Single-crystal analysis of La-doped pyromorphite [Pb5(PO4)3Cl]
-
Julia Sordyl
 ,
John Rakovan
,
John Rakovan

Abstract
Rare earth elements (REE) in calcium apatite have been widely described in the literature. Based on the investigations of minerals and their synthetic analogs, the mechanism of substitution of REE3+ for Ca2+ and their structural positions are well established. Although the presence of REE in natural pyromorphite has been reported, the structural response of substitution of REE3+ for Pb2+ is not established. A better understanding of REE-rich Pb-apatite may facilitate the potential use of this mineral in industrial processes. Two La-doped pyromorphite analogs [Pb5(PO4)3Cl] and two control pyromorphite analogs (with the absence of La) were synthesized from aqueous solutions at 25 °C. Na+ and K+ were used as charge-compensating ions to facilitate the incorporation of trivalent REE cations (La3+ + Na+ ↔ 2Pb2+ and La3+ + K+ ↔ 2Pb2+). Microprobe analysis, scanning electron microscopy, and Raman spectroscopy were used to confirm the purity of obtained phases. High-precision crystal structure refinements (R1 = 0.0140–0.0225) of all four compounds were performed from single-crystal X-ray diffraction data. The La content varied from 0.12(1) to 0.19(1) atoms per formula unit with the counter ions of K+ and Na+, respectively. Both substituting ions were accommodated at the Pb1 site only. By comparing the La-doped pyromorphite analogs with their control samples, it was possible to detect small changes in bond distances and polyhedral volumes caused by the La substitution. Variations in individual and mean interatomic distances reflected the cumulative effect of both the amount of substitution and ionic radii of substituting ions (La3+, Na+, and K+).
Introduction
Pyromorphite [Pb5(PO4)3Cl] belongs to the apatite supergroup of minerals and occurs in the oxidation zone of polymetallic deposits (Pasero et al. 2010). The pyromorphite structure was discussed in detail in Dai and Hughes (1989) and Okudera (2013). In the unit cell, Pb occupies four Pb1 sites (bonding to nine O atoms: 3 × O1, 3 × O2, and 3 × O3) and six Pb2 sites (bonding to six O atoms [O1, O2, and 4 × O3] and two Cl atoms located on the hexad at 0,0,0 and 0,0,1/2). Two distinct Pb polyhedrons are linked through O atoms shared with phosphate tetrahedra and are hexagonally distributed about a central [001] anion column.
As Pb-phosphates are highly stable under environmental conditions found in the critical zone of the Earth (Nriagu 1974), pyromorphite is a natural weathering product in Pb-contaminated soils and a product of in situ immobilization by precipitation induced by phosphate amendments (Laperche et al. 1997; Ma et al. 1993, 1995; Manecki et al. 2000, 2020; Tang et al. 2013; Manecki 2019). The presence of rare earth elements (REE) has been reported in natural pyromorphite (Markl et al. 2014). However, the mechanisms of incorporation of REE, their structural position in Pb-apatite, and charge compensation mechanisms are poorly understood, as compared to widely described calcium apatite (Borisov and Klevcova 1963; Mackie and Young 1973; Hughes et al. 1991; Fleet and Pan 1995, 1997a, 1997b; Rakovan and Reeder 1996; Fleet et al. 2000). This is because the REE content in Ca-apatite plays a role in petrological and genetic interpretations of mineral deposit formation and petrogenesis (Papike et al. 1984; Sha and Chappell 1999; Belousova et al. 2002; Harlov 2015; Bouzari et al. 2016; Jonsson et al. 2016; O’Sullivan et al. 2018; Andersson at al. 2019; Zhang et al. 2021). Nevertheless, the presence of REE in Pb-apatite is of interest because of its potential use in industrial processes.
Laboratory experiments for the synthesis of Pb-apatite analogs containing REE were described by Newby (1981) in an unpublished Ph.D. dissertation, which demonstrated differences in the amount of incorporated REE under different pH conditions. Owing to the similarity in their structures (White et al. 2005), the substitution mechanisms of REE in Ca- and Pb-apatites are likely to exhibit similarities. However, given the larger unit-cell size and the different preference of Pb over Ca in occupying the cationic position, as well as the presence of a free electron pair on the Pb2+ ion (Kim et al. 2000; Baikie et al. 2014), the mechanism and magnitude of substitution may be different.
Exchange of REE3+ for Ca2+ requires a coupled substitution. The two most common mechanisms of REE substitution in Ca, P-bearing apatite supergroup members are (Rønsbo 1989; Pan and Fleet 2002):
In natural Ca-apatites, Me+ is most frequently Na, and
In this study, pyromorphite Pb5(PO4)3Cl was chosen as the model Pb-apatite, and La was chosen as the model REE, as its chemical and physical properties well represent the entire group of light rare earth elements. Moreover, it is easily available in high-purity reagent form for synthesis [La(NO3)3·6H2O]. Na+ and K+ were used as model charge-compensating ions. It was hypothesized that the substitution occurs through the following reactions:
The research methodology involved the synthesis of pyromorphite crystals from aqueous solutions, which is most relevant to natural environmental conditions and to natural REE-enriched pyromorphite found in the oxidation zones of deposits. Therefore, in the present study, we report a single-crystal X-ray diffraction study of four synthetic Pb5(PO4)3Cl pyromorphite analogs containing La and Na or La and K substitutions and control samples synthesized from aqueous solutions under the same conditions in the absence of La. The synthesized samples were characterized using scanning electron microscopy (SEM), microprobe analysis, Raman spectroscopy, and single-crystal X-ray diffraction. We aimed to provide an explanation of the site occupancy and substitution mechanism, to determine the effect of Na+ and K+ substitution on the magnitude of La3+ substitution, and to capture similarities and differences with the better-studied Ca-apatite. Our findings contribute to a better understanding of light REE substitution in Pb-apatite, which is particularly important because REE are considered critical metals and REE-rich Pb-apatite may prove to be useful in future industrial applications.
Experimental procedure
Four pyromorphite analogs were synthesized via precipitation from aqueous solution at room temperature, namely two La-doped samples: LaxNaxPb5–2x(PO4)3Cl (La-Na-Pym), LaxKxPb5–2x(PO4)3Cl (La-K-Pym), and two control samples without La: Na2xPb5–x(PO4)3Cl (Na-Pym) and K2xPb5–x(PO4)3Cl (K-Pym). The starting mixtures of Pb(NO3)2 with La(NO3)3·6H2O in a molar proportion of 4:1 (or without La(NO3)3·6H2O for control samples) were dissolved in double-distilled water and added slowly (10 mL/h) by dripping through a glass funnel into the still solution column of dissolved
Quantitative chemical analyses were performed via electron microprobe (EMP) analysis using a JEOL SuperProbe JXA-8230 located at the Laboratory of Critical Elements at the Faculty of Geology, Geophysics and Environmental Protection, AGH UST Krakow. The EMP operated at an accelerating voltage of 15 kV, a probe current of 15 nA, a peak count time of 20 s, and background count time of 10 s, with a beam diameter of 1–5 μm, depending on individual crystal sizes. Standards [analytical lines and wavelength-dispersive spectrometry (WDS) diffracting crystals] included fluorite for F (Kα, LDE1), fluorapatite for P (Kα, PET), albite for Na (Kα, TAP) and Si (Kα, TAP), sanidine for K (Kα, PET), crocoite for Pb (Mα, PET) tugtupite for Cl (Kα, PET), and synthetic LaPO4 phosphate for La (Lα, LiF) (Jarosewich and Boatner 1991). Durango apatite was used as secondary reference material for monitoring the Cl analysis. Fluorapatite was not used as the F reference material because of time-dependent orientation effects of fluorapatite on the X-ray emission. During EMP measurement of the NaKα, KKα, and ClKα lines of all synthetic Pb-apatite crystals, no time-dependent intensity loss effects were recorded. For sample preparation, a dry precipitate was mixed with epoxy resin, placed in a standard ring, and polished.
A Thermo Scientific DXR Raman confocal microscope was used to collect Raman spectra of samples, which were excited with a green laser (532 nm, with a power maintained at 10 mW and a slit aperture of 50 μm) in the range of 100–3580 cm–1. Deconvolution of spectra was done using OMNIC for Dispersive Raman software (Thermo Fisher Scientific) in the ranges of 350–600 and 800–1200 cm–1 using the Gaussian/Lorentzian function, high-sensitivity factor, and a constant baseline.
Single-crystal X-ray diffraction measurements were performed on a Bruker APEXII Quazar CCD X-ray diffractometer with MoKα, λ = 0.71073 Å, for two control samples (Na-Pym and K-Pym) at 273 K. A Rigaku XtaLAB Synergy-S diffractometer equipped with a full 4-circle kappa goniometer, a Hypix6000E detector (hybrid), and MoKα X-ray radiation from a microfocus sealed tube was used for collection of the X-ray diffraction data at 180 K for La-doped samples (La-Na-Pym and La-K-Pym). One sample (La-K-Pym) was analyzed at both temperatures, 273(2) and 180(2) K, showing that final parameters did not change significantly due to cooling (Online Materials[1] Appendix A). The results of the measurements at these two temperatures are the same within the experimental error. The reason for the absence of thermal expansion in this temperature range is unknown. It is possible, though, that in a larger range of temperatures such a tendency would be observed. According to previous studies for Pb-apatite, pure pyromorphite shows thermal expansion in the temperature range 298–1373K (Hovis et al. 2015; Knyazev et al. 2015; Gu et al. 2020). However, there is no similar data for REE-substituted pyromorphite. The La-Na-Pym sample was measured only at 180 K. A data collection strategy was calculated with the CrysAlisPro software. The crystal structures were refined using the Bruker SHELXTL v.6.14 package of programs (Sheldrick 2015).
Results and discussion
Morphology of the crystals
La-doped samples yielded elongated needle-like crystals with hexagonal cross sections in the size range of 100–500 μm (Figs. 1a and 1b), whereas those containing no La yielded two generations of crystals: needle-like crystals elongated parallel to the [001] of 200 μm and hexagonal rods of <10 μm (Figs. 1c and 1d). EDS and microprobe analyses confirmed the intended chemical compositions for both types of crystals habit.

Scanning electron micrographs (BSE) of synthesized analogs: (a) La-Na-Pym; (b) La-K-Pym; (c) control Na-Pym; and (d) control K-Pym.
Chemistry
Chemical compositions of each phase were analyzed using 13–21 individual spots. For each sample, two results with totals furthest from 100% were discarded. Therefore, the calculation of the chemical formulas was based on the average of the 11–18 superior spot analyses for each sample. The microprobe analysis results (Table 1) showed that the average La2O3 concentrations were 1.19 wt% (σ = 0.68) and 0.63 wt% (σ = 0.28) for La-Na-Pym and La-K-Pym, respectively. The relatively high standard deviations may be attributed to heterogeneity among different crystals.
Wavelength-dispersive analyses for all analyzed samples (see Online Materials[1] Appendix B for all WDS data)
| La-Na-Pym [wt%] | Na-Pym [wt%] | La-K-Pym [wt%] | K-Pym [wt%] | |
|---|---|---|---|---|
| P2O5 | 16.03(24) | 15.63(37) | 15.53(27) | 15.41(41) |
| PbO | 79.42(90) | 81.27(71) | 79.20(58) | 81.09(51) |
| Na2O | 0.04(4) | 0.02(4) | b.d. | b.d. |
| K2O | b.d. | b.d. | 0.05(9) | 0.03(3) |
| La2O3 | 1.19(68) | b.d. | 0.63(28) | b.d. |
| Cl | 2.34(19) | 2.61(20) | 2.97(24) | 2.96(24) |
| O=Cl | –0.53 | –0.59 | –0.67 | –0.67 |
| Total | 98.49 | 98.94 | 97.69 | 98.82 |
| apfu based upon 13 anions | ||||
| P | 3.03(1) | 3.01(2) | 3.01(2) | 2.99(3) |
| Pb | 4.77(9) | 4.97(6) | 4.88(7) | 5.01(8) |
| Na | 0.02(2) | 0.01(2) | b.d. | b.d. |
| K | b.d. | b.d. | 0.01(3) | 0.01(1) |
| La | 0.10(6) | b.d. | 0.05(2) | b.d. |
| Cl | 0.89(8) | 1.01(9) | 1.15(10) | 1.15(10) |
| Empirical formula | Pb4.77(9)La0.10(6)Na0.02(2)(PO4)3.03(1)Cl0.89(8) | Pb4.97(6)Na0.01(2)(PO4)3.01(2)Cl1.01(9) | Pb4.88(7)La0.05(2)K0.01(3)(PO4)3.01(2)Cl1.15(10) | Pb5.01(8)K0.01(1)(PO4)2.99(3)Cl1.15(10) |
Note: b.d. = below detection.
Owing to the difficulties in the preparation of small crystals, there were several problems in the quantitative analysis of the chemical composition, mainly in the determination of the Cl content in small or porous crystals (due to the crystal habit). The epoxy infiltrated porosity, causing an artificial Cl content within the samples. Crystal structure refinement, as discussed below, confirmed no vacancies at the column anion X-site. Furthermore, no other monovalent anions were available in the system, and Raman spectroscopy did not reveal the presence of OH in the samples (Fig. 2). This indicates that the X-site is fully occupied by the Cl–. Therefore, we interpret EMP data that yield Cl concentrations different from 1 apfu suspect.
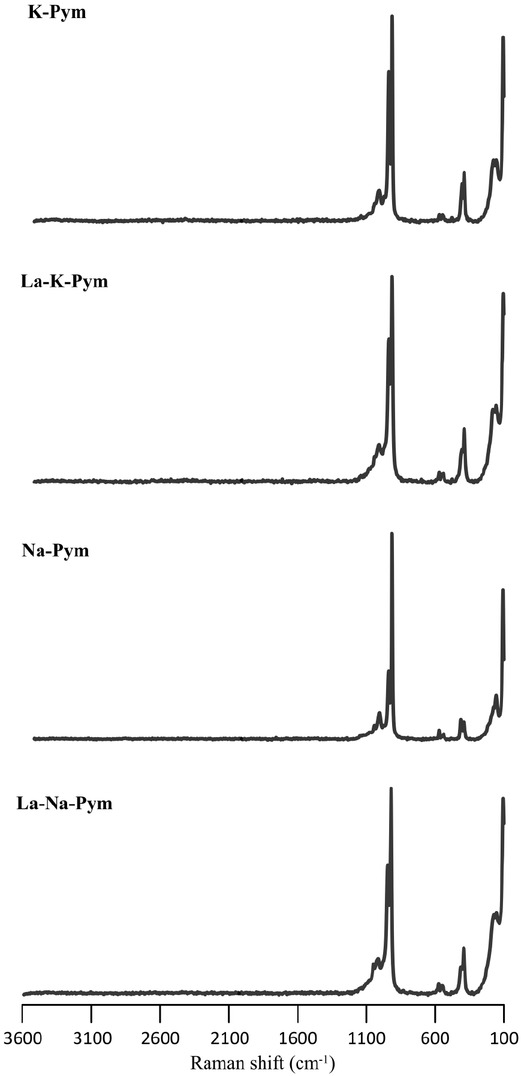
Raman spectra of synthesized analogs of La-doped pyromorphites and control samples in the 100–3600 cm–1 region.
Quantitative analysis of Na2O and K2O in Pb-rich apatite (the average amount of PbO in all samples is 80.2 wt%) was also challenging due to the low contents of these in the pyromorphites and the molar mass differences between the Pb and Na or K ions. The inconsistency of Na and K contents between WDS and structure refinement likely results from the detection limit of the electron microprobe analysis under the conditions applied. Therefore, all average empirical formulas based on electron microprobe analysis exhibit slight cation deficiency in charge balance.
Purity confirmation by Raman spectroscopy
The aim of this study was the structural analysis of Ladoped pyromorphite free from impurities such as carbonate or hydroxyl ions. Therefore, to verify the absence of potential impurities of

Raman spectra of synthesized analogs of La-doped pyromorphites and control samples in the 100–1200 cm–1 region with deconvoluted phosphate bands at the 300–600 and 800–1100 cm–1 regions.
The carbonate ion can be incorporated into an apatite structure in three positions: as a substitution for the anion in the hexagonal channel (two type-A substitutions) and for
In the pyromorphite spectra, the most intense bands at 919–947 cm–1 along with the bands in the 974–1048 cm–1 region are attributed to the stretching vibrations of the P–O bond (ν1 and ν3) (Levitt and Condrate 1970; Bartholomäi and Klee 1978; Botto et al. 1997; Frost and Palmer 2007; Bajda et al. 2011). A very weak band was apparent in all deconvoluted spectra at 1091 or 1092 cm–1, which may simply be a part of the background or the result of degeneration of the Pb-apatite structure (Kwaśniak-Kominek et al. 2017). The bands in the 541–586 cm–1 region correspond to the ν4 scissor vibrations of the O–P–O angle and may be resolved into three bands for control pyromorphites, and into four bands for La-doped pyromorphites. Splitting was also observed for the ν2 scissor vibrations of the O–P–O angle in the 392–439 cm–1 region. These, however, were not systematic, as the bands for control samples were split into three bands and those for La-doped pyromorphites into two (La-Na-Pym) or four (La-K-Pym). Such splitting is attributed to the reduction of the symmetry of the ideal (PO4)3– tetrahedron (Adler 1964). A broad profile from 100 to 250 cm–1 was attributed to lattice vibrations. However, the presence or absence of the strongest band at 107 cm–1 is strongly related to the crystal orientation. Therefore, the lattice vibrations are of little use for phase identification. All band positions resolved in the spectra collected for pyromorphite crystals are consistent with the previously described Raman spectra for pyromorphite (Levitt and Condrate 1970; Bartholomäi and Klee 1978; Botto et al. 1997; Frost and Palmer 2007; Bajda et al. 2011). Owing to the small amount of La substituting for Pb, no significant shifts were observed in the positions of the bands for doped samples. However, the intensities of the different bands varied. This is influenced by the crystal orientation relative to the incident beam (Frost and Palmer 2007).
Structure
Refinement of the structure. The crystallographic characteristics and conditions for data collection are shown in Table 2. The crystallographic information files (CIF) can be found in the Online Materials[1] Appendix C. Crystal structures were solved and refined in space group P63/m. No reflections characteristic of a monoclinic superstructure were noted (Mackie et al. 1972; Elliott et al. 1973; Bauer and Klee 1993). All structures were refined to R1 = 0.0140–0.0225. The positional parameters, equivalent isotropic displacement parameters, and site occupancies can be found in Online Materials[1] Appendix D.
Experimental details and crystallographic characteristics of the analyzed samples
| La-Na-Pym | Na-Pym | La-K-Pym | K-Pym | |
|---|---|---|---|---|
| Diffractometer | Rigaku Oxford Diffraction | Bruker AXS | Rigaku Oxford Diffraction | Bruker AXS |
| X-ray radiation | MoKα (λ = 0.71073 Å) | |||
| Temperature (K) | 180(2) | 273(2) | 180(2) | 273(2) |
| Space group | P63/m | |||
| Unit-cell parameters | ||||
| a (Å) | 9.98367(9) | 9.996(2) | 10.00129(8) | 9.9944(4) |
| c (Å) | 7.30631(10) | 7.319(2) | 7.29865(8) | 7.3142(4) |
| V (Å3) | 630.679(14) | 633.3(3) | 632.244(12) | 632.72(6) |
| Z | 2 | 2 | 2 | 2 |
| Absorption coefficient (mm–1) | 66.522 | 66.225 | 64.024 | 66.303 |
| F(000) | 1035 | 1129 | 1113 | 1130 |
| θ range | 2.36 to 29.03° | 2.35 to 29.01° | 2.35 to 33.78° | 2.35 to 28.95° |
| Index ranges | –13 < = h < = 13, | –13 < = h < = 13 | –15 < = h < = 15, | –13 < = h < = 13, |
| –13 < = k < = 13, | –13 < = k < = 12, | –15 < = k < = 15, | –13 < = k < = 13, | |
| –9 < = l < = 9, | –9 < = l < = 9 | –10 < = l < = 11 | –9 < = l < = 9 | |
| Collected reflections/unique reflections | 43286/591 | 7606/593 | 45590/893 | 15516/586 |
| Refinement method | Full-matrix least-squares on F2 | |||
| Refined parameters | 41 | 42 | 43 | 41 |
| R1, Fo >4σ(Fo) | 0.0140 | 0.0215 | 0.0225 | 0.0196 |
| R1, all unique data | 0.0149 | 0.0222 | 0.0238 | 0.0201 |
| wR2 | 0.0285 | 0.0509 | 0.0448 | 0.0432 |
| GooF | 1.194 | 1.214 | 1.189 | 1.205 |
| Extinction coefficient | 0.00021(6) | 0.00102(14) | 0.00006(7) | 0.00018(8) |
| Largest difference peaks (e– Å–3) | 1.476 and –0.802 | 2.831 and –1.093 | 4.679 and –1.960 | 2.829 and –1.008 |
Site occupancy. In all the refined structures, the Pb2, P, and Cl sites were fully occupied by a single constituent. However, the scattering from the Pb1 site indicated the substitution of lighter elements. Based on this, the La, K, and Na ions were assigned exclusively to the Pb1 site. The occupancies were refined with the constraints of Pb1 + La + X = 1 (X = Na or K) for La-doped samples and Pb1 + X = 1 for control samples. The following chemical formulas were derived from the refinements: Pb4.62(1) La0.19(1)Na0.19(1)(PO4)3Cl (La-Na-Pym), Pb4.76(1)La0.12(1)K0.12(2) (PO4)3Cl (La-K-Pym), Pb4.90(3)Na0.10(4)(PO4)3Cl (Na-Pym), and Pb4.88(3)K0.12(4)(PO4)3Cl (K-Pym). Small deficiencies in positive charge are probably easily compensated by vacancies among anionic positions, which are below the detection limit of the structural and microprobe analysis.
Various structural studies of La-doped apatites have been reported (Cockbain and Smith 1967; Mayer et al. 1980; Mayer and Swissa 1985; Hughes et al. 1991; Fleet et al. 2000). The site preference for La is strongly related to the chemical composition of apatite. In an X-ray study of a synthetic lanthanum calcium silicate apatite Ca4La6(SiO4)6(OH)2 (space group P63/m), Cockbain and Smith (1967) concluded that the La atoms are randomly distributed between the two Ca sites. Hughes et al. (1991) described the observed site preference of light REE for the Ca2 site based on the single-crystal analysis of four natural apatites, Ca5(PO4)3(F,OH), with REE substitutions (space group P63/m). In contrast, the Ca1 site is favored to host La in the structure of chlorapatite, Ca5(PO4)3Cl (Fleet et al. 2000). In this case, the substitution of Cl for (F, OH) results in the distortion and large increase in size of the Ca2O6X polyhedron, which is consistent with this unusual Ca-apatite site preference. However, these crystals of REE-doped chlorapatite were monoclinic with the space group P21/b. Numerous studies on REE-doped Ca-apatite have shown that the volatile anion components (F, OH, Cl) are a significant factor in the selectivity of apatite for REE. This is due to their marked influence on the stereochemical environment and the effective size of the Ca2 site (Fleet and Pan 1997b).
The first attempts to refine the site preference of light REE in Pb-bearing apatite were made by Mayer et al. (1980) and Mayer and Swissa (1985). Their conclusions, however, were based only on the lattice parameter changes, mainly the c/a ratios, calculated from X-ray powder data. They described some structural differences in F- and Cl-end-members that potentially led to different site preferences for La, but in all cases, La preference for Pb1 was observed. Partial ordering was noted, i.e., the Pb ions mainly occupied the 6h position (Pb2 site), while the smaller rare earth cations occupied in the 4f column positions (Pb1 site). These findings are consistent with our refinements. Pb has a strong preference for the Me(2) (6h) site in the apatite structure because it better accommodates the stereoactive 6s2 electron lone pair on the Pb2+ cation. Moreover, the avoidance of the Me2 site by REE in Cl−bearing apatite is caused by the off-centering of Pb at the Me2 site, helped by the Cl at the channel. Therefore, the Me1 (4f) site is more susceptible to substitution of other ions (Rouse et al. 1984). Similar preferences were found in Pb-substituted hydroxylapatite, Ca5(PO4)3(OH), phosphohedyphane, Ca2Pb3(PO4)3Cl, and fluorphosphohedyphane, Ca2Pb3(PO4)3F (Bigi et al. 1991; Kampf et al. 2006; Kampf and Housley 2011), where Pb2+ always fills the Me2 site first.
Bond-valence calculations for the substitution of La and Na (or K) for Pb are either inconclusive or not fully consistent with the structural findings (Table 3). Based on the bond valence-sum rule, La3+ should prefer the Pb2 site. However, in the case of apatite, ambiguity may occur in the interpretation of bond valence: calculations of bond valence values are based on the relationship between bond distance and bond strength and include a component attributed to variation in the size of structural positions that can cause ambiguity in interpretation (Fleet and Pan 1995).
Bond valence sums calculated for Pb, La, Na, and K in Pb1 and Pb2 structural sites in pyromorphite
| La-Na-Pym | Na-Pym | La-K-Pym | K-Pym | |
|---|---|---|---|---|
| Pb | ||||
| Pb1 | 2.03 | 2.02 | 2.01 | 2.01 |
| Pb2 | 1.98 | 1.97 | 1.99 | 1.98 |
| La | ||||
| Pb1 | 2.20 | – | 2.16 | – |
| Pb2 | 2.26 | – | 2.26 | – |
| Na | ||||
| Pb1 | 0.85 | 0.84 | – | – |
| Pb2 | 0.82 | 0.82 | – | – |
| K | ||||
| Pb1 | – | – | 1.77 | 1.78 |
| Pb2 | – | – | 1.86 | 1.86 |
Notes: Bond-valence parameters are taken from the studies by: Brown and Altermatt (1985) for K+–Cl–, Brese and O’Keeffe (1991) for Na+–Cl– and La3+–Cl–; Hu (2007) for Pb2+–Cl–; Gagné and Hawthorne (2015) for all cations –O2–.
Structure variation. Table 4 shows the variations in selected bond lengths, bond angles, polyhedral volumes, and twist angles for all analyzed samples. The volume of each Pb polyhedron was calculated using the software program VOLCAL (Hazen and Finger 1982). Minor substitution of Na and K ions in the control samples slightly affected the mean Pb1–O distances, resulting in the increase in the Pb1 polyhedral volume from 38.688 Å3 in Na-Pym to 38.729 Å3 in K-Pym. This is consistent with the smaller ionic radius of Na than that of K, which for ninefold coordination are 1.24 and 1.55 Å, respectively (values taken from Shannon 1976). The mean Pb1–O distances for La-doped samples increased by 0.005 Å for La-K-Pym compared to those of La-Na-Pym, reflecting the polyhedral volume changes. The most sensitive bond for substitution was the Pb1–O1 bond, which varied from 2.572(3) Å (La-Na-Pym) to 2.581(4) Å (La-K-Pym). The Pb1 polyhedral volume for La-Na-Pym decreased by 0.173 Å3 compared to the control sample, whereas the corresponding volume in La-K-Pym increased in comparison to the control sample by 0.048 Å3. This variation may be caused by the differences in ionic radii. In the case of Na and La substitutions for Pb, both substituting ions are smaller than Pb [ionic radii are: 1.216 Å (La3+) <1.24 Å (Na+) <1.35 Å (Pb2+)], whereas in the case of K and La substitutions for Pb, the K ion is larger than Pb and the La ion is smaller [1.216 Å (La3+) <1.35 Å (Pb2+) <1.55 Å (K+)]. However, the average ionic radii of La and K is very close to that of Pb, which may be the reason for the small volume change in this polyhedron compared to the control sample (only by 0.048 Å3). Variations in individual and mean interatomic distances in Pb1 polyhedron reflect the cumulative effect of both the amount of substitution and ionic radii of substituting ions.
Selected bond lengths (Å), bond angles (°), and twist angles (°) for all analyzed samples
| La-Na-Pym | Na-Pym | La-K-Pym | K-Pym | |
|---|---|---|---|---|
| Pb1–O1 (×3) | 2.572(3) | 2.580(5) | 2.581(4) | 2.578(5) |
| –O2 (×3) | 2.671(3) | 2.672(5) | 2.674(4) | 2.675(5) |
| –O3 (×3) | 2.877(3) | 2.878(5) | 2.881(5) | 2.879(5) |
| Mean | 2.707 | 2.710 | 2.712 | 2.711 |
| Polyhedral volume (Å3) | 38.515 | 38.688 | 38.777 | 38.729 |
| Pb2–O1 | 3.065(3) | 3.086(7) | 3.079(4) | 3.08619(11) |
| –O2 | 2.357(4) | 2.359(7) | 2.356(6) | 2.352(6) |
| –O3 (×2) | 2.630(3) | 2.633(5) | 2.625(4) | 2.634(5) |
| –O3 (×2) | 2.637(3) | 2.637(5) | 2.636(5) | 2.635(5) |
| Cl (×2) | 3.10891(18) | 3.1109(6) | 3.1131(2) | 3.1103(3) |
| Mean | 2.772 | 2.776 | 2.773 | 2.774 |
| Polyhedral volume (Å3) | 36.941 | 37.138 | 37.019 | 37.085 |
| P–O1 | 1.528(4) | 1.523(7) | 1.519(6) | 1.522(7) |
| –O2 | 1.555(4) | 1.559(7) | 1.557(6) | 1.562(7) |
| –O3 (×2) | 1.534(3) | 1.538(5) | 1.539(4) | 1.537(5) |
| Mean | 1.538 | 1.540 | 1.536 | 1.540 |
| Polyhedral volume (Å3) | 1.862 | 1.869 | 1.866 | 1.869 |
| O1–P–O2 | 110.6(3) | 110.5(4) | 110.5(4) | 110.3(4) |
| O1–P–O3 (×2) | 112.19(16) | 112.3(2) | 112.3(2) | 112.2(2) |
| O2–P–O3 (×2) | 107.29(16) | 107.2(3) | 107.3(2) | 107.4(2) |
| O3–P–O3 | 107.0(3) | 107.0(4) | 106.9(4) | 107.0(4) |
| Twist angle | 17.19 | 17.89 | 17.91 | 17.73 |
Changes in the Pb1 polyhedron slightly affected the mean Pb2–O distances. The largest difference was visible for the weakest bond, Pb2–O1, which was shorter in both the La-doped pyromorphites compared to the control samples. A similar trend was observed for the Pb2 polyhedral volume. Substitution in Pb1 did not affect the PO4 tetrahedron, with volumes differing by only 0.007 Å3, which is within uncertainties. Moreover, all O–P–O angles within each PO4 tetrahedron were identical in all refined structures.
Changes in distances also affected the unit-cell parameters and, consequently, their volumes (Table 2). The unit-cell parameter c appears to be more affected by the substitution and decreased in La-doped samples compared to the control samples. In La-Na-Pym, the unit-cell parameter a also decreased compared to the control sample. However, in La-K-Pym, a increased compared to K-Pym. This affected the volumes of the unit cell, and within the samples the smallest unit-cell volume, 630.680 Å3, was observed for La-Na-Pym. This is due to the substitution of two smaller ions (Na+ and La3+) for Pb2+. In the La-K-Pym sample, the La content is lower than in La-Na-Pym, and the K ion is larger than Na, which resulted in an overall increase in the unit-cell volume.
Implications
The restricted chemistry of synthetic analogs of La-doped pyromorphite allowed us to determine the effect of La substitution on the structure. From our single-crystal X-ray diffraction study, the following implications emerged. The mechanisms controlling La substitution in Pb-apatite are somewhat different from those in Ca-apatite. In both types of apatite, owing to the charge difference, heterovalent substitution of La3+ for Me2+ requires counter ions, which, in this case, were K+ and Na+. However, in contrast to natural Ca-apatite, in Pb-apatite La occupies the Me1 site and not the Me2 site. This is at least the case in pyromorphites in which the counter ion is K or Na. It is possible that a greater size of Ca2 in Ca-chlorapatite diminishes the selectivity of this position for REE relative to that of Ca1 (Fleet et al. 2000). However, considering the present stage of knowledge, it is not possible to accurately compare the results obtained for pyromorphite with its calcium chlorapatite counterpart Ca5(PO4)3Cl due to the difficulty in obtaining such apatite by simple synthesis from aqueous solutions. Therefore, it is difficult to clearly assess how much of this difference in substitution is due to the crystallization process and how much is due to a slight difference in structural properties, the influence of the lone-electron pair in Pb2+ ions, or the preference of Pb to occupy the Me2 site. It is not yet clear as to how the charges are balanced in REE substitutions in natural pyromorphite. Nevertheless, the magnitude of La substitution in the synthetic pyromorphites described here was larger than that reported in natural ones (see for example, Markl et al. 2014). This substitution model appears to be the model for all REE in Pb-apatite (or at least for all LREE).
Our preliminary tests with REE heavier than La indicate that the fractionation of REE during the crystallization of pyromorphite from aqueous solutions at ambient temperatures is nearly negligible. It is necessary to determine the structure of pyromorphite containing heavy REE (e.g., Yb, Lu) formed under identical conditions to determine the possible differences and similarities in the substitution mechanisms. However, only the determination of the structures of pyromorphite containing successively (but separately) other REE, with different charge compensation mechanisms, will enable the determination of possible systematic similarities and discrepancies in the structures and substitution mechanisms depending on the ionic size and mass of the REE.
Acknowledgments and Funding
Fernando Camara and an anonymous reviewer provided thoughtful reviews that are very gratefully acknowledged as they improved the present paper. This research was partly funded by the Polish NCN grant no. 2019/35/B/ST10/03379 and partly by program “Excellence initiative–research university” for the AGH University of Science and Technology.
-
Declaration of Competing Interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References cited
Adler, H.H. (1964) Infrared spectra of phosphate minerals: Symmetry and substitutional effects in the pyromorphite series. American Mineralogist. Journal of Earth and Planetary Materials, 49, 1002–1015.Search in Google Scholar
Andersson, S.S., Wagner, T., Jonsson, E., Fusswinkel, T., and Whitehouse, M.J. (2019) Apatite as a tracer of the source, chemistry and evolution of ore-forming fluids: The case of the Olserum-Djupedal REE-phosphate mineralisation, SE Sweden. Geochimica et Cosmochimica Acta, 255, 163–187, https://doi.org/10.1016/j.gca.2019.04.014Search in Google Scholar
Baikie, T., Schreyer, M., Wei, F., Herrin, J.S., Ferraris, C., Brink, F., Topolska, J., Piltz, R.O., Price, J., and White, T. J. (2014) The influence of stereochemically active lone-pair electrons on crystal symmetry and twist angles in lead apatite-2H type structures. Mineralogical Magazine, 78, 325–345, https://doi.org/10.1180/minmag.2014.078.2.07Search in Google Scholar
Bajda, T., Mozgawa, W., Manecki, M., and Flis, J. (2011) Vibrational spectroscopic study of mimetite–pyromorphite solid solutions. Polyhedron, 30, 2479–2485, https://doi.org/10.1016/j.poly.2011.06.034Search in Google Scholar
Bartholomäi, G. and Klee, W.E. (1978) The vibrational spectra of pyromorphite, vanadinite and mimetite. Spectrochimica Acta. Part A: Molecular Spectroscopy, 34, 831–843, https://doi.org/10.1016/0584-8539(78)80038-5Search in Google Scholar
Bauer, M. and Klee, W.E. (1993) The monoclinic-hexagonal phase transition in chlorapatite. European Journal of Mineralogy, 5, 307–316, https://doi.org/10.1127/ejm/5/2/0307Search in Google Scholar
Belousova, E.A., Griffin, W.L., O’Reilly, S.Y., and Fisher, N.I. (2002) Apatite as an indicator mineral for mineral exploration: Trace-element compositions and their relationship to host rock type. Journal of Geochemical Exploration, 76, 45–69, https://doi.org/10.1016/S0375-6742(02)00204-2Search in Google Scholar
Bigi, A., Gandolfi, M., Gazzano, M., Ripamonti, A., Roveri, N., and Thomas, S.A. (1991) Structural modifications of hydroxyapatite induced by lead substitution for calcium. Journal of the Chemical Society, Dalton Transactions: Inorganic Chemistry, 11, 2883–2886, https://doi.org/10.1039/dt9910002883Search in Google Scholar
Borisov, S.V. and Klevcova, R.F. (1963) The crystal structure of RE-Sr apatite. Zhurnal Strukturnoi Khimii, 4, 629–631.Search in Google Scholar
Botto, I.L., Barone, V.L., Castiglioni, J.L., and Schalamuk, I.B. (1997) Characterization of a natural substituted pyromorphite. Journal of Materials Science, 32, 6549–6553, https://doi.org/10.1023/A:1018667428762Search in Google Scholar
Bouzari, F., Hart, C.J., Bissig, T., and Barker, S. (2016) Hydrothermal alteration revealed by apatite luminescence and chemistry: A potential indicator mineral for exploring covered porphyry copper deposits. Economic Geology and the Bulletin of the Society of Economic Geologists, 111, 1397–1410, https://doi.org/10.2113/econgeo.111.6.1397Search in Google Scholar
Brese, N.E. and O’Keeffe, M. (1991) Bond-valence parameters for solids. Acta Crystallographica, B47, 192–197, https://doi.org/10.1107/S0108768190011041Search in Google Scholar
Brown, I.D. and Altermatt, D. (1985) Bond-valence parameters obtained from a systematic analysis of the Inorganic Crystal Structure Database. Acta Crystallographica, B41, 244–247, https://doi.org/10.1107/S0108768185002063Search in Google Scholar
Cockbain, A.G. and Smith, G.V. (1967) Alkaline-earth-rare-earth silicate and germanate apatites. Mineralogical Magazine and Journal of the Mineralogical Society, 36, 411–421, https://doi.org/10.1180/minmag.1967.036.279.11Search in Google Scholar
Dai, Y. and Hughes, J.M. (1989) Crystal structure refinements of vanadinite and pyromorphite. Canadian Mineralogist, 27, 189–192.Search in Google Scholar
Elliott, J.C., Mackie, P.E., and Young, R.A. (1973) Monoclinic hydroxyapatite. Science, 180, 1055–1057.Search in Google Scholar
Fleet, M.E. and Pan, Y. (1995) Site preference of rare earth elements in fluorapatite. American Mineralogist, 80, 329–335.Search in Google Scholar
Fleet, M.E. and Pan, Y. (1997a) Site preference of rare earth elements in fluorapatite: Binary (LREE+HREE)-substituted crystals. American Mineralogist, 82, 870–877, https://doi.org/10.2138/am-1997-9-1004Search in Google Scholar
Fleet, M.E. and Pan, Y. (1997b) Rare earth elements in apatite: Uptake from H2O-bearing phosphate fluoride melts and the role of volatile components. Geochimica et Cosmochimica Acta, 61, 4745–4760, https://doi.org/10.1016/S0016-7037(97)00292-5Search in Google Scholar
Fleet, M.E., Liu, X., and Pan, Y. (2000) Rare-earth elements in chlorapatite [Ca10(PO4)6Cl2]: Uptake, site preference, and degradation of monoclinic structure. American Mineralogist, 85, 1437–1446, https://doi.org/10.2138/am-2000-1012Search in Google Scholar
Fleet, M.E., Liu, X., and King, P.L. (2004) Accommodation of the carbonate ion in apatite: An FTIR and X-ray structure study of crystals synthesized at 2–4 GPa. American Mineralogist, 89, 1422–1432, https://doi.org/10.2138/am-2004-1009Search in Google Scholar
Frost, R.L. and Palmer, S.J. (2007) A Raman spectroscopic study of the phosphate mineral pyromorphite Pb5(PO4)3Cl. Polyhedron, 26, 4533–4541, https://doi.org/10.1016/j.poly.2007.06.004Search in Google Scholar
Gagné, O.C. and Hawthorne, F.C. (2015) Comprehensive derivation of bond-valence parameters for ion pairs involving oxygen. Acta Crystallographica, B71, 562–578, https://doi.org/10.1107/S2052520615016297Search in Google Scholar
Gu, T., Qin, S., and Wu, X. (2020) Thermal behavior of pyromorphite (Pb10(PO4)6Cl2): In situ high temperature powder X-ray diffraction study. Crystals, 10, 1070, https://doi.org/10.3390/cryst10121070Search in Google Scholar
Harlov, D.E. (2015) Apatite: A fingerprint for metasomatic processes. Elements (Quebec), 11, 171–176, https://doi.org/10.2113/gselements.11.3.171Search in Google Scholar
Hazen, R.M. and Finger, L.W. (1982) Comparative Crystal Chemistry: Temperature, pressure, composition, and the variation of crystal structure, 248 p. Wiley.Search in Google Scholar
Hovis, G., Abraham, T., Hudacek, W., Wildermuth, S., Scott, B., Altomare, C., Medford, A., Conlon, M., Morris, M., Leaman, A., and others. (2015) Thermal expansion of F-Cl apatite crystalline solutions. American Mineralogist, 100, 1040–1046, https://doi.org/10.2138/am-2015-5176Search in Google Scholar
Hu, S.-Z. (2007) A new approach to bond valence parameters for Pb(II)-halide bonds. Wuli Huaxue Xuebao, 23, 786–789.Search in Google Scholar
Hughes, J.M., Cameron, M., and Mariano, A.N. (1991) Rare-earth-element ordering and structural variations in natural rare-earth-bearing apatites. American Mineralogist, 76, 1165–1173.Search in Google Scholar
Jarosewich, E. and Boatner, L.A. (1991) Rare-earth element reference samples for electron microprobe analysis. Geostandards and Geoanalytical Research, 15, 397–399, https://doi.org/10.1111/j.1751-908X.1991.tb00115.xSearch in Google Scholar
Jonsson, E., Harlov, D.E., Majka, J., Högdahl, K., and Persson-Nilsson, K. (2016) Fluorapatite-monazite-allanite relations in the Grängesberg apatite-iron oxide ore district, Bergslagen, Sweden. American Mineralogist, 101, 1769–1782, https://doi.org/10.2138/am-2016-5655Search in Google Scholar
Kampf, A.R. and Housley, R.M. (2011) Fluorphosphohedyphane, Ca2Pb3(PO4)3F, the first apatite supergroup mineral with essential Pb and F. American Mineralogist, 96, 423–429, https://doi.org/10.2138/am.2011.3586Search in Google Scholar
Kampf, A.R., Steele, I.M., and Jenkins, R.A. (2006) Phosphohedyphane, Ca2Pb3(PO4)3Cl, the phosphate analog of hedyphane: Description and crystal structure. American Mineralogist, 91, 1909–1917, https://doi.org/10.2138/am.2006.2268Search in Google Scholar
Kim, J. Y., Fenton, R.R., Hunter, B.A., and Kennedy, B.J. (2000) Powder diffraction studies of synthetic calcium and lead apatites. Australian Journal of Chemistry, 53, 679–686, https://doi.org/10.1071/CH00060Search in Google Scholar
Knyazev, A.V., Bulanov, E.N., and Korokin, V.Z. (2015) Thermal expansion of solid solutions in apatite binary systems. Materials Research Bulletin, 61, 47–53, https://doi.org/10.1016/j.materresbull.2014.09.089Search in Google Scholar
Kwaśniak-Kominek, M., Manecki, M., Matusik, J., and Lempart, M. (2017) Carbonate substitution in lead hydroxyapatite Pb5(PO4)3OH. Journal of Molecular Structure, 1147, 594–602, https://doi.org/10.1016/j.molstruc.2017.06.111Search in Google Scholar
Laperche, V., Logan, T.J., Gaddam, P., and Traina, S.J. (1997) Effect of apatite amendments on plant uptake of lead from contaminated soil. Environmental Science & Technology, 31, 2745–2753, https://doi.org/10.1021/es961011oSearch in Google Scholar
Levitt, S.R. and Condrate, R.A. Sr. (1970) The vibrational spectra of lead apatites. American Mineralogist. Journal of Earth and Planetary Materials, 55, 1562–1575.Search in Google Scholar
Ma, Q.Y., Traina, S.J., Logan, T.J., and Ryan, J.A. (1993) In situ lead immobilization by apatite. Environmental Science & Technology, 27, 1803–1810, https://doi.org/10.1021/es00046a007Search in Google Scholar
Ma, Q.Y., Logan, T.J., and Traina, S.J. (1995) Lead immobilization from aqueous solutions and contaminated soils using phosphate rocks. Environmental Science & Technology, 29, 1118–1126, https://doi.org/10.1021/es00004a034Search in Google Scholar
Mackie, P.E. and Young, R.A. (1973) Location of Nd dopant in fluorapatite, Ca5(PO4)3F: Nd. Journal of Applied Crystallography, 6, 26–31, https://doi.org/10.1107/S0021889873008009Search in Google Scholar
Mackie, P.E., Elliot, J.C., and Young, R.A. (1972) Monoclinic structure of synthetic Ca5(PO4)3Cl, chlorapatite. Acta Crystallographica, B28, 1840–1848, https://doi.org/10.1107/S0567740872005114Search in Google Scholar
Manecki, M. (2019) Lead in Water and Soil: Speciation, Toxicity, and Treatment Technologies. In P.A. Maurice, Ed., Encyclopedia of Water: Science, Technology, and Society, p. 1713–1727. Wiley.Search in Google Scholar
Manecki, M., Maurice, P.A., and Traina, S.J. (2000) Kinetics of aqueous Pb reaction with apatites. Soil Science, 165, 920–933, https://doi.org/10.1097/00010694-200012000-00002Search in Google Scholar
Manecki, M., Kwaśniak-Kominek, M., Majka, J.M., and Rakovan, J. (2020) Model of interface-coupled dissolution-precipitation mechanism of pseudomorphic replacement reaction in aqueous solutions based on the system of cerussite PbCO3–pyromorphite Pb5(PO4)3Cl. Geochimica et Cosmochimica Acta, 289, 1–13, https://doi.org/10.1016/j.gca.2020.08.015Search in Google Scholar
Markl, G., Marks, M.A., Holzäpfel, J., and Wenzel, T. (2014) Major, minor, and trace element composition of pyromorphite-group minerals as recorder of supergene weathering processes from the Schwarzwald mining district, SW Germany. American Mineralogist, 99, 1133–1146, https://doi.org/10.2138/am.2014.4789Search in Google Scholar
Mayer, I. and Swissa, S. (1985) Lead and strontium phosphate apatites substituted by rare earth and silver ions. Journal of the Less Common Metals, 110, 411–414, https://doi.org/10.1016/0022-5088(85)90350-9Search in Google Scholar
Mayer, I., Semadja, A., and Weiss, V. (1980) Lead phosphate apatites substituted by rare earth, sodium, and potassium ions. Journal of Solid State Chemistry, 34, 223–229, https://doi.org/10.1016/0022-4596(80)90225-XSearch in Google Scholar
Newby, H.P. (1981) Rare-earth elements in pyromorphite-group minerals. Ph.D. dissertation, University of London Reactor Centre, Department of Mechanical Engineering, Imperial College of Science and Technology.Search in Google Scholar
Nriagu, J.O. (1974) Lead orthophosphates—IV Formation and stability in the environment. Geochimica et Cosmochimica Acta, 38, 887–898, https://doi.org/10.1016/0016-7037(74)90062-3Search in Google Scholar
Okudera, H. (2013) Relationships among channel topology and atomic displacements in the structures of Pb5(BO4)3Cl with B = P (pyromorphite), V (vanadinite), and As (mimetite). American Mineralogist, 98, 1573–1579, https://doi.org/10.2138/am.2013.4417Search in Google Scholar
O’Sullivan, G.J., Chew, D.M., Morton, A.C., Mark, C., and Henrichs, I.A. (2018) An integrated apatite geochronology and geochemistry tool for sedimentary provenance analysis. Geochemistry, Geophysics, Geosystems, 19, 1309–1326, https://doi.org/10.1002/2017GC007343Search in Google Scholar
Pan, Y. and Fleet, M.E. (2002) Compositions of the apatite-group minerals: Substitution mechanisms and controlling factors. Reviews in Mineralogy and Geochemistry, 48, 13–49, https://doi.org/10.2138/rmg.2002.48.2Search in Google Scholar
Papike, J.J., Jensen, M., Shearer, C.K., Simon, S.B., Walker, R.J., and Laul, J.C. (1984) Apatite as a recorder of pegmatite petrogenesis. Geological Society of America Abstracts with Programs, 16, 617.Search in Google Scholar
Pasero, M., Kampf, A.R., Ferraris, C., Pekov, I.V., Rakovan, J., and White, T.J. (2010) Nomenclature of the apatite supergroup minerals. European Journal of Mineralogy, 22, 163–179, https://doi.org/10.1127/0935-1221/2010/0022-2022Search in Google Scholar
Rakovan, J. and Reeder, R.J. (1996) Intracrystalline Rare Earth Element distributions in apatite: Surface structural influences on incorporation during growth. Geochimica et Cosmochimica Acta, 60, 4435–4445, https://doi.org/10.1016/S0016-7037(96)00244-XSearch in Google Scholar
Rønsbo, J.G. (1989) Coupled substitutions involving REEs and Na and Si in apatites in alkaline rocks from the Ilimaussaq intrusion, South Greenland, and the petrological implications. American Mineralogist, 74, 896–901.Search in Google Scholar
Rouse, R.C., Dunn, P. J., and Peacor, D.R. (1984) Hedyphane from Franklin, New Jersey and Långban, Sweden: Cation ordering in an arsenate apatite. American Mineralogist, 69, 920–927.Search in Google Scholar
Sha, L.K. and Chappell, B.W. (1999) Apatite chemical composition, determined by electron microprobe and laser-ablation inductively coupled plasma mass spectrometry, as a probe into granite petrogenesis. Geochimica et Cosmochimica Acta, 63, 3861–3881, https://doi.org/10.1016/S0016-7037(99)00210-0Search in Google Scholar
Shannon, R.D. (1976) Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallographica, 32, 751–767, https://doi.org/10.1107/S0567739476001551Search in Google Scholar
Sheldrick, G.M. (2015) Crystal structure refinement with SHELXL. Acta Crystallographica, C71, 3–8, https://doi.org/10.1107/S2053229614024218Search in Google Scholar
Tang, P., Zhou, Y.C., and Xie, Z.M. (2013) Immobilization of heavy metals in sludge using phosphoric acid and monobasic calcium phosphate. Journal of Zhejiang University. Science A, 14, 177–186, https://doi.org/10.1631/jzus.A1200263Search in Google Scholar
Vignoles, M., Bonel, G., Holcomb, D.W., and Young, R.A. (1988) Influence of preparation conditions on the composition of type B carbonated hydroxyapatite and on the localization of the carbonate ions. Calcified Tissue International, 43, 33–40, https://doi.org/10.1007/BF02555165Search in Google Scholar
White, T., Ferraris, C., Kim, J., and Madhavi, S. (2005) Apatite–an adaptive framework structure. Reviews in Mineralogy and Geochemistry, 57, 307–401, https://doi.org/10.2138/rmg.2005.57.10Search in Google Scholar
Zhang, L., Chen, Z., Wang, F., and Zhou, T. (2021) Apatite geochemistry as an indicator of petrogenesis and uranium fertility of granites: A case study from the Zhuguangshan batholith, South China. Ore Geology Reviews, 128, 103886, https://doi.org/10.1016/j.oregeorev.2020.103886Search in Google Scholar
© 2023 Julia Sordyl, John Rakovan, Peter C. Burns, Justyna Topolska, Adam Włodek, Jennifer E.S. Szymanowski, Ginger E. Sigmon, Jarosław Majka, Maciej Manecki, published by Mineralogical Society of America
This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
Articles in the same Issue
- Evidence for abundant organic matter in a Neoarchean banded iron formation
- Electrical properties of iron sulfide-bearing dunite under pressure: Effect of temperature, composition, and annealing time
- Gas-mediated trace element incorporation into rhyolite-hosted topaz: A synchrotron microbeam XAS study
- 10.2138/am-2023-8927
- A dunite fragment in meteorite Northwest Africa (NWA) 11421: A piece of the Moon’s mantle
- 10.2138/am-2023-9054
- Hydrogen bond symmetrization and high-spin to low-spin transition of ε-FeOOH at the pressure of Earth’s lower mantle
- CURIES: Compendium of uranium Raman and infrared experimental spectra
-
- Efect of faceting on olivine wetting properties
- The obscuring efect of magma recharge on the connection of volcanic-plutonic rocks
- In-situ study of microstructures induced by the olivine to wadsleyite transformation at conditions of the 410 km depth discontinuity
- Effect of pre-existing crystals and melt homogeneity on the decompression-induced crystallization of hydrous rhyodacite magma
- Origin of clinopyroxene-ilmenite symplectites in mafic granulites from eastern parts of the Chotanagpur granite gneissic complex, East Indian shield
- Single-crystal analysis of La-doped pyromorphite [Pb5(PO4)3Cl]
- Crystal structure of calcium-ferrite type NaAlSiO4 up to 45 GPa
- Revision of the CaMgSi2O6-CO2 P-T phase diagram at 3–6 GPa
Articles in the same Issue
- Evidence for abundant organic matter in a Neoarchean banded iron formation
- Electrical properties of iron sulfide-bearing dunite under pressure: Effect of temperature, composition, and annealing time
- Gas-mediated trace element incorporation into rhyolite-hosted topaz: A synchrotron microbeam XAS study
- 10.2138/am-2023-8927
- A dunite fragment in meteorite Northwest Africa (NWA) 11421: A piece of the Moon’s mantle
- 10.2138/am-2023-9054
- Hydrogen bond symmetrization and high-spin to low-spin transition of ε-FeOOH at the pressure of Earth’s lower mantle
- CURIES: Compendium of uranium Raman and infrared experimental spectra
-
- Efect of faceting on olivine wetting properties
- The obscuring efect of magma recharge on the connection of volcanic-plutonic rocks
- In-situ study of microstructures induced by the olivine to wadsleyite transformation at conditions of the 410 km depth discontinuity
- Effect of pre-existing crystals and melt homogeneity on the decompression-induced crystallization of hydrous rhyodacite magma
- Origin of clinopyroxene-ilmenite symplectites in mafic granulites from eastern parts of the Chotanagpur granite gneissic complex, East Indian shield
- Single-crystal analysis of La-doped pyromorphite [Pb5(PO4)3Cl]
- Crystal structure of calcium-ferrite type NaAlSiO4 up to 45 GPa
- Revision of the CaMgSi2O6-CO2 P-T phase diagram at 3–6 GPa