Repositioning of fungal-based peptides as modulators of angiotensin-converting enzyme-related carboxypeptidase, SARS-coronavirus HR2 domain, and coronavirus disease 2019 main protease
-
Babatunde Joseph Oso


Abstract
Background and Objectives
Angiotensin-converting enzyme-related carboxypeptidase, SARS-Coronavirus HR2 Domain, and COVID-19 main protease are essential for the cellular entry and replication of coronavirus in the host. This study investigated the putative inhibitory action of peptides form medicinal mushrooms, namely Pseudoplectania nigrella, Russula paludosa, and Clitocybe sinopica, towards selected proteins through computational studies.
Materials and Methods
The respective physicochemical properties of selected peptides were predicted using ProtParam tool, while the binding modes and binding free energy of selected peptides toward proteins were computed through HawkDock server. The structural flexibility and stability of docked protein–peptide complexes were assessed through iMODS server.
Results
The peptides showed an optimum binding afinity with the molecular targets; plectasin from P. nigrella showed the highest binding free energy compared to peptides from R. paludosa and C. sinopica. Besides, molecular dynamic simulations showed all fungal-based peptides could influence the flexibility and stability of selected proteins.
Conclusion
The study revealed fungal-based peptides could be explored as functional modulators of essential proteins that are involved in the cellular entry of coronavirus.
Introduction
The coronavirus disease 2019 (COVID-19), an outbreak caused by Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2), which are enveloped, non-segmented, single-stranded, positive-sense RNA viruses of the family Coronaviridae, subfamily Orthocoronaviridae, and genera Betacoronavirus, has been regarded as a global pandemic disease.[1,2] The COVID-19 outbreak emerged in Wuhan, Hubei Province, China in December 2019, where it was initially named as the 2019-novel coronavirus (2019-nCoV) on January 12, 2020 by the World Health Organization (WHO). The WHO officially named the disease as COVID-19, and the Coronavirus Study Group (CSG) of the International Committee proposed to name the new coronavirus as SARS-CoV-2, both issued on February 11, 2020.[3,4] It uncontrollably spread to North America, Oceania, Europe, Africa, and South America during January– February 2020.[5] Since then, the COVID-19 outbreak has created a challenging period in many geographic locations. It has suspended socioeconomic activities, caused a setback in the world economy, and created public fear and panic, with psychological instability across individuals. Active surveillance, contact tracing, quarantine, and early strong social distancing efforts were reported as the non-pharmacological strategies to limit transmission of the virus.[6] Most countries affected by COVID-19 adopted temporary measures like isolation of suspected individuals, quarantine of people from risk countries, lockdown or curfew in cities or countries, use of hand sanitizer, wearing of face mask in the public, and social distancing as the basic measures to curtail the spread of the virus. With these unconstitutional preventive measures, COVID-19 has spread to 216 countries with 4,619,477 confirmed cases and a death toll of 311,847 as of May 18, 2020.[7] Yet, no vaccine or drug has been substantiated to cure the disease, but there are several ongoing clinical trials of different drugs (vaccines).
Computational investigation of natural bioactive compounds could be a potential pathway to discover novel drugs (vaccines) to end the global pandemic infection. Bioactive proteins and peptides from different sources have various bioactivity attributes like antiviral, antibacterial, antifungal, antiparasitic, antihypertensive, antisporadical, and anticancer. These biological activities are exhibited by peptide molecules with different specificities and potencies depending on their structure and amino acid composition.[8] The use of peptides, especially short peptide fragments, to engineer the induction-targeted immune responses has been adopted as an alternative approach in treating various infectious diseases, and thus has proven to be more effective in reducing mortality and morbidity.[9] As peptides alone are weakly immunogenic due to their relatively small size, they require carrier molecules to stabilize and act as an adjuvant in order to achieve a strong immune response as well as toward appropriate health delivery systems.[10]
To increase the complexity of peptide–peptide/protein with regard to drug design, repositioning of active ingredients is performed to produce a cost-effective drug capable of activating receptors and acting indirectly or directly to regulate a series of enzymatic activities including ion transportation, transcription of nucleic acids, and various post-translational modifications of translated protein.[11,12] Drug repositioning is an emergent approach to new drug development from compounds that have already gone through some level of clinical testing and thus examined to have efficacy against diseases divergent from their original application.[13] This can be accelerated by using molecular software to screen existing chemical databases to identify suitable drug candidates. The development of identified small-molecule drugs that target a specific receptor by exploring the conformational binding space of peptide ligands could be effective efficacy against diseases divergent.[14] Computational methods enhance drug discovery when appropriate molecular applications such as cheminformatics, bioinformatics, network biology, and systems biology are adopted. Hence, this serves as a pathway to establish novel methods towards achieving crucial clinical trials against recent diseases.[15,16] Isolation and identification of drug molecules of natural origin (fungi) with potentially tolerable side effects may, therefore, help to create modulators of SARS-CoV-2 enzymes.
Medicinal mushrooms have a high content of proteins and other bioactive peptides that have immense therapeutic applications.[17] The fruiting bodies of Pseudoplectania nigrella contain plectasin, a peptide (fungal defensin) with antiviral potential and strong antibacterial action against various Gram-positive and negative bacteria.[18] Peptide from Russula paludosa (PRP) has a length of 10 amino acids and a molecular mass of 4.5 kDa.[19] PRP has shown effective inhibition against HIV-1 reverse transcriptase.[19] Moreover, a peptide from Clitocybe sinopica (PCS) with a molecular weight of 44 kDa, composed of two subunits of protein with N-terminal amino acid sequence, has been found to possess strong pharmacological effects against microbial agents.[20] This study investigated the putative influence of peptides of wild medicinal mushrooms on angiotensin-converting enzyme-related carboxypeptidase (ACE2), SARS-Coronavirus HR2 Domain (HRD), and COVID-19 main protease (CMP).
Materials and methods
Selection and modeling of peptides
Peptides from edible mushrooms, namely P. nigrella, R. paludosa, and C. sinopica, were selected for this hypothetical study based on their reported antiviral and other medicinal activities.[17] The 3D structures of selected peptides were predicted by submitting the respective FASTA amino acid sequence of peptides into I-TASSER modeling server (https://zhanglab.ccmb.med.umich.edu/I-TASSER/).[21,22] The best model for each peptide was selected based on its C-scores and was further validated using Structure Analysis and Verification server SAVES v5.0 (https://servicesn.mbi.ucla.edu/SAVES/).[23]
Physicochemical characterization
Physicochemical characterization of the peptides was predicted using ProtParam tool on the SIB Expasy server ProtParam tool (https://www.expasy.org/tools/) using mammalian as defined organism.[24]
Molecular docking
The structures of ACE2, HRD, and CMP were retrieved from the Protein Data Bank (https://www.rcsb.org/) with their PDB IDs being 1R42, 2FXP, and 6LU7, respectively, and presented as receptors. Structural modeling analysis of selected peptides with the receptors was performed using the HawkDock server (http://cadd.zju.edu.cn/hawkdock/) integrated with the ATTRACT docking algorithm, the HawkRank scoring function, and Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) free energy decomposition analysis.[25,26] The interactions were visualized using PyMOL ver. 1.1eval (De Lano Scientific LLC, CA, USA). All peptides and the protein were converted into Auto Dock Pdbqt format. The binding energy between the peptides and the protein was computed.
Molecular dynamics simulation
The conformational stability of protein–ligand interactions was evaluated using molecular dynamics simulation analysis performed through iMODS server (http://imods.chaconlab.org) by normal mode analysis (NMA), predicting properties such as deformability, mobility profiles, eigenvalues, variance, covariance map, and elastic network of the protein–ligand interactions.[27]
Results
In this study, a total of three fungal-based peptide sequences were selected from edible and medicinal mushrooms. The selected peptides from P. nigrella (plectasin), R. paludosa (PRP), and C. sinopica (PCS) with their corresponding FASTA sequence, amino acid length, and sources are presented in Table 1.
FASTA sequence, length of amino acid residues, and sources of selected fungal-based peptides
| Assigned name | FASTA sequence | Number of amino acids | Source |
|---|---|---|---|
| Plectasin | GFGCNGPWDEDDMQCHNHCKSIKGYKGGYCAKGGFVCKCY | 40 | Pseudoplectania nigrella |
| PRP | KREHGQHCEF | 10 | Russula paludosa |
| PCS | SVQATVNGDKML | 12 | Clitocybe sinopica |
PCS: peptide from Clitocybe sinopica; PRP: peptide from Russula paludosa
Table 2 shows the physicochemical properties of fungal-based peptides, which include molecular weight, theoretical isoelectric point, instability index, aliphatic index, and grand average hydropathy of selected peptides predicted through Expasy’s ProtParam server. The results indicated that plectasin had the highest isoelectric point and estimated half-life, while PRP had the highest instability index. The computational study also revealed PCS had the highest aliphatic index and grand average of hydropathicity compared to other selected peptides.
Physicochemical characterization of selected fungal-based peptides
| Peptides | Molecular weight (Da) | Theoretical isoelectric point | Estimated half-life (h) | Instability index (II) | Aliphatic index | Grand average of hydropathicity (GRAVY) |
|---|---|---|---|---|---|---|
| Plectasin | 4407.99 | 7.77 | 30.0 | 13.82 | 19.50 | -0.695 |
| PRP | 1270.39 | 6.92 | 1.3 | 23.69 | 0.00 | -2.040 |
| PCS | 1262.44 | 5.55 | 1.9 | 16.73 | 89.17 | -0.033 |
PCS: peptide from Clitocybe sinopica; PRP: peptide from Russula paludosa
The outcome of the molecular docking is presented in Table 3. The results reveal the energy scores for different models. The first model of each computation with the lowest energy score was selected as the score for each interaction. Plectasin had the highest affinity with all the selected proteins, as indicated by the least value of each binding energy and assessment by MM/GBSA free energy computation through an approximation of solvent electrostatic contribution in the Poisson–Boltzmann model. The molecular interactions of peptides with the selected proteins are shown in Figure 1, while the interacting residues of the protein and peptide interface are presented in Table 4.
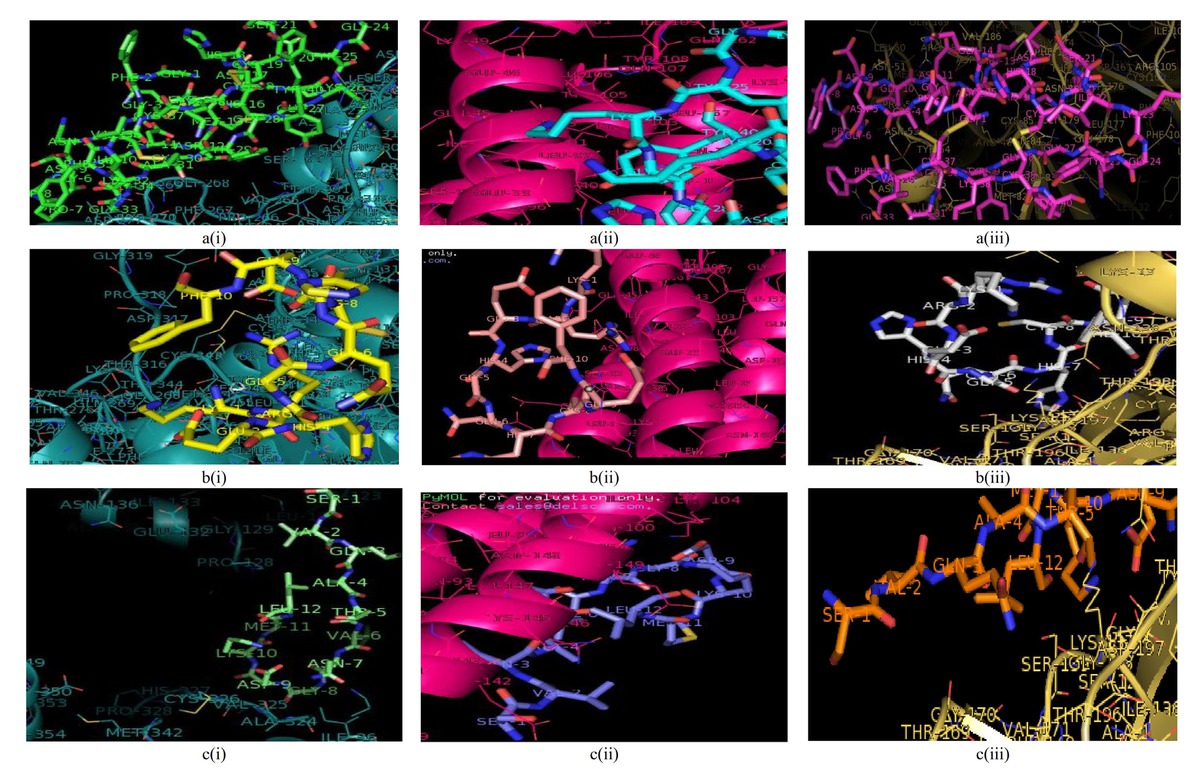
3D illustration of the interactions between the complexes of (A) angiotensin-converting enzyme-related carboxypeptidase, (B) SARS-Coronavirus HR2 Domain, and (C) COVID-19 main protease with (i) plectasin, (ii) PRP, and (iii) PCS. PCS: peptide from Clitocybe sinopica; PRP: peptide from Russula paludosa.
Binding score of docked complexes and binding energy (kcal/mol) of docked complexes by MM/GBSA
| ACE2 | HRD | CMP | ||
|---|---|---|---|---|
| Plectasin | Binding score | -3890.64 | -4131.60 | -3871.87 |
| Binding energy | -31.97 | -36.38 | -30.46 | |
| PRP | Binding score | -2563.52 | -2709.79 | -2556.80 |
| Binding energy | -11.92 | -17.59 | -3.02 | |
| PCS | Binding score | -2641.87 | -2647.73 | -2596.95 |
| Binding energy | -9.07 | -19.58 | -12.77 |
ACE2: angiotensin-converting enzyme-related carboxypeptidase; HRD: SARS-coronavirus HR2 domain; CMP: COVID-19 main protease; MM/GBSA: molecular mechanics/generalized born surface area; PCS: peptide from Clitocybe sinopica; PRP: peptide from Russula paludosa
Interacting amino acid residues of protein and selected peptides
| ACE2 | HRD | CMP | ||
|---|---|---|---|---|
| Plectasin | Protein | ASN-232, ALA-233, SER-262, GLY-268 | LEU-105, TYR-105 | ASN-53, MET-82, GLN-83, ASN-84, GLY-179, ASN-180, PHE-181, VAL-186 |
| Peptide | GLY-24, LYS-26, GLY-28, TRY-29 | HIS-16, LYS-20, TYR-25, GLY-27 | GLY-6, ASP-11, ASN-17, SER-21, GLY-24, PHE-35, TYR-40 | |
| PRP | Protein | THR-34, ASP-317, CYS-343 | ASP-28, GLN-45, ASN-91, SER-95 | ASN-238 |
| Peptide | LYS-1, ARG-2 | LYS-1, CYS-8, GLU-9 | GLU-9, PHE-10 | |
| PCS | Protein | GLU-122, GLY-129, CYS-326 | VAL-143, ASN-146, GLU- 149 | ASN-238 |
| Peptide | SER-1, VAL-2, ASP-9 | GLN-3, THR-5, ASN-7 | ASN-7, ASP-9 |
ACE2: angiotensin-converting enzyme-related carboxypeptidase; HRD: SARS-coronavirus HR2 domain; CMP: COVID-19 main protease; PCS: peptide from Clitocybe sinopica; PRP: peptide from Russula paludosa.
The interactions of the selected peptides with the proteins evaluated via molecular dynamics simulations analysis to examine the flexibility and dynamics of each of the proteins are presented in Figure 2 and Table 5. Significant changes were noticed in the structural flexibility of all the selected proteins based on main chain deformability, B-factor, variance associated with each normal mode, covariance matrix, elastic network model, and Eigenvalues upon binding of the peptides comparable to wild types, which were without bound peptides.
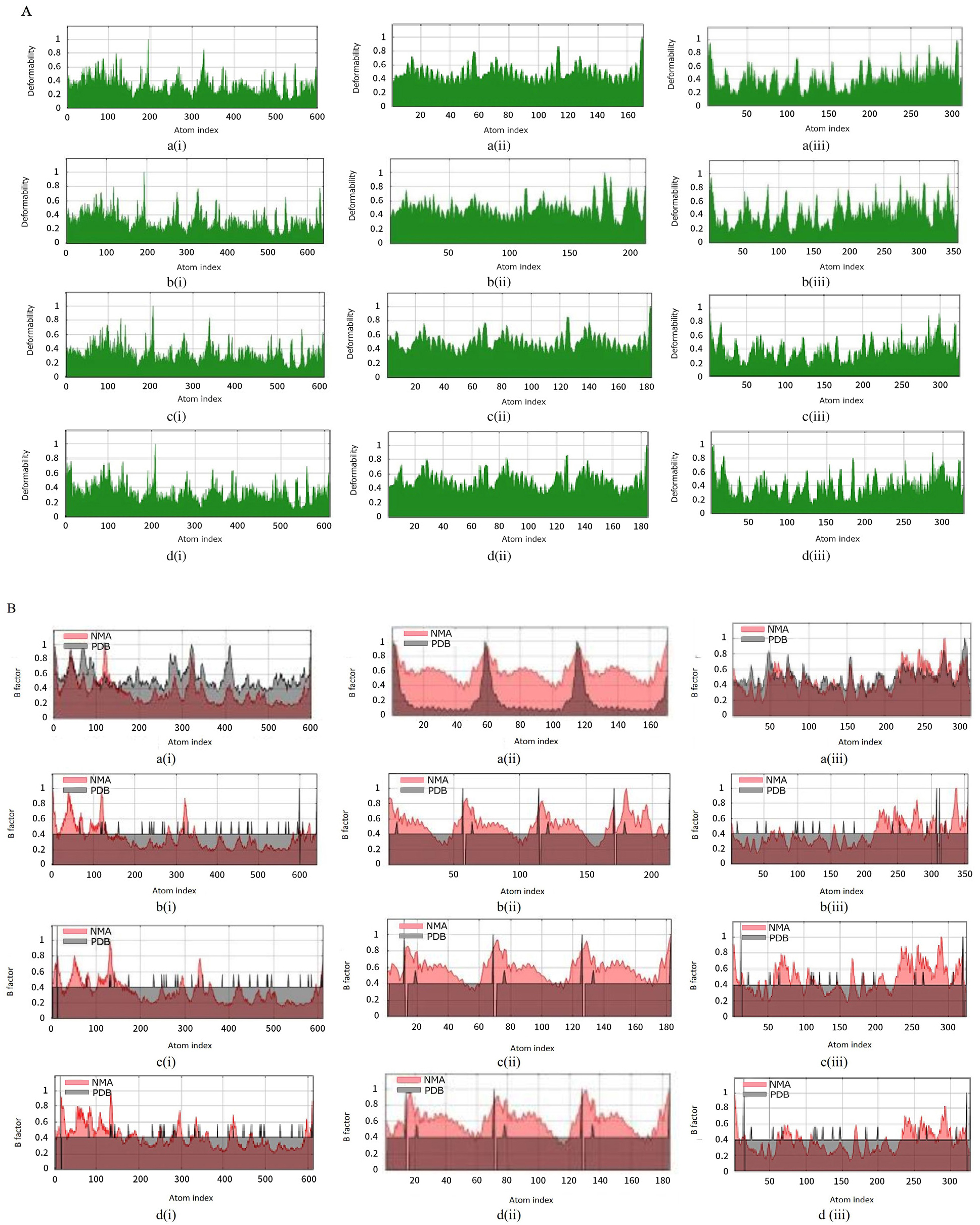
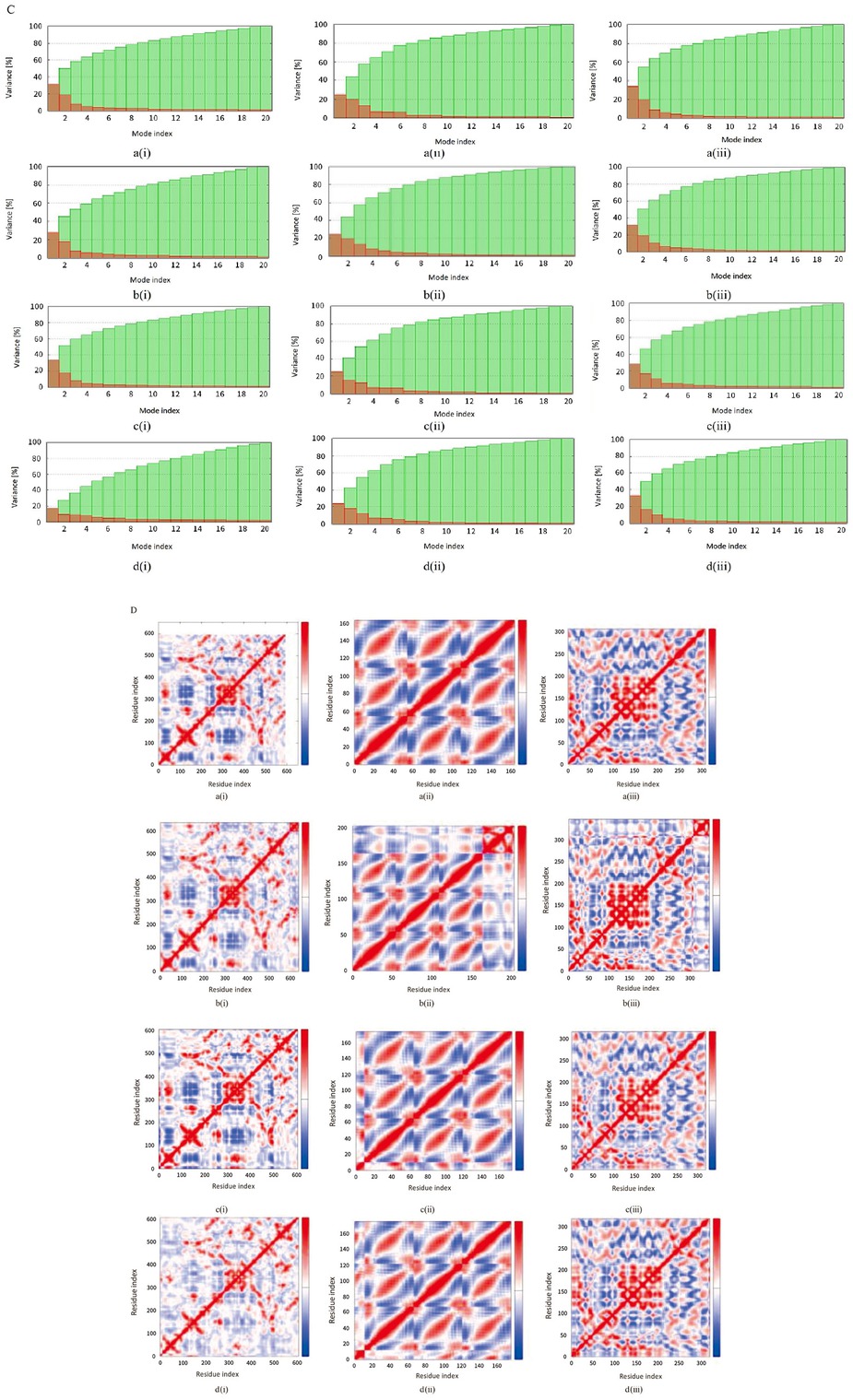
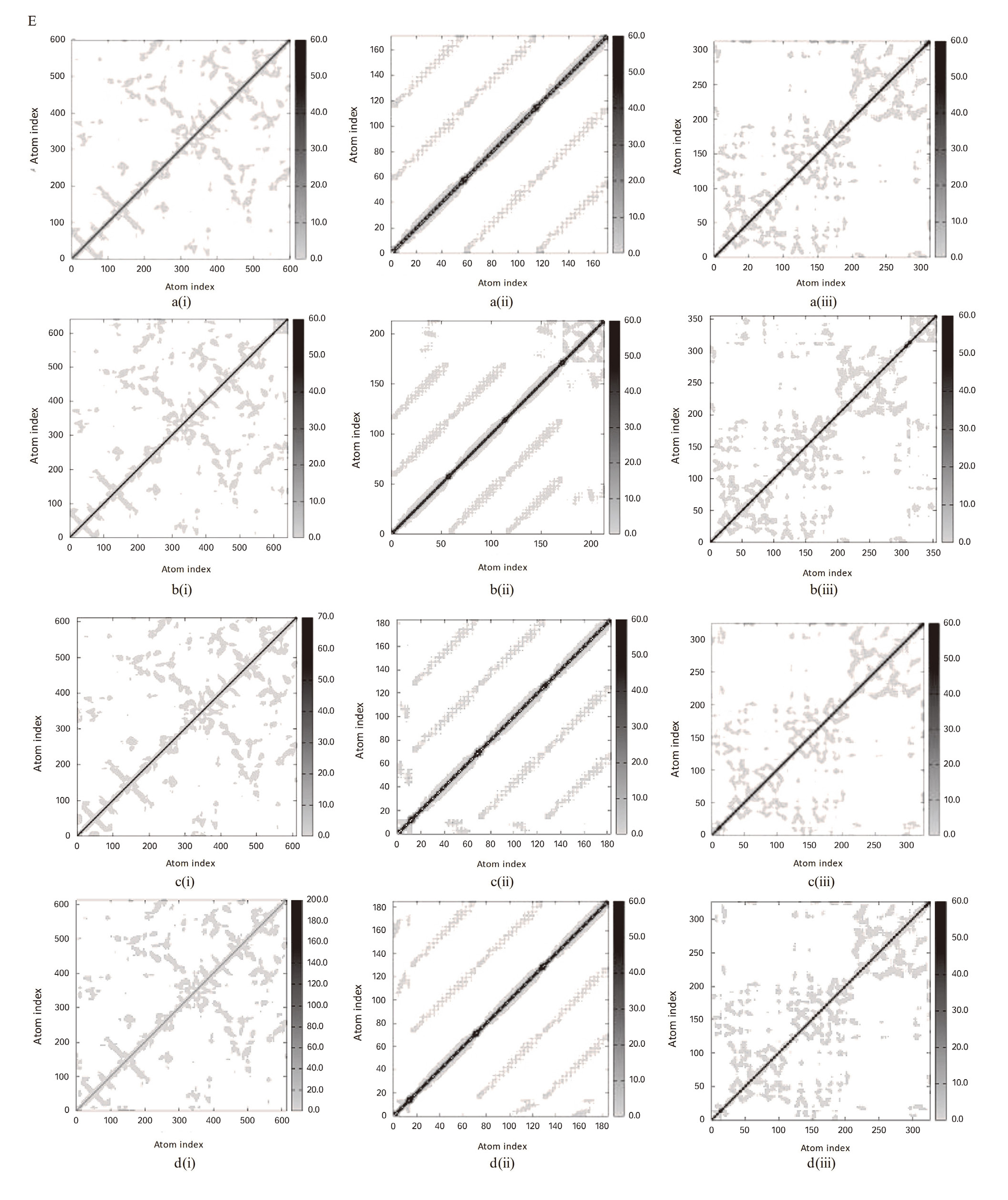
Molecular dynamics simulation: (A) main chain deformability, (B) experimental B-factor, (C) variance associated with each normal mode, (D) covariance matrix and (E) the elastic network model of (a) wild types and bound complexes of (i) plectasin, (ii) PRP, and (iii) PCS with (b) angiotensin-converting enzyme-related carboxypeptidase, (c) SARS-Coronavirus HR2 Domain, and (d) COVID-19 main protease. PCS: peptide from Clitocybe sinopica; PRP: peptide from Russula paludosa.
Eigenvalues of wild-type receptor and bound complexes
| ACE2 | HRD | CMP | |
|---|---|---|---|
| Wild type | 5.63 × 10-5 | 2.15 × 10-4 | 1.21 × 10-4 |
| Plectasin | 7.73 × 10-5 | 1.54 × 10-4 | 1.03 × 10-4 |
| PRP | 5.21 × 10-5 | 1.90 × 10-4 | 1.67 × 10-4 |
| PCS | 1.69 × 10-4 | 1.91 × 10-4 | 1.19 × 10-4 |
ACE2: angiotensin-converting enzyme-related carboxypeptidase; HRD: SARS-coronavirus HR2 domain; CMP: COVID-19 main protease; PCS: peptide from Clitocybe sinopica; PRP: peptide from Russula paludosa.
Discussion
Therapeutic use of bioactive peptides is growing significantly as they are a popular candidate for production of various drugs. The state-of-the-art technology for using peptides as drugs for different clinical applications with advanced activity in therapeutic development for cancer and infection has a great prospect that has attracted billion dollar markets.[28] Mushrooms produce a large number of pharmaceutically active proteins (lectins, laccases, ribosome-inactivating proteins [RIPs], nucleases, and fungal immunomodulatory proteins [FIPs]) as a natural source of antiviral, antitumor, antimicrobial, immune-enhancing agents.[29] Many medicinal mushrooms have attracted increasing attention as functional foods or medicines for the prevention and treatment of several chronic diseases.[30] The bioactive peptides in mushrooms could be considered as functional products with substantial health benefits beyond basic nutrients. The diversity of bioactive compounds in medicinal mushrooms with new action of mechanisms has attracted the attention of scientists to use them as potential options of treatment against current life-threatening diseases.[31]
Physicochemical characterization of selected peptides predicted through the Expasy’s ProtParam server showed the molecular weight, theoretical isoelectric point, and half-life of each of the peptides corresponding to the amino acid length of selected peptides. These factors were deduced from the corresponding peptide sequence and could affect the stability and optimal actions of the peptides. The theoretical isoelectric points and half-life revealed charge stability and estimated half-life of each of the peptides. The half-life predicts the time it takes for half of the amount of each peptide to degrade in a cell. Plectasin was predicted to have an optimal in vivo half-life, which indicates its bioavailability could be essential for optimal function.[32] The corresponding estimated half-life in mammalian reticulocytes through in vitro evaluation correlated with the length of their amino acid composition. The instability index, which is an estimate of stability of the peptides in a test tube, revealed that the peptides were hypothetically stable when smaller than 40.[33] The respective aliphatic index of peptides, which was determined by the volume occupied by aliphatic side chains, showed that plectasin and PCS were thermally stable over a wide temperature range. The low grand average of hydropathicity indicates the provable hydrophilicity of peptides.[24] These properties demonstrate that the putative therapeutic application of the selected peptides would depend on their respective sequence-derived physicochemical characteristics.
The interactions between the selected peptides and the coronavirus-associated proteins were assessed through molecular docking analysis. This study predicted the putative molecular mechanism of action of these compounds through computational analyses. The first model of each molecular interaction was selected as the best docked complex based on the c-scores.[26] The binding affinity obtained through the semi-empirical docking scoring function for the complex of plectasin and HRD was found to be the lowest among other complexes, indicating an interaction with the highest binding affinity. Plectasin showed a relatively high affinity toward all the selected proteins. This putative computation positively correlates with the free energy of binding of the complexes computed through molecular mechanics combined with MM/GBSA, which involved minimization of the docked complexes.[34] The docking studies showed that the peptides could have reasonable biological effect that could interfere with the target proteins and limit the activities of the ACE2, HRD, and CMP.
Further investigations were carried out on the interactions between the selected peptides and proteins during a timeframe through molecular dynamics simulation studies. These studies are helpful for predicting the effect of binding of ligands on the biological processes involving proteins.[35] Predominantly, a notable increase in deformity of all complexes of ACE2 and HRD with the peptides is found in simulation studies (Figure 2A), when compared to the unbound ACE2. This shows that binding of selected peptides could influence the biological function of these proteins and their interactions with their respective targets.[36] However, the binding of all the peptides affected the experimental B-factor, which is an atomic displacement parameter computed from the PDB field and the NMA (Figure 2B). This substantiated the effect of peptides on the flexibility and rigidity of proteins, which are critical due to their role in the cellular entry and subsequent replication of coronavirus.[37] PCS increases the variance associated with each normal mode of the amino acid residues in all the studied proteins (Figure 2C). The covariance matrix shows the connection between the pairs of amino acid residues in the proteins; it showed that plectasin induces correlated motions among the amino acids, which are indicated by an increase in the intensity of red coloration (Figure 2D). Correspondingly, plectasin increased the elasticity of HRD and rigidity in CMP, as revealed by the pattern map of the elastic network (Figure 2E). The associated eigenvalue, which is directly correlated to the energy required to deform proteins, is enhanced by the binding of PCS to all the proteins (Table 5). This implies that PCS could increase the motion stiffness of the proteins and be useful as a better therapeutic agent over small molecules, based on its high target specificity and potency. This observation could substantiate the use of peptides as better therapeutic agents over small molecules based on their high target specificity and potency.[38] Furthermore, this observation is inversely related to the hypothetical influence of peptides on the associated variance to each normal mode.
Conclusion
This study has shown that bioactive peptides from medicinal mushrooms could be developed and explored for their use in peptide-based drugs against coronavirus infection through their modulatory effects on ACE2, HRD, and CMP and probable effect in preventing the cellular entrance and replication of coronavirus. Further experimental studies are, therefore, recommended to validate the putative theoretical potential of the selected peptides.
-
Conflict of Interests
The authors declare no competing interests.
References
1 Astuti I, Ysrafil. Severe Acute Respiratory Syndrome Coronavirus 2 (SARSCoV-2): an overview of viral structure and host response. Diabetes Metab Syndr 2020; 14: 407-12.10.1016/j.dsx.2020.04.020Search in Google Scholar PubMed PubMed Central
2 Li Q, Guan X, Wu P, Wang X, Zhou L, Tong Y. Early transmission dynamics in Wuhan, China, of novel coronavirus–infected pneumonia. N Engl J Med 2020; 382:1199–207.10.1056/NEJMoa2001316Search in Google Scholar PubMed PubMed Central
3 Guo YR, Cao QD, Hong ZS, Tan YY, Chen SD, Jin HJ, et al. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak – an update on the status. Mil Med Res. 2020;7, 11.10.1186/s40779-020-00240-0Search in Google Scholar PubMed PubMed Central
4 Li H, Liu SM, Yu XH, Tang SL, Tang CK. Coronavirus disease 2019 (COVID-19): current status and future perspectives. Int J Antimicrob Agents 2020; 55: 105951.10.1016/j.ijantimicag.2020.105951Search in Google Scholar PubMed PubMed Central
5 Plapp F. The COVID-19 Pandemic: A Summary. The pathologist 2020; 1-7Search in Google Scholar
6 Sanche S, Lin YT, Xu C, Romero-Severson E, Hengartner N, Ke R. High contagiousness and rapid spread of severe acute respiratory syndrome coronavirus 2. Emerg Infect Dis 2020; 267.10.3201/eid2607.200282Search in Google Scholar PubMed PubMed Central
7 W.H.O. Coronavirus disease (COVID-19) WHO; 2020. Available at: https://www.who.int/emergencies/diseases/novel-coronavirus-2019.Accessed on June 1, 2020.Search in Google Scholar
8 Sánchez A, Vázquez A. Bioactive peptides: A review. Food Qual Saf 2017; 1: 29–46.10.1093/fqs/fyx006Search in Google Scholar
9 Li W, Oshi MD, Singhania S, Ramsey KH, Murthy AK. Peptide vaccine: progress and challenges. Vaccines 2014; 2: 515-36.10.3390/vaccines2030515Search in Google Scholar PubMed PubMed Central
10 Purcell AW, McCluskey J, Rossjohn J. More than one reason to rethink the use of peptides in vaccine design. Nat Rev Drug Discov 2007; 6: 404 -14.10.1038/nrd2224Search in Google Scholar PubMed
11 Yan Z, Wang J. Specificity quantification of biomolecular recognition and its implication for drug discovery. Sci Rep 2012; 2:309.10.1038/srep00309Search in Google Scholar PubMed PubMed Central
12 Lee AC, Harris JL, Khanna KK, Hong JH. A comprehensive review on current advances in peptide drug development and design. Intern J Mol Sci 2019; 20: 2383.10.3390/ijms20102383Search in Google Scholar PubMed PubMed Central
13 Xue H, Li J, Xie H, Wang Y. Review of drug repositioning approaches and resources. Intern J Biol Sci 2018; 14: 1232-44.10.7150/ijbs.24612Search in Google Scholar PubMed PubMed Central
14 King MD, Long T, Pfalmer DL, Andersen TL, McDougal OM. SPIDR: small-molecule peptide-influenced drug repurposing. BMC Bioinform 2018; 19:138.10.1186/s12859-018-2153-ySearch in Google Scholar PubMed PubMed Central
15 Romano JD, Tatonetti NP. Informatics and computational methods in natural product drug discovery: a review and perspectives. Front Genet 2019; 10: 368.10.3389/fgene.2019.00368Search in Google Scholar PubMed PubMed Central
16 Ryall KA, Tan AC Systems biology approaches for advancing the discovery of effective drug combinations. J Cheminform 2015; 7: 7.10.1186/s13321-015-0055-9Search in Google Scholar PubMed PubMed Central
17 Lavanya J, Subhashini S. Therapeutic proteins and peptides from edible and medicinal mushrooms-review. Euro Sci J 2013; 9: 162-76.Search in Google Scholar
18 Mygind PH, Fischer RL, Schnorr KM, Hansen MT, Sönksen CP, Ludvigsen S, et al. Plectasin is a peptide antibiotic with therapeutic potential from a saprophytic fungus. Nature 2005; 437:975–80.10.1038/nature04051Search in Google Scholar PubMed
19 Wang J, Wang HX, Ng TB. A peptide with HIV-1 reverse transcriptase inhibitory activity from the medicinal mushroom Russula paludosa. Peptides 2007; 28:560-5.10.1016/j.peptides.2006.10.004Search in Google Scholar PubMed
20 Zheng S, Liu Q, Zhang G-Q, Wang H-X, Ng T-B. Purification and characterization of an antibacterial protein from dried fruiting bodies of the wild mushroom Clitocybe sinopica. Acta biochimica Polonica 2010; 57:43-8.10.18388/abp.2010_2370Search in Google Scholar
21 Yang J, Zhang Y. I-TASSER server: new development for protein structure and function predictions, Nucleic Acids Res 2015; 43: W174-8110.1093/nar/gkv342Search in Google Scholar PubMed PubMed Central
22 Zhang C, Freddolino PL, Zhang. Y COFACTOR: improved protein function prediction by combining structure, sequence and protein– protein interaction information. Nucleic Acids Res 2017; 45: W291-9.10.1093/nar/gkx366Search in Google Scholar PubMed PubMed Central
23 Pontius J, Richelle J, Wodak SJ. Deviations from Standard Atomic Volumes as a Quality Measure for Protein Crystal Structures. J Mol Biol 1996; 264: 121-36.10.1006/jmbi.1996.0628Search in Google Scholar PubMed
24 Artimo P, Jonnalagedda M, Arnold K, Baratin D, Csardi G, de Castro E, et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012; 40:597-603.10.1093/nar/gks400Search in Google Scholar PubMed PubMed Central
25 Feng T, Chen F, Kang Y, Sun HY, Liu H, Li D, et al. HawkRank: A new scoring function for protein-protein docking based on weighted energy terms. J Cheminform 2017; 9:66.10.1186/s13321-017-0254-7Search in Google Scholar PubMed PubMed Central
26 Weng GQ, Wang EC, Wang Z, Liu H, Li D, Zhu F, et al. HawkDock: a web server to predict and analyze the structures of protein-protein complexes based on computational docking and MM/GBSA. Nucleic Acids Res 2019; 47: W322-30.10.1093/nar/gkz397Search in Google Scholar PubMed PubMed Central
27 López-Blanco, J.R., Aliaga, J.I., Quintana-Ortí,E.S, Chacón, P. iMODS: Internal coordinates normal mode analysis server. Nucleic Acids Res 2014; 42: W271-6.10.1093/nar/gku339Search in Google Scholar PubMed PubMed Central
28 Falciani C, Pini A, Bracci L. Peptide Drugs in: Hughes AB. Amino acids, peptides and proteins in organic chemistry: protection reactions, medicinal chemistry, combinatorial synthesis. Wiley Online Library. 2011; 4. Available at: https://onlinelibrary.wiley.com/doi/10.1002/9783527631827.ch7. Access on June 1, 2020.10.1002/9783527631827.ch7Search in Google Scholar
29 Zhou R, Liu ZK, Zhang YN, Wong JH, Ng TB, Liu F. Research progress of bioactive proteins from the edible and medicinal mushrooms. Curr Protein Pept Sci 2019;20:196‐219.10.2174/1389203719666180613090710Search in Google Scholar PubMed
30 Zhang JJ, Li Y, Zhou T, Xu DP, Zhang P, Li S, et al. Bioactivities and health benefits of mushrooms mainly from China. Molecules 2016; 21: 938.10.3390/molecules21070938Search in Google Scholar PubMed PubMed Central
31 Elkhateeb WA, Daba GM, Thomas PW, Wen TC. Medicinal mushrooms as a new source of natural therapeutic bioactive compounds. Egypt Pharm J 2019; 18: 88-101.Search in Google Scholar
32 Jenssen H, Aspmo SI. Serum Stability of Peptides. In: Otvos L. (eds) Peptide-Based Drug Design. Methods In Molecular Biology™, vol 494. Humana Press 2008; 177-186.10.1007/978-1-59745-419-3_10Search in Google Scholar PubMed
33 Guruprasad K, Reddy BV, Pandit MW. Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng 1990; 4: 155 -61.10.1093/protein/4.2.155Search in Google Scholar PubMed
34 Hou TJ, Wang JM, Li YY, Wang W. Assessing the performance of the MM/PBSA and MM/GBSA methods: I. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem Info Mod 2011; 51:69-82.10.1021/ci100275aSearch in Google Scholar PubMed PubMed Central
35 Boehr DD, Nussinov R, Wright PE. The role of dynamic conformational ensembles in biomolecular recognition. Nat Chem Biol 2009; 5: 789 -96.10.1038/nchembio.232Search in Google Scholar
36 Kovacs J, Chacón P, Abagyan R. Predictions of Protein Flexibility: First Order Measures. PROTEINS: Structure, Function, and Bioinformatics. Proteins 2004; 56(4):661-8.10.1002/prot.20151Search in Google Scholar
37 Chapados BR, Hosfield DJ, Han S, Qiu J, Yelent B, Shen B, et al. Structural basis for FEN-1 substrate specificity and PCNA-mediated activation in DNA replication and repair. Cell 2004; 9;16:39-50.10.1016/S0092-8674(03)01036-5Search in Google Scholar
38 Craik DJ, Fairlie DP, Liras S, Price D. The future of peptide-based drugs. Chem Biol Drug Des 2013; 81; 136–47.10.1111/cbdd.12055Search in Google Scholar PubMed
© 2021 Babatunde Joseph Oso, Clement Olusola Ogidi, published by Sciendo
This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
Articles in the same Issue
- Editorial
- Advances in clinical research in chronic kidney disease
- Commentary
- Repair of damaged pancreatic β cells: New hope for a type 2 diabetes reversal?
- Review Article
- Cardiovascular involvement in patients with 2019 novel coronavirus disease
- Traditional Chinese medicine in treating IgA nephropathy: from basic science to clinical research
- Endoscopic transmural drainage and necrosectomy in acute necrotizing pancreatitis: A review
- Original Article
- Joint predictive value of cTnI and NT-proBNP on mortality in patients with coronavirus disease 2019: A retrospective research in Wuhan, China
- Acute bronchiolitis: Why put an IV line?
- Repositioning of fungal-based peptides as modulators of angiotensin-converting enzyme-related carboxypeptidase, SARS-coronavirus HR2 domain, and coronavirus disease 2019 main protease
- The lifetime cost estimation of human papillomavirus-related diseases in China: a modeling study
- The prognostic value of Krebs von den Lungen-6 and surfactant protein-A levels in the patients with interstitial lung disease
- Letter to Editor
- Malnutrition and venous thromboembolism are specific issues in elderly patients with coronavirus disease 2019
Articles in the same Issue
- Editorial
- Advances in clinical research in chronic kidney disease
- Commentary
- Repair of damaged pancreatic β cells: New hope for a type 2 diabetes reversal?
- Review Article
- Cardiovascular involvement in patients with 2019 novel coronavirus disease
- Traditional Chinese medicine in treating IgA nephropathy: from basic science to clinical research
- Endoscopic transmural drainage and necrosectomy in acute necrotizing pancreatitis: A review
- Original Article
- Joint predictive value of cTnI and NT-proBNP on mortality in patients with coronavirus disease 2019: A retrospective research in Wuhan, China
- Acute bronchiolitis: Why put an IV line?
- Repositioning of fungal-based peptides as modulators of angiotensin-converting enzyme-related carboxypeptidase, SARS-coronavirus HR2 domain, and coronavirus disease 2019 main protease
- The lifetime cost estimation of human papillomavirus-related diseases in China: a modeling study
- The prognostic value of Krebs von den Lungen-6 and surfactant protein-A levels in the patients with interstitial lung disease
- Letter to Editor
- Malnutrition and venous thromboembolism are specific issues in elderly patients with coronavirus disease 2019