Structure and function of HECT E3 ubiquitin ligases and their role in oxidative stress
-
Hao Qian
 and
Yingxian Sun
and
Yingxian Sun

Abstract
Ubiquitination is a modification after protein transcription that plays a vital role in maintaining the homeostasis of the cellular environment. The Homologous to E6AP C-terminus (HECT) family E3 ubiquitin ligases are a kind of E3 ubiquitin ligases with a C-terminal HECT domain that mediates the binding of ubiquitin to substrate proteins and a variable-length N-terminal extension. HECT-ubiquitinated ligases can be divided into three categories: NEDD4 superfamily, HERC superfamily, and other HECT superfamilies. HECT ubiquitin ligase plays an essential role in the development of many human diseases. In this review, we focus on the physiological and pathological processes involved in oxidative stress and the role of E3 ubiquitin ligase of the HECT family.
UBIQUITIN–PROTEASOME SYSTEM
The ubiquitin–proteasome system is an important mechanism that regulates the protein levels in the body as well as intracellular protein levels. It is used to degrade misfolded, damaged, or useless proteins[1]; 80%–90% of the proteins in cells are degraded by this pathway.[2] However, more and more evidence now shows that ubiquitination and even the combination of ubiquitinated protein and proteasome may not lead to protein degradation.[3] Ubiquitin is a protein consisting of 76 amino acids. It is typically linked to substrates through an isopeptide bond between the ɛ-amino group of a substrate lysine residue and the carboxyl terminus of ubiquitin, making it as a substrate for the proteasome.[4, 5, 6] The main feature of ubiquitin is its seven lysine residues, and all of these residues can be ubiquitinated, resulting in a ubiquitin chain linked to an isopeptide. There are several different types of ubiquitination. Some substrate proteins can only be monoubiquitinated or multi-monoubiquitinated, and substrate proteins can form polyubiquitin chains at a single lysine site. This polyubiquitin chain can be divided into single, mixed, and dendritic structures according to the lysine site connecting the ubiquitin chain. When ubiquitin is linked to the N-terminus of the second ubiquitin, an eighth chain type is produced.[7] In the ubiquitin chain, the ubiquitin moiety can bind through one of its lysine residues (K6, K11, K27, K29, K33, K48, and K63) or the N-terminal methionine residue, providing countless possibilities of assembling specific polymers.[8] Because the tools and techniques for detecting posttranslational modifications are still lacking, the cellular function of K6-connected ubiquitin chains is still unclear. There are literature reports that HUWE1 can be modified by K6 to connect ubiquitin chains.[9] The linear ubiquitin chain assembly complex is the only known mammalian ubiquitin ligase that makes methionine 1 (Met1)-linked polyubiquitin. Nowadays, evidence suggests that Met1-linked polyubiquitin is inextricably linked to NF-ϰB signaling, cell death, inflammation, immunity, and cancer. K11 is a powerful degradation signal.[10, 11] K11-linked chains can drive proteasome degradation and mitotic exit. K11-linked chains are the product of the human E3 anaphase-promoting complex (APC/C), which can regulate cell division. When APC/C is activated during mitosis, K11-linked chains increase substantially.[12] K27-linked chains participate in the DNA damage response. According to reports, RNF168-dependent chromatin ubiquitination requires K27-linked chain residues, which is an important ubiquitin-based modification marking chromatin upon DNA damage.[13] K29-linked chains can negatively regulate Wnt signaling pathway.[14] Deletion of the Really Interesting New Gene/U-box (RING)-type E3 ligase Cbl-b and the Homologous to E6AP C-terminus (HECT)-type E3 ligase Itch resulted in an increase in T-cell activation and an autoimmune response. The two E3 ligases cooperate to induce K33-linked polyubiquitination of TCR-ζ, functionally altering receptor phosphorylation and protein binding.[15] K48-K63–linked chains can also regulate NF-ϰB signaling.[16] Protein ubiquitination requires three enzymes: E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzyme, and an E3 ubiquitin ligase.[17] First, the E1 enzyme forms a thioester bond between its active site Cys and the C-terminal Gly of ubiquitin in an ATP-dependent manner. Ubiquitin is then transferred on to the Cys residue in the active site of the E2 enzyme; E2 can bind to E3-ubiquitinated ligase after activation. E3-ubiquitinated ligase recognizes degraded proteins and links ubiquitin to the substrate (Figure 1).[7, 18, 19] E3-ubiquitinated ligases are divided into three categories: the largest class is the RING-type E3s, followed by the HECT-type E3s and the RING between RING (RBR)-type E3s.[20] The majority of the 600 E3s present in humans belong to the RING family, which are characterized by a cross-brace structure with two zinc ions coordinated by cysteine and histidine residues.[19] In the past 10 years, the ubiquitin–proteasome system has been extensively studied in the cardiovascular field, including atherosclerosis, familial cardiac protein disease, idiopathic dilated cardiomyopathy, and myocardial ischemia.[21, 22, 23, 24] At the same time, there are related reports that the ubiquitin–proteasome system can also promote the metabolism of toxins, fats, and cancer cells, and the energy generated by metabolism can stimulate cells to self-replicate and undergo self-metabolic repair (Figure 2).[25, 26, 27, 28]
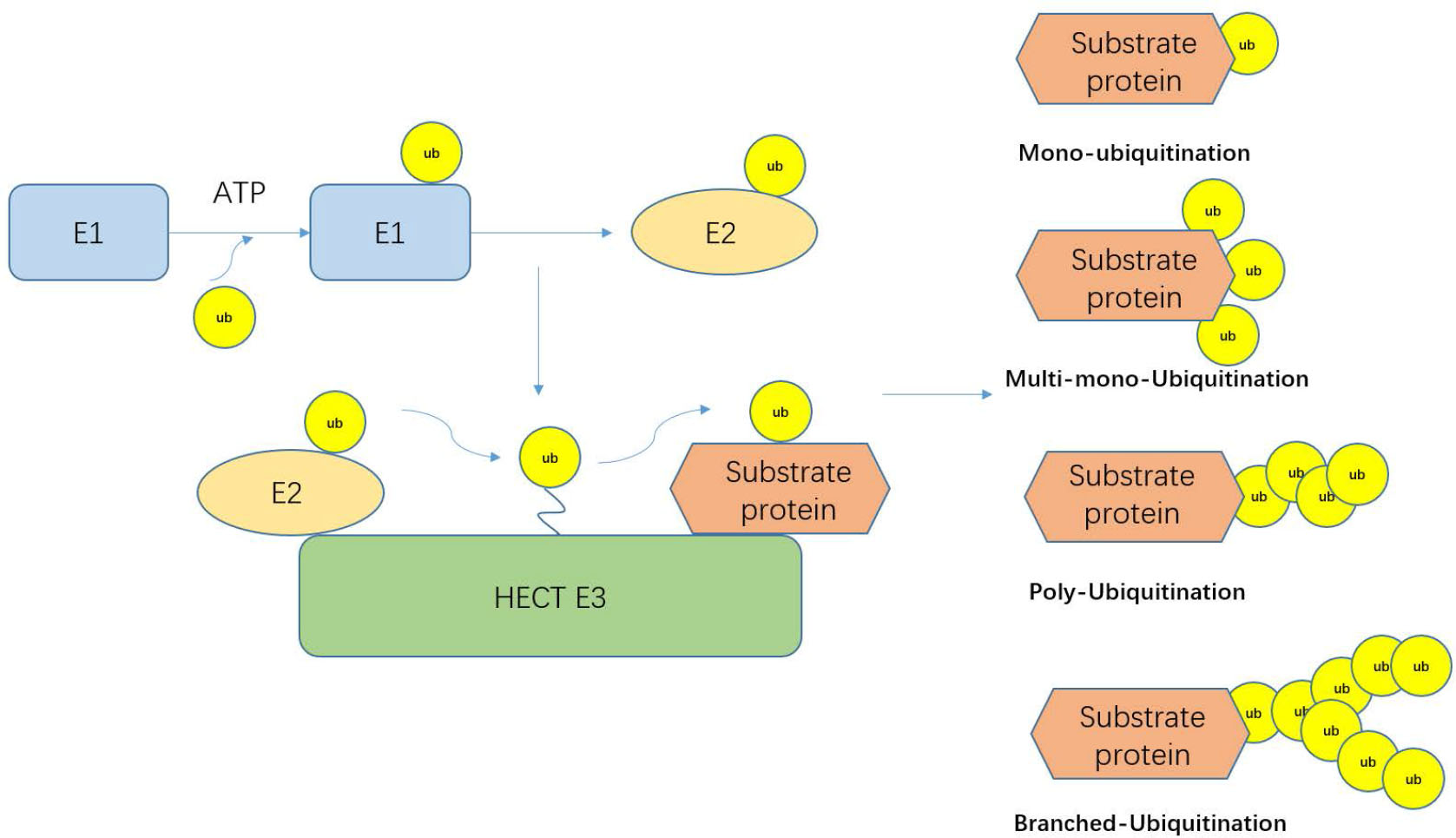
The ubiquitination cascade. The E1 enzyme forms a thioester bond between its active site Cys and the C-terminal Gly of ubiquitin in an ATP-dependent manner. Ubiquitin is then transferred on to the Cys residue in the active site of the E2 enzyme. E2 can bind to E3-ubiquitinated ligase. After activation, E3-ubiquitinated ligase recognizes degraded proteins and links ubiquitin to the substrate.
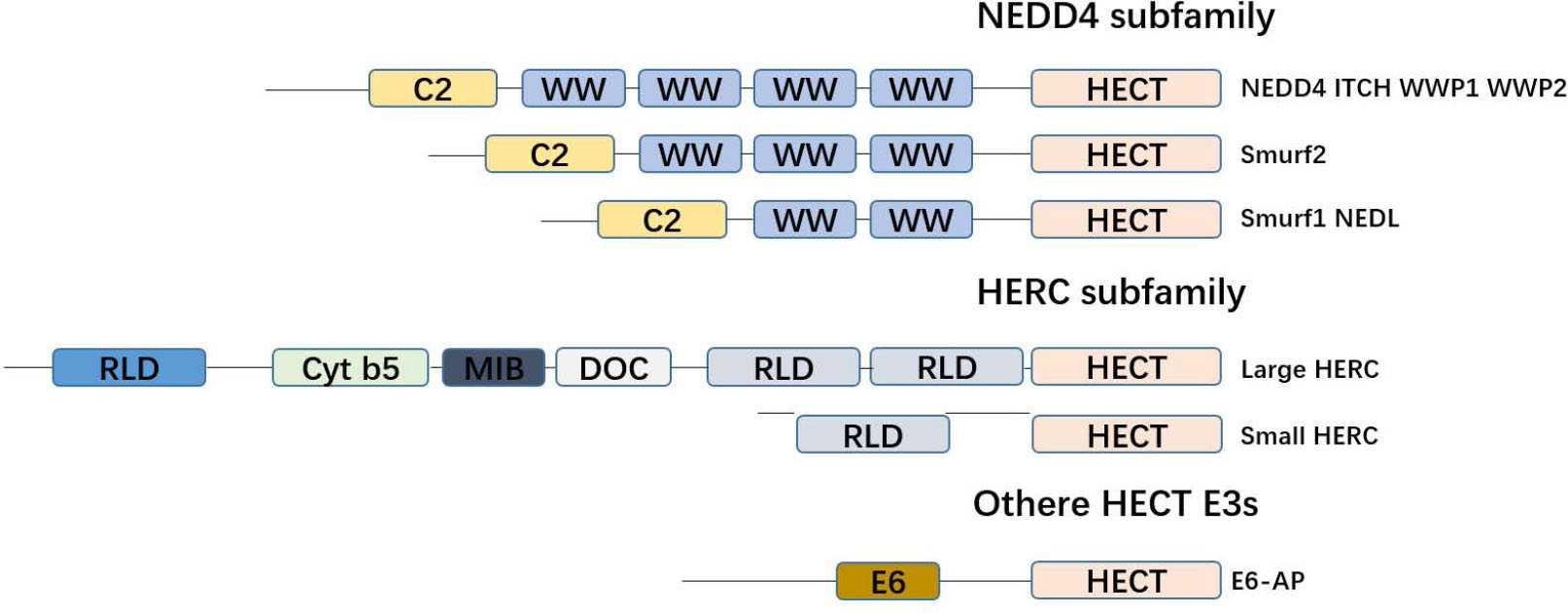
Classification of the HECT E3 family. The family of HECT E3. All members of the HECT E3 family are characterized by the C-terminal HECT domain, which consists of approximately 350 amino acid residues and represents the catalytic domain. The NEDD4 subfamily has nine members that are characterized by an N-terminal C2 domain and the presence of several WW domains. The HERC family comprises six members, which are characterized by the presence of one or several RLD domains. Members of other HECT E3 subfamily are characterized by the notion that they contain neither RLDs nor WW domains.
Proteasomes are widely distributed in the cytoplasm and nucleus, have multiple proteolytic enzyme activities, and are ubiquitin dependent. Polyubiquitin-labeled proteins are often degraded by the 26s proteasome. The 26S proteasome can be divided into two subcomplexes: the 19S regulatory particle (RP) and the 20S core particle (CP). The function of 19s is to recognize, expand, and deubiquitinate and translocate substrates into the 20s proteasome, which contains the proteolytic site.[29] The dyad-symmetric CP is a highly conservative complex composed of four stacked heptameric rings, and the two inner rings create an internal chamber that houses the proteolytic active sites responsible for protein cleavage; these rings are each formed by seven β subunits.[30] The 20S proteasomes degrade unfolded or loosely folded proteins and peptides in a manner independent of ATP.[5]
HECT FAMILY E3 UBIQUITIN LIGASE
The difference between HECT family E3 ubiquitin ligases and other ubiquitin E3 ligases is that they have an active site for cysteine, which forms an intermediate thioester bond with ubiquitin before it is linked to its substrate.[31] HECT family E3 has a key domain, the HECT domain (homologous to the C-terminus of E6AP), which is a conservative carboxy-terminal catalytic domain composed of about 350 amino acids.[32] The N-terminus of the HECT family E3 is generally not conserved, but the N-terminus can specifically recognize the substrate.[33] The HECT domain consists of two lobes: a larger N-terminal lobe (N-lobe) and a smaller C-terminal lobe (C-lobe). Structural studies show that the two lobes are connected by a flexible hinge region, which is essential for the catalytic Cys residues (HECT domains) incorporated into E2 and E3 during ubiquitin transfer.[34] Based on the existence of different amino acid sequence motifs in these N-terminal extensions, the human HECT E3 family consists of 28 members, of which 15 members can be divided into two subfamilies based on the commonness of the N-terminal domain. The human NEDD4 subfamily has nine members characterized by the existence of a WW domain and a C2 domain, and is the most famous and researched. Another family is the six-member HERC E3 ligase with two characteristic domains in its sequence: the HECT domain and the RCC1-like domain (RLD). Other HECT E3 families lack WW or RLD domains and have various N-terminal domains. Yeast has five HECT E3 ligases: Rsp5, Ufd4, Hul4, Hul5, and Tom1. Rsp5 is a member of the NEDD4 family, while the other four yeast HECT E3s do not belong to any family (Figure 3).[35, 36, 37, 38]
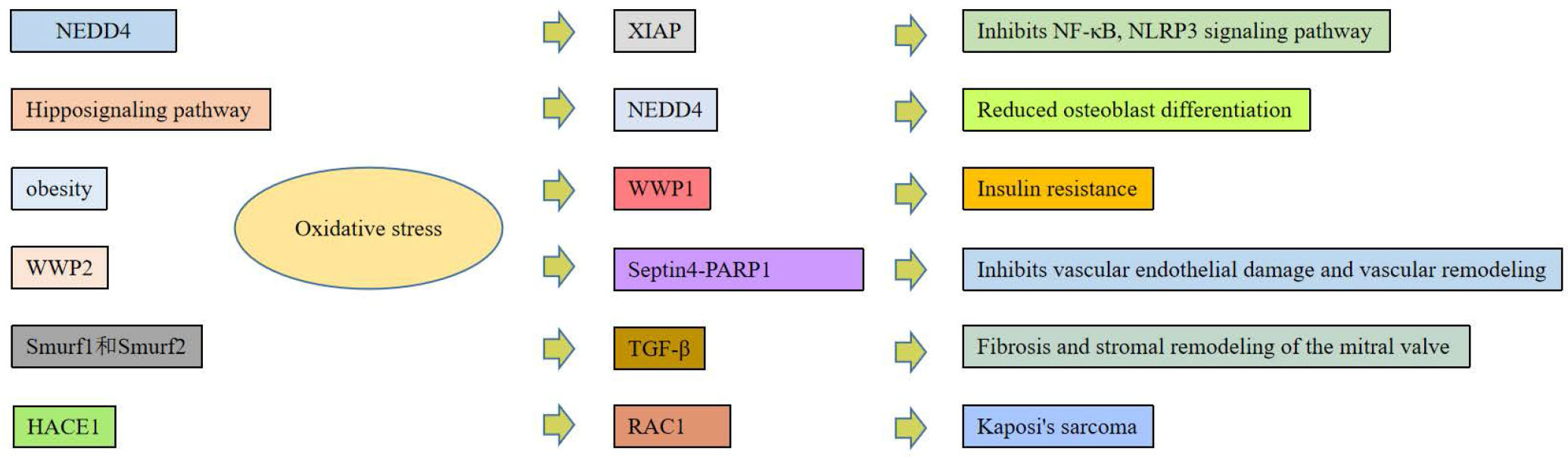
Overview of related functions of different HECT E3 ubiquitin ligases under oxidative stress.
NEDD4 subfamily
NEDD4 is a founding member of the NEDD4 family of ubiquitin ligases, a highly evolved and conserved protein from yeast to humans. It is one of the earliest discovered HECT E3 ubiquitin ligases.[39] It includes an N-terminal C2 domain, three to four WW domains, and a C-terminal catalytic HECT domain for ligation of ubiquitin proteins.[40] The C2 domain is a calcium-dependent lipid-binding domain with a length of about 116 amino acids. The C2 domain not only binds to lipid membranes, but has also been shown to bind proteins.[41] The WW domain consists of 35–40 amino acids and has two conserved tryptophan residues. These domains usually bind to PY (PPxY) motifs in substrates and regulatory proteins. Multiple WW domains in NEDD4 members suggest they may interact with multiple proteins simultaneously.[42] In humans, NEDD4 includes nine members: NEDD4, NEDD4-2/NEDD4L, ITCH, Smad ubiquitin regulators1,2 (Smurf1, Smurf2), WWP1, WWP2, NEDL1, and NEDL2. NEDD4 was originally thought to be a developmental regulatory gene in the central nervous system.[43] Smurf1 and Smurf2 are related members of the NEDD4 family, both of which contain three WW structures. Smurf1 is involved in many important biological functions, including the bone morphogenetic protein, cell growth, and morphogenesis.[44] Smurf2 was originally thought to negatively regulate the BMP/TGF-β signaling pathway and play a vital role in the pathogenesis of embryogenesis, adult tissue homeostasis, tumors, and various human diseases.[45, 46] WWP1 contains four WW domains. WWP1 regulates a variety of cellular biological processes, including transcription, degradation, and protein transport. WWP1 is linked to many diseases such as cancer, acute myeloid leukemia, and immune-related diseases.[47] Like other NEDD4 family members, ITCH is ubiquitous in patients with inflammatory skin diseases and patients with systemic and neurological diseases, and it can seriously affect the quality of life.[48]
HERC subfamily
The HERC subfamily has six members. Based on the molecular weight, it can be further divided into “large” HERCs (molecular weights greater than 500 kDa, HEC1 and HEC2) and “small” HERCs (molecular weights about 100–120 kDa), which are characterized by having HECT domain and one or more RLDs of ubiquitin ligase activity.[49] The large HERC family contains multiple RCC-like domains, but the small HERC family usually carries only one.[50] HERC protein is mainly located in the cytoplasm, from the cytoplasm to the membrane or vesicular structure.[36] The RLD of HERC E3s usually consists of seven repeats of 50–70 amino acids. Structural studies show that RCC1 is composed of seven β blades resembling the shape of a propeller. Each repeat sequence corresponds to a blade.[51]
Other HECT E3 subfamilies
Ube3A (E6 associated protein [E6AP]) has a conserved N-terminal domain (residues 24–87), a novel Zn-binding fold called amino-terminal Zn-finger of Ube3a ligase (AZUL).[52] E6AP can form a complex with human papillomavirus E6 oncoprotein and target the degradation of tumor suppressor p53, thereby promoting canceration.[53, 54] Other HECT subfamilies include HUWE1, UBR5, and TRIP12. HUWE1 contains two N-terminal unknown functional domains, DUF908 and DUF913, similar to the domain in Saccharomyces cerevisiae HECT ligase Tom1, followed by the ubiquitin-related UBA domain.[55] HACE1 can be targeted to bind to p53 and many other substrates involved in tumorigenesis[56]; it has received widespread attention as a target for anticancer drugs.[57] UBR5 is widely expressed in a variety of cell types. The catalytically active HECT domain of UBR5 is made up of N- and C-lobes separated by a linker sequence. UBR5 is highly conserved in metazoans and involved in many biological functions, including DNA damage response, metabolism, transcription, and apoptosis. UBR5 is a cellular signal regulator related to many cancer biological processes. However, the mechanisms of UBR5 involved in tumorigenesis and development are not clear.[58]
OXIDATIVE STRESS
Oxidative stress injury is the basis of the occurrence and development of many diseases. In order to maintain the stability of the internal environment, there is a balance between oxidation and antioxidants; but when this balance is broken by various factors, it will have a series of negative effects.[59] Reactive oxygen species (ROS) is a type of one-electron reduction product of oxygen in the body, including superoxide anion (O2−), one-electron reduction product of oxygen; hydrogen peroxide (H2O2), two-electron reduction product; hydroxyl radical (•OH); nitric oxide, etc.[60]
Mitochondrial metabolism is the main source of ROS, including singlet oxygen (O2), superoxide anion (O2•−), hydrogen peroxide (H2O2), nitric oxide (NO•), hydroxyl radical (OH•), and hydroxyl ion (OH−).[61] It is generally believed that most of the mitochondrial ROS are formed in complex I (NADH-CoQ reductase) and complex III (cytochrome C oxidase), where electrons can react with oxygen to generate O2−.[62, 63, 64] In addition, mitochondrial flavin proteases, especially pyruvate dehydrogenase and α-ketoglutarate dehydrogenase, are also considered sources of O2−.[65]
The main ROS production in the cell membrane is mainly NADPH oxidase.[66] The heterodimer transmembrane part of the classic NADPH oxidase complex from phagocytes consists of NOx2 and p22phox. The NADPH-dependent oxidoreductase (NOx enzyme) family is mainly divided into four types (NOx1, NOx2, NOx4, and NOx5). NOx1 is mainly expressed in smooth muscle cells, but is also found in endothelial cells. NOx2 is expressed in endothelial cells, adventitial fibroblasts, inflammatory cells, platelets, and microvascular smooth muscle cells. NOx4 is widely expressed in vascular cells and is the most abundant NOx homolog.[67] NOx4 is different in that it can quickly convert O2− to H2O2, while H2O2 does not interact with NO signals and degrade NO. NOx4 in the vascular system has been reported to promote angiogenesis and reduce inflammation [68]. At the same time, NOx4 can reduce inflammation, fibrosis, and improve endothelial function without affecting dyslipidemia in atherosclerosis.[69, 70]. In the vessel wall, ROS production systems include NADPH oxidase, xanthine oxidase, mitochondrial electron transport chain, and uncoupled endothelial nitric oxide.[71, 72] Under physiological conditions, low concentration of ROS has the function of signal transduction. However, excessive or sustained production of ROS can lead to oxidative stress injury.[73] In the arterial wall, oxidative stress not only causes direct and irreversible oxidative damage, but also destroys the key redox-dependent signal transduction process. The literature indicates that the mechanism by which oxidative stress can promote vascular disease is through the destruction of vascular protective NO signaling pathway.[74] In addition to NO inactivation, ROS can also directly promote vascular inflammation and remodeling. Indeed, many adverse effects of ROS on the arterial wall are attributable to the oxidation of key signaling proteins and activation of the pro-inflammatory redox-dependent transcription factor NF-ϰB; this leads to the expression of adhesion molecules on the endothelium and the proliferation and migration of vascular smooth muscle cells.[75]
The endoplasmic reticulum contains various oxygenases and oxidases (e.g. cytochrome P450 enzymes, flavin monooxygenases, prolyl and lysyl hydroxylases), and these enzymes are important sources of ROS formation. Especially during protein folding, ROS and glutathione disulfide (GSSG) form by-products under the action of endoplasmic oxidoreductase 1 (Er1) thiol oxidase. The same endoplasmic reticulum and nucleus are also related to the production of RO.[76, 77]
HECT SUBFAMILY AND OXIDATIVE STRESS
NEDD4 subfamily and oxidative stress
Dexmedetomidine (Dex) is a widely used anesthetic and has some anti-inflammatory effects.[78] Dex significantly inhibits the production of pro-inflammatory factors such as interleukin (IL)-6 and tumor necrosis factor alpha (TNF-α) in endotoxemia.[79] In addition, there are also reports in literature demonstrating the protective effect of Dex on lipopolysaccharide (LPS)-induced acute lung injury through HMGB1-mediated TLR4/NF-ϰB and PI3K/Akt/mTOR pathways.[80] It has been reported that upregulation of XIAP inhibits chondrocyte apoptosis in degenerated nucleus pulposus and osteoarthritis.[81, 82] XIAP is a member of the apoptosis inhibitory protein (IAP) and represents a family of endogenous caspase inhibitors. Upregulating XIAP can block degenerative nucleus pulposus and osteoarthritis chondrocyte apoptosis.[82] It is well known that the NF-ϰB signaling pathway is a key molecular switch for cells to respond to oxidative stress.[83] In a model of degenerative disk disease, Dex blocked the activation of NF-ϰB signaling pathway by H2O2. Dex inhibits NLRP3 by inhibiting the NF-ϰB signaling pathway. Activated NLRP3 recruits ASC and caspase-1 to form a protein complex that is essential for caspase-1 activation. Activated caspase-1 leads to the maturation of pre-IL-1β and pre-IL-18. XIAP not only directly inhibits initiation and execution of the caspase cascade during programmed cell death, but also regulates a series of cellular activities that enhance survival signals, including NFϰB activity, in a caspase-independent manner.[84, 85] In a model of H2O2-induced chondrocyte death and degeneration, H2O2 stimulation increased the expression of NEDD4. Co-treatment with Dex and H2O2 caused an increase in XIAP protein level. Dex can reduce the amount of NEDD4 bound to XIAP, thereby protecting the XIAP protein from NEDD4-mediated ubiquitination and degradation. Dex destroys the effects of H2O2 on NF-ϰB/NLRP3, JNK/NLRP3, and NEDD4/XIAP, thus preventing the death and degradation of chondrocytes.[86]
The Hippo signaling pathway was originally discovered in the genetic study of Drosophila. The core components of the Hippo signaling pathway (MST1/2, LATS1/2, YAP, and TAZ) are involved in many important biological functions.[87] Its main function is to restrict the growth of adult tissues and regulate cell proliferation, differentiation, and migration in developmental organs.[88] Besides, dysregulation of the Hippo pathway leads to abnormal cell growth and tumor formation.[89] During apoptosis, PARP1 is cleaved by caspase-3,[90] so that PARP1 loses its enzyme activity, and then completes the biological process of apoptosis.[91] Under high concentrations of oxidative stress, NEDD4 is cleaved by various activated caspases. Interestingly, the kinetics of NEDD4 cleavage is similar to that of PARP1.[91] In human bone marrow–derived stem cells (hBMSCs), low-concentration H2O2 stimulation can increase the expression of NEDD4 and activate the Hippo signaling pathway. NEDD4 can regulate the differentiation of osteoblasts in hBMSCs under oxidative stress and further reduce the differentiation of osteoblasts.[92]
Thioredoxin-interacting protein (TXNIP) is expressed in most eukaryotic cells; it remains stable under oxidative stress and acts as a ROS scavenger [93]. Under conditions of high ROS levels and oxidative stress, TXNIP dissociates from thioredoxin to associate with NLRP3.[94] It can activate inflammatory cells and mediate inflammatory signals.[95] It has been reported that the E3-ubiquitinated ligase ITCH degrades TXNIP through the ubiquitin–proteasome pathway to maintain cellular homeostasis and response to ROS. ITCH increases proteasomal TXNIP degradation and augments thioredoxin activity, leading to inhibition of ROS generation, p38 MAPK, and p53, improvement of oxidative stress response in ROS-induced cardiotoxicity, and improvement of survival in ROS-induced cardiotoxicity and myocardial infarction.[96]
WWP1 also belongs to the E3 ubiquitinated ligase HECT family. It has been reported that WWP1 can degrade p53 through the ubiquitin–proteasome pathway.[97] It is also reported that deletion of the WWP1 gene inhibits the growth of liver cancer cells and leads to apoptosis of liver cancer cells. Knockout of WWP1 can promote the expression of caspase-3 protein and p53 in liver cancer cells.[98] In obesity, excessive supply of energy substrate leads to increased ROS level, which leads to inflammation and insulin resistance. Mitochondrial antioxidant enzymes such as superoxide dismutase and glutathione peroxidase are impaired in obese patients with type 2 diabetes.[99]In vivo, WWP1 is a new obesity-inducing protein and p53-dependent E3 ubiquitin ligase that can help protect fat cells from oxidative stress.[101]
Endothelial dysfunction caused by oxidative stress is the initial event and main cause of cardiovascular system diseases such as atherosclerosis and hypertension vascular disease.[100, 101] WWP2 is involved in the cell cycle.[102] At the same time, in endometrial cancer, there may be a correlation between increased WWP2 transcription levels and loss of PTEN protein.[103] Septin4 is a member of the GTP-binding protein family and is involved in the formation of the cytoskeleton during mitotic, apoptotic, fibrotic, and other cellular processes.[104] WWP2, a member of NEDD4 family, is involved in endothelial cell injury and vascular remodeling as a new regulator. In addition, WWP2 ubiquitinates the Septin4-K174 site by interacting with the Septin4-GTP domain, and degrades Septin4 via the ubiquitin–proteasome pathway. This inhibits the formation of Septin4–PARP1 complex to suppress endothelial injury and vascular remodeling after endothelial injury.[105]
The NADPH (NOx) family is an important source of ROS. The literature shows that NOxs regulates the growth and death of hepatocytes through the TGF-β signaling pathway of hepatocytes and the activation of hepatic stellate cells on myofibroblasts, which are the key to the fibrotic process.[106] In myxomatous mitral valve disease, multiple TGF-β receptors and their ligands are increased, and NOx2 and NOx4 aggravate oxidative stress in the mucus mitral valve, which in turn increases fibrosis and matrix remodeling. At the same time, the expression of E3 ubiquitin ligases Smurf1 and Smurf2 is upregulated, but the expression of TGF-β1 and phosphorylation of SMAD2/3 are also increased. Increased expression of Smurf1/2 plays a role in limiting the increase in typical Smad signals, but not sufficient to eliminate the rise in profibrosis or matrix-remodeling genes.[107]
HERC subfamily and oxidative stress
Little is known about the HECR superfamily under oxidative stress. Only one literature mentions it. Engineered fullerenes (C60) is used in many clinical and industrial routes. For example, its lipophilicity is used for potential antibacterial activity, and various chemical modifications (−OH, −COOH, −NH2) are easily made, making it suitable as a pharmacological agent. This study reports that tris-C60 can inhibit apoptosis and cell proliferation. Studies have shown that apoptotic cells are mainly distributed in the S cell cycle, while cells that have stalled are mainly distributed in the G1/M and G2/M cell cycle. H2O2 pretreatment can induce G1 cell cycle arrest and apoptosis. Increased expression of p16, p21, and p53 proteins in cells exposed to tris-C60, which are related to cell aging.[108, 109] Despite evidence that ROS plays a role in triggering aging.[110, 111] no increase in intracellular ROS levels was observed in tris-C60–treated cells. In contrast, tris-C60 appears to reduce total ROS levels. At the same time, HERC5 gene and protein levels are significantly reduced.[108]. HERC5 belongs to the HECT E3 family of RCC-like domains. HERC5 may be regulated by p53 and Rb, which can interact with cyclins. However, to date, there is no evidence that HERC5 is involved in cellular aging.[112] The interconnection between HERC and oxidative stress needs further exploration.
Other HECT subfamily and oxidative stress
Kaposi's sarcoma (KS) remains one of the most common malignancies in patients with human immunodeficiency virus (HIV) infection.[113] KS-associated herpesvirus infection also induces ROS in endothelial cells to facilitate its entry and amplify the infection.[114] The E3 ubiquitination protein ligase HACE1 protein can act on Rac1 protein and degrade through ubiquitin–proteasome, and block the production of ROS by Rac1-dependent NADPH oxidase. HACE1 can also promote Nrf2 activity of endothelial cells and play an essential role in regulating KSHV-induced oxidative stress. ROS in HACE1-deficient mouse embryonic fibroblasts are dependent on glutamine uptake. Cells lacking HACE1 show uncontrolled ROS production, and cell death further increases after glutamine withdrawal. In KSHV-infected cells, knocking out HACE1 results in high RAC1 and NOx 1 activity, increased ROS, increased cell death, and decreased KSHV gene expression. HACE1 slows oxidative stress in KSHV infection mediated by Nrf2.[115]
It has been reported in the literature that the absence of HACE1 can cause abnormalities in the zebrafish heart, especially a circular or “inverted” circular defect in the heart. Knockout of Rac1 in myocardial cells of mice showed that cardiac NOx activity and cardiac hypertrophy in these animals were reduced, suggesting that Rac1 is also critical for cardiac hypertrophy.[116] Knockout of zebrafish HACE1 can lead to cardiac malformations, especially circulation defects, and increased Rac1 expression. Importantly, this phenotype appears to be directly related to NOx enzyme-dependent ROS production, as both genetic inhibition by NOx1 and NOx2 morpholinos or pharmacologic rescue using ROS scavenging agents restore normal cardiac structure.[117] There is a strong relationship between the loss of function of HACE1 and oxidative stress; especially, the ROS level in myocardial cells is significantly increased. An increase in ROS levels can promote the development of tumors.[118, 119] HACE1 expression can negatively regulate NOx-dependent ROS production (Figure 3).
Conflict of Interest
None declared.
REFERENCE
1 Thibaudeau TA, Smith DM. A Practical Review of Proteasome Pharmacology. Pharmacol Rev 2019; 71: 170–97.10.1124/pr.117.015370Search in Google Scholar PubMed PubMed Central
2 Soave CL, Guerin T, Liu J, Dou QP. Targeting the ubiquitin-proteasome system for cancer treatment: discovering novel inhibitors from nature and drug repurposing. Cancer Metastasis Rev 2017; 36:717–36.10.1007/s10555-017-9705-xSearch in Google Scholar PubMed PubMed Central
3 Xie F, Zhang Z, van Dam H, Zhang L, Zhou F. Regulation of TGF-beta Superfamily Signaling by SMAD Mono-Ubiquitination. Cells 2014; 3: 981–93.10.3390/cells3040981Search in Google Scholar PubMed PubMed Central
4 Finley D, Ulrich HD, Sommer T, Kaiser P. The ubiquitin-proteasome system of Saccharomyces cerevisiae. Genetics 2012; 192: 319–60.10.1534/genetics.112.140467Search in Google Scholar PubMed PubMed Central
5 Nandi D, Tahiliani P, Kumar A, Chandu D. The ubiquitin-proteasome system. J Biosci 2006; 31:137–55.10.1007/BF02705243Search in Google Scholar PubMed
6 Saeki Y. Ubiquitin recognition by the proteasome. J Biochem 2017; 161: 113–24.10.1093/jb/mvw091Search in Google Scholar PubMed
7 Swatek KN, Komander D. Ubiquitin modifications. Cell Res 2016; 26: 399–422.10.1038/cr.2016.39Search in Google Scholar PubMed PubMed Central
8 Akutsu M, Dikic I, Bremm A. Ubiquitin chain diversity at a glance. J Cell Sci 2016; 129: 875–80.10.1242/jcs.183954Search in Google Scholar PubMed
9 Michel MA, Swatek KN, Hospenthal MK, Komander D. Ubiquitin Linkage-Specific Affimers Reveal Insights into K6-Linked Ubiquitin Signaling. Mol Cell 2017; 68: 233–46 e235.10.1016/j.molcel.2017.08.020Search in Google Scholar PubMed PubMed Central
10 Hrdinka M, Gyrd-Hansen M. The Met1-Linked Ubiquitin Machinery: Emerging Themes of (De)regulation. Mol Cell 2017; 68: 265–80.10.1016/j.molcel.2017.09.001Search in Google Scholar PubMed
11 Fiil BK, Gyrd-Hansen M. Met1-linked ubiquitination in immune signalling. FEBS J 2014; 281: 4337–50.10.1111/febs.12944Search in Google Scholar PubMed PubMed Central
12 Wickliffe KE, Williamson A, Meyer HJ, Kelly A, Rape M. K11-linked ubiquitin chains as novel regulators of cell division. Trends Cell Biol 2011; 21: 656–63.10.1016/j.tcb.2011.08.008Search in Google Scholar PubMed PubMed Central
13 Gatti M, Pinato S, Maiolica A, Rocchio F, Prato MG, Aebersold R, et al. RNF168 promotes noncanonical K27 ubiquitination to signal DNA damage. Cell Rep 2015; 10: 226–38.10.1016/j.celrep.2014.12.021Search in Google Scholar PubMed
14 Fei C, Li Z, Li C, Chen Y, Chen Z, He X, et al. Smurf1-mediated Lys29-linked nonproteolytic polyubiquitination of axin negatively regulates Wnt/beta-catenin signaling. Mol Cell Biol 2013; 33: 4095–105.10.1128/MCB.00418-13Search in Google Scholar PubMed PubMed Central
15 Huang H, Jeon MS, Liao L, Yang C, Elly C, Yates JR, 3rd, et al. K33-linked polyubiquitination of T cell receptor-zeta regulates proteolysis-independent T cell signaling. Immunity 2010; 33: 60–70.10.1016/j.immuni.2010.07.002Search in Google Scholar PubMed PubMed Central
16 Ohtake F, Saeki Y, Ishido S, Kanno J, Tanaka K. The K48–K63 Branched Ubiquitin Chain Regulates NF-kappaB Signaling. Mol Cell 2016; 64: 251–66.10.1016/j.molcel.2016.09.014Search in Google Scholar PubMed
17 Ebner P, Versteeg GA, Ikeda F. Ubiquitin enzymes in the regulation of immune responses. Crit Rev Biochem Mol Biol 2017; 52: 425–60.10.1080/10409238.2017.1325829Search in Google Scholar PubMed PubMed Central
18 Sluimer J, Distel B. Regulating the human HECT E3 ligases. Cell Mol Life Sci 2018; 75: 3121–41.10.1007/s00018-018-2848-2Search in Google Scholar PubMed PubMed Central
19 Ye Y, Rape M. Building ubiquitin chains: E2 enzymes at work. Nat Rev Mol Cell Biol 2009; 10:755–64.10.1038/nrm2780Search in Google Scholar PubMed PubMed Central
20 Weber J, Polo S, Maspero E. HECT E3 Ligases: A Tale With Multiple Facets. Front Physiol 2019; 10: 370.10.3389/fphys.2019.00370Search in Google Scholar PubMed PubMed Central
21 Wilck N, Ludwig A. Targeting the ubiquitin-proteasome system in atherosclerosis: status quo, challenges, and perspectives. Antioxid Redox Signal 2014; 21: 2344–63.10.1089/ars.2013.5805Search in Google Scholar PubMed
22 Powell SR, Herrmann J, Lerman A, Patterson C, Wang X. The ubiquitinproteasome system and cardiovascular disease. Prog Mol Biol Transl Sci 2012; 109: 295–346.10.1016/B978-0-12-397863-9.00009-2Search in Google Scholar PubMed PubMed Central
23 Calise J, Powell SR. The ubiquitin proteasome system and myocardial ischemia. Am J Physiol Heart Circ Physiol 2013; 304: H337–49.10.1152/ajpheart.00604.2012Search in Google Scholar PubMed PubMed Central
24 Spanig S, Kellermann K, Dieterlen MT, Noack T, Lehmann S, Borger MA, et al. The Ubiquitin Proteasome System in Ischemic and Dilated Cardiomyopathy. Int J Mol Sci 2019; 20: 6354.10.3390/ijms20246354Search in Google Scholar PubMed PubMed Central
25 Yoshizawa T, Karim MF, Sato Y, Senokuchi T, Miyata K, Fukuda T, et al. SIRT7 controls hepatic lipid metabolism by regulating the ubiquitinproteasome pathway. Cell Metab 2014; 19: 712–21.10.1016/j.cmet.2014.03.006Search in Google Scholar PubMed
26 Tai H, Wang X, Zhou J, Han X, Fang T, Gong H, et al. Protein kinase Cbeta activates fat mass and obesity-associated protein by influencing its ubiquitin/proteasome degradation. FASEB J 2017; 31: 4396–406.10.1096/fj.201601159RRSearch in Google Scholar PubMed
27 Zhang GW, Cai HC, Shang XJ. [Ubiquitin-proteasome system and sperm DNA repair: An update]. Zhonghua Nan Ke Xue 2016; 22: 834–7.Search in Google Scholar
28 Caron P, Pankotai T, Wiegant WW, Tollenaere MAX, Furst A, Bonhomme C, et al. WWP2 ubiquitylates RNA polymerase II for DNA-PK-dependent transcription arrest and repair at DNA breaks. Genes Dev 2019; 33: 684–704.10.1101/gad.321943.118Search in Google Scholar PubMed PubMed Central
29 Budenholzer L, Cheng CL, Li Y, Hochstrasser M. Proteasome Structure and Assembly. J Mol Biol 2017; 429: 3500–24.10.1016/j.jmb.2017.05.027Search in Google Scholar PubMed PubMed Central
30 Kunjappu MJ, Hochstrasser M. Assembly of the 20S proteasome. Biochim Biophys Acta 2014; 1843:2–12.10.1016/j.bbamcr.2013.03.008Search in Google Scholar PubMed PubMed Central
31 Scheffner M, Nuber U, Huibregtse JM. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature 1995; 373: 81–3.10.1038/373081a0Search in Google Scholar PubMed
32 Bernassola F, Karin M, Ciechanover A, Melino G. The HECT family of E3 ubiquitin ligases: multiple players in cancer development. Cancer Cell 2008; 14: 10–21.10.1016/j.ccr.2008.06.001Search in Google Scholar
33 Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell 1994; 79: 13–21.10.1016/0092-8674(94)90396-4Search in Google Scholar
34 Verdecia MA, Joazeiro CA, Wells NJ, Ferrer JL, Bowman ME, Hunter T, et al. Conformational flexibility underlies ubiquitin ligation mediated by the WWP1 HECT domain E3 ligase. Mol Cell 2003; 11: 249–59.10.1016/S1097-2765(02)00774-8Search in Google Scholar
35 Ingham RJ, Gish G, Pawson T. The Nedd4 family of E3 ubiquitin ligases: functional diversity within a common modular architecture. Oncogene 2004; 23:1972–84.10.1038/sj.onc.1207436Search in Google Scholar PubMed
36 Garcia-Gonzalo FR, Rosa JL. The HERC proteins: functional and evolutionary insights. Cell Mol Life Sci 2005; 62: 1826–38.10.1007/s00018-005-5119-ySearch in Google Scholar PubMed
37 Rotin D, Kumar S. Physiological functions of the HECT family of ubiquitin ligases. Nat Rev Mol Cell Biol 2009; 10: 398–409.10.1038/nrm2690Search in Google Scholar PubMed
38 Senft D, Qi J, Ronai ZA. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat Rev Cancer 2018; 18: 69–88.10.1038/nrc.2017.105Search in Google Scholar PubMed PubMed Central
39 Boase NA, Kumar S. NEDD4: The founding member of a family of ubiquitin-protein ligases. Gene 2015; 557: 113–22.10.1016/j.gene.2014.12.020Search in Google Scholar PubMed PubMed Central
40 Dunn R, Klos DA, Adler AS, Hicke L. The C2 domain of the Rsp5 ubiquitin ligase binds membrane phosphoinositides and directs ubiquitination of endosomal cargo. J Cell Biol 2004; 165: 135–44.10.1083/jcb.200309026Search in Google Scholar PubMed PubMed Central
41 Scheffner M, Kumar S. Mammalian HECT ubiquitin-protein ligases: biological and pathophysiological aspects. Biochim Biophys Acta 2014; 1843: 61–74.10.1016/j.bbamcr.2013.03.024Search in Google Scholar PubMed
42 Hofmann K, Bucher P. The rsp5-domain is shared by proteins of diverse functions. FEBS Lett 1995; 358: 153–7.10.1016/0014-5793(94)01415-WSearch in Google Scholar
43 Kumar S, Harvey KF, Kinoshita M, Copeland NG, Noda M, Jenkins NA. cDNA cloning, expression analysis, and mapping of the mouse Nedd4 gene. Genomics 1997; 40: 435–43.10.1006/geno.1996.4582Search in Google Scholar PubMed
44 Cao Y, Zhang L. A Smurf1 tale: function and regulation of an ubiquitin ligase in multiple cellular networks. Cell Mol Life Sci 2013; 70: 2305–17.10.1007/s00018-012-1170-7Search in Google Scholar PubMed
45 Srivastava D, Chakrabarti O. Ubiquitin in regulation of spindle apparatus and its positioning: implications in development and disease. Biochem Cell Biol 2015; 93: 273–81.10.1139/bcb-2015-0011Search in Google Scholar PubMed
46 Kumari N, Jaynes PW, Saei A, Iyengar PV, Richard JLC, Eichhorn PJA. The roles of ubiquitin modifying enzymes in neoplastic disease. Biochim Biophys Acta Rev Cancer 2017; 1868: 456–83.10.1016/j.bbcan.2017.09.002Search in Google Scholar PubMed
47 Zhi X, Chen C. WWP1: a versatile ubiquitin E3 ligase in signaling and diseases. Cell Mol Life Sci 2012; 69: 1425–34.10.1007/s00018-011-0871-7Search in Google Scholar PubMed
48 Yosipovitch G, Rosen JD, Hashimoto T. Itch: From mechanism to (novel) therapeutic approaches. J Allergy Clin Immunol 2018; 142: 1375–90.10.1016/j.jaci.2018.09.005Search in Google Scholar PubMed
49 Mao X, Sethi G, Zhang Z, Wang Q. The Emerging Roles of the HERC Ubiquitin Ligases in Cancer. Curr Pharm Des 2018; 24:1676–81.10.2174/1381612824666180528081024Search in Google Scholar PubMed
50 Garcia-Cano J, Martinez-Martinez A, Sala-Gaston J, Pedrazza L, Rosa JL. HERCing: Structural and Functional Relevance of the Large HERC Ubiquitin Ligases. Front Physiol 2019; 10: 1014.10.3389/fphys.2019.01014Search in Google Scholar PubMed PubMed Central
51 Renault L, Nassar N, Vetter I, Becker J, Klebe C, Roth M, et al. The 1.7 A crystal structure of the regulator of chromosome condensation (RCC1) reveals a seven-bladed propeller. Nature 1998; 392: 97–101.10.1038/32204Search in Google Scholar PubMed
52 Lemak A, Yee A, Bezsonova I, Dhe-Paganon S, Arrowsmith CH. Zn-binding AZUL domain of human ubiquitin protein ligase Ube3A. J Biomol NMR 2011; 51: 185–90.10.1007/s10858-011-9552-ySearch in Google Scholar PubMed
53 Martinez-Zapien D, Ruiz FX, Poirson J, Mitschler A, Ramirez J, Forster A, et al. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature 2016; 529: 541–5.10.1038/nature16481Search in Google Scholar PubMed PubMed Central
54 Sailer C, Offensperger F, Julier A, Kammer KM, Walker-Gray R, Gold MG, et al. Structural dynamics of the E6AP/UBE3A-E6-p53 enzyme-substrate complex. Nat Commun 2018; 9: 4441.10.1038/s41467-018-06953-0Search in Google Scholar PubMed PubMed Central
55 Liu Z, Oughtred R, Wing SS. Characterization of E3Histone, a novel testis ubiquitin protein ligase which ubiquitinates histones. Mol Cell Biol 2005; 25: 2819–31.10.1128/MCB.25.7.2819-2831.2005Search in Google Scholar PubMed PubMed Central
56 Yang D, Cheng D, Tu Q, Yang H, Sun B, Yan L, et al. HUWE1 controls the development of non-small cell lung cancer through down-regulation of p53. Theranostics 2018; 8: 3517–29.10.7150/thno.24401Search in Google Scholar PubMed PubMed Central
57 Gong X, Du D, Deng Y, Zhou Y, Sun L, Yuan S. The structure and regulation of the E3 ubiquitin ligase HUWE1 and its biological functions in cancer. Invest New Drugs 2020; 38: 515–24.10.1007/s10637-020-00894-6Search in Google Scholar PubMed
58 Shearer RF, Iconomou M, Watts CK, Saunders DN. Functional Roles of the E3 Ubiquitin Ligase UBR5 in Cancer. Mol Cancer Res 2015; 13: 1523–32.10.1158/1541-7786.MCR-15-0383Search in Google Scholar PubMed
59 Sack MN, Fyhrquist FY, Saijonmaa OJ, Fuster V, Kovacic JC. Basic Biology of Oxidative Stress and the Cardiovascular System: Part 1 of a 3-Part Series. J Am Coll Cardiol 2017; 70: 196–211.10.1016/j.jacc.2017.05.034Search in Google Scholar PubMed PubMed Central
60 Neri M, Fineschi V, Di Paolo M, Pomara C, Riezzo I, Turillazzi E, et al. Cardiac oxidative stress and inflammatory cytokines response after myocardial infarction. Curr Vasc Pharmacol 2015; 13: 26–36.10.2174/15701611113119990003Search in Google Scholar PubMed
61 Kudryavtseva AV, Krasnov GS, Dmitriev AA, Alekseev BY, Kardymon OL, Sadritdinova AF, et al. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016; 7: 44879–905.10.18632/oncotarget.9821Search in Google Scholar PubMed PubMed Central
62 Berry BJ, Trewin AJ, Amitrano AM, Kim M, Wojtovich AP. Use the Protonmotive Force: Mitochondrial Uncoupling and Reactive Oxygen Species. J Mol Biol 2018; 430: 3873–91.10.1016/j.jmb.2018.03.025Search in Google Scholar PubMed PubMed Central
63 Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 2009; 417: 1–13.10.1042/BJ20081386Search in Google Scholar PubMed PubMed Central
64 Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol 2003; 552: 335–44.10.1113/jphysiol.2003.049478Search in Google Scholar PubMed PubMed Central
65 Cardoso AR, Chausse B, da Cunha FM, Luevano-Martinez LA, Marazzi TB, Pessoa PS, et al. Mitochondrial compartmentalization of redox processes. Free Radic Biol Med 2012; 52: 2201–8.10.1016/j.freeradbiomed.2012.03.008Search in Google Scholar PubMed
66 Brown DI, Griendling KK. Nox proteins in signal transduction. Free Radic Biol Med 2009; 47: 1239–53.10.1016/j.freeradbiomed.2009.07.023Search in Google Scholar PubMed PubMed Central
67 Stanic B, Pandey D, Fulton DJ, Miller FJ, Jr. Increased epidermal growth factor-like ligands are associated with elevated vascular nicotinamide adenine dinucleotide phosphate oxidase in a primate model of atherosclerosis. Arterioscler Thromb Vasc Biol 2012; 32: 2452–60.10.1161/ATVBAHA.112.256107Search in Google Scholar PubMed PubMed Central
68 Zhang M, Brewer AC, Schroder K, Santos CX, Grieve DJ, Wang M, et al. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci U S A 2010; 107: 18121–6.10.1073/pnas.1009700107Search in Google Scholar PubMed PubMed Central
69 Craige SM, Kant S, Reif M, Chen K, Pei Y, Angoff R, et al. Endothelial NADPH oxidase 4 protects ApoE−/− mice from atherosclerotic lesions. Free Radic Biol Med 2015; 89: 1–7.10.1016/j.freeradbiomed.2015.07.004Search in Google Scholar PubMed PubMed Central
70 Langbein H, Brunssen C, Hofmann A, Cimalla P, Brux M, Bornstein SR, et al. NADPH oxidase 4 protects against development of endothelial dysfunction and atherosclerosis in LDL receptor deficient mice. Eur Heart J 2016; 37: 1753–61.10.1093/eurheartj/ehv564Search in Google Scholar PubMed PubMed Central
71 Mohazzab KM, Kaminski PM, Wolin MS. NADH oxidoreductase is a major source of superoxide anion in bovine coronary artery endothelium. Am J Physiol 1994; 266: H2568–72.10.1152/ajpheart.1994.266.6.H2568Search in Google Scholar PubMed
72 Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res 1994; 74: 1141–8.10.1161/01.RES.74.6.1141Search in Google Scholar PubMed
73 Nair N, Gongora E. Oxidative Stress and Cardiovascular Aging: Interaction Between NRF-2 and ADMA. Curr Cardiol Rev 2017; 13: 183–8.10.2174/1573403X13666170216150955Search in Google Scholar PubMed PubMed Central
74 Forstermann U. Nitric oxide and oxidative stress in vascular disease. Pflugers Arch 2010; 459: 923–39.10.1007/s00424-010-0808-2Search in Google Scholar PubMed
75 Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov 2011; 10: 453–71.10.1038/nrd3403Search in Google Scholar PubMed PubMed Central
76 Kurz T, Eaton JW, Brunk UT. Redox activity within the lysosomal compartment: implications for aging and apoptosis. Antioxid Redox Signal 2010; 13: 511–23.10.1089/ars.2009.3005Search in Google Scholar PubMed
77 Cai C, Ching A, Lagace C, Linsenmayer T. Nuclear ferritin-mediated protection of corneal epithelial cells from oxidative damage to DNA. Dev Dyn 2008; 237: 2676–83.10.1002/dvdy.21494Search in Google Scholar PubMed
78 Taniguchi T, Kurita A, Kobayashi K, Yamamoto K, Inaba H. Dose- and time-related effects of dexmedetomidine on mortality and inflammatory responses to endotoxin-induced shock in rats. J Anesth 2008; 22: 221–8.10.1007/s00540-008-0611-9Search in Google Scholar PubMed
79 Qiao H, Sanders RD, Ma D, Wu X, Maze M. Sedation improves early outcome in severely septic Sprague Dawley rats. Crit Care 2009; 13: R136.10.1186/cc8012Search in Google Scholar PubMed PubMed Central
80 Meng L, Li L, Lu S, Li K, Su Z, Wang Y, et al. The protective effect of dexmedetomidine on LPS-induced acute lung injury through the HMGB1-mediated TLR4/NF-kappaB and PI3K/Akt/mTOR pathways. Mol Immunol 2018; 94: 7–17.10.1016/j.molimm.2017.12.008Search in Google Scholar PubMed
81 Bohm B, Hess S, Krause K, Schirner A, Ewald W, Aigner T, et al. ADAM15 exerts an antiapoptotic effect on osteoarthritic chondrocytes via up-regulation of the X-linked inhibitor of apoptosis. Arthritis Rheum 2010; 62: 1372–82.10.1002/art.27387Search in Google Scholar PubMed
82 Cheng X, Zhang L, Zhang K, Zhang G, Hu Y, Sun X, et al. Circular RNA VMA21 protects against intervertebral disc degeneration through targeting miR-200c and X linked inhibitor-of-apoptosis protein. Ann Rheum Dis 2018; 77: 770–9.10.1136/annrheumdis-2017-212056Search in Google Scholar PubMed PubMed Central
83 Sies H, Berndt C, Jones DP. Oxidative Stress. Annu Rev Biochem 2017; 86: 715–48.10.1146/annurev-biochem-061516-045037Search in Google Scholar PubMed
84 Lu M, Lin SC, Huang Y, Kang YJ, Rich R, Lo YC, et al. XIAP induces NF-kappaB activation via the BIR1/TAB1 interaction and BIR1 dimerization. Mol Cell 2007; 26: 689–702.10.1016/j.molcel.2007.05.006Search in Google Scholar PubMed PubMed Central
85 Lewis J, Burstein E, Reffey SB, Bratton SB, Roberts AB, Duckett CS. Uncoupling of the signaling and caspase-inhibitory properties of X-linked inhibitor of apoptosis. J Biol Chem 2004; 279: 9023–9.10.1074/jbc.M312891200Search in Google Scholar PubMed
86 Zhou L, Zhou J, Sheng B, Li X, Yuan Y. Dexmedetomidine exerts dual effects on human annulus fibrosus chondrocytes depending on the oxidative stress status. Biosci Rep 2019; 39: BSR20190419.10.1042/BSR20190419Search in Google Scholar PubMed PubMed Central
87 Taha Z, Janse van Rensburg HJ, Yang X. The Hippo Pathway: Immunity and Cancer. Cancers (Basel) 2018; 10: 94.10.3390/cancers10040094Search in Google Scholar PubMed PubMed Central
88 Bae SJ, Kim M, Kim SH, Kwon YE, Lee JH, Kim J, et al. NEDD4 controls intestinal stem cell homeostasis by regulating the Hippo signalling pathway. Nat Commun 2015; 6: 6314.10.1038/ncomms7314Search in Google Scholar PubMed
89 Meng Z, Moroishi T, Guan KL. Mechanisms of Hippo pathway regulation. Genes Dev 2016; 30: 1–17.10.1101/gad.274027.115Search in Google Scholar PubMed PubMed Central
90 Henning RJ, Bourgeois M, Harbison RD. Poly(ADP-ribose) Polymerase (PARP) and PARP Inhibitors: Mechanisms of Action and Role in Cardiovascular Disorders. Cardiovasc Toxicol 2018; 18: 493–506.10.1007/s12012-018-9462-2Search in Google Scholar PubMed
91 Harvey KF, Harvey NL, Michael JM, Parasivam G, Waterhouse N, Alnemri ES, et al. Caspase-mediated cleavage of the ubiquitin-protein ligase Nedd4 during apoptosis. J Biol Chem 1998; 273: 13524–30.10.1074/jbc.273.22.13524Search in Google Scholar PubMed
92 Jeon SA, Kim DW, Cho JY. Neural precursor cell-expressed, developmentally down-regulated 4 (NEDD4) regulates hydrogen peroxide-induced cell proliferation and death through inhibition of Hippo signaling. FASEB J 2019; 33: 14772–83.10.1096/fj.201901404RSearch in Google Scholar PubMed
93 Zhang X, Zhang JH, Chen XY, Hu QH, Wang MX, Jin R, et al. Reactive oxygen species-induced TXNIP drives fructose-mediated hepatic inflammation and lipid accumulation through NLRP3 inflammasome activation. Antioxid Redox Signal 2015; 22: 848–70.10.1089/ars.2014.5868Search in Google Scholar PubMed PubMed Central
94 Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011; 469: 221–5.10.1038/nature09663Search in Google Scholar PubMed
95 Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol 2010; 11: 136–40.10.1038/ni.1831Search in Google Scholar PubMed
96 Otaki Y, Takahashi H, Watanabe T, Funayama A, Netsu S, Honda Y, et al. HECT-Type Ubiquitin E3 Ligase ITCH Interacts With Thioredoxin-Interacting Protein and Ameliorates Reactive Oxygen Species-Induced Cardiotoxicity. J Am Heart Assoc 2016; 5: e002485.10.1161/JAHA.115.002485Search in Google Scholar PubMed PubMed Central
97 Laine A, Ronai Z. Regulation of p53 localization and transcription by the HECT domain E3 ligase WWP1. Oncogene 2007; 26: 1477–83.10.1038/sj.onc.1209924Search in Google Scholar PubMed PubMed Central
98 Cheng Q, Cao X, Yuan F, Li G, Tong T. Knockdown of WWP1 inhibits growth and induces apoptosis in hepatoma carcinoma cells through the activation of caspase3 and p53. Biochem Biophys Res Commun 2014; 448: 248–54.10.1016/j.bbrc.2014.04.117Search in Google Scholar PubMed
99 Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest 2009; 119: 573–81.10.1172/JCI37048Search in Google Scholar PubMed PubMed Central
100 Gimbrone MA, Jr., Garcia-Cardena G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ Res 2016; 118: 620–36.10.1161/CIRCRESAHA.115.306301Search in Google Scholar PubMed PubMed Central
101 Konukoglu D, Uzun H. Endothelial Dysfunction and Hypertension. Adv Exp Med Biol 2017; 956: 511–40.10.1007/5584_2016_90Search in Google Scholar PubMed
102 Choi BH, Che X, Chen C, Lu L, Dai W. WWP2 is required for normal cell cycle progression. Genes Cancer 2015; 6: 371–7.10.18632/genesandcancer.83Search in Google Scholar PubMed PubMed Central
103 Clements AE, Bravo V, Koivisto C, Cohn DE, Leone G. WWP2 and its association with PTEN in endometrial cancer. Gynecol Oncol Rep 2015; 13: 26–9.10.1016/j.gore.2015.05.004Search in Google Scholar PubMed PubMed Central
104 Sun X, Yang Y, Zhu D, Qian H, Duan Y, He X, et al. Expression of Septin4 in human hepatic stellate cells LX-2 stimulated by LPS. Inflammation 2013; 36: 539–48.10.1007/s10753-012-9575-xSearch in Google Scholar PubMed
105 Zhang N, Zhang Y, Wu B, You S, Sun Y. Role of WW domain E3 ubiquitin protein ligase 2 in modulating ubiquitination and Degradation of Septin4 in oxidative stress endothelial injury. Redox Biol 2020; 30: 101419.10.1016/j.redox.2019.101419Search in Google Scholar PubMed PubMed Central
106 Crosas-Molist E, Bertran E, Fabregat I. Cross-Talk Between TGF-beta and NADPH Oxidases During Liver Fibrosis and Hepatocarcinogenesis. Curr Pharm Des 2015; 21: 5964–76.10.2174/1381612821666151029112126Search in Google Scholar PubMed
107 Hagler MA, Hadley TM, Zhang H, Mehra K, Roos CM, Schaff HV, et al. TGF-beta signalling and reactive oxygen species drive fibrosis and matrix remodelling in myxomatous mitral valves. Cardiovasc Res 2013; 99: 175–84.10.1093/cvr/cvt083Search in Google Scholar PubMed PubMed Central
108 He S, Sharpless NE. Senescence in Health and Disease. Cell 2017; 169: 1000–11.10.1016/j.cell.2017.05.015Search in Google Scholar PubMed PubMed Central
109 Regulski MJ. Cellular Senescence: What, Why, and How. Wounds 2017; 29: 168–74.Search in Google Scholar
110 Baranov VS, Baranova EV. Aging and Ambiguous ROS. System Genetics Analysis. Curr Aging Sci 2017; 10: 6–11.10.2174/1874609809666160921114504Search in Google Scholar PubMed
111 Davalli P, Mitic T, Caporali A, Lauriola A, D’Arca D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxid Med Cell Longev 2016; 2016: 3565127.10.1155/2016/3565127Search in Google Scholar PubMed PubMed Central
112 Gao J, Wang HL, Shreve A, Iyer R. Fullerene derivatives induce premature senescence: a new toxicity paradigm or novel biomedical applications. Toxicol Appl Pharmacol 2010; 244: 130–43.10.1016/j.taap.2009.12.025Search in Google Scholar PubMed
113 Hoffmann C, Sabranski M, Esser S. HIV-Associated Kaposi's Sarcoma. Oncol Res Treat 2017; 40: 94–8.10.1159/000455971Search in Google Scholar PubMed
114 Mesri EA, Cavallin LE, Ashlock BM, Leung HJ, Ma Q, Goldschmidt-Clermont PJ. Molecular studies and therapeutic targeting of Kaposi's sarcoma herpesvirus (KSHV/HHV-8) oncogenesis. Immunol Res 2013; 57: 159–65.10.1007/s12026-013-8458-zSearch in Google Scholar PubMed
115 Kumar B, Roy A, Asha K, Sharma-Walia N, Ansari MA, Chandran B. HACE1, an E3 Ubiquitin Protein Ligase, Mitigates Kaposi's Sarcoma-Associated Herpesvirus Infection-Induced Oxidative Stress by Promoting Nrf2 Activity. J Virol 2019; 93: e01812–18.10.1128/JVI.01812-18Search in Google Scholar PubMed PubMed Central
116 Leung C, Lu X, Liu M, Feng Q. Rac1 signaling is critical to cardiomyocyte polarity and embryonic heart development. J Am Heart Assoc 2014; 3: e001271.10.1161/JAHA.114.001271Search in Google Scholar PubMed PubMed Central
117 Razaghi B, Steele SL, Prykhozhij SV, Stoyek MR, Hill JA, Cooper MD, et al. hace1 Influences zebrafish cardiac development via ROS-dependent mechanisms. Dev Dyn 2018; 247: 289–303.10.1002/dvdy.24600Search in Google Scholar PubMed
118 Prasad S, Gupta SC, Tyagi AK. Reactive oxygen species (ROS) and cancer: Role of antioxidative nutraceuticals. Cancer Lett 2017; 387: 95–105.10.1016/j.canlet.2016.03.042Search in Google Scholar PubMed
119 Yang Y, Karakhanova S, Hartwig W, D’Haese JG, Philippov PP, Werner J, et al. Mitochondria and Mitochondrial ROS in Cancer: Novel Targets for Anticancer Therapy. J Cell Physiol 2016; 231: 2570–81.10.1002/jcp.25349Search in Google Scholar PubMed
© 2020 Hao Qian et al., published by Sciendo
This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 License.
Articles in the same Issue
- Editorial
- Should the high-flow nasal oxygen therapy be used or avoided in COVID-19?
- Highlight
- COVID-19 and tuberculosis
- The application of corticosteroids in COVID-19: A two-edged sword
- Review Article
- Structure and function of HECT E3 ubiquitin ligases and their role in oxidative stress
- The role of mitochondria in vascular calcification
- Original Article
- Detection of early cytokine storm in patients with septic shock after abdominal surgery
- Metabolic syndrome and atherogenic indices in rheumatoid arthritis and their relationship with disease activity: A hospital-based study from northeast India
- Autoimmune hepatitis: Clinical characteristics and predictors of biochemical response to treatment
- Case Report
- Enlightenments of asymptomatic cases of SARS-CoV-2 infection
Articles in the same Issue
- Editorial
- Should the high-flow nasal oxygen therapy be used or avoided in COVID-19?
- Highlight
- COVID-19 and tuberculosis
- The application of corticosteroids in COVID-19: A two-edged sword
- Review Article
- Structure and function of HECT E3 ubiquitin ligases and their role in oxidative stress
- The role of mitochondria in vascular calcification
- Original Article
- Detection of early cytokine storm in patients with septic shock after abdominal surgery
- Metabolic syndrome and atherogenic indices in rheumatoid arthritis and their relationship with disease activity: A hospital-based study from northeast India
- Autoimmune hepatitis: Clinical characteristics and predictors of biochemical response to treatment
- Case Report
- Enlightenments of asymptomatic cases of SARS-CoV-2 infection