Different substituent effects on the supramolecular arrays in some (E)-halo- and nitro-benzaldehyde oximes: confirmation of attractive π(C=N)···π(phenyl) interactions
-
Ligia R. Gomes
 ,
Tanja van Mourik
,
Tanja van Mourik

Abstract
The crystal structures and Hirshfeld surface analyses are reported for four aldoximes, (E)-X–C6H4CH=N–OH [X = 3-Cl (1), 4-F (2), 2-O2N (3) and 4-O2N (4)]. The strong classical O–H · · · N hydrogen bonds involving the oxime group generate C(3) chains in compound 1, in contrast to the R22(6) dimers formed in compounds 2–4; such arrangements have been shown to be the most frequently found for oximes other than salicylaldoximes (2-hydroxybenzaldehyde oximes). In general, weaker intermolecular interactions involving the X substituents, as well as C–H · · · O and π · · · π interactions have significant effects on the supramolecular arrays generated in the aggegation. A further important interaction in compound 1, and to a lesser extent in compound 4, is a π(C=N) · · · π(phenyl) molecular stacking. A data base search has indicated that short Cg(C=N) · · · Cg(phenyl) distances, <3.3 Å (Cg = centre of gravity), have been found in various compounds, including other oximes. A theoretical study was carried out starting from the crystal structure data of compound 1, with optimisation at the BLYP-D3/def2-DZVP level, as well as at the higher PBE0/ma-def2-TZVP level. Breakdown of the interaction energy into separate contributions was achieved using SAPT (using the jun-cc-pvdz basis set). Overall, the calculations indicate that the π(C=N) ·· · π(phenyl) interaction is attractive, with a magnitude of 14–18 kJ mol−1.
1 Introduction
The oxime group, R1R2C=NOH, is found in many biologically active compounds [1], [2], with a wide range of uses including as antitumor agents [3], [4], [5], [6], acaricides and insecticides [7], thymidine phosphorylase inhibitors [8], anti-microbial agents [9], bactericides [10] and anti-inflammatory agents [11], as well as in the treatment of nerve-gas poisoning [12], [13], [14], [15]. In the plant kingdom, oximes play vital metabolic roles [16]. As an important group of organic compounds, it is not surprising that crystal structures of aldoximes have attracted attention [16], [17], [18], [19], with well over 60 structures listed in the CCDC data base.
Benzaldehyde oximes, ArCH=NOH, with their -CH=N–OH functional group are ideally arranged for classical O–H···O and/ or O–H···N hydrogen bonding. The last survey of the classical hydrogen bonding patterns in benzaldehyde oximes reported in 2010 [19] confirmed the most frequently found arrangements, with the exception of salicylaldoxines, were

Illustrations of the C3 chains and
Hydrogen bonds are considered as the strongest and most directional of intermolecular interactions available in molecules [22] and thus play the major role in determining the overall supramolecular structures. However, the involvement of weaker intermolecular interactions, such as C–H···O hydrogen bonds, π···π interactions and interactions involving the substituents can have a significant influence on the supramolecular arrays generated in the aggregation.
In a further study of benzaldehyde oximes, we have determined the crystal structures of four compounds, namely 3-chlorobenzaldehyde oxime, 1, 4-fluorobenzaldehyde oxime, 2, 2-nitrobenzaldehyde oxime, 3, and 4-nitrobenzaldehyde oxime, 4, from data collected at T=100 K, see Scheme 1. The crystal structure of 4 has previously been determined from data collected at room temperature, and the same P21/c phase was found in each case [23], [24], [25]. These earlier studies made no mention of the intermolecular interactions, aside from the classical hydrogen bonds involving the oxime atoms. Aims of this study were to further investigate the occurrence of

Synthesis of 1–4.
2 Results and Discussion
2.1 General
The asymmetric unit of each compound, 1–4, contains a single molecule. All four compounds crystallize in the monoclinic system: compounds 1, 2 and 4 in space group P21/c and compound 3 in P21/n, all with Z=4. The geometry around the oxime moieties is (E) in all four molecules. Figure 2 illustrates the atom arrangements and numbering schemes. The interplanar angles between the substituents and their attached phenyl group are all less than 10° in compounds 1, 2 and 4, but in the ortho-substituted compound, 3, all such angles are greater than 26°, due to steric hindrance between the nitro and oxime groups (Table 1). Details of the hydrogen bonding and other intermolecular interactions are provided in Table 2. The major intermolecular interactions in 1–4 involve the O13–H13···N12 hydrogen bonds. In 1 these generate C(3) chains which run parallel to the b axis formed by the action of the screw axis at (1/2, y, 1/4) (Fig. 3), whilst in 2–4, the O13–H13···N12 hydrogen bonds form
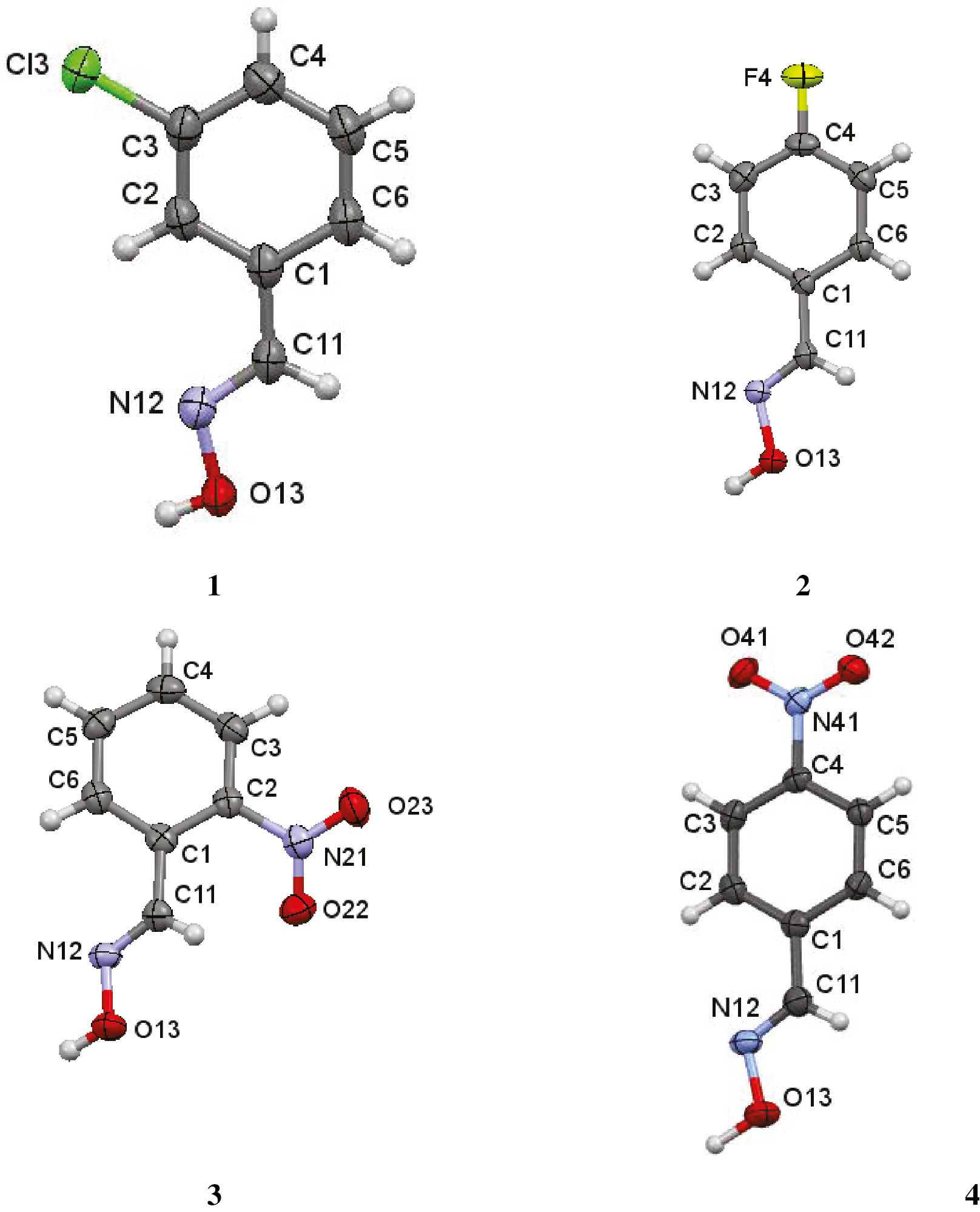
Views of the asymmetric units of 1–4 with the atom numbering schemes. Displacement ellipsoids are drawn at the 80% probability level, hydrogen atoms as spheres with arbitrary radii.
Interplanar angles (deg) for compounds 1–4.
| Between oxime group and attached phenyl ring | Between nitro group and attached phenyl ring | Between oxime and nitro groups | |
|---|---|---|---|
| 1 | 7.60(1) | ||
| 2 | 9.63(1) | ||
| 3 | 37.82(1) | 26.2(1) | 39.26(1) |
| 4 | 8.25(1) | 3.86(1) | 4.86(1) |
Geometric parameters and symmetry operations for hydrogen bonds and intermolecular interactions (Å, deg).a
| Intermolecular hydrogen bonds | ||||||
|---|---|---|---|---|---|---|
| D–H···A | D–H | H···A | D···A | D–H···A | Symmetry code | |
| 1 | O13–H13···N12 | 0.85(3) | 1.96(3) | 2.8007(18) | 177(3) | –x+1, y+1/2, –z+1/2 |
| 2 | O13–H13···O13 | 1.00(2) | 2.585(19) | 3.2002(10) | 119.7(13) | –x+1, –y, –z+1 |
| 2 | O13–H13···N12 | 1.00(2) | 1.88(2) | 2.7991(11) | 151.8(16) | –x+1, –y, –z+1 |
| 2 | C3–H3···F4 | 0.95 | 2.58 | 3.4670(13) | 156 | –x, y–1/2, –z+1/2 |
| 2 | C5–H5···F4 | 0.95 | 2.65 | 3.4416(13) | 142 | –x, –y, –z |
| 2 | C11–H11···O13 | 0.95 | 2.50 | 3.4229(13) | 165 | –x+1, y+1/2, –z+1/2 |
| 3 | O13–H13···O13 | 0.921(19) | 2.636(18) | 3.2451(17) | 124.3(13) | –x+2, –y+1, –z+1 |
| 3 | O13–H13···N12 | 0.921(19) | 1.937(19) | 2.8016(13) | 155.5(15) | –x+2, –y+1, –z+1 |
| 3 | C3–H3···O13 | 0.95 | 2.47 | 3.3132(15) | 148 | –x+3/2, y–1/2, –z+1/2 |
| 3 | C6–H6···O23 | 0.95 | 2.60 | 3.1744(15) | 119 | –x–3/2, y–1/2, –z–3/2 |
| 4 | O13—H13···O13 | 0.89 (1) | 2.68 (2) | 3.2559(19) | 124 (2) | –x+1, –y–1, –z+1 |
| 4 | O13—H13···N12 | 0.89 (1) | 1.99 (1) | 2.8191(15) | 154(2) | –x+1, –y–1, –z+1 |
| 4 | C6—H6···O41 | 0.95 | 2.53 | 3.3032(16) | 139 | x+1, y–1, z |
| 4 | C11—H11···O41 | 0.95 | 2.42 | 3.1577(15) | 134 | x+1, y–1, z |
| Y–X···π interactions | |||||||
|---|---|---|---|---|---|---|---|
| Y–X···Cg | X···Cg | Xperp | γ | Y–X···Cg | Y···Cg | Symmetry code | |
| 4 | N41–O41···Cg1 | 3.8471(10) | 3.480 | 25.25 | 65.05(6) | 3.5105(11)(15) | x, 1+y, z |
| 4 | N42–O41···Cg1 | 3.3858(10) | 3.345 | 8.89 | 85.57(7) | 3.5105(11) | x, 1+y, z |
| π···π interactionsa | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| CgI···Cg(J) | Cg···Cg | α | β | γ | CgIperp | CgJperp | Slippage | Symmetry code | |
| 2 | Cg···Cg | 3.7672(7) | 0 | 24.8 | 24.8 | 3.4186(5) | 3.4186(5) | 1.583 | x, –1+y, z |
| 2 | Cg···Cg | 3.7672(7) | 0 | 24.8 | 24.8 | 3.4186(5) | 3.4185(5) | 1.583 | x, 1+y, z |
| 3 | Cg···Cg | 3.6405(7) | 0 | 15.6 | 15.6 | 3.5064(4) | 3.5063(4) | 0.979 | –1+x, y, z |
| 3 | Cg···Cg | 3.6405(7) | 0 | 15.6 | 15.6 | 3.5063(4) | 3.5064(4) | 0.979 | 1+x, y, z |
aα=Dihedral angle between planes I and J (deg); β=angle Cg(I)→Cg(J); γ=angle Cg(I)→Cg(J) vector and normal to plane J (deg); Cg···Cg=distance between ring centroids (Å); CgIPerp=perpendicular distance of Cg(I) on ring J (Å); CgJperp=perpendicular distance of Cg(J) on ring I (Å).

The C3 chain in compound 1, formed from C13–H13···N12 hydrogen bonds. Table 3 lists the symmetry operations.

Two views of the
Table 3 lists the percentage atom-atom contacts for all compounds.
Percentages of atom-atom contacts.
| Compound | H···H | H···O/O···H | H···C/C···H | H···N/N···H | C···C | O···C/C···O | N···O/O···N | N···C/C···N | O···O |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 26.3 | 10.5 | 16.1 | 8.0 | 5.2 | 2.3 | – | 2.6 | |
| 2 | 32.0 | 11.3 | 13.4 | 10.3 | 9.1 | 1.0 | 0.7 | ||
| 3 | 27.5 | 38.2 | 4.7 | 7.5 | 10.8 | 2.0 | 2.8 | 2.9 | 3.4 |
| 4 | 29.7 | 29.1 | 7.5 | 7.5 | 4.0 | 9.1 | 2.4 | 5.2 | 5.1 |
| Compound | H···Cl/Cl···H | Cl···C/C···Cl | F···C/C···F | F···F | H···F/F···H |
|---|---|---|---|---|---|
| 1 | 24.3 | 2.3 | |||
| 2 | 2.1 | 2.1 | 17.6 |
2.2 Other intermolecular interactions in individual compounds
2.2.1 The chain-forming compound 1
Interactions in 1, in addition to the C(3) chain forming O13–H13···N12 hydrogen bond, are (i) short Cl···Cli intermolecular contacts, which link the C3 chains: the Cl···Cl distance is 3.418(6) Å, just within the sum of the contact radii of 3.50 Å (symmetry code: i=–x, –y, –z), and (ii) π(C=N)···π(phenyl) interactions, with a Cg(C=N)···Cg(phenyl)ii distance of 3.290(1) Å (Cg being the center of gravity of the phenyl ring): symmetry code: ii=x, −1+y, z) (Fig. 5). The π(C=N)···π(phenyl) interaction is more fully discussed in the Section: π(C=N)···π(phenyl) contacts below.

Compound 1. (a) Part of a stack of molecules formed from π(C=N) ···π(phenyl) interactions. The Cg(C=N)···Cgi (phenyl) distance is 3.290(1) Å (symmetry code: i=x, –1+y, z; (b) the overlap of the C=N and phenyl π systems in successive rows of the π(C=N)···π(phenyl).
The Hirshfeld surface and Fingerprint (FP) plots [29], [30] for compound 1 are shown in Fig. 6. The C···C, Cl···Cl and N···H close contacts are designated as are the site of the π···π contacts. In the FP plot, the two spikes pointing southwest relate to N···H contacts including those making the C(3) chains; the high density of pixels near di≈de≈1.8 Å is due to C···C contacts, and that close to the di≈de≈1.8–2.0 Å region is due to Cl···Cl contacts, and the wings ending approximately at (di; de)≈(1.2; 1.8) Å are due to H···Cl contacts.

Compound 1. (a) View of the Hirshfeld surface mapped over dnorm: the set of two intense red area spots relate to N···H close contacts in forming the C3 chain; also designated are the sites of the C···C and π···π contacts; (b) the FP plot, the two spikes pointing southwest relate to N···H contacts including those making the C(3) chains; the high density of pixels near di≅de≅1.8 Å are due to C···C and those close to di≅de≅1.8–2.0 Å interval are due to Cl···Cl contacts, wings ending approximately at (di; de)≅(1.2; 1.8) Å are due to H···Cl contacts.
Compound 1 has the highest percentage of H···C/C···H contacts and the lowest percentage of H···H and H···O/O···H contacts of all four compounds studied here, see Table 3. There is also a high percentage of H···Cl/Cl···H contacts in compound 1
2.3 The compounds forming R 2 2 ( 6 )
2.3.1 Compound 2
In 2, in addition to the O13–H13···N12 and O13–H13···O13 hydrogen bonds, there are weaker C11–H11···O13, C3–H3···F4 and C5–H5···F4 hydrogen bonds, and π···π interactions. The overall structure can be conveniently considered to be formed from two sub-structures. Firstly, the

Compound 2. (a) Part of a sheet formed from linking the
Compound 2 has the highest percentage of H···H contacts, 32.0%, significantly higher than the sum, 21.8%, of fluorine contacts (H···F/F···H, F···C/C···F and F···F). The H···O/O···H, H···C/C···H, and H···N/N···H contacts all are ca 10%.
The three other 4-halobenzaldehyde oximes, (E)-4-XC6H4CH=N–OH (X=Cl [31], Br [28] and I [20]) were also shown to form
2.3.2 Compound 3
The intermolecular interactions in compound 3, in addition to the dimer forming O13–H13···N12 and C3–H3···O13 hydrogen bonds, are C6–H6···O23(nitro) hydrogen bonds and π···π interactions. The R22(6) dimers are linked by the C3–H3···O13 and C6–H6···O23 hydrogen bonds into sheets. These sheets are composed of a network of

Compound 3. (a) Part of a sheet of molecules formed from combination of O13–H13···N12, C3–H3···O13 and C6–H6···O23 hydrogen bonds; (b) part of a double π···π stack, formed from the
2.3.3 Compound 4
There are major differences between the contacts found in compounds 3 and 4, which are both nitro phenyl derivatives. In compound, 4, in addition to the O13–H13···N12 and C3–H3···O13 hydrogen bonds, which form the

Compound 4. (a) Part of a two-molecular wide tilted sheet formed from linking the
2.4 Comparison of the structures of the three isomeric nitrobenzaldehyde oximes
Comparisons of the structures of the three isomeric mono-nitrobenzaldehyde oximes can be made, as the structure of 3-nitrobenzaldehyde oxime, 5, has been briefly reported from data collected at room temperature [33]. The ortho- and meta-isomers, compounds 3 and 5, have similar sets of intermolecular interactions, which are significantly different from those of the para-isomer, 4. While only the classical hydrogen bonds forming the
No π(C=N)···π(phenyl) interaction is apparent in 5, as the relevant Cg···Cg distance is >4.4 Å.
2.5 Hirshfeld surfaces and FP plots for compounds, 2–4
Views of the Hirshfeld surface [29], [30] for compounds, 2–4, which form

Views of the Hirshfeld surface mapped over dnorm for compounds (a) 2, (b) 3 and (c) 4, forming

FP plots for compounds (a) 2, (b) 3 and (c) 4, which form the

Partial FP plots for 2 showing pixels due to (a) F···H, (b) H···H, (c) N···H and (d) O···H contacts.
2.6 π(C=N)···π(phenyl) interactions
The clear indication of a π(C=N)···π(phenyl) interaction in compound 1, and the less clear indication in compound 4, led to an investigation of similar interactions in other oximes with Cg(C=N)···Cg(phenyl) distances up to 3.30 Å. Among the compounds found were the high pressure P21/n phase of salicylaldoxime [SALOXM-08] [34], the high pressure I2/a phase of 3-t-butylsalicylaldoxime [NIRJII-07] [35] and benzene-1,3,5-tris(N-hydroxy-methanimine) [ADOJEK] [36]. Oxime ethers, such as, 1-(salicylidene-oxy)-2-(3-methoxysalicylidenesmino-oxy)ethane [FOJROM] [37] were also found with short Cg(C=N)···Cg((phenyl) distances. A search on 12-06-2018 of the CCDC data base of compounds possessing C=N–X fragments (X=OH, OR, NR1R2 etc) with Cg(C=N)···Cg(phenyl) distances less than 3.30 Å, revealed more than 80 hits. Extending the Cg(C=N)···Cg(phenyl) distance beyond 3.30 to 4.0 Å brought in many more compounds. Short Cg(C=N)···Cg(phenyl) distances were found to be very much less common for compounds with -CH=N–OR (R=H or organyl) fragments, compared to compounds with C=N–NXY fragments.
2.6.1 Theoretical calculations on the potential of π(C=N) ···π(phenyl) interactions in compound 1
A theoretical study of the potential π(C=N) ···π(phenyl) interactions was carried out on compound 1, using initially a stacked dimer, extracted from the CIF (structure A in Fig. 13). Figure 13 shows the various structures and sub-structures, and their associated calculated interaction energies. To separate the two possible intermolecular interactions in the dimer A – the π(CN)···π(phenyl) interaction and the OH···N hydrogen bond – two additional dimers were constructed: replacement of the CH=N(OH) moiety by a hydrogen atom in the upper molecule removes the OH···N interaction (structure B) and replacement of the chlorophenyl unit by a hydrogen atom in both molecules removes the potential π(CN) ···π(phenyl) interaction (structure C). The interaction energy of structure A (−22.9 kJ mol−1) shows that there is a considerable and attractive interaction between the two molecules at the experimental geometry. However, only part of this attraction can be attributed to the π(CN)···π(phenyl) interaction, as there is also an OH···N interaction in the dimer. When optimizing the geometry (structure D), this interaction makes the structure less parallel. Structure E, where one of the molecules is rotated to avoid this OH···N interaction, remains parallel and has an interaction energy of −29.6 kJ mol−1. Assuming that this interaction energy results from two CN···π interactions, one such interaction is estimated to be −14.8 kJ mol−1.

Stuctures, used in the theoretical calculations on the π(CN)···π(phenyl) interaction in compound 1, and their energies.
Structures B and C aim to eliminate the π(CN)···π(phenyl) and OH···N interactions. The sum of the interaction energies of B and C (−23.9 kJ mol−1) is not far off from that of the full dimer (−22.9 kJ mol−1), so it seems reasonable to assume that the interaction energy of the full dimer is a sum of the π(CN)···π(phenyl) and OH···N interactions. These results suggest that the π(CN)···π(phenyl) interaction accounts for −19.3 kJ mol−1. This is a bit higher than the −14.8 kJ mol−1 estimated above, but is probably an overestimation, as there are more attractive interactions in this dimer than just the π(CN)···π(phenyl) interaction (for example, with the hydrogen atom of the upper molecule above the ring of the lower molecule in structures A and B). In summary, the calculations support that the CN···π interaction is attractive, with a considerable magnitude estimated to be in the range of −15 to −18 kJ mol−1.
Table 4 shows a breakdown of the interaction energy into separate contributions using sSAPTO. The sSAPT0 interaction energies are in reasonable agreement with the PBE0 values. The results show that dispersion is relatively more important in A and B compared to C, and electrostatics are more important in C.
Crystal data.
| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| Crystal data | ||||
| Chemical formula | C7H6ClNO | C7H6FNO | C7H6N2O3 | C7H6N2O3 |
| Mr | 155.58 | 139.13 | 166.14 | 166.14 |
| Crystal system, space group | Monoclinic, P21/c | Monoclinic, P21/c | Monoclinic, P21/n | Monoclinic, P21/c |
| Temperature, K | 100 | 100 | 100 | 100 |
| a, b, c, Å | 12.1214(7), 4.4332(2), 13.6946(8) | 14.2137(5), 3.7672(1), 11.9364(4) | 3.6405(1), 13.4466(6), 14.8993(7) | 6.2083(2), 4.8431(2), 24.3013(8) |
| β, deg | 109.283(6) | 99.210(3) | 95.086(4) | 94.986(3) |
| V, Å3 | 694.62(7) | 630.90(4) | 726.48(5) | 727.91(5) |
| Z | 4 | 4 | 4 | 4 |
| Radiation type | MoKα | MoKα | MoKα | MoKα |
| μ, mm−1 | 0.5 | 0.1 | 0.1 | 0.1 |
| Crystal size, mm3 | 0.20×0.05×0.03 | 0.20×0.10×0.02 | 0.15×0.05×0.03 | 0.06×0.05×0.01 |
| Data collection | ||||
| Diffractometer | (a) | (a) | (a) | (b) |
| Absorption correction | (c) | (c) | (c) | (c) |
| No. of measured/independent/observed [I>2 σ(I)] reflections | 14977/1591/1381 | 13530/1443/1349 | 16152/1669/1549 | 8656/1670/1462 |
| Rint | 0.068 | 0.029 | 0.064 | 0.021 |
| (sin θ/λ)max, Å––1 | 0.649 | 0.649 | 0.649 | 0.649 |
| Refinement | ||||
| No. of reflections | 1591 | 1443 | 1669 | 1670 |
| No. of parameters | 95 | 95 | 113 | 113 |
| No. of restraints | 0 | 0 | 0 | 1 |
| H atom treatment | (d) | (d) | (d) | (d) |
| R[F2>2 σ(F2)], wR(F2), S | 0.041, 0.118, 1.08 | 0.033, 0.101, 1.09 | 0.033, 0.089, 1.07 | 0.036, 0.102, 1.08 |
| Δρmax/min, e Å–3 | 0.53, –0.37 | 0.35, –0.21 | 0.31, –0.16 | 0.38, –0.23 |
| CCDC No | 1846653 | 1846654 | 1846655 | 1856711 |
(a) Rigaku FRE+equipped with VHF Varimax confocal mirrors and an AFC12 goniometer and HyPix 6000 detector diffractometer; (b) XtaLAB AFC12 (RCD3): Kappa single diffractometer; (c) Multi-scan CrysAlis Pro 1.171.39.30d (Rigaku Oxford Diffraction, 2017) Empirical absorption correction using spherical harmonics, implemented in SCALE3 ABSPACK scaling algorithm; (d) H atoms treated by a mixture of independent and constrained refinement. Computer programs used: CrysAlis PRO 1.171.39.30d [38], Oscail [39], Shelxt [40], ShelXle [41], Shelxl2017/1 [42], Mercury [43], Platon [44].
3 Experimental
3.1 Synthesis and crystallization
The compounds, 1–4, were prepared by refluxing the corresponding aldehyde with hydroxyamine in an aqueous solution containing potassium carbonate, rather than pyridine, following a published procedure [45]. They were purified by recrystallization from methanol solutions.
3-Chlorobenzaldehyde oxime, 1: m.p. 64–65°C [46]. 4-Fluorobenzaldehyde oxime, 2:. m.p. 85–87°C (lit [http://synquestlabs.com/product/id/53507.html]: m.p. 85–86°C). 2-Nitrobenzaldehyde oxime, 3: m.p. 97–98°C (lit. [https://www.alfa.com/en/catalog/A14565/]: m.p. 98–102°C). 4-Nitrobenzaldehyde oxime, 4: m.p. 123–124°C (lit. [https://www.alfa.com/en/catalog/L09144/]: m.p. 122–125°C).
3.2 Crystallography
Crystal data, data collection and structure refinement details are summarized in Table 5 [38], [39], [40], [41], [42], [43], [44]. In each case the oxime group has an (E) geometry. In all compounds the oxime hydroxyl hydrogen atoms were refined and their positions checked on a final difference map where all atom positions lie within the maximum contour level. All other hydrogen atoms were refined as riding atoms at a distance of 0.95Å.
Breakdown of the interaction energies of the different dimers using sSAPT0
| Structure | Electrostatics | Exchange | Induction | Dispersion | Total |
|---|---|---|---|---|---|
| A | –21.0 | 50.6 | –4.5 | –50.7 | –25.7 |
| B | –19.7 | 50.0 | –4.5 | –47.3 | –21.5 |
| C | –2.6 | 4.6 | –0.7 | –3.8 | –2.6 |
| D | –24.36 | 45.02 | –6.26 | –45.37 | –30.98 |
| E | –19.07 | 43.41 | –4.05 | –53.27 | –32.98 |
The Hirshfeld surfaces and two-dimensional fingerprint (FP) plots [29], [30] were generated using Crystal Explorer 3.1 [29], [30]. The Hirshfeld surface mapped over dnorm is scaled in the range −0.68 to 1.17.
3.3 Theoretical study of the π(C=N)···π(phenyl) interaction in compound 1
Hydrogen geometries were optimised with the BLYP-D3 density functional (BLYP [47], [48] augmented with Grimme’s D3 dispersion correction [49] in conjunction with the def2-SVP basis set [50] using Orca [51], keeping all other atoms at the experimental geometry. Interaction energies were computed with PBE0-D3 (the hybrid PBE0 functional [52] augmented with D3) and the ma-def2-TZVP basis set [50], [53]. The counterpoise procedure [54] was used to eliminate BSSE. As the heavy atoms are at the crystallographic positions, deformation energies were omitted. The interaction energies were thus computed as follows:
Here, the superscript {AB} denotes that all calculations employ the dimer basis set, and the attributes in round brackets, (AB), denote that all calculations employ the dimer geometry.
Additional calculations at the PBE0-D3/ma-def2-TZVP level employing the counterpoise procedure. The structures were additionally analyzed with Symmetry-Adapted Perturbation Theory (SAPT) [55], which calculates the interaction energy directly without computing the dimer and monomer energies. We employed a scaled version, sSAPT0 [56], of the simplest truncation of the SAPT expansion (SAPT0), with a truncated aug-cc-pVDZ basis set labelled jun-cc-pVDZ (previously called aug-cc-pVDZ’ [57]), using Psi4 [58]. This level of theory was recommended as a good-performing, efficient level of SAPT [56].
4 Conclusions
The crystal structures and the results of the Hirshfeld surface analysis of four aldoximes, XC6H4CH=N–OH (X=3-Cl (1), 4-F (2), 2-O2N (3) and 4-O2N (4), are reported. The strong classical O13–H13···N12 hydrogen bonds involving the oxime moiety generate C3 chains in compound 1, in contrast to the
5 Supporting information
CCDC 1846653–1846655 and 1856711 for compounds 1–4, respectively, contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre viawww.ccdc.cam.ac.uk/data_request/cif.
The crystallographic CIFs are also given as supplementary material available online (DOI: 10.1515/znb-2018-0222).
Acknowledgements
The authors thank the National Crystallographic Service, University of Southampton, UK, for the data collection, and for their help and advice. LRG thanks the Portuguese Foundation for Science and Technology (FCT) UID/Multi/04546/2013.for support.
References
[1] E. Abele, R. Abele, E. Lukevics, Chem. Heterocycl. Compds. 2018, 44, 769.10.1007/s10593-008-0110-9Search in Google Scholar
[2] A. Nikitjuka, A. Jirgensons, Chem. Heterocycl. Compds.2014, 49, 1544.10.1007/s10593-014-1407-5Search in Google Scholar
[3] R. Martinez-Pascual, S. Meza-Reyes, J. L. Vega-Baez, P. Merino-Montiel, J. M. Padron, A. Mendoza, S. Montiel-Smith, Steroids2017, 122, 24.10.1016/j.steroids.2017.03.008Search in Google Scholar PubMed
[4] H. L. Qin, J. Leng, B. G. M. Youssif, M. W. Amjad, M. A. G. Raja, M. A. Hussain, Z. Hussain, S. N. Kazmi, S. N. A. Bukhari, Chem. Bio. Drug Des. 2017, 90, 443.10.1111/cbdd.12964Search in Google Scholar PubMed
[5] C. Canario, S. Silvestre, A. Falcao, G. Alves, Curr. Med. Chem. 2018, 25, 660.10.2174/0929867324666171003115400Search in Google Scholar PubMed
[6] G. Huang, H. R. Zhao, Q. Q. Meng, Q. J. Zhang, J. Y. Dong, B. Q. Zhu, S. S. Li, Eur. J. Med. Chem. 2018, 143, 166.10.1016/j.ejmech.2017.11.031Search in Google Scholar PubMed
[7] H. Dai, J. Chen, G. Li, S. S. Ge, Y. J. Shi, Y. Fang, Y. Ling, Bioorg. Med. Chem. Lett.2017, 27, 950.10.1016/j.bmcl.2016.12.083Search in Google Scholar PubMed
[8] S. Y. Zhao, K. Li, Y. Jin, J. Lin, Eur. J. Med. Chem. 2018, 144, 41.10.1016/j.ejmech.2017.12.016Search in Google Scholar PubMed
[9] P. Yadav, K. Lal, P. Rani, S. Mor, A. Kumar, Med. Chem. Res. 2017, 26, 1469.10.1007/s00044-017-1845-6Search in Google Scholar
[10] J. Kozlowska, B. Potaniec, B. Zarowska, M. Aniol, Molecules2017, 22, 1485.10.3390/molecules22091485Search in Google Scholar PubMed PubMed Central
[11] A. M. Mohassab, H. A. Hassan, D. Abdelhamid, M. Abdel-Aziz, K. N. Dalby, T. S. Kaoud, Bioorg. Chem. 2017, 75, 242.10.1016/j.bioorg.2017.09.018Search in Google Scholar PubMed
[12] D. E. Lorke, H. Kalasz, G. A. Petroianu, K. Tekes, Curr. Med. Chem. 2008, 15, 743.10.2174/092986708783955563Search in Google Scholar PubMed
[13] V. A. Voicu, H. Thiermann, F. S. Radulescu, C. Mircioiu, D. S. Miron, Basic. Clin. Pharmacol. Toxicol. 2010, 106, 73.10.1111/j.1742-7843.2009.00486.xSearch in Google Scholar PubMed
[14] M. Katalinic, A. Zandona, A. Ramic, T. Zorbaz, I. Primozic, Z. Kovarik, Molecules2017, 22, 1234.10.3390/molecules22071234Search in Google Scholar PubMed PubMed Central
[15] Z. Radic, T. Dale, Z. Kovarik, S. Berend, E. Garcia, L. Zhang, G. Amitais, C. Green, B. Radi, B. M. Duggan, D. Ajami, P. Taylor, Biochem. J. 2013, 450, 231.10.1042/BJ20121612Search in Google Scholar PubMed PubMed Central
[16] M. Sorensen, E. H. J. Neilson, B. L. Moller, Mol. Plant, 2018, 11, 95.10.1016/j.molp.2017.12.014Search in Google Scholar PubMed
[17] V. Bertolasi, G. Gilli, A. C. Veronese, Acta Crystallogr.1982, B38, 502.10.1107/S0567740882003288Search in Google Scholar
[18] E. A. Bruton, L. Brammer, F. C. Pigge, C. B. Aakeröy, D. S. Leinen, New J. Chem. 2003, 27, 1084.10.1039/B301045GSearch in Google Scholar
[19] J. N. Low, L. M. N. B. F. Santos, C. F. R. A. C. Lima, P. Brandão, L. R. Gomes, Eur. J. Chem. 2010, 61.10.5155/eurjchem.1.2.61-66.76Search in Google Scholar
[20] C. B. Aakeroy, A. S. Sinha, K. N. Epa, P. D. Chopada, M. M. Smit, J. Desper, Cryst. Growth Des. 2013, 13, 2687.10.1021/cg4005246Search in Google Scholar
[21] J. N. Low, J. L. Wardell, C. F. da Costa, M. V. N. de Souza, L. R. Gomes, Eur. J. Chem. 2018, 9, 151.10.5155/eurjchem.9.3.151-160.1734Search in Google Scholar
[22] M. C. Etter, Acc. Chem. Res. 1990, 23, 120.10.1021/ar00172a005Search in Google Scholar
[23] M. Tonogaki, T. Kawata, S. Ohba, Y. Iwata, I. Shibuya, Acta Crystallogr. 1993, B49, 1031.10.1107/S0108768193005130Search in Google Scholar
[24] V. Sharutin, CSD Communication code: 1045607, 2016.Search in Google Scholar
[25] L. Brehm, K. J. Watson, Acta Crystallogr. 1972, B28, 3646.10.1107/S0567740872008507Search in Google Scholar
[26] F. Bachechi, L. Zambonelli, Acta Crystallogr. 1972, B28, 2489.10.1107/S056774087200634XSearch in Google Scholar
[27] E. Pindelska, T. M. Krygowski, R. Anulewicz-Ostrowska, M. K.Cyranski, J. Nowacki, J. Phys. Org. Chem. 2001, 14, 764.10.1002/poc.429Search in Google Scholar
[28] A. D. Ward, V. R. Ward, E. R. T. Tiekink, Z. Kristallogr. NCS2001, 216, 583.10.1524/ncrs.2001.216.14.597Search in Google Scholar
[29] S. K. Wolff, D. I. Grimwood, J. J. McKinnon, M. J. Turner, D. Jayatilaka, M. A. Spackman, Crystal Explorer (version 3.1), The University of Western Australia, Perth (Australia) 2012.Search in Google Scholar
[30] J. J. McKinnon, M. A. Spackman, A. S. Mitchell, Acta Crystallogr. 2004, B60, 627.10.1107/S0108768104020300Search in Google Scholar PubMed
[31] J. T. Mague, Y. El Bakri, CSD private Communication, CBALOS02: 1506791, 2016.Search in Google Scholar
[32] M. Daszkiewicz, CrystEngComm2013, 15, 10427.10.1039/c3ce41788cSearch in Google Scholar
[33] A. Abbas, S. R. Hussain, N. Hafeez, A. Badshah, A. Hasan, K. M. Lo, Acta Crystallogr. 2010, E66, o1130.10.1107/S1600536810013978Search in Google Scholar
[34] P. A. Wood, R. S. Forgan, D. Henderson, S. Parsons, E. Pidcock, P. A. Tasker, J. E. Warren, Acta Crystallogr. 2006, B62, 1099.10.1107/S0108768106031752Search in Google Scholar PubMed
[35] P. A. Wood, R. S. Forgan, A. R. Lennie, S. Parsons, E. Pidcock, P. A. Tasker, J. E. Warren, CrystEngComm, 2008, 10, 239.10.1039/B712397CSearch in Google Scholar
[36] C. B. Aakeroy, A. S. Sinha, K. N. Epa, C. L. Spartz, J. Desper, Chem. Commun. 2012, 48, 11289.10.1039/c2cc36315aSearch in Google Scholar PubMed
[37] S. Akine, T. Taniguchi, W. Dong, S. Masubuchi, T. Nabeshima, J. Org. Chem.2005, 70, 1704.10.1021/jo048030ySearch in Google Scholar PubMed
[38] Rigaku Oxford Diffraction, Rigaku Corporation, Tokyo, Japan, 2017.Search in Google Scholar
[39] P. McArdle, K. Gilligan, D. Cunningham, R. Dark, M. Mahon, CrystEngComm2004, 6, 303.10.1039/B407861FSearch in Google Scholar
[40] G. M. Sheldrick, Acta Crystallogr.2015, A71, 3.10.1107/S2053273314026370Search in Google Scholar
[41] C. B. Hübschle, G. M. Sheldrick, B. Dittrich, J. Appl. Crystallogr.2011, 44, 1281.10.1107/S0021889811043202Search in Google Scholar PubMed PubMed Central
[42] G. M. Sheldrick, Shelxl-2017/1, University of Göttingen, Göttingen (Germany) 2017.Search in Google Scholar
[43] Mercury, Crystal Structure Visualisation, Exploration and Analysis made Easy, CCDC, 2018.Search in Google Scholar
[44] A. L. Spek, Acta Crystallogr.2009, D65, 148.10.1107/S090744490804362XSearch in Google Scholar
[45] Vogel´s Textbook of Practical Organic Chemitry, 5th edition, revised by B. S. Furniss, A. J. Hannaford, P. W. G. Smith, A. R. Tatchell, Longman Group UK Limited, London, 1989, p. 1259.Search in Google Scholar
[46] L. Zhang, J. C. Chung, T. D. Costello, I. Valvis, P. Ma, S. Kauffman, R. Ward, J. Org. Chem. 1997, 62, 2466.10.1021/jo9612537Search in Google Scholar PubMed
[47] A. D. Becke, Phys. Rev. 1988, A38, 3098.10.1103/PhysRevA.38.3098Search in Google Scholar
[48] C. Lee, W. Yang, R. G. Parr, Phys. Rev. 1988, B37, 785.10.1103/PhysRevB.37.785Search in Google Scholar
[49] S. Grimme, J. Antony, S. Ehrlich, H. Krieg, J. Chem. Phys. 2010, 132, 154104.10.1063/1.3382344Search in Google Scholar PubMed
[50] F. Weigend, R. Ahlrichs, Phys. Chem. Chem. Phys. 2005, 7, 3297.10.1039/b508541aSearch in Google Scholar PubMed
[51] F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci.2012, 2, 73.10.1002/wcms.81Search in Google Scholar
[52] C. Adamo, V. Barone, J. Chem. Phys. 1999, 110, 6158.10.1063/1.478522Search in Google Scholar
[53] J. Zheng, X. Xu, D. G. Truhlar, Theor. Chem. Acc. 2011, 128, 295.10.1007/s00214-010-0846-zSearch in Google Scholar
[54] S. F. Boys, F. Bernardi, Mol. Phys. 1970, 19, 553.10.1080/00268977000101561Search in Google Scholar
[55] B. Jeziorski, R. Moszynski, K. Szalewicz, Chem. Rev. 1994, 94, 1887.10.1021/cr00031a008Search in Google Scholar
[56] T. M. Parker, L. A. Burns, R. M. Parrish, A. G. Ryno, C. D. Sherrill, J. Chem. Phys. 2014, 140, 094106.10.1063/1.4867135Search in Google Scholar PubMed
[57] E. G. Hohenstein, R. M. Parrish, C. D. Sherrill, J. M. Turney, H. F. Schaefer, J. Chem. Phys. 2011, 35, 174107.10.1063/1.3656681Search in Google Scholar PubMed
[58] J. M. Turney, A. C. Simmonett, R. M. Parrish, E. G. Hohenstein, F. Evangelista, J. T. Fermann, B. J. Mintz, L. A. Burns, J. J. Wilke, M. L. Abrams, N. J. Russ, M. L. Leininger, C. L. Janssen, E. T. Seidl, W. D. Allen, H. F. Schaefer, R. A. King, E. F. Valeev, C. D. Sherrill, T. D. Crawford, Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 556.10.1002/wcms.93Search in Google Scholar
Supplementary Material
The online version of this article offers supplementary material (https://doi.org/10.1515/znb-2018-0222).
©2019 Walter de Gruyter GmbH, Berlin/Boston
Articles in the same Issue
- Frontmatter
- In this Issue
- Review
- Coloring in the ZrBeSi-type structure
- Research Articles
- Different substituent effects on the supramolecular arrays in some (E)-halo- and nitro-benzaldehyde oximes: confirmation of attractive π(C=N)···π(phenyl) interactions
- Synthesis, crystal structure, and magnetic properties of a multicage compound: [(Me)2EtNH][Mn(N3)3] with a perovskite-related structure
- A new one-dimensional Cd(II) coordination polymer with a two-dimensional supramolecular architecture: synthesis, structural characterization and fluorescence properties
- Cycloaddition reactions of acetylenic iminium salts and diazoacetates leading to pyrazole iminium salts
- High-pressure synthesis and crystal structure of the mixed-cation borate Ga4In4B15O33(OH)3
- Evaluation of benzthiazolidine-based formamidinium salts for the synthesis of penem-type β-lactams by uncatalysed carbonylation of acyclic diaminocarbenes
- Temperature- and solvate-dependent disorder in the crystal structure of [PNP]+[HSO4]−
- Serendipitous formation and characterization of K2[Pd(NO3)4]·2HNO3
- Phosphanchalkogenide und ihre Metallkomplexe. V. Derivate von [2.2]Paracyclophanylphosphanena
Articles in the same Issue
- Frontmatter
- In this Issue
- Review
- Coloring in the ZrBeSi-type structure
- Research Articles
- Different substituent effects on the supramolecular arrays in some (E)-halo- and nitro-benzaldehyde oximes: confirmation of attractive π(C=N)···π(phenyl) interactions
- Synthesis, crystal structure, and magnetic properties of a multicage compound: [(Me)2EtNH][Mn(N3)3] with a perovskite-related structure
- A new one-dimensional Cd(II) coordination polymer with a two-dimensional supramolecular architecture: synthesis, structural characterization and fluorescence properties
- Cycloaddition reactions of acetylenic iminium salts and diazoacetates leading to pyrazole iminium salts
- High-pressure synthesis and crystal structure of the mixed-cation borate Ga4In4B15O33(OH)3
- Evaluation of benzthiazolidine-based formamidinium salts for the synthesis of penem-type β-lactams by uncatalysed carbonylation of acyclic diaminocarbenes
- Temperature- and solvate-dependent disorder in the crystal structure of [PNP]+[HSO4]−
- Serendipitous formation and characterization of K2[Pd(NO3)4]·2HNO3
- Phosphanchalkogenide und ihre Metallkomplexe. V. Derivate von [2.2]Paracyclophanylphosphanena