematosus Blood. J Exp Med, 2003;197(6):711–723.10.1084/jem.20021553Search in Google Scholar PubMed PubMed Central
[24] Crow MK, Kirou KA, Wohlgemuth J, Microarray Analysis of Interferon-regulated Genes in SLE. Autoimmunity, 2003;36(8):481–490.10.1080/08916930310001625952Search in Google Scholar PubMed
[25] Kirou KA, Lee C, George S, et al. Coordinate Overexpression of Interferon-Alpha-Induced Genes in Systemic Lupus Erythematosus. Arthritis Rheum, 2004;50(12):3958–3967.10.1002/art.20798Search in Google Scholar PubMed
[26] Kirou KA, Lee C, George S, et al. Activation of the Interferon-alpha Pathway Identifies a Subgroup of Systemic Lupus Erythematosus Patients with Distinct Serologic Features and Active Disease. Arthritis Rheum, 2005;52(5):1491–1503.10.1002/art.21031Search in Google Scholar PubMed
[27] Feng X, Wu H, Grossman JM, et al. Association of Increased Interferon-inducible Gene Expression with Disease Activity and Lupus Nephritis in Patients with Systemic Lupus Erythematosus. Arthritis Rheum, 2006;54(9):2951–2962.10.1002/art.22044Search in Google Scholar PubMed
[28] Higgs BW, Liu Z, White B, et al. Patients with Systemic Lupus Erythematosus, Myositis, Rheumatoid Arthritis and Scleroderma Share Activation of a Common Type I Interferon Pathway. Ann Rheum Dis, 2011;70(11):2029–2036.10.1136/ard.2011.150326Search in Google Scholar PubMed
[29] Nikpour M, Dempsey AA, Urowitz MB, et al. Association of a Gene Expression Profile from Whole Blood with Disease Activity in Systemic Lupus Erythaematosus. Ann Rheum Dis, 2008;67(8):1069–1075.10.1136/ard.2007.074765Search in Google Scholar PubMed
[30] Petri M, Singh S, Tesfasyone H, et al. Longitudinal Expression of Type I Interferon Responsive Genes in Systemic Lupus Erythematosus. Lupus, 2009;18(11):980–989.10.1177/0961203309105529Search in Google Scholar PubMed PubMed Central
[31] Preble OT, Black RJ, Friedman RM, et al., Systemic Lupus Erythematosus: Presence in Human Serum of an Unusual Acid-Labile Leukocyte Interferon. Science, 1982;216(4544):429–431.10.1126/science.6176024Search in Google Scholar PubMed
[32] Ronnblom L, Alm GV. Systemic Lupus Erythematosus and the Type I Interferon System. Arthritis Res Ther, 2003;5(2):68–75.10.1002/art.21571Search in Google Scholar PubMed
[33] Ronnblom L, Alm GV. The Natural Interferon-alpha Producing Cells in Systemic Lupus Erythematosus. Hum Immunol, 2002;63(12): 1181–1193.10.1016/S0198-8859(02)00757-7Search in Google Scholar
[34] Meller S, Winterberg F, Gilliet M, et al. Ultraviolet Radiation-induced Injury, Chemokines, and Leukocyte Recruitment: An Amplification Cycle Triggering Cutaneous Lupus Erythematosus. Arthritis Rheum, 2005;52(5):1504–1516.10.1002/art.21034Search in Google Scholar PubMed
[35] Der E, Ranabothu S, Suryawanshi H, et al. Single Cell RNA Sequencing to Dissect the Molecular Heterogeneity in Lupus Nephritis. JCI Insight, 2017;2(9).10.1172/jci.insight.93009Search in Google Scholar PubMed PubMed Central
[36] Der E, Suryawanshi H, Morozov P, et al. Tubular Cell and Keratinocyte Single-cell Transcriptomics Applied to Lupus Nephritis Reveal Type I IFN and Fibrosis Relevant Pathways. Nat Immunol, 2019;20(7):915–927.10.1038/s41590-019-0386-1Search in Google Scholar PubMed PubMed Central
[37] Niewold TB, Swedler WI. Systemic Lupus Erythematosus Arising During Interferon-alpha Therapy for Cryoglobulinemic Vasculitis Associated with Hepatitis C. Clin Rheumatol, 2005;24(2): 178–181.10.1007/s10067-004-1024-2Search in Google Scholar PubMed
[38] Ronnblom LE, Alm GV, Oberg KE. Possible Induction of Systemic Lupus Erythematosus by Interferon-alpha Treatment in a Patient with a Malignant Carcinoid Tumour. J Intern Med, 1990;227(3): 207–210.10.1111/j.1365-2796.1990.tb00144.xSearch in Google Scholar PubMed
[39] Elkon KB, Wiedeman A. Type I IFN System in the Development and Manifestations of SLE. Curr Opin Rheumatol, 2012;24(5): 499–505.10.1097/BOR.0b013e3283562c3eSearch in Google Scholar PubMed
[40] Mustelin T, Lood C, Giltiay NV. Sources of Pathogenic Nucleic Acids in Systemic Lupus Erythematosus. Front Immunol, 2019;10:1028.10.3389/fimmu.2019.01028Search in Google Scholar PubMed PubMed Central
[41] Elkon KB. Review: Cell Death, Nucleic Acids, and Immunity: Inflammation Beyond the Grave. Arthritis Rheumatol, 2018;70(6): 805–816.10.1002/art.40452Search in Google Scholar PubMed PubMed Central
[42] Lood C, Blanco LP, Purmalek MM, et al. Neutrophil Extracellular Traps Enriched in Oxidized Mitochondrial DNA are Interferogenic and Contribute to Lupus-like Disease. Nat Med, 2016;22(2):146–153.10.1038/nm.4027Search in Google Scholar PubMed PubMed Central
[43] Volkman HE, Stetson DB. The Enemy Within: Endogenous Retroelements and Autoimmune Disease. Nat Immunol, 2014;15(5): 415–422.10.1038/ni.2872Search in Google Scholar PubMed PubMed Central
[44] Mireles-Canales MP, Gonzalez-Chavez SA, Quinonez-Flores CM, et al. DNA Damage and Deficiencies in the Mechanisms of Its Repair: Implications in the Pathogenesis of Systemic Lupus Erythematosus. J Immunol Res, 2018;2018:8214379.10.1155/2018/8214379Search in Google Scholar PubMed PubMed Central
[45] Honda K, Taniguchi T. IRFs: Master Regulators of Signalling by Toll-like Receptors and Cytosolic Pattern-recognition Receptors. Nat Rev Immunol, 2006;6(9):644–658.10.1038/nri1900Search in Google Scholar PubMed
[46] Honda K, Taniguchi T. Toll-like Receptor Signaling and IRF Transcription Factors. IUBMB Life, 2006;58(5–6):290–295.10.1080/15216540600702206Search in Google Scholar PubMed
[47] Avalos AM, Busconi L, Marshak-Rothstein A. Regulation of Autoreactive B Cell Responses to Endogenous TLR Ligands. Autoimmunity, 2010;43(1):76–83.10.3109/08916930903374618Search in Google Scholar PubMed PubMed Central
[48] Paradowska-Gorycka A, Wajda A, Stypinska B, et al. Variety of Endosomal TLRs and Interferons (IFN-alpha, IFN-beta, IFN-gamma) Expression Profiles in Patients with SLE, SSc and MCTD. Clin Exp Immunol, 2021;204(1):49–63.10.1111/cei.13566Search in Google Scholar PubMed PubMed Central
[49] McWhirter SM, Jefferies CA. Nucleic Acid Sensors as Therapeutic Targets for Human Disease. Immunity, 2020;53(1):78–97.10.1016/j.immuni.2020.04.004Search in Google Scholar PubMed
[50] Mabbott NA, Baillie JK, Brown H, et al. An Expression Atlas of Human Primary Cells: Inference of Gene Function from Coexpression Networks. BMC Genomics, 2013;14(1):632.10.1186/1471-2164-14-632Search in Google Scholar PubMed PubMed Central
[51] Watkin LB, Jessen B, Wiszniewski W, et al. COPA Mutations Impair ER-Golgi Transport and Cause Hereditary Autoimmune-mediated Lung Disease and Arthritis. Nat Genet, 2015;47(6):654–660.10.1038/ng.3279Search in Google Scholar PubMed PubMed Central
[52] Rivara S, Ablasser A. COPA Silences STING. J Exp Med, 2020;217(11).10.1084/jem.20201517Search in Google Scholar PubMed PubMed Central
[53] Zhang X, Wu J, Du F, et al. The Cytosolic DNA Sensor cGAS Forms an Oligomeric Complex with DNA and Undergoes Switch-like Conformational Changes in the Activation Loop. Cell Rep, 2014;6(3):421–430.10.1016/j.celrep.2014.01.003Search in Google Scholar PubMed PubMed Central
[54] Xiao TS, Fitzgerald KA. The cGAS-STING Pathway for DNA Sensing. Mol Cell, 2013;51(2):135–139.10.1016/j.molcel.2013.07.004Search in Google Scholar PubMed PubMed Central
[55] Ishikawa H, Ma Z, Barber GN, STING Regulates Intracellular DNA-mediated, Type I Interferon-dependent Innate Immunity. Nature, 2009;461(7265):788–792.10.1038/nature08476Search in Google Scholar PubMed PubMed Central
[56] Wu J, Sun L, Chen X, et al. Cyclic GMP-AMP is an Endogenous Second Messenger in Innate Immune Signaling by Cytosolic DNA. Science, 2013;339(6121):826–830.10.1126/science.1229963Search in Google Scholar PubMed PubMed Central
[57] Ishikawa H, Barber GN. STING is an Endoplasmic Reticulum Adaptor that Facilitates Innate Immune Signalling. Nature, 2008;455(7213):674–678.10.1038/nature07317Search in Google Scholar PubMed PubMed Central
[58] Tanaka Y, Chen ZJ. STING Specifies IRF3 Phosphorylation by TBK1 in the Cytosolic DNA Signaling Pathway. Sci Signal, 2012;5(214):ra20.10.1126/scisignal.2002521Search in Google Scholar PubMed PubMed Central
[59] Zhang C, Shang G, Gui X, et al. Structural Basis of STING Binding with and Phosphorylation by TBK1. Nature, 2019;567(7748): 394–398.10.1038/s41586-019-1000-2Search in Google Scholar PubMed PubMed Central
[60] Mukai K, Konno H, Akiba T, et al. Activation of STING Requires Palmitoylation at the Golgi. Nat Commun, 2016;7(1):11932.10.1038/ncomms11932Search in Google Scholar PubMed PubMed Central
[61] Abe T, Barber GN. Cytosolic-DNA-mediated, STING-dependent Proinflammatory Gene Induction Necessitates Canonical NF-kappaB Activation through TBK1. J Virol, 2014;88(10):5328–5341.10.1128/JVI.00037-14Search in Google Scholar PubMed PubMed Central
[62] Fitzgerald KA, McWhirter SM, Faia KL, et al. IKKepsilon and TBK1 are Essential Components of the IRF3 Signaling Pathway. Nat Immunol, 2003;4(5):491–496.10.1038/ni921Search in Google Scholar PubMed
[63] Hopfner KP, Hornung V. Molecular Mechanisms and Cellular Functions of cGAS-STING Signalling. Nat Rev Mol Cell Biol, 2020;21(9):501–521.10.1038/s41580-020-0244-xSearch in Google Scholar PubMed
[64] Wan D, Jiang W, Hao J. Research Advances in How the cGAS-STING Pathway Controls the Cellular Inflammatory Response. Front Immunol, 2020;11:615.10.3389/fimmu.2020.00615Search in Google Scholar PubMed PubMed Central
[65] Barnett KC, Coronas-Serna JM, Zhou W, et al. Phosphoinositide Interactions Position cGAS at the Plasma Membrane to Ensure Efficient Distinction between Self- and Viral DNA. Cell, 2019;176(6): 1432–1446.10.1016/j.cell.2019.01.049Search in Google Scholar PubMed PubMed Central
[66] Volkman HE, Cambier S, Gray EE, et al. Tight Nuclear Tethering of cGAS is Essential for Preventing Autoreactivity. Elife, 2019;8:e47491.10.7554/eLife.47491Search in Google Scholar PubMed PubMed Central
[67] Yang H, Wang H, Ren J, et al. cGAS is Essential for Cellular Senescence. Proc Natl Acad Sci U S A, 2017;114(23):E4612–E4620.10.1073/pnas.1705499114Search in Google Scholar PubMed PubMed Central
[68] Gentili M, Lahaye X, Nadalin F, et al. The N-Terminal Domain of cGAS Determines Preferential Association with Centromeric DNA and Innate Immune Activation in the Nucleus. Cell Rep, 2019; 26(13):3798.10.1016/j.celrep.2019.03.049Search in Google Scholar PubMed PubMed Central
[69] Deng Z, Chong Z, Law CS, et al. A Defect in COPI-mediated Transport of STING Causes Immune Dysregulation in COPA Syndrome. J Exp Med, 2020;217(11).10.1084/jem.20201045Search in Google Scholar PubMed PubMed Central
[70] Kato T, Yamamoto M, Honda Y, et al. Augmentation of STING-induced Type I Interferon Production in COPA Syndrome. Arthritis Rheumatol, 2021.10.1002/art.41790Search in Google Scholar PubMed
[71] Lepelley A, Martin-Niclos MJ, Le Bihan M, et al. Mutations in COPA Lead to Abnormal Trafficking of STING to the Golgi and Interferon Signaling. J Exp Med, 2020;217(11).10.1084/jem.20200600Search in Google Scholar PubMed PubMed Central
[72] Volpi S, Tsui J, Mariani M, et al. Type I Interferon Pathway Activation in COPA Syndrome. Clin Immunol, 2018;187:33–36.10.1016/j.clim.2017.10.001Search in Google Scholar PubMed
[73] Srikanth S, Woo JS, Wu B, et al. The Ca(2+) Sensor STIM1 Regulates the Type I Interferon Response by Retaining the Signaling Adaptor STING at the Endoplasmic Reticulum. Nat Immunol, 2019;20(2):152–162.10.1038/s41590-018-0287-8Search in Google Scholar PubMed PubMed Central
[74] Son A, de Jesus AA, Schwartz DM. STIM1 Holds a STING in its (N-terminal) Tail. Cell Calcium, 2019;80: 192–193.10.1016/j.ceca.2019.03.003Search in Google Scholar PubMed PubMed Central
[75] Bridgeman A, Maelfait J, Davenne T, et al. Viruses Transfer the Antiviral Second Messenger cGAMP between Cells. Science, 2015;349(6253):1228–1232.10.1126/science.aab3632Search in Google Scholar PubMed PubMed Central
[76] Gentili M, Kowal J, Tkach M, et al. Transmission of Innate Immune Signaling by Packaging of cGAMP in Viral Particles. Science, 2015;349(6253):1232–1236.10.1126/science.aab3628Search in Google Scholar PubMed
[77] Luteijn RD, Zaver SA, Gowen BG, et al. SLC19A1 Transports Immunoreactive Cyclic Dinucleotides. Nature, 2019;573(7774):434–438.10.1038/s41586-019-1553-0Search in Google Scholar PubMed PubMed Central
[78] Ritchie C, Cordova AF, Hess GT, et al. SLC19A1 Is an Importer of the Immunotransmitter cGAMP. Mol Cell, 2019;75(2):372–381.10.1016/j.molcel.2019.05.006Search in Google Scholar PubMed PubMed Central
[79] Rupasree Y, Naushad SM, Rajasekhar L, et al. Epigenetic Modulation of RFC1, MHC2TA and HLA-DR in Systemic Lupus Erythematosus: Association with Serological Markers and Six Functional Polymorphisms of One-carbon Metabolic Pathway. Gene, 2014;536(1): 45–52.10.1016/j.gene.2013.11.094Search in Google Scholar PubMed
[80] Zhou Y, Fei M, Zhang G, et al. Blockade of the Phagocytic Receptor MerTK on Tumor-Associated Macrophages Enhances P2X7R-Dependent STING Activation by Tumor-Derived cGAMP. Immunity, 2020;52(2):357–373.10.1016/j.immuni.2020.01.014Search in Google Scholar PubMed
[81] Zhou C, Chen X, Planells-Cases R, et al. Transfer of cGAMP into Bystander Cells via LRRC8 Volume-Regulated Anion Channels Augments STING-Mediated Interferon Responses and Anti-viral Immunity. Immunity, 2020;52(5):767–781.10.1016/j.immuni.2020.03.016Search in Google Scholar PubMed
[82] Li L, Yin Q, Kuss P, et al. Hydrolysis of 2′3′-cGAMP by ENPP1 and Design of Nonhydrolyzable Analogs. Nat Chem Biol, 2014;10(12):1043–1048.10.1038/nchembio.1661Search in Google Scholar PubMed PubMed Central
[83] Carozza JA, Brown JA, Bohnert V, et al. Structure-Aided Development of Small-Molecule Inhibitors of ENPP1, the Extracellular Phosphodiesterase of the Immunotransmitter cGAMP. Cell Chem Biol, 2020;27(11):1347–1358.10.1016/j.chembiol.2020.07.007Search in Google Scholar PubMed PubMed Central
[84] Liu H, Zhang H, Wu X, et al. Nuclear cGAS Suppresses DNA Repair and Promotes Tumorigenesis. Nature, 2018;563(7729): 131–136.10.1038/s41586-018-0629-6Search in Google Scholar PubMed
[85] Seo GJ, Yang A, Tan B, et al. Akt Kinase-Mediated Checkpoint of cGAS DNA Sensing Pathway. Cell Rep, 2015;13(2):440–449.10.1016/j.celrep.2015.09.007Search in Google Scholar PubMed PubMed Central
[86] Zhong L, Hu MM, Bian LJ, et al. Phosphorylation of cGAS by CDK1 Impairs Self-DNA Sensing in Mitosis. Cell Discov, 2020; 6(1):26.10.1038/s41421-020-0162-2Search in Google Scholar PubMed PubMed Central
[87] Li T, Huang T, Du M, et al. Phosphorylation and Chromatin Tethering Prevent cGAS Activation During Mitosis. Science, 2021; 371(6535).10.1126/science.abc5386Search in Google Scholar PubMed PubMed Central
[88] Seo GJ, Kim C, Shin WJ, et al. TRIM56-mediated Monoubiquitination of cGAS for Cytosolic DNA Sensing. Nat Commun, 2018; 9(1):613.10.1038/s41467-018-02936-3Search in Google Scholar PubMed PubMed Central
[89] Wang Q, Huang L, Hong Z, et al. The E3 Ubiquitin Ligase RNF185 Facilitates the cGAS-mediated Innate Immune Response. PLoS Pathog, 2017;13(3):e1006264.10.1371/journal.ppat.1006264Search in Google Scholar PubMed PubMed Central
[90] Chen M, Meng Q, Qin Y, et al. TRIM14 Inhibits cGAS Degradation Mediated by Selective Autophagy Receptor p62 to Promote Innate Immune Responses. Mol Cell, 2016;64(1):105–119.10.1016/j.molcel.2016.08.025Search in Google Scholar PubMed
[91] Wu Y, Li S. Role of Post-Translational Modifications of cGAS in Innate Immunity. Int J Mol Sci, 2020;21(21):7842.10.3390/ijms21217842Search in Google Scholar PubMed PubMed Central
[92] Halkom A, Wu H, Lu Q. Contribution of Mouse Models in our Understanding of Lupus. Int Rev Immunol, 2020;39(4):174–187.10.1080/08830185.2020.1742712Search in Google Scholar PubMed
[93] Gall A, Treuting P, Elkon KB, et al. Autoimmunity Initiates in Nonhematopoietic Cells and Progresses via Lymphocytes in an Interferon-dependent Autoimmune Disease. Immunity, 2012;36(1): 120–131.10.1016/j.immuni.2011.11.018Search in Google Scholar PubMed PubMed Central
[94] Hemphill WO, Simpson SR, Liu M, et al. TREX1 as a Novel Immunotherapeutic Target. Front Immunol, 2021;12:660184.10.3389/fimmu.2021.660184Search in Google Scholar PubMed PubMed Central
[95] Mazur DJ, Perrino FW. Structure and Expression of the TREX1 and TREX2 3′ --> 5′ Exonuclease Genes. J Biol Chem, 2001;276(18):14718–14727.10.1074/jbc.M010051200Search in Google Scholar PubMed
[96] Mazur DJ, Perrino FW. Identification and Expression of the TREX1 and TREX2 cDNA Sequences Encoding Mammalian 3′-->5′ Exonucleases. J Biol Chem, 1999;274(28):19655–19660.10.1074/jbc.274.28.19655Search in Google Scholar PubMed
[97] Rice GI, Rodero MP, Crow YJ. Human Disease Phenotypes Associated with Mutations in TREX1. J Clin Immunol, 2015;35(3): 235–243.10.1007/s10875-015-0147-3Search in Google Scholar PubMed
[98] Lee-Kirsch MA, Gong M, Chowdhury D, et al. Mutations in the Gene Encoding the 3′-5′ DNA Exonuclease TREX1 are Associated with Systemic Lupus Erythematosus. Nat Genet, 2007;39(9): 1065–1067.10.1038/ng2091Search in Google Scholar PubMed
[99] Grieves JL, Fye JM, Harvey S, et al. Exonuclease TREX1 Degrades Double-stranded DNA to Prevent Spontaneous Lupus-like Inflammatory Disease. Proc Natl Acad Sci U S A, 2015;112(16): 5117–5122.10.1073/pnas.1423804112Search in Google Scholar PubMed PubMed Central
[100] Lee-Kirsch MA, Chowdhury D, Harvey S, et al. A Mutation in TREX1 that Impairs Susceptibility to Granzyme A-mediated Cell Death Underlies Familial Chilblain Lupus. J Mol Med (Berl), 2007;85(5): 531–537.10.1007/s00109-007-0199-9Search in Google Scholar PubMed
[101] Lehtinen DA, Harvey S, Mulcahy MJ, et al. The TREX1 Double-stranded DNA Degradation Activity is Defective in Dominant Mutations Associated with Autoimmune Disease. J Biol Chem, 2008;283(46):31649–31656.10.1074/jbc.M806155200Search in Google Scholar PubMed PubMed Central
[102] Rice G, Newman WG, Dean J, et al. Heterozygous Mutations in TREX1 Cause Familial Chilblain Lupus and Dominant Aicardi-Goutieres Syndrome. Am J Hum Genet, 2007;80(4):811–815.10.1086/513443Search in Google Scholar PubMed PubMed Central
[103] Crow YJ, Chase DS, Schmidt JL, et al. Characterization of Human Disease Phenotypes Associated with Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A, 2015;167(2):296–312.10.1002/ajmg.a.36887Search in Google Scholar PubMed PubMed Central
[104] Crow YJ, Hayward BE, Parmar R, et al. Mutations in the Gene Encoding the 3′-5′ DNA Exonuclease TREX1 Cause Aicardi-Goutieres Syndrome at the AGS1 Locus. Nat Genet, 2006;38(8): 917–920.10.1038/ng1845Search in Google Scholar PubMed
[105] Rice GI, Forte GM, Szynkiewicz M, et al. Assessment of Interferon-related Biomarkers in Aicardi-Goutieres Syndrome Associated with Mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: A Case-control Study. Lancet Neurol, 2013;12(12):1159–1169.10.1016/S1474-4422(13)70258-8Search in Google Scholar PubMed PubMed Central
[106] Morita M, Stamp G, Robins P, et al. Gene-targeted Mice Lacking the Trex1 (DNase III) 3′-->5′ DNA Exonuclease Develop Inflammatory Myocarditis. Mol Cell Biol, 2004;24(15):6719–6727.10.1128/MCB.24.15.6719-6727.2004Search in Google Scholar PubMed PubMed Central
[107] Ahn J, Ruiz P, Barber GN. Intrinsic Self-DNA Triggers Inflammatory Disease Dependent on STING. J Immunol, 2014;193(9): 4634–4642.10.4049/jimmunol.1401337Search in Google Scholar PubMed PubMed Central
[108] Gao D, Li T, Li XD, et al. Activation of Cyclic GMP-AMP Synthase by Self-DNA Causes Autoimmune Diseases. Proc Natl Acad Sci U S A, 2015;112(42):E5699–E5705.10.1073/pnas.1516465112Search in Google Scholar PubMed PubMed Central
[109] Xiao N, Wei J, Xu S, et al. cGAS Activation Causes Lupus-like Autoimmune Disorders in a TREX1 Mutant Mouse Model. J Autoimmun, 2019;100:84–94.10.1016/j.jaut.2019.03.001Search in Google Scholar PubMed
[110] Gray EE, Treuting PM, Woodward JJ, et al. Cutting Edge: cGAS Is Required for Lethal Autoimmune Disease in the Trex1-Deficient Mouse Model of Aicardi-Goutieres Syndrome. J Immunol, 2015;195(5):1939–1943.10.4049/jimmunol.1500969Search in Google Scholar PubMed PubMed Central
[111] Yang YG, Lindahl T, Barnes DE. Trex1 Exonuclease Degrades ssDNA to Prevent Chronic Checkpoint Activation and Autoimmune Disease. Cell, 2007;131(5):873–886.10.1016/j.cell.2007.10.017Search in Google Scholar PubMed
[112] Zhu XW, Wang Y, Wei YH, et al. Comprehensive Assessment of the Association Between FCGRs Polymorphisms and the Risk of Systemic Lupus Erythematosus: Evidence from a Meta-Analysis. Sci Rep, 2016;6(1):31617.10.1038/srep31617Search in Google Scholar PubMed PubMed Central
[113] Bolland S, Yim YS, Tus K, et al. Genetic Modifiers of Systemic Lupus Erythematosus in FcgammaRIIB(−/−) Mice. J Exp Med, 2002;195(9):1167–1174.10.1084/jem.20020165Search in Google Scholar PubMed PubMed Central
[114] Morel L. Genetics of SLE: Evidence from Mouse Models. Nat Rev Rheumatol, 2010;6(6):348–357.10.1038/nrrheum.2010.63Search in Google Scholar PubMed
[115] Thim-Uam A, Prabakaran T, Tansakul M, et al. STING Mediates Lupus via the Activation of Conventional Dendritic Cell Maturation and Plasmacytoid Dendritic Cell Differentiation. iScience, 2020;23(9):101530.10.1016/j.isci.2020.101530Search in Google Scholar PubMed PubMed Central
[116] Martin GR, Henare K, Salazar C, et al. Expression of a Constitutively Active Human STING Mutant in Hematopoietic Cells Produces an Ifnar1-Dependent Vasculopathy in Mice. Life Sci Alliance, 2019;2(3).10.26508/lsa.201800215Search in Google Scholar PubMed PubMed Central
[117] Motwani M, McGowan J, Antonovitch J, et al. cGAS-STING Pathway Does Not Promote Autoimmunity in Murine Models of SLE. Front Immunol, 2021;12:605930.10.3389/fimmu.2021.605930Search in Google Scholar PubMed PubMed Central
[118] Sharma S, Campbell AM, Chan J, et al. Suppression of Systemic Autoimmunity by the Innate Immune Adaptor STING. Proc Natl Acad Sci U S A, 2015;112(7):E710–E717.10.1073/pnas.1420217112Search in Google Scholar PubMed PubMed Central
[119] Scumpia PO, Botten GA, Norman JS, et al. Opposing Roles of Toll-like Receptor and Cytosolic DNA-STING Signaling Pathways for Staphylococcus aureus Cutaneous Host Defense. PLoS Pathog, 2017;13(7):e1006496.10.1371/journal.ppat.1006496Search in Google Scholar PubMed PubMed Central
[120] Kuechle MK, Elkon KB. Shining Light on Lupus and UV. Arthritis Res Ther, 2007;9(1):101.10.1186/ar2100Search in Google Scholar PubMed PubMed Central
[121] Besaratinia A, Synold TW, Chen HH, et al. DNA Lesions Induced by UV A1 and B Radiation in Human Cells: Comparative Analyses in the Overall Genome and in the p53 Tumor Suppressor Gene. Proc Natl Acad Sci U S A, 2005;102(29):10058–10063.10.1073/pnas.0502311102Search in Google Scholar PubMed PubMed Central
[122] Kunisada M, Sakumi K, Tominaga Y, et al. 8-Oxoguanine Formation Induced by Chronic UVB Exposure Makes Ogg1 Knockout Mice Susceptible to Skin Carcinogenesis. Cancer Res, 2005;65(14): 6006–6010.10.1158/0008-5472.CAN-05-0724Search in Google Scholar PubMed
[123] Gehrke N, Mertens C, Zillinger T, et al. Oxidative Damage of DNA Confers Resistance to Cytosolic Nuclease TREX1 Degradation and Potentiates STING-dependent Immune Sensing. Immunity, 2013;39(3):482–495.10.1016/j.immuni.2013.08.004Search in Google Scholar PubMed
[124] Skopelja-Gardner S, An J, Tai J, et al. The Early Local and Systemic Type I Interferon Responses to Ultraviolet B Light Exposure are cGAS Dependent. Sci Rep, 2020;10(1):7908.10.1038/s41598-020-64865-wSearch in Google Scholar PubMed PubMed Central
[125] Skopelja-Gardner S, Tai J, Sun X, et al. Acute Skin Exposure to Ultraviolet Light Triggers Neutrophil-mediated Kidney Inflammation. Proc Natl Acad Sci U S A, 2021;118(3).10.1073/pnas.2019097118Search in Google Scholar PubMed PubMed Central
[126] Tumurkhuu G, Chen S, Montano EN, et al. Oxidative DNA Damage Accelerates Skin Inflammation in Pristane-Induced Lupus Model. Front Immunol, 2020;11:554725.10.3389/fimmu.2020.554725Search in Google Scholar PubMed PubMed Central
[127] Liu Y, Jesus AA, Marrero B, et al. Activated STING in a Vascular and Pulmonary Syndrome. N Engl J Med, 2014;371(6):507–518.10.1056/NEJMoa1312625Search in Google Scholar PubMed PubMed Central
[128] Fremond ML, Hadchouel A, Berteloot L, et al. Overview of STING-Associated Vasculopathy with Onset in Infancy (SAVI) Among 21 Patients. J Allergy Clin Immunol Pract, 2021;9(2):803–818.10.1016/j.jaip.2020.11.007Search in Google Scholar PubMed
[129] Liu X, Wang C. The Emerging Roles of the STING Adaptor Protein in Immunity and Diseases. Immunology, 2016;147(3): 285–291.10.1111/imm.12561Search in Google Scholar PubMed PubMed Central
[130] Picard C, McCarl CA, Papolos A, et al. STIM1 Mutation Associated with a Syndrome of Immunodeficiency and Autoimmunity. N Engl J Med, 2009;360(19):1971–1980.10.1056/NEJMoa0900082Search in Google Scholar PubMed PubMed Central
[131] Fremond ML, Nathan N. COPA Syndrome, 5 Years After: Where are we? Joint Bone Spine, 2021;88(2):105070.10.1016/j.jbspin.2020.09.002Search in Google Scholar PubMed
[132] Kumrah R, Mathew B. Genetics of COPA Syndrome. Appl Clin Genet, 2019;12:11–18.10.2147/TACG.S153600Search in Google Scholar PubMed PubMed Central
[133] Psarianos P, Kwan JY, Dell S, et al. COPA Syndrome (Ala-239Pro) Presenting with Isolated Follicular Bronchiolitis in Early Childhood: Case Report. J Clin Immunol, 2021.10.1007/s10875-021-01082-8Search in Google Scholar PubMed
[134] Tsui JL, Estrada OA, Deng Z, et al. Analysis of Pulmonary Features and Treatment Approaches in the COPA Syndrome. ERJ Open Res, 2018;4(2).10.1183/23120541.00017-2018Search in Google Scholar PubMed PubMed Central
[135] McCauley ME, O’Rourke JG, Yanez A, et al. C9orf72 in Myeloid Cells Suppresses STING-induced Inflammation. Nature, 2020;585(7823):96–101.10.1038/s41586-020-2625-xSearch in Google Scholar PubMed PubMed Central
[136] Kato Y, Park J, Takamatsu H, et al. Apoptosis-derived Membrane Vesicles Drive the cGAS-STING Pathway and Enhance Type I IFN Production in Systemic Lupus Erythematosus. Ann Rheum Dis, 2018;77(10):1507–1515.10.1136/annrheumdis-2018-212988Search in Google Scholar PubMed PubMed Central
[137] Murayama G, Chiba A, Kuga T, et al. Inhibition of mTOR Suppresses IFNalpha Production and the STING Pathway in Monocytes from Systemic Lupus Erythematosus Patients. Rheumatology (Oxford), 2020;59(10):2992–3002.10.1093/rheumatology/keaa060Search in Google Scholar PubMed
[138] An J, Woodward JJ, Sasaki T, et al. Cutting Edge: Antimalarial Drugs Inhibit IFN-beta Production Through Blockade of Cyclic GMP-AMP Synthase-DNA Interaction. J Immunol, 2015;194(9): 4089–4093.10.4049/jimmunol.1402793Search in Google Scholar PubMed
[139] Decout A, Katz JD, Venkatraman S, et al. The cGAS-STING Pathway as a Therapeutic Target in Inflammatory Diseases. Nat Rev Immunol, 2021.10.1038/s41577-021-00524-zSearch in Google Scholar PubMed PubMed Central
[140] Lama L, Adura C, Xie W, et al. Development of Human cGAS-specific Small-molecule Inhibitors for Repression of dsDNA-triggered Interferon Expression. Nat Commun, 2019;10(1):2261.10.1038/s41467-019-08620-4Search in Google Scholar PubMed PubMed Central
[141] Wong W. Activating cGAS at the Right Time and Place. Science Signaling, 2019;12(574):eaax4336.10.1126/scisignal.aax4336Search in Google Scholar
[142] Jiang H, Xue X, Panda S, et al. Chromatin-bound cGAS is an Inhibitor of DNA Repair and Hence Accelerates Genome Destabilization and Cell Death. EMBO J, 2019;38(21):e102718.10.15252/embj.2019102718Search in Google Scholar PubMed PubMed Central
[143] Gentili M, Lahaye X, Nadalin F, et al. The N-Terminal Domain of cGAS Determines Preferential Association with Centromeric DNA and Innate Immune Activation in the Nucleus. Cell Rep, 2019;26(9):2377–2393.10.1016/j.celrep.2019.01.105Search in Google Scholar PubMed PubMed Central
[144] Haag SM, Gulen MF, Reymond L, et al. Targeting STING with Covalent Small-molecule Inhibitors. Nature, 2018;559(7713): 269–273.10.1038/s41586-018-0287-8Search in Google Scholar PubMed
[145] Namjou B, Kothari PH, Kelly JA, et al. Evaluation of the TREX1 Gene in a Large Multi-ancestral Lupus Cohort. Genes Immun, 2011;12(4):270–279.10.1038/gene.2010.73Search in Google Scholar PubMed PubMed Central
© 2021 Audrey M. Hagiwara et al., published by Sciendo
This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
Articles in the same Issue
- Review
- Systemic lupus erythematosus and immunodeficiency
- Telerheumatology: A Narrative Review
- CD8+ T regulatory cells in lupus
- Cardiovascular disease in systemic lupus erythematosus
- Regulation of cGAS-STING pathway - Implications for systemic lupus erythematosus
- Original Article
- Nanoparticle-mediated delivery of IL-2 to T follicular helper cells protects BDF1 mice from lupus-like disease
- Long-term outcomes of patients with systemic lupus erythematosus: A Multicenter Cohort Study from CSTAR registry
- Case Report
- A 31-year-old woman with painful black palms: Thrombosis or vasculitis?
- Images
- Subcutaneous calcinosis in a patient with anti-MDA5 positive dermatomyositis
Articles in the same Issue
- Review
- Systemic lupus erythematosus and immunodeficiency
- Telerheumatology: A Narrative Review
- CD8+ T regulatory cells in lupus
- Cardiovascular disease in systemic lupus erythematosus
- Regulation of cGAS-STING pathway - Implications for systemic lupus erythematosus
- Original Article
- Nanoparticle-mediated delivery of IL-2 to T follicular helper cells protects BDF1 mice from lupus-like disease
- Long-term outcomes of patients with systemic lupus erythematosus: A Multicenter Cohort Study from CSTAR registry
- Case Report
- A 31-year-old woman with painful black palms: Thrombosis or vasculitis?
- Images
- Subcutaneous calcinosis in a patient with anti-MDA5 positive dermatomyositis