Synthesis, crystal structure and Raman spectrum of Ba7[BO3]3Br(O1.33F1.33)
-
Olaf Reckeweg
 ,
Armin Schulz
,
Armin Schulz

Abstract
In addition to amorphous material and Ba7[BO3]4−x F2−3x , air and moisture sensitive single crystals of Ba7[BO3]3Br(O1.33F1.33) were formed from H3BO3, Ba(OH)2, BaF2 and BaBr2·2 H2O in alumina crucibles open to the atmosphere at 1300 K for 13 h. Ba7[BO3]3Br(O1.33F1.33) crystallizes in the hexagonal space group P63mc (no. 186, Z=2) with the lattice parameters a=1118.1(2) and c=723.93(13) pm. The Raman spectrum of the title compounds was also acquired and is compared to literature data.
1 Introduction
In 2010 we reported the structure of some orthoborates with the general composition M5[BO3]3X (M=Eu, Ba; X=Cl, Br) [1]. At that time, we also performed experiments with fluoride borates, but our results were inconclusive to us. So we just mentioned these compounds with the tentative compositions “Ba7[BO3]3F(Y)4” and “Ba7[BO3]3Br(Z)4”. Some bromide borates such as Ba2[BO3]Br and Ba3[BO3]Br3 [2] and a whole class of fluoride borates with the general composition Ba7[BO3]4−x F2−3x have been reported recently [3, 4]. One of the of latter compounds showed lattice parameters and crystallographic coordinates which were nearly identical with our Ba7[BO3]3F(Y)4. A disorder between oxygen and fluoride and isomorphous replacement of the borate anion by three fluoride ions to were used to explain the somewhat unexpected stoichiometry and geometries.
After applying this knowledge to Ba7[BO3]3Br(Z)4, we can now report the synthesis, the structural characterization by X-ray single crystal methods and the Raman spectrum of Ba7[BO3]3Br(O1.33F1.33).
2 Experimental section
2.1 Synthesis
All manipulations were carried out under normal atmosphere. The stoichiometry of the starting mixture was according to that of the initial target compound “Ba7[BO3]3BrF4”. The overall mass of the educts was 1 g. Ba7[BO3]3Br(O1.33F1.33) was obtained as minority product by reacting mixtures of H3BO3 (Fisher Scientific, Analytical Grade), Ba(OH)2 (Fisher Scientific, Analytical Grade), BaF2 (Alfa Aesar, powder, ultra dry, 99.995%) and BaBr2·2 H2O (Fisher Scientific, Analytical Grade). The ground educts were placed in an alumina crucible and heated over 13 h from room temperature to 1300 K. This temperature was held for an additional 13 h. After that, the furnace was shut off and allowed to cool to room temperature. Most of the sintered reaction cake was found to be amorphous, but next to some brick-shaped blocks of Ba5[BO3]3Br [2], a few crystals of Ba2[BO3]Br [2] appeared as hexagonal prisms as the barium fluoride borates do [3, 4] while the few specimens of Ba7[BO3]3Br(O1.33F1.33) had needle like morphologies.
The remaining crystals that were not used for X-ray or Raman measurements were heated with a few drops of concentrated sulfuric acid in a small test tube showing the etching reaction typical for a fluoride containing compound reacting with glass surfaces.
Ba7[BO3]3Br(O1.33F1.33) reacts with air and/or moisture after a few days and yields either X-ray amorphous products or BaCO3.
2.2 Raman spectroscopy
Single crystals of the title compounds were sealed under a protective argon atmosphere inside Mark capillaries and used for the Raman investigations (microscope laser Raman spectrometer: Jobin Yvon, 1 mW, excitation line at λ=632.817 nm (HeNe laser), 20× magnification, 360 s accumulation time, Fig. 1).
![Fig. 1: Raman spectrum of Ba7[BO3]3Br(O1.33F1.33). Raman intensity is displayed on the vertical axis in arbitrary units.](/document/doi/10.1515/znb-2016-0243/asset/graphic/j_znb-2016-0243_fig_001.jpg)
Raman spectrum of Ba7[BO3]3Br(O1.33F1.33). Raman intensity is displayed on the vertical axis in arbitrary units.
2.3 Crystallographic studies
Needle-shaped crystals were chipped off the sintered transparent colorless mass adhering to the crucible and immersed in polybutene oil (Aldrich, Mn~320, isobutylene>90%). Suitable single crystals were selected under a polarization microscope, mounted in a drop of polybutene sustained in a plastic loop, and placed onto the goniometer. A cold stream of nitrogen (T=173(2)K) froze the polybutene oil, thus keeping the crystal stationary and protected from oxygen and moisture. Preliminary examination and subsequent data collection were performed on a Bruker X8 Apex II diffractometer equipped with a 4 K CCD detector and graphite-monochromatized MoKα radiation (λ=71.073 pm). The intensity data were handled with the program package that came with the diffractometer [5]. An empirical absorption correction was applied using Sadabs [6]. The program Shelxs-97 [7, 8] found the positions of barium and bromine with the help of Direct Methods. The positions of the light atoms were apparent from the positions of the highest electron density on the difference Fourier maps resulting from the first refinement cycles by full-matrix least-squares calculations on F2 in Shelxl-97 [9, 10]. The stoichiometry would have been “Ba7[BO3]3BrF4” at this stage, but the atoms F1 and F2 showed unusually large displacement parameters. The free refinement of the site occupation factor yielded “Ba7[BO3]3BrF2.6”, but this composition did not concur with the electroneutrality of the compound demanded by its transparency and lack of color. Since oxides and fluorides often show similar crystal chemical behavior when stabilized by a suitable host structure, a 1:1 split occupancy of these positions with F and O was introduced leading to balanced ionic charges following (Ba2+)7([BO3]3−)3(Br−)(O2−)1.33(F−)1.33. No surplus electron densities were found in the hollow F2/O4 tetrahedron which indicates the absence of additional [BO3]3− moieties. For the final refinement cycles the positions with the split occupancies were fixed to the previously obtained occupation factors. The refinement converged into a stable model for the crystal structure. Additional crystallographic details are given in Table 1. Atomic coordinates and equivalent isotropic displacement coefficients are shown in Table 2, Table 3 displays the ranges of the bond lengths.
Summary of single-crystal X-ray diffraction structure determination data of Ba7[BO3]3Br(O1.33F1.33).
| Compound | Ba7[BO3]3Br(O1.33F1.33) |
|---|---|
| Mr | 1264.44 |
| Crystal color | Transparent colorless |
| Crystal shape | Hexagonal needle |
| Crystal size, mm3 | 0.03×0.03×0.17 |
| Crystal system | Hexagonal |
| Space group (no.) | P63mc (186) |
| Z | 2 |
| Lattice parameters a, pm b, pm c, pm |
1118.1(2) a 723.93(13) |
| V, Å3 | 783.7(3) |
| Dcalcd, gcm−3 | 5.36 |
| F(000); e | 1073 |
| Diffractometer | Bruker X8 Apex II diffractometer equipped with a 4 K CCD detector |
| Radiation/λ, pm/monochromator | MoKα /λ=71,073 pm/graphite |
| Scan mode | φ and ω scans |
| T, K | 173(2) |
| Ranges, 2θmax, deg | 60.97 |
| hkl range | ±13, –15→9, –9→10 |
| Data correction | LP, Sadabs [6] |
| μ(MoKα ), mm−1 | 19.9 |
| Transmission: min/max | 0.5126/0.7465 |
| Reflections: measured/unique | 6005/899 |
| Unique reflections with Fo>4σ(Fo) | 859 |
| |E2–1| | 0.782 |
| Rint/Rσ | 0.041/0.028 |
| Refined parameters | 45 |
| Flack parameter [11, 12] | 0.02(7) |
| R1a/wR2b/GoFc (all refl.) | 0.037/0.083/1.096 |
| Max. shift/esd, last refinement cycle | <0.00005 |
| ∆ρfin (max/min), e−/Å3 | 1.9/–3.8 |
| CSD number | 431987 |
aR1=Σ||Fo|–|Fc||/Σ|Fo|; bwR2=[Σw(Fo2–Fc2)2/Σ(wFo2)2]1/2; w=1/[σ2(Fo2)+(xP)2+yP] with P=[(Fo2)+2Fc2]/3; cGooF(S)=[Σw(Fo2–Fc2)2/(n–p)]1/2, with n being the number of reflections and p being the number of refined parameters.
Atomic coordinates and equivalent isotropica displacement parameters (pm2) of Ba7[BO3]3Br(O1.33F1.33).
| Atom | Wyckoff site | s.o.f. | x | y | z | Ueq |
|---|---|---|---|---|---|---|
| Ba1 | 6c | 1 | 0.14583(4) | 2x | 0.00835(12) | 165(2) |
| Ba2 | 6c | 1 | 0.52915(4) | 2x | 0.16675(11) | 166(2) |
| Ba3 | 2b | 1 | 1/3 | 2/3 | 0.2818(3) | 192(3) |
| Br | 2a | 1 | 0 | 0 | 0.2810(3) | 190(5) |
| O1 | 12d | 1 | 0.0910(8) | 0.3940(8) | 0.3622(11) | 196(15) |
| O2 | 6c | 1 | 0.3218(16) | –x | 0.1250(17) | 307(30) |
| O3/F1 | 2b | 1/3:1/3 | 1/3 | 2/3 | 0.666(5) | 281(61) |
| O4/F2 | 6c | 1/3:1/3 | 0.5928(11) | –x | 0.475(3) | 476(52) |
| B | 6c | 1 | 0.1847(7) | 2x | 0.452(2) | 94(25) |
aUeq is defined as a third of the orthogonalised Uij tensor.
Range of atomic distances for Ba7[BO3]3Br(O1.33F1.33) compared with the respective sum of their ionic radii for the coordination numbers VI to X for Ba2+ according to Shannon [13].
| Ranges of distances for Ba7[BO3]3Br(O1.33F1.33) | Ranges of ionic radii sums for different C.N. [13] | |
|---|---|---|
| Ba2+–F– | 255(2)–278(4) | 266–283 |
| Ba2+–O2− | 255(2)–310(1) | 275–292 |
| Ba2+–Br− | 326.9(2);344.6(2) | 331–348 |
| B3+–O2− | 134(2);137(1) | 140 |
Distances are given in pm.
Further details of the crystal structure investigation may be obtained from FIZ Karlsruhe, 76344 Eggenstein-Leopoldshafen, Germany [fax: (+49)7247-808-666; e-mail: crysdata@fiz-karlsruhe.de] on quoting the deposition number CSD-431987 for Ba7[BO3]3Br(O1.33F1.33).
3 Results and discussion
3.1 Raman spectra of Ba7[BO3]3Br(O1.33F1.33)
The Raman spectroscopic data (Fig. 1 and Table 4) show clearly the presence of the orthoborate [BO3]3− anion when compared to other compounds containing this anion. The symmetric stretching mode is clearly detectable in the Raman spectrum of the title compound at a frequency typical of many orthoborates. The symmetric stretching mode of carbonate moieties was found at 1035 cm−1 – barely visible with <4% of the intensity of analog mode of the orthoborate anion. In Ba2[BO3]0.9[CO3]0.1Cl1.1 [14] the relation of the aforementioned intensities is 1:1/3. Therefore it can be assumed, that virtually no carbonate is present in the title compound.
Optical frequencies (given in cm−1) for selected compounds containing the [BO3]3− orthoborate anion.
| Compound | νsym | νsym (CO32–) | νasym | δ | γ |
|---|---|---|---|---|---|
| Ba2[BO3]0.9[CO3]0.1Cl1.1 [14] | 913 | 1063 | 1180 | 768/787 | 573 |
| Ba5[BO3]3Br [1] | 906 | – | – | 745 | 581/599 |
| Ba2[BO3]Br [2] | 910 | – | 1173 | 768/787 | 571 |
| Ba3[BO3]Br3 [2] | 900/915 | – | 1215 | 755/789 | 600 |
| Ba7[BO3]3Br(O1.33F1.33) | 914 | 1035 | 1166 | 769 | 605 |
Frequencies given were obtained by Raman measurements only.
3.2 The crystal structure of Ba7[BO3]3Br(O1.33F1.33)
The crystal structure of Ba7[BO3]3Br(O1.33F1.33) can be seen as complex 3D structure forming channels parallel to the crystallographic c axis which are filled by bromide anions rattling inside these large pores (Fig. 2). The borate anions are close to D3h symmetry with familiar bond lengths (around 136 pm) and angles (approximately 120°) and it is surrounded in a familiar fashion by a tricapped trigonal prism of barium atoms (Fig. 3). The plane of these anions is parallel to the crystallographic c axis. Fig. 4a shows the F1/O3 position which is approximately in the centre of a distorted tetrahedron of Ba2+, while Fig. 4b displays the F2/O4 position which is in an environment suitable for a borate unit [3, 4], but there are no indications of any surplus electron density inside this tetrahedron that would indicate disordered [BO3]3− moieties. All bond lengths are in a familiar range (Table 4) when compared to the sum of the ionic radii suggested by Shannon [13].
![Fig. 2: View of the unit cell of Ba7[BO3]3Br(O1.33F1.33) along the crystallographic c axis. The borate anion is displayed as cyan colored triangle, Ba–F/O bonds are shown as black lines.](/document/doi/10.1515/znb-2016-0243/asset/graphic/j_znb-2016-0243_fig_002.jpg)
View of the unit cell of Ba7[BO3]3Br(O1.33F1.33) along the crystallographic c axis. The borate anion is displayed as cyan colored triangle, Ba–F/O bonds are shown as black lines.
![Fig. 3: Coordination polyhedron around the [BO3]3− anion. The same color code as in Fig. 2 is used. Ba–O bonds are shown as broken lines and B–O bonds as black lines.](/document/doi/10.1515/znb-2016-0243/asset/graphic/j_znb-2016-0243_fig_003.jpg)
Coordination polyhedron around the [BO3]3− anion. The same color code as in Fig. 2 is used. Ba–O bonds are shown as broken lines and B–O bonds as black lines.
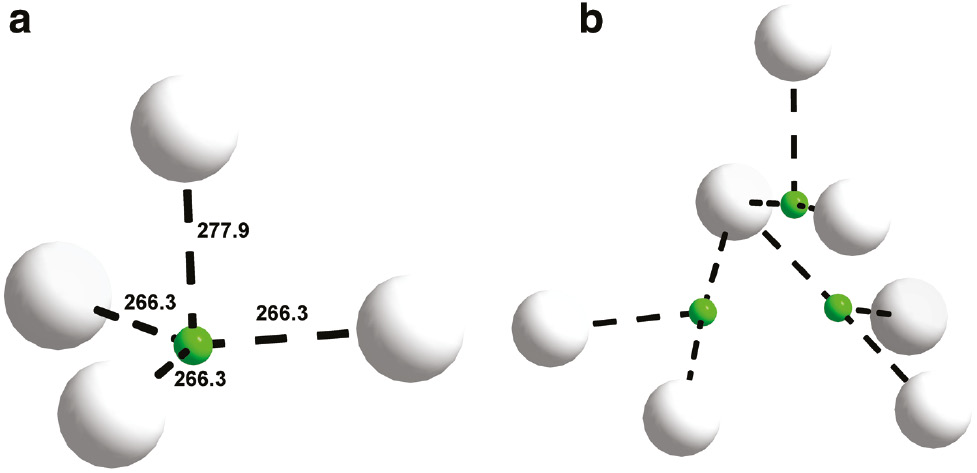
Coordination polyhedra around the F1/O3 and F2/O4 positions. The same color code as in Fig. 3 is used.
4 Conclusion
Ba7[BO3]3Br(O1.33F1.33) is the first borate containing two different halides. Its crystal structure was solved and refined sometime after its synthesis due to structure solutions and refinements in recently reported borates. The Raman spectrum of Ba7[BO3]3Br(O1.33F1.33) indicates the absence of carbonate and the presence of borate anions in the structure. The channel structure seems to be quite stable being able to harbor quite different anions such as bromide ions as the title compound demonstrates or fluoride anions as Ba7[BO3]4−x F2−3x [3, 4] shows.
References
[1] O. Reckeweg, A. Schulz, F. J. DiSalvo, Z. Naturforsch.2011, 66b, 359–365.10.1515/znb-2011-0404Search in Google Scholar
[2] J. Zhao, R. K. Li, Solid State Sci.2013, 24, 54–57.10.1016/j.solidstatesciences.2013.07.009Search in Google Scholar
[3] T. B. Bekker, V. Rashchenko, V. V. Bakakin, Yu. V. Seryotkin, P. P. Fedorov, A. E. Kokh, S. Yu Stonoga, Cryst. Eng. Comm.2012, 14, 6910–6915.10.1039/c2ce26122gSearch in Google Scholar
[4] S. V. Rashchenko, T. B. Bekker, V. V. Bakakin, Yu. V. Seryotkin, A. E. Kokh, A. Gille, A. I. Popov, P. P. Fedorov, J. Appl. Crystallogr.2013, 46, 1081–1084.10.1107/S0021889813015756Search in Google Scholar
[5] Apex2 (version 1.22), Saint Plus,Xprep (version 6.14), Software for the CCD system, Bruker Axs Inc., Madison, WI (USA) 2004.Search in Google Scholar
[6] G. M. Sheldrick, Sadabs, University of Göttingen, Göttingen (Germany) 2003.Search in Google Scholar
[7] G. M. Sheldrick, Shelxs-97, Program for the Solution of Crystal Structures, University of Göttingen, Göttingen (Germany) 1997.Search in Google Scholar
[8] G. M. Sheldrick, Acta Crystallogr.1990, A46, 467–473.10.1107/S0108767390000277Search in Google Scholar
[9] G. M. Sheldrick, Shelxl-97, Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen (Germany) 1997.Search in Google Scholar
[10] G. M. Sheldrick, Acta Crystallogr.2008, A64, 112–122.10.1107/S0108767307043930Search in Google Scholar PubMed
[11] H. D. Flack, Acta Crystallogr.1983, A39, 876–881.10.1107/S0108767383001762Search in Google Scholar
[12] H. D. Flack, G. Bernardinelli, Acta Crystallogr.1999, A55, 908–915.10.1107/S0108767399004262Search in Google Scholar PubMed
[13] R. D. Shannon, Acta Crystallogr.1976, A32, 751–767.10.1107/S0567739476001551Search in Google Scholar
[14] J. Zhao, R. K. Li, Inorg. Chem.2012, 51, 4568–457110.1021/ic3005135Search in Google Scholar PubMed
©2017 Walter de Gruyter GmbH, Berlin/Boston
Articles in the same Issue
- Frontmatter
- In this Issue
- Synthesis, crystal structure and magnetic properties of the complex [C(NH2)3]2[Mn(N3)4] with a polynuclear azido-bridged chain anion
- A new furan carboxamide and two potential precursors from a terrestrial streptomycete
- A Co(II) complex based on a mixed N- and O-donor: synthesis, structural characterization, and properties
- Synthesis, structural characterization, and fluorescence of a Cd(II) coordination polymer with N-donor ligands
- Note
- Synthesis, crystal structure and Raman spectrum of Ba7[BO3]3Br(O1.33F1.33)
- A route to new colorimetric pH sensors
- Electronic structure and chemical bonding in LaIrSi-type intermetallics
- Synthesis and characterization of the alkali borate-nitrates Na3–x Kx[B6O10]NO3 (x=0.5, 0.6, 0.7)
- Synthesis, single-crystal structure determination and Raman spectrum of Ca2.57(4)Sr0.43(4)Cl2[CBN]
Articles in the same Issue
- Frontmatter
- In this Issue
- Synthesis, crystal structure and magnetic properties of the complex [C(NH2)3]2[Mn(N3)4] with a polynuclear azido-bridged chain anion
- A new furan carboxamide and two potential precursors from a terrestrial streptomycete
- A Co(II) complex based on a mixed N- and O-donor: synthesis, structural characterization, and properties
- Synthesis, structural characterization, and fluorescence of a Cd(II) coordination polymer with N-donor ligands
- Note
- Synthesis, crystal structure and Raman spectrum of Ba7[BO3]3Br(O1.33F1.33)
- A route to new colorimetric pH sensors
- Electronic structure and chemical bonding in LaIrSi-type intermetallics
- Synthesis and characterization of the alkali borate-nitrates Na3–x Kx[B6O10]NO3 (x=0.5, 0.6, 0.7)
- Synthesis, single-crystal structure determination and Raman spectrum of Ca2.57(4)Sr0.43(4)Cl2[CBN]