Alkylation of tetrathiotungstate anions: crystal structures of the alkylthiolatotrithiotungstate complexes [PPh4]2[WS3(Sn Pr)][WS3(Sn Bu)]·½C6H6 and [PPh4][WS3(SCH2C6H4CH2Cl-4)]
-


Abstract
The reaction of [PPh4]2[WS4] in CH3CN with excess n-propylbromide or n-butylbromide gave alkylthiolatotrithiotungstate complexes, [PPh4][WS3(SR)] (1: R=n-propyl; 2: R=n-butyl). The analogous reaction with 1-bromomethyl-4-chloromethyl-benzene afforded a benzylthiolatotrithiotungstate complex, [PPh4][WS3(SCH2C6 H4CH2Cl-4)] (3), whereas the reaction with 1,4-bis-bromomethyl-benzene led to isolation of a dinuclear complex [PPh4]2[WS3(μ-SCH2C6H4CH2S)WS3] (4). Complexes 1–4 were characterized spectroscopically and the crystal structures of 1/2·½C6H6 and 3 have been determined by X-ray diffraction.
1 Introduction
Since Boorman et al. reported an alkylation reaction between [PPh4]2[WS4] and EtBr under carefully controlled conditions to give [PPh4][WS3(SEt)] [1], the preparation and structural characterization of other alkylated complexes, [PPh4][WS3(SR)] (R=i Pr, i Bu, t Bu, benzyl, allyl), were studied by the same group [2]. Later, Tatsumi et al. used [PPh4][WS3(SEt)] as starting material to react it with phosphine-containing ligands, and they obtained two five-coordinated trithiotungsten complexes (PPh4)[(dmsp)WS3] and (PPh4)[(dpsp)WS3] (dmsp: Me2PCH2CH2S-; dpsp: Ph2PCH2CH2S-), which could serve as potential synthons for various heterometallic sulfide clusters [3], [4], [5], [6]. Meanwhile, Tatsumi et al. also reported that interactions of [PPh4]2[WS4] with excess n-hexylbromide, 1,4-dichlorobutane, 2-(bromomethyl)tetrahydro-2H-pyran) (bmthp), and 2-(bromoethyl)-1,3-dioxane (bedo) gave a series of new alkylthiolatotrithiotungstate complexes [PPh4][WS3(SR)] (R=n-hexyl, ClCH2CH2CH2CH2-, mthp, edo) [7]. We have been particularly interested in developing the chemistry of tetrathiometallate complexes [8], especially with respect to their alkylation. Herein we report the synthesis of four alkylthiolatotrithiotungstate complexes, [PPh4][WS3(Sn Pr)], [PPh4][WS3(Sn Bu)], [PPh4][WS3(SCH2C6H4CH2 Cl-4)], and [PPh4]2[WS3(μ-SCH2C6H4CH2S)WS3].
2 Experimental section
2.1 General
All syntheses were performed in oven-dried glassware under a purified nitrogen atmosphere using standard Schlenk techniques. The solvents were purified by conventional methods and degassed prior to use. (NH4)2[WS4], n-propylbromide, n-butylbromide, 1-bromomethyl-4-chloromethyl-benzene, and 1,4-bis-bromomethyl-benzene were purchased from Alfa Aesar Ltd. and used as supplied. [Ph4P]2[WS4] was prepared by the addition of (NH4)2[WS4] in water solution to an aqueous solution of Ph4PBr. The resulting solid was collected by filtration; washed with water, ethanol, and diethyl ether; and dried in vacuo. All elemental analyses were carried out using a Perkin-Elmer 2400 CHN analyzer. NMR spectra were recorded on a Bruker ALX 400 spectrometer operating at 400 MHz for 1H. Chemical shifts (δ in ppm) are reported with reference to SiMe4 (1H).
2.2 Synthesis of [PPh4][WS3(SnPr)] (1)
To a solution of [Ph4P]2[WS4] (250 mg, 0.25 mmol) in CH3CN (20 mL) was added n-propylbromide (0.15 mL, 1.60 mmol) in CH3CN (5 mL) with stirring at 5°C. The resulting mixture was stirred at room temperature for 1 h, during which the color gradually changed from yellow to red. The solvent was removed in vacuo, and the residue was treated with THF (10 mL) and filtered. The filtrate was layered with 20 mL of Et2O and allowed to stand at ca. 4°C. Red needle-shaped micro-crystals of 1 were obtained after 5 days, and were isolated by filtration, washed with Et2O, and dried in vacuo. Yield: 77 mg (46%). – Anal. for C27H27PS4W: calcd. C 46.69, H 3.92; found C 46.47, H 3.90. – 1H NMR (CDCl3, ppm): δ=1.12 (t, 3H, CH3), 1.65 (m, 2H, SCH2CH2), 3.21 (t, 2H, SCH2), 7.59–7.86 (m, 20H, Ph4P).
2.3 Synthesis of [PPh4][WS3(SnBu)] (2)
Compound 2 was synthesized by a procedure similar to that used for 1 except that n-butylbromide (0.63 mL, 1.60 mmol) was added and the mixture was stirred for 3 h before the solvent and volatiles were removed in vacuo. After the residue was extracted with THF (2×5 mL) and the solution concentrated to ca. 5 mL, 20 mL Et2O was introduced to precipitate 2 as an orange-red solid. Yield: 98 mg (57%). – Anal. for C28H29PS4W: calcd. C 47.46, H 4.12; found C 47.21, H 4.17. – 1H NMR (CDCl3, ppm): δ=0.97 (t, 3H, CH3), 1.47 (m, 2H, SCH2CH2), 1.94 (m, 2H, SCH2CH2CH2), 3.42 (t, 2H, SCH2), 7.63–7.92 (m, 20H, Ph4P).
2.4 Synthesis of [PPh4][WS3(SCH2C6H4 CH2Cl-4)] (3)
A solution of 1-bromomethyl-4-chloromethyl-benzene (220 mg, 1.00 mmol) in CH3CN (5 mL) was added to a CH3CN solution (20 mL) of [Ph4P]2[WS4] (250 mg, 0.25 mmol), and the mixture was stirred overnight at room temperature, during which the color of the solution slowly changed from yellow to red. From a workup similar to that used for the isolation of 1, red block crystals of 3 were obtained in 3 days. Yield: 100 mg (51%). – Anal. for C32H28PClS4W: calcd. C 48.58, H 3.57; found C 48.33, H 3.62. – 1H NMR (CDCl3, ppm): δ=3.64 (s, 2H, SCH2), 4.78 (s, 2H, CH2Cl), 7.01–7.18 (m, 4H, C6H4 ), 7.61–7.95 (m, 20H, Ph4P).
2.5 Synthesis of [PPh4]2[WS3 (μ-SCH2C6H4CH2S)WS3] (4)
A solution of 1,4-bis-bromomethyl-benzene (66.5 mg, 0.25 mmol) in CH3CN (5 mL) was added to a CH3CN solution (20 mL) of [Ph4P]2[WS4] (250 mg, 0.25 mmol), and the mixture was stirred for 24 h at room temperature, during which the color of the solution gradually changed from yellow to dark red. From a workup similar to that used for the isolation of 3, dark red micro-crystals of 4 were obtained in 2 days. Yield: 85 mg (62%). – Anal. for C56H48P2S8W2: calcd. C 47.80, H 3.44; found C 48.02, H 3.49. – 1H NMR (CDCl3, ppm): δ=3.76 (s, 4H, SCH2), 6.86–6.93 (m, 4H, C6H4 ), 7.52–7.91 (m, 40H, Ph4P).
2.6 X-ray crystallography
As high-quality single crystals of complexes 1 and 2 were difficult to obtain for X-ray diffraction, recrystallization of the mixture of 1 and 2 in a 1:1 ratio (equimolar ratio) from THF-benzene-Et2O (1:1:2) gave single crystals of 1/2·½C6H6 suitable for X-ray diffraction analysis. Crystallographic data and experimental details for [PPh4]2[WS3(Sn Pr)][WS3(Sn Bu)]·C6H6 (1/2·½C6H6) and [PPh4][WS3(SCH2C6H4CH2Cl-4)] (3) are summarized in Table 1. Intensity data were collected on a Bruker SMART APEX 2000 CCD diffractometer using graphite-monochromatized MoKα radiation (λ=0.71073 Å) at 296(2) K. The collected frames were processed with the software Saint [9]. The data were corrected for absorption using the program Sadabs [10]. The structures were solved by Direct Methods and refined by full-matrix least squares on F2 using the shelxtl software package [11], [12], [13]. All non-hydrogen atoms were refined anisotropically. The positions of all hydrogen atoms were generated geometrically (Csp3−H=0.97 and Csp2−H=0.93 Å), assigned isotropic displacement parameters, and allowed to ride on their respective parent carbon or nitrogen atoms before the final cycle of least-squares refinement. The largest peaks in the final difference maps that had heights of 2.02 (for 1/2·½C6H6) and 2.09 e Å−3 (for 3) are in the vicinity of the tungsten atoms. The selected bond lengths and bond angles of complexes 1–3 were listed in Table 2 for the comparison.
Crystallographic data and experimental details for [PPh4]2[WS3(Sn Pr)][WS3(Sn Bu)]·C6H6 (1/2·½C6H6) and [PPh4][WS3(SCH2C6H4CH2Cl-4)] (3).
| Complex | 1/2·½C6H6 | 3 |
|---|---|---|
| Empirical formula | C58H59P2S8W2 | C32H28PClS4W |
| Formula weight | 1441.16 | 791.05 |
| Crystal system | Triclinic | Triclinic |
| Space group | P1̅ | P1̅ |
| a, Å | 10.933(9) | 9.621(3) |
| b, Å | 15.186(13) | 10.751(3) |
| c, Å | 19.152(17) | 16.368(5) |
| α, deg | 93.916(11) | 95.454(4) |
| β, deg | 103.882(12) | 104.768(4) |
| γ, deg | 101.512(11) | 92.986(4) |
| V, Å3 | 3002(5) | 1624.6(8) |
| Z | 2 | 2 |
| Dcalcd., g cm−3 | 1.59 | 1.62 |
| Temperature, K | 296(2) | 296(2) |
| F(000), e | 1424 | 780 |
| μ(MoKα ), mm−1 | 4.2 | 4.0 |
| Total reflns. | 8686 | 9862 |
| Independent reflns. | 8684 | 7126 |
| Rint | 0.0562 | 0.0512 |
| Parameters | 613 | 352 |
| R1a/wR2b [I>2σ(I)] | 0.0504/0.1270 | 0.0726/0.1651 |
| R1a/wR2b (all data) | 0.0682/0.1363 | 0.1353/0.2000 |
| Goodness-of-fit (GoF)c | 0.929 | 0.951 |
| Final max/min difference peaks, e Å−3 | +2.02/−1.69 | +2.09/−2.45 |
aR1=Σ||Fo|−|Fc||/Σ|Fo|.
bwR2=[Σw(Fo2−Fc2)2/Σw(Fo2)2]1/2, w=[σ2(Fo2)+(AP)2+BP]−1, where P=(Max(Fo2, 0)+2Fc2)/3.
cGoF=S=[Σw(Fo2−Fc2)2/(nobs−nparam)]1/2.
Comparison of selected bond lengths (Å) and angles (deg) for 1–3a.
| 1 | 2 | 3 |
|---|---|---|
| W(2)–S(5) 2.148(4) | W(1)–S(1) 2.150(3) | W(1)–S(1) 2.148(4) |
| W(2)–S(6) 2.126(3) | W(1)–S(2) 2.134(3) | W(1)–S(2) 2.147(4) |
| W(2)–S(7) 2.132(3) | W(1)–S(3) 2.135(3) | W(1)–S(3) 2.152(4) |
| W(2)–S(8) 2.309(4) | W(1)–S(4) 2.310(3) | W(1)–S(4) 2.324(3) |
| S(8)–C(5) 1.834(19) | S(4)–C(1) 1.820(9) | S(4)–C(1) 1.875(14) |
| S(6)–W(2)–S(7) 111.49(11) | S(2)–W(1)–S(3) 112.23(11) | S(2)–W(1)–S(1) 111.77(17) |
| S(6)–W(2)–S(5) 112.53(15) | S(2)–W(1)–S(1) 112.28(13) | S(1)–W(1)–S(3) 111.85(16) |
| S(6)–W(2)–S(8) 109.74(12) | S(3)–W(1)–S(1) 110.89(11) | S(2)–W(1)–S(3) 109.98(18) |
| S(7)–W(2)–S(8) 109.67(12) | S(3)–W(1)–S(4) 108.36(10) | S(2)–W(1)–S(4) 109.37(14) |
| S(5)–W(2)–S(8) 110.30(16) | S(1)–W(1)–S(4) 109.55(11) | S(3)–W(1)–S(4) 109.99(17) |
| S(6)–W(2)–S(8) 102.91(15) | S(2)–W(1)S(4) 103.18(12) | S(1)–W(1)–S(4) 103.70(13) |
| C(5)–S(8)–W(2) 103.7(7) | C(1)–S(4)–W(1) 105.8(4) | C(1)–S(4)–W(1) 104.0(4) |
aThe structural parameters of 1 and 2 are taken from the structure determination of 1/2·½C6H6.
CCDC 1495648 and 1495649 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
3 Results and discussion
Synthetic reactions of alkylthiolatotrithiotungstate complexes 1–4 are shown in Scheme 1. The interaction of [PPh4]2[WS4] with excess n-propylbromide or n-butylbromide in acetonitrile afforded complexes [PPh4][WS3(SR)] (1: R=n-propyl; 2: R=n-butyl) in moderate yields. Previously, the benzylthiolatotrithiotungstate complex [PPh4][WS3(SCH2Ph)] was prepared by the reaction of [PPh4]2[WS4] and benzyl chloride [2]. A similar reaction using 1-bromomethyl-4-chloromethyl-benzene instead of benzyl chloride gave a benzylthiolatotrithiotungstate complex, [PPh4][WS3(SCH2C6H4CH2Cl-4)] (3), indicating that benzyl bromide is obviously more reactive than benzyl chloride. Bearing this in mind, we tried the reaction of [PPh4]2[WS4] and 1,4-bis-bromomethyl-benzene, and obtained a dinuclear complex [PPh4]2[WS3(μ-SCH2C6H4CH2S)WS3] (4) as confirmed by the proton NMR data and elemental analyses. All the 1H NMR spectra for complexes 1–4 measured in CDCl3 gave signal shifts and intensities consistent with the proposed formulation. For example, complex 3 showed two singlets ascribed to SCH2 (δ=3.64 ppm) and CH2Cl (δ=4.78 ppm), respectively, whereas complex 4 exhibited one singlet for SCH2 (δ=3.76 ppm) protons.
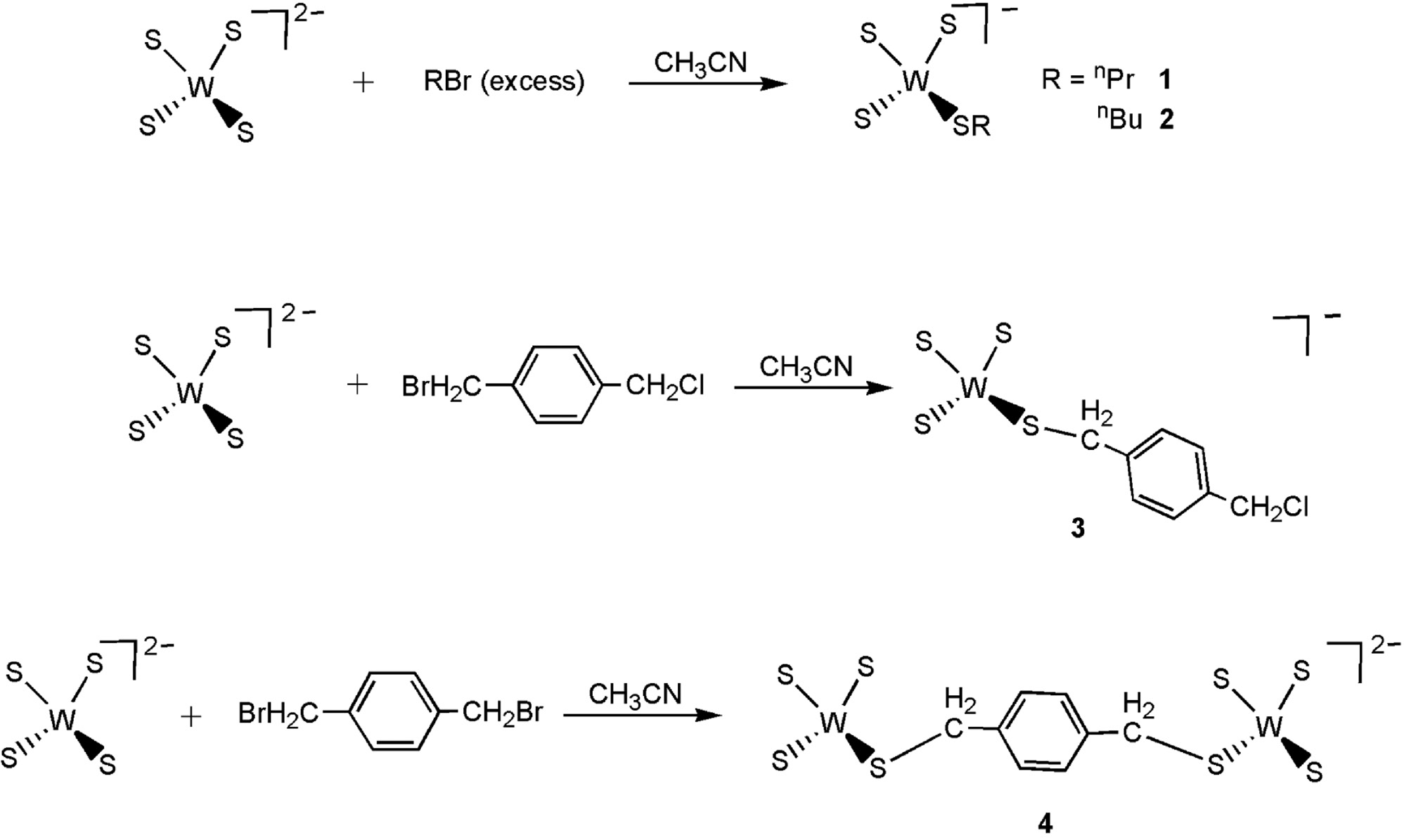
Synthetic reactions of alkylthiolatotrithiotungstate complexes 1–4.
It was not easy to obtain high-quality crystals of complexes 1 and 2 for single-crystal X-ray diffraction by recrystallization of complexes 1 and 2 in a separate way. However, with luck it was possible to isolate single crystals of complex 1/2·½C6H6 by recrystallization of the mixture of the two complexes in a molar ratio of 1:1 from THF–benzene–Et2O (1:1:2). Although an effort to obtain crystals of complex 4 for single-crystal X-ray diffraction failed, this complex has been well characterized using spectroscopic and microanalytical methods. The structures of 1/2·½C6H6 and 3 were established by X-ray diffraction. Complex 1/2·½C6H6 crystallizes in space group P1̅ with Z=2, with independent [WS3(Sn Pr)]− and [WS3(Sn Bu)]− anions, two [PPh4]+ cations, and half benzene molecules, as shown in Fig. 1a. The n-propyl and n-butyl groups of [WS3(SR)]− are clearly shown in Fig. 1b. C–H···S···H–C hydrogen-bond interactions were also observed in 1/2·½C6H6, as shown in Fig. 1c. The bond distances of W–Sterminal range from 2.126(3) to 2.148(4) Å in 1, whereas in 2, they range from 2.134(3) to 2.150(3) Å. The W–Sthiolate bond lengths are 2.309(4) and 2.310(3) Å in 1 and 2, respectively, comparable to those in the related alkylthiolatotrithiotungstate complexes [PPh4][WS3(Si Pr)] (2.326(3) Å) and [PPh4][WS3(Si Bu)] (2.3231(10) Å) [2]. The S–C bond distances are 1.834(19) Å in 1 and 1.820(9) Å in 2, similar to those in [PPh4][WS3(Si Pr)] (1.833(9) Å) and [WS3(Si Bu)] (1.825(5) Å) [2]. The bond angles S–W–S range from 102.91(15)° to 112.53(15)° in 1, similar to those in 2 (103.18(12)°–112.28(13)°). Complex 3 crystallizes in space group P1̅ with Z=2, and its structure is shown in Fig. 2a. The packing mode of 3 shows C–H···S and C–H···Cl hydrogen-bond interactions, as shown in Fig. 2b. The average W–Sterminal bond distance is 2.149(4) Å, similar to those in 1 and 2. The W–Sthiolate bond length is 2.324(3) Å, almost the same as that in [WS3(Si Bu)] (2.3231(10) Å) [2]. The bond angles of S–W–S range from 103.70(13)° to 111.85(16)° in 3, similar to those in 1 and 2, indicating that the substituent has little influence on the geometry of alkylthiolatotrithiotungstate complexes.
![Fig. 1: (a) The content of the asymmetric unit in 1/2·½C6H6, showing the atom labeling scheme. Displacement ellipsoids are drawn at the 40% probability level. (b) Structures of the two anions [WS3(Sn Pr)]− and [WS3(Sn Bu)]−. (c) A packing diagram of 1/2·½C6H6; hydrogen bonds are shown as dashed lines.](/document/doi/10.1515/znb-2016-0181/asset/graphic/j_znb-2016-0181_fig_001.jpg)
(a) The content of the asymmetric unit in 1/2·½C6H6, showing the atom labeling scheme. Displacement ellipsoids are drawn at the 40% probability level. (b) Structures of the two anions [WS3(Sn Pr)]− and [WS3(Sn Bu)]−. (c) A packing diagram of 1/2·½C6H6; hydrogen bonds are shown as dashed lines.
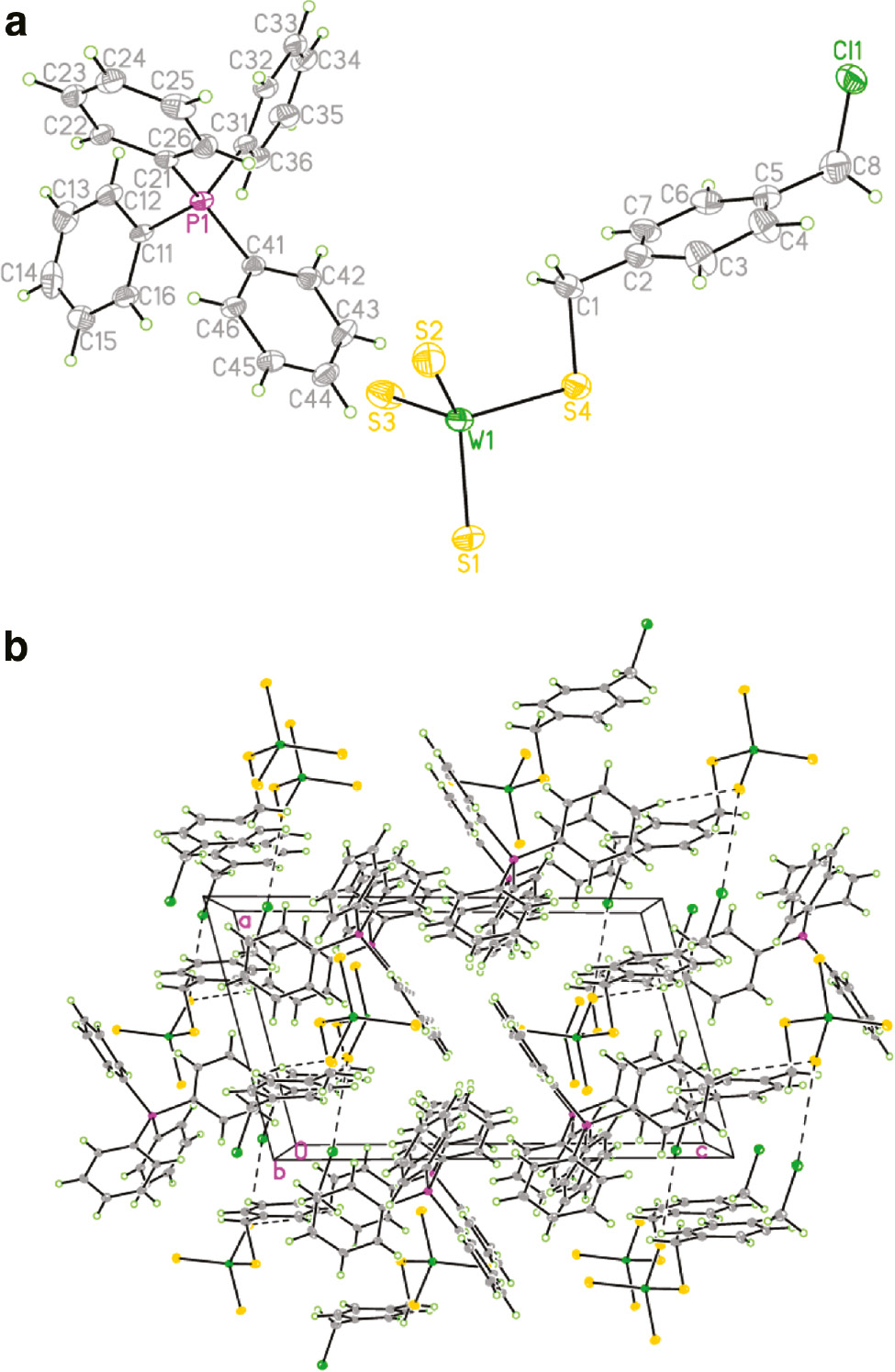
(a) The content of the asymmetric unit in 3, showing the atom labeling scheme. Displacement ellipsoids are drawn at the 40% probability level. (b) A packing diagram of 3; hydrogen bonds are shown as dashed lines.
Alkylation reactions of [PPh4]2[WS4] with various substituted haloalkanes have been investigated. We were successful in isolating a series of alkylated complexes, [PPh4][WS3(RS)] (R=n-Pr, n-Bu, CH2C6H4CH2Cl-4) and [PPh4]2[WS3(μ-SCH2C6H4CH2S)WS3], in moderate yields, which will be used as effective synthons for further construction of new heterobimetallic clusters.
Acknowledgments
This project was supported by the Natural Science Foundation of China (21471003 and 21372007).
References
[1] P. M. Boorman, M.-P. Wang, M. Parrez, J. Chem. Soc.Chem.Commun.1995, 999.10.1039/c39950000999Suche in Google Scholar
[2] N. L. Kruhlak, M. Wang, P. M. Boorman, M. Parvez, Inorg. Chem.2001, 40, 3141.10.1021/ic000970nSuche in Google Scholar PubMed
[3] Y. Arikawa, H. Kawaguchi, K. Kashiwabara, K. Tatsumi, Inorg. Chem.2002, 41, 513.10.1021/ic010921qSuche in Google Scholar PubMed
[4] H.-C. Zhou, W. Su, C. Achim, P. V. Rao, R. H. Holm, Inorg. Chem.2002, 41, 3191.10.1021/ic0201250Suche in Google Scholar PubMed
[5] P. Lin, X. Wu, Q. Huang, Q. Wang, T. Sheng, W. Zhang, J. Guo, J. Lu, Inorg. Chem.1998, 37, 5672.10.1021/ic9704142Suche in Google Scholar PubMed
[6] P. Lin, X. Wu, W. Zhang, J. Guo, T. Sheng, Q. Wang, J. Lu, Chem. Commun.1997, 1349.10.1039/a703468gSuche in Google Scholar
[7] J.-P. Lang, H. Kawaguchi, K. Tatsumi, J. Chem. Soc.Dalton Trans.2002, 2573.10.1039/b106572fSuche in Google Scholar
[8] H.-T. Shi, C. Xu, A.-Q. Jia, X.-H. Huang, Q.-F. Zhang, Inorg. Chim. Acta2014, 419, 55.10.1016/j.ica.2014.04.026Suche in Google Scholar
[9] smart and Saint+ for Windows NT (version 6.02a), Bruker AXS Inc., Madison, WI (USA) 1998.Suche in Google Scholar
[10] G. M. Sheldrick, Sadabs, University of Göttingen, Göttingen (Germany) 1996.Suche in Google Scholar
[11] G. M. Sheldrick, shelxtl (version 5.1) Software Reference Manual, Bruker AXS Inc., Madison, WI (USA) 1997.Suche in Google Scholar
[12] A. Thorn, G. M. Sheldrick, Acta Crystallogr.2008, A64, C221.10.1107/S0108767308092891Suche in Google Scholar
[13] G. M. Sheldrick, Acta Crystallogr.2008, A64, 112.10.1107/S0108767307043930Suche in Google Scholar PubMed
©2017 Walter de Gruyter GmbH, Berlin/Boston
Artikel in diesem Heft
- Frontmatter
- In this Issue
- Crystal and molecular structure of 1-picryl-2-phenyl-2-(4-picrylamidophenyl)-diazenium betaine: analogy between a picramido group and an oxygen atom
- Synthesis and crystal structure of the rare earth borogermanate EuGeBO5
- Alkylation of tetrathiotungstate anions: crystal structures of the alkylthiolatotrithiotungstate complexes [PPh4]2[WS3(Sn Pr)][WS3(Sn Bu)]·½C6H6 and [PPh4][WS3(SCH2C6H4CH2Cl-4)]
- Two isostructural fluorinated metal-organic frameworks with rare rod-packing architecture: syntheses, structures and luminescent properties
- Synthesis and characterization of a macrocyclic copper complex containing the 14-membered 1,3,5,8,10,12-hexaazacyclotetradecane unit
- Synthesis of lignin model compound containing a β-O-4 linkage
- Crystal structures and third-order optical properties of three manganese(II) complexes constructed from N-heterocyclic and polycarboxylate ligands
- Trigonal dodecahedral sodium coordination in a trinuclear copper(II)-sodium complex incorporating a salen-type compartmental Schiff base
- X-ray and NQR studies of bromoindate(III) complexes: [C2H5NH3]4InBr7, [C(NH2)3]3InBr6, and [H3NCH2C(CH3)2CH2NH3]InBr5
- Synthesis and structural characterization of Li3Y(BO3)2
- About the air- and water-stable copper(I) dicyanamide: synthesis, crystal structure, vibrational spectra and DSC/TG analysis of Cu[N(CN)2]
- Note
- Synthesis, crystal structure, and photoluminescence of a dumbbell-like sodium dicyanamide compound with 15-crown-5
Artikel in diesem Heft
- Frontmatter
- In this Issue
- Crystal and molecular structure of 1-picryl-2-phenyl-2-(4-picrylamidophenyl)-diazenium betaine: analogy between a picramido group and an oxygen atom
- Synthesis and crystal structure of the rare earth borogermanate EuGeBO5
- Alkylation of tetrathiotungstate anions: crystal structures of the alkylthiolatotrithiotungstate complexes [PPh4]2[WS3(Sn Pr)][WS3(Sn Bu)]·½C6H6 and [PPh4][WS3(SCH2C6H4CH2Cl-4)]
- Two isostructural fluorinated metal-organic frameworks with rare rod-packing architecture: syntheses, structures and luminescent properties
- Synthesis and characterization of a macrocyclic copper complex containing the 14-membered 1,3,5,8,10,12-hexaazacyclotetradecane unit
- Synthesis of lignin model compound containing a β-O-4 linkage
- Crystal structures and third-order optical properties of three manganese(II) complexes constructed from N-heterocyclic and polycarboxylate ligands
- Trigonal dodecahedral sodium coordination in a trinuclear copper(II)-sodium complex incorporating a salen-type compartmental Schiff base
- X-ray and NQR studies of bromoindate(III) complexes: [C2H5NH3]4InBr7, [C(NH2)3]3InBr6, and [H3NCH2C(CH3)2CH2NH3]InBr5
- Synthesis and structural characterization of Li3Y(BO3)2
- About the air- and water-stable copper(I) dicyanamide: synthesis, crystal structure, vibrational spectra and DSC/TG analysis of Cu[N(CN)2]
- Note
- Synthesis, crystal structure, and photoluminescence of a dumbbell-like sodium dicyanamide compound with 15-crown-5