Consecutive three- and four-component coupling-Bagley-Bohlmann-Rahtz syntheses of tri- and tetrasubstituted pyridines
-
Janis Dohe


Abstract
The concatenation of the modified Sonogashira alkynone synthesis and the Bagley-Bohlmann-Rahtz pyridine synthesis gives novel consecutive three- and four-component coupling-Bagley-Bohlmann-Rahtz (cBBR) syntheses of tri- and tetrasubstituted pyridines in a one-pot fashion. With these processes 15 differently substituted 3-ethoxycarbonyl 2-methylpyridines can be readily obtained in modest to moderate yields.
1 Introduction
The synthesis of pyridines [1, 2] and their derivatization is an ongoing important topic in synthetic chemistry. In addition numerous compounds containing the pyridine core exhibit highly interesting properties. Besides their application as building blocks in synthesis [3–8], supramolecular and coordination chemistry [9–16], and as functional constituents in materials science [17–22] many derivatives have raised particular interest due to their high biological activity [23–35].
Today pyridines are commonly found in drugs and agrochemicals. In particular, they are important active ingredients in herbicides, fungicides and insecticides [36–39], and furthermore in several potent pharmaceuticals against, e.g. asthma, inflammation and coronary diseases (Scheme 1) [40]. Therefore, the development of novel syntheses of substituted pyridines is particularly important [41, 42].

Examples of biological active compounds containing a pyridine core.
Pyridines can be accessed either by ring functionalization and transformation or by de novo syntheses [43, 44]. Among de novo pyridine formations one-pot multicomponent syntheses by Tschitschibabin [45] (name spelled as in the original publication [45]; engl. Chichibabin) and Hantzsch [46] have become most attractive (Scheme 2). However, a major drawback of the Tschitschibabin pyridine synthesis lies in the high pressure which leads to the formation of many byproducts. Although the Hantzsch (dihydro)pyridine synthesis has become a useful tool for the synthesis of many pyridines, among them the dihydropyridine Nifedipine, a powerful medication in the treatment of coronary diseases [47, 48], the downside of this one-pot process are the necessity of an oxidation step to the pyridine target and the inherent low degree of diversity.

Classical pseudo four-component syntheses of pyridines by Tschitschibabin (top) and Hantzsch (bottom).
For overcoming these limitations, a couple of years ago we have disclosed versatile three- and four-component syntheses of unsymmetrically substituted [49, 50] and anellated pyridines [51, 52] initiated by a Pd/Cu-catalyzed coupling-isomerization domino reaction [53–56]. Interestingly, already in 1957, Bohlmann and Rahtz reported a two-step pyridine synthesis by cyclization of an alkynone with an enamine (Scheme 3) [57].
The amino-butadienone intermediate had to be isolated and the employed high temperatures for the cyclocondensation step made the original protocol less attractive for application until Bagley and coworkers used the Bohlmann-Rahtz synthesis in 1998 as an elegant key step in the synthesis of promothiocin A for quickly establishing the 2,3,6-trisubstituted pyridine core (Scheme 4) [58–60].

Bohlmann-Rahtz pyridine synthesis.

Bohlmann-Rahtz pyridine synthesis as a key step in the synthesis of promothiocin A.
Later, Bagley’s group undertook major efforts to develop general two-component processes that circumvent the isolation of the amino-butadienone intermediate by adjusting the reaction conditions, using microwave irradiation, Brønsted or Lewis acid catalysis as well as ethanol as a favorable solvent in the cyclization step (Scheme 5) [61–63].

Bagley’s one-pot Bohlmann-Rahtz pyridine synthesis.
Another major achievement was the in-situ generation of the enamine by reaction of a β-ketoester with an ammonia source furnishing a three-component variation of the Bohlmann-Rahtz pyridine synthesis (Scheme 6) [64–66].
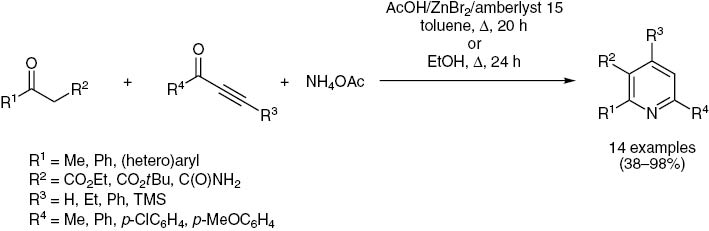
Bagley’s three-component Bohlmann-Rahtz pyridine synthesis.
Having expanded the cross-coupling avenue to Michael systems to catalytic ynone formation as an entry to consecutive multicomponent syntheses of heterocycles [67–70], we became interested in developing a coupling-Bagley-Bohlmann-Rahtz (cBBR) pyridine synthesis initiated by catalytic ynone generation. Here, we report studies on consecutive three- and four-component pyridine syntheses via the Bagley-Bohlmann-Rahtz avenue.
2 Results and discussion
Starting from the outset of modified Sonogashira coupling as an entry to ynone formation from acid chlorides and terminal alkynes [71, 72], we first set out to probe the reaction conditions of the three-component process with 2-thienoyl chloride (1a), 1-hexyne (2a), and ethyl 3-aminocrotonate (3) as a model system to give ethyl 2-methyl-4-n-butyl-6-(thiophen-2-yl)pyridine-3-carboxylate (4a) (Scheme 7, Table 1).

Optimization of the three-component cBBR synthesis of pyridine 4a.
Optimization of the three-component cBBR synthesis of ethyl 2-methyl-4-n-butyl-6-(thiophen-2-yl)pyridine-3-carboxylate (4a).
| Entry | Solvent | Ethyl 3-amino-crotonate (3) | Cosolvent | Acid | T (°C) | t (h) | Yield of pyridine 4a (%)a |
|---|---|---|---|---|---|---|---|
| 1 | THF (5.0 mL) | 1.0 equiv | – | AcOH (17.5 equivs) | 100 | 3 | n.i.b |
| 2 | THF (5.0 mL) | 1.0 equiv | – | AcOH (17.5 equivs) | 100 | 24 | 49 |
| 3 | Toluene (5.0 mL) | 1.0 equiv | – | AcOH (17.5 equivs) | 50 | 24 | 24 |
| 4 | 1,4-dioxane (5.0 mL) | 1.0 equiv | – | AcOH (17.5 equivs) | 120 | 24 | 14 |
| 5 | THF (5.0 mL) | 2.0 equivs | – | AcOH (17.5 equivs) | 100 | 24 | 29 |
| 6 | THF (5.0 mL) | 2.0 equivs | – | AcOH (17.5 equivs) | 120c | 4 | 11 |
| 7 | THF (5.0 mL) | 1.0 equiv | EtOH (2.0 mL) | AcOH (2.0 equivs) | 100 | 24 | 36 |
| 8 | THF (5.0 mL) | 2.0 equivs | EtOH (5.0 mL) | – | 100 | 74 | 39 |
| 9 | Toluene (2.0 mL) | 2.0 equivs | – | AcOH (7.0 equivs) | 170c | 14 | n.i.b |
| 10 | Toluene (5.0 mL) | 1.0 equiv | – | ZnBr2 (0.18 equivs) | 140 | 24 | 6 |
| 11d | Toluene (5.0 mL) | 1.0 equiv | – | BF3.OEt2 (1.25 equivs) | 170c | 2 | 12 |
| 12 | 1,4-dioxane (5.0 mL) | 1.0 equiv | – | Amberlyst 15 (100 mg) | 120 | 24 | n.i.b |
| 13 | 1,4-dioxane (5.0 mL) | 1.0 equiv | – | PTSA (5.0 equivs) | 120 | 24 | – |
| 14 | MeCN (5.0 mL) | 2.0 equivs | – | PTSA (2.0 equivs) | 140c MW | 6 | – |
aIsolated yield after flash chromatography; bn.i. (not isolated due to incomplete conversion or decomposition according to TLC); chold temperature in the microwave cavity; dcoupling at 70°C, 17 h. Bold face refers to the optimal conditions in this study.
With a reaction temperature of 100°C for the Bagley-Bohlmann-Rahtz (BBR) step, a reaction time of 3 h was not sufficient (Table 1, entry 1) and full conversion was achieved after 24 h (Table 1, entry 2). Increasing ethyl-3-aminocrotonate (3) from 1 to 2 equivalents caused a significant loss in yield of pyridine 4a (Table 1, entry 5). A further increase of the reaction temperature, for instance by microwave irradiation, either gave substantially lower yield or increased decomposition (Table 1, entries 6, 9, 11, and 14). Interestingly, the absence of acetic acid led to considerably longer reaction times to achieve full conversion (Table 1, entry 8). Unfortunately, Bagley’s best original conditions only furnished 24% of compound 4a (Table 1, entry 3). Neither 1,4-dioxane, toluene or acetonitrile as solvents (Table 1, entries 3, 4, 9–14) nor the employment of different Brønsted or Lewis acids, such as PTSA (Table 1, entries 13 and 14), amberlyst 15 (Table 1, entry 12), zinc bromide or boron trifluoride etherate (Table 1, entries 10, 11) gave satisfying yields. In summary, the conditions of entry 2 employing THF as a solvent and addition of 17.5 equivalents of acetic acid for the BBR step performed at 100°C for 24 h turned out to give the highest yields of model compound 4a.
With these conditions in hand, the stage was set to probe the scope at two points of diversity, namely from the acid chloride 1 and the alkyne 2, while ethyl 3-aminocrotonate (3) was kept constant as a substrate in this study. Upon coupling acid chloride 1 and alkyne 2 in the presence of catalytic amounts of Pd(PPh3)2Cl2 and CuI and triethylamine as a base, the formed ynone was subsequently reacted with ethyl 3-aminocrotonate (3a) and acetic acid in the same reaction vessel at 100°C for 24 h to give after purification 3-ethoxycarbonyl 2-methylpyridines 4 in low to moderate yields in the sense of a consecutive three-component coupling-BBR sequence (Scheme 8, Table 2). The structures of all compounds were unambiguously assigned by extensive 1H and 13C NMR and IR spectroscopy, mass spectrometry and combustion analysis or HRMS.
The isolated yields of this novel one-pot sequence fall in a range between 19–55%. Taking into account that three new bonds are being formed, the yield per bond forming step counts to 57–82%.
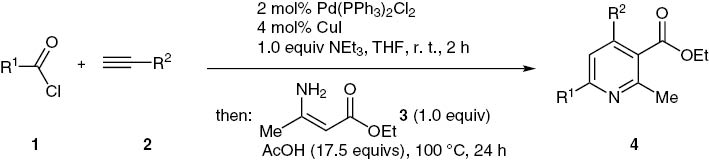
Three-component cBBR synthesis of 3-ethoxycarbonyl 2-methylpyridines 4.
Consecutive three-component cBBR synthesis of 3-ethoxycarbonyl-2-methylpyridines 4.
| Entry | Acid chloride 1 | Alkyne 2 | 3-Ethoxycarbonyl-2-methylpyridine 4 (%)a |
|---|---|---|---|
| 1 | R1 = 2-thienyl (1a) | R2 = n-butyl (2a) |  |
| 2 | R1 = p-MeOC6H4 (1b) | 2a |  |
| 3 | R1 = p-ClC6H4 (1c) | 2a |  |
| 4 | R1 = p-O2NC6H4 (1d) | 2a |  |
| 5 | 1a | R2 = Ph (2b) |  |
| 6 | R1 = p-MeC6H4 (1e) | 2b |  |
| 7 | 1c | 2b |  |
| 8 | R1 = o-FC6H4 (1f) | 2b |  |
| 9 | 1a | R2 = H (2c)b |  |
| 10 | 1b | 2c |  |
| 11 | 1c | 2c |  |
| 12 | 1d | 2c |  |
| 13 | 1e | 2c |  |
| 14 | 1f | 2c |  |
| 15 | R1 = Ph (1g) | 2c |  |
aIsolated yield after flash chromatography; bthe employed alkyne 2c is (trimethylsilyl)acetylene.
In general, the yields for the trisubstituted pyridines 4 (Table 2, entries 9–15) are higher than for the tetrasubstituted derivatives (Table 2, entries 1–8). The variation of electron withdrawing and electron donating groups in the acid chloride 1 is generally possible and these polar functional groups are tolerated. As previously reported for other one-pot synthesis initiated by catalytic ynone formation [67–70], when (trimethylsilyl)acetylene is employed as alkyne the silyl group is concomitantly cleaved upon Michael addition/cyclocondensation and, therefore, trisubstituted pyridines are the obvious reaction products. For the synthesis of tetrasubstituted pyridines n-butyl and phenyl substituents as representatives of alkyl and aryl substitution are introduced in the 4-position of the pyridines 4.
Furthermore, we became interested in probing Bagley’s three-component Bohlmann-Rahtz pyridine synthesis, i.e. the in-situ formation of ethyl 3-aminocrotonate (3) by condensation of ethyl acetoacetate and an ammonia source after the cross-coupling step, and developing it into a four-component cBBR pyridine synthesis. Therefore, we chose the previous model system (Scheme 7), however, now employing ethyl acetoacetate (5) and ammonium acetate as third and fourth components for finding the optimal conditions for the formation of model pyridine 4a (Scheme 9, Table 3).

Optimization of the four-component cBBR synthesis of pyridine 4a.
Optimization of the four-component cBBR synthesis of ethyl 2-methyl-4-n-butyl-6-(thiophen-2-yl)pyridine-3-carboxylate (4a).
| Entry | NH4OAc (equivs) | Ethyl acetoacetate (5) (equivs) | Cosolvent | Acid (equivs) | T (°C) | t (h) | Yield of pyridine 4a (%)a |
|---|---|---|---|---|---|---|---|
| 1 | 2.0 | 1.0 | – | – | 100 | 74 | 24 |
| 2 | 10 | 1.0 | EtOH (3.0 mL) | – | 100 | 74 | 44 |
| 3 | 10 | 1.0 | EtOH (3.0 mL) | – | 100 | 24 | 29 |
| 4 | 1.0 | 1.0 | EtOH (3.0 mL) | – | 100 | 24 | 12 |
| 5 | 10 | 2.0 | EtOH (3.0 mL) | – | 100 | 24 | 18 |
| 6 | 1.0 | 1.0 | EtOH (3.0 mL) | AcOH (0.5) | 100 | 24 | 23 |
| 7 | 10 | 1.0 | EtOH (5.0 mL) | AcOH (1.0) | 100 | 24 | 43 |
| 8 | 2.0 | 1.0 | EtOH (2.0 mL) | AcOH (2.0) | 100 | 24 | 48 |
| 9 | 2.0 | 1.0 | EtOH (2.0 mL) | AcOH (2.0) | 70 | 24 | 12 |
| 10 | 2.0 | 1.0 | EtOH (2.0 mL) | AcOH (35.0) | 100 | 24 | 36 |
| 11 | 3.0 | 1.5 | EtOH (2.0 mL) | AcOH (2.0) | 100 | 24 | 36 |
| 12 | 2.0 | 1.0 | EtOH (2.0 mL) | PTSA (2.0) | 100 | 24 | 10 |
| 13 | 2.0 | 1.0 | – | AcOH (70.0) | 100 | 24 | 20 |
| 14 | 2.0 | 1.5 | – | AcOH (17.5) | 100 | 24 | 14 |
| 15 | 17 | 1.7 | EtOH (10.0 mL) | – | 100 | 24 | 3 |
| 16 | 17 | 1.7 | EtOH (10.0 mL) | AcOH (52.5) | 100 | 24 | 24 |
| 17 | 2.0 | 1.0 | MeOCH2CH2OH (2.0 mL) | AcOH (2.0) | 140 | 24 | 7 |
| 18 | 2.0 | 1.0 | tBuOH (2.0 mL) | AcOH (2.0) | 140 | 24 | 10 |
| 19b | 10 | 1.0 | Toluene (5.0 mL) | AcOH (35.0) | 140 | 24 | 24 |
| 20 | 2.0 | 1.0 | EtOH (2.0 mL) | AcOH (2.0) | 170 (MW) | 0.5 | 7 |
| 21 | 2.0 | 1.0 | – | AcOH (17.5) | 120 (MW) | 4 | 8 |
| 22 | 2.0c | 1.0 | EtOH (2.0 mL) | AcOH (2.0) | 100 | 24 | 27 |
| 23 | 2.0d | 1.0 | EtOH (2.0 mL) | AcOH (2.0) | 100 | 24 | n.i.e |
| 24 | 2.0d | 1.0 | – | AcOH (8.75) | 100 | 24 | n.i.e |
aIsolated yield after flash chromatography; btoluene was also employed for the coupling step instead of THF; c(NH4)2CO3 was employed as an ammonia source; d(NH4)Cl was employed as an ammonia source; en.i. (not isolated due to incomplete conversion or decomposition according to TLC). Bold face refers to the optimal conditions in this study.
Without the addition of a cosolvent and an acid the BBR step took 74 h to full conversion of the intermediate alkynone at 100°C and only 24% of product 4a could be isolated (Table 3, entry 1). Ethanol as a cosolvent and increasing the reaction time from 24 to 74 h (Table 3, entries 2 and 3) led to an increase in the yield of pyridine 4a. However, upon reducing the amount of ammonium acetate to one equivalent caused a significant drop in yield (Table 3, entry 4). Surprisingly, the increase in ethyl acetoacetate to two equivalents had a detrimental effect on the yield (Table 3, entry 5). Expectedly, the addition of acetic acid as a cocatalyst (Table 3, entries 6–11) caused an increase in the yield of the desired product, indicating the crucial importance of buffered conditions. In this optimization series the highest yield could be obtained with equimolar amounts of two equivalents of ammonium acetate and acetic acid, respectively, at 100°C for 24 h (Table 3, entry 8). Excessive acetic acid (Table 3, entries 10 and 13), unbalanced amounts of ethyl acetoacetate as well as omitting ethanol as a cosolvent (Table 3, entries 13–16) and lower reaction temperatures (Table 3, entry 9) always causes a drop in yield. It is also noteworthy to mention that the stronger acid PTSA is not an effective catalyst (Table 3, entry 12) and 2-methoxyethanol and tert-butanol turned out to be poor cosolvents in the BBR step, even at elevated reaction temperatures (Table 3, entries 17 and 18) and toluene did not reach yields comparable to those in ethanol (Table 3, entry 19). Dielectric heating with or without ethanol as a cosolvent in the BBR step only gave poor yields of the desired product (Table 3, entries 20 and 21). The more basic ammonium carbonate as an ammonia source did not prove to be as suitable as ammonium acetate (Table 3, entry 22) and the more acidic ammonium chloride turned out to be detrimental (Table 3, entries 23 and 24). In summary, the conditions of entry 8 employing equimolar amounts of ammonium acetate and acetic acid and ethanol as a cosolvent for the BBR step performed at 100°C for 24 h turned out to give the highest yields of model compound 4a. These conditions were employed to the one-pot four-component synthesis of three selected examples furnishing the desired 3-ethoxycarbonyl-2-methylpyridines 4a, 4i, and 4j in low to moderate yields (Scheme 10). Interestingly, the comparison of the yields of the four-component process reveals that they are equal (compound 4a), lower (compound 4i), or higher (compound 4j) than the corresponding three-component reaction (Table 2, entries 1, 9, and 10). However, taking into account that four new bonds are being formed in this novel consecutive four-component process the yield per bond forming step counts to 68–83%, the sequence can be considered to be quite efficient.

Four-component cBBR synthesis of selected 3-ethoxycarbonyl-2-methylpyridines 4 (a(trimethylsilyl)acetylene (2c) was employed as an alkyne).
In conclusion we could expand the two- and three-component Bagley variation of the Bohlmann-Rahtz pyridine synthesis into three- and four-component cBBR sequences in a one-pot fashion furnishing the targeted 3-ethoxycarbonyl-2-methylpyridines in modest to moderate yields, however, with high average bond forming yields. This novel process not only tolerates various electronic modulations in the 4- and 6-positions of the pyridyl core as introduced in the initial coupling step. In particular, the extension from the three- to the four-component process indicated that the in-situ formation of the relevant 3-aminocrotonate is delicate and only narrow variations in the solvents, catalysts, ammonia sources, and stoichiometries are permitted. Further studies directed to expand the coupling-cyclization concept to the formation of other six-membered hetero- and carbocycles are currently underway.
3 Experimental details
3.1 General considerations
All cross coupling reactions were carried out in oven-dried Schlenk or microwave tubes using septa and syringes under a nitrogen atmosphere. Dry tetrahydrofuran and 1,4-dioxane were supplied by MBraun system MB-SPS-800. Chemicals were either commercially obtained from ABCR GmbH & Co KG, Acros Organics, Alfa Aesar GmbH & Co KG, Fluka, Merck KGaA, Riedel-de Haën, Sigma-Aldrich Co and used as supplied or were already available in the research group. All products were purified via column chromatography on silica gel 60 M (0.04–0.063 mm) from Macherey-Nagel using flash technique under a pressure of 2 bar. The crude mixtures were absorbed on Celite® 545 (0.02–0.10 mm) from Carl Roth Gmbh (Karslruhe, Germany) before chromatographic purification. The reaction progress was observed qualitatively using TLC Silica gel 60 F254 aluminum sheets obtained by Merck KGaA. The spots were detected with UV light at 254 nm and with aqueous potassium permanganate solution. 1H, 13C, and 135 DEPT spectra were recorded on Bruker AVIII-300 and Bruker AVIII-600 spectrometers. CDCl3 was used as solvent. The resonance of CDCl3 was locked as internal standard (CDCl3: 1H δ = 7.26 ppm, 13C δ = 77.0 ppm). The multiplicities of signals were abbreviated as follows: s, singlet; d, doublet; dd, doublet of doublets; ddd, doublet of doublets of doublets; t, triplet; dt, doublet of triplets; td, triplet of doublets; tt, triplet of triplets; q, quartet; m, multiplet. The type of carbon atom was determined on the basis of 135-DEPT NMR spectra. For the description of the 13C NMR spectra primary carbon atoms are abbreviated as CH3, secondary carbon atoms as CH2, tertiary carbon atoms as CH and quaternary carbon atoms as Cquat. EI mass spectra were measured on a Finnigan MAT TSQ 7000 spectrometer. HRMS ((+)-ESI) was measured on a Bruker maXis 4G instrument. IR spectra were obtained on a Shimadzu IRAffinity spectrometer. The intensity of the signals was abbreviated as follows: s (strong), m (medium), w (weak). The melting points were measured on a Büchi Melting Point B-540 apparatus. Combustion analyses were carried out on a Perkin Elmer Series II Analyser 2400 and an Elementar vario MICRO CUBE in the microanalytical laboratory of the Institut für Pharmazeutische und Medizinische Chemie of Heinrich-Heine-Universität Düsseldorf.
3.2 General procedure (GP1) for the consecutive three-component cBBR synthesis of 3-ethoxycarbonyl-2- methylpyridines 4
In a screw-cap Schlenk vessel Pd(PPh3)2Cl2 (14 mg, 0.02 mmol, 2 mol%) and CuI (7 mg, 0.04 mmol, 4 mol%) were placed and dry THF (5 mL) was added under a nitrogen atmosphere. The yellow suspension was degassed with a stream of nitrogen through a syringe for 2 min (for experimental details see Table 4). Successively, triethylamine (0.14 mL, 1.0 mmol), the acid chloride 1 (1.0 mmol), and the terminal alkyne 2 (1.0 mmol) were added and the reaction mixture was stirred at room temperature for 2 h to complete the conversion (monitored by TLC). Then ethyl 3-aminocrotonate (3) (0.13 mL, 1.0 mmol) and acetic acid (1 mL) were added under nitrogen and the solution was heated at 100°C for 24 h. After cooling to room temperature the solvents were removed in vacuo and the residue was dissolved in ethyl acetate (30 mL) and extracted with a saturated aqueous NaHCO3 solution (30 mL). The aqueous layer was extracted with ethyl acetate (20 mL) and the combined organic layers were washed with saturated aqueous NaHCO3 solution (20 mL) and brine (20 mL) and dried with anhydrous magnesium sulfate. The crude product was adsorbed on Celite® and purified by column chromatography on silica gel.
Experimental details of the consecutive three-component cBBR synthesis of 3-ethoxycarbonyl-2-methylpyridines 4.
| Entry | Acid chloride 1 | Alkyne 2 | 3-Ethoxycarbonyl-2-methylpyridine 4a |
|---|---|---|---|
| 1 | 110 μL (1.0 mmol) of 1a | 118 μL (1.0 mmol) of 2a | 149 mg (49%) of 4a |
| 2 | 138 μL (1.0 mmol) of 1b | 118 μL (1.0 mmol) of 2a | 79 mg (24%) of 4b |
| 3 | 132 μL (1.0 mmol) of 1c | 118 μL (1.0 mmol) of 2a | 123 mg (37%) of 4c |
| 4 | 186 mg (1.0 mmol) of 1d | 118 μL (1.0 mmol) of 2a | 99 mg (29%) of 4d |
| 5 | 110 μL (1.0 mmol) of 1a | 113 μL (1.0 mmol) of 2b | 64 mg (20%) of 4e |
| 6 | 134 μL (1.0 mmol) of 1e | 113 μL (1.0 mmol) of 2b | 81 mg (24%) of 4f |
| 7 | 132 μL (1.0 mmol) of 1c | 113 μL (1.0 mmol) of 2b | 133 mg (38%) of 4g |
| 8 | 122 μL (1.0 mmol) of 1f | 113 μL (1.0 mmol) of 2b | 112 mg (33%) of 4h |
| 9 | 110 μL (1.0 mmol) of 1a | 144 μL (1.0 mmol) of 2cb | 88 mg (36%) of 4i |
| 10 | 138 μL (1.0 mmol) of 1b | 144 μL (1.0 mmol) of 2cb | 52 mg (19%) of 4j |
| 11 | 132 μL (1.0 mmol) of 1c | 144 μL (1.0 mmol) of 2cb | 130 mg (47%) of 4k |
| 12 | 186 mg (1.0 mmol) of 1d | 144 μL (1.0 mmol) of 2cb | 150 mg (52%) of 4l |
| 13 | 134 μL (1.0 mmol) of 1e | 144 μL (1.0 mmol) of 2cb | 77 mg (30%) of 4m |
| 14 | 122 μL (1.0 mmol) of 1f | 144 μL (1.0 mmol) of 2cb | 142 mg (55%) of 4n |
| 15 | 116 μL (1.0 mmol) of 1g | 144 μL (1.0 mmol) of 2cb | 67 mg (28%) of 4o |
aIsolated yield after flash chromatography; bcompound 2c is (trimethylsilyl)acetylene.
3.3 General procedure (GP2) for the consecutive four-component cBBR synthesis of 3-ethoxycarbonyl-2- methylpyridines 4
In a screw-cap Schlenk vessel Pd(PPh3)2Cl2 (14 mg, 0.02 mmol, 2 mol%) and CuI (7 mg, 0.04 mmol, 4 mol%) were placed and dry THF (5 mL) was added under a nitrogen atmosphere. The yellow suspension was degassed with a stream of nitrogen through a syringe for 2 min (for experimental details see Table 5). Successively, triethylamine (0.14 mL, 1.0 mmol), the acid chloride 1 (1.0 mmol), and the terminal alkyne 2 (1.0 mmol) were added and the reaction mixture was stirred at room temperature for 2 h to complete the conversion (monitored by TLC). Then ammonium acetate (154 mg, 2.0 mmol), ethyl acetoacetate (5) (0.13 mL, 1.0 mmol), acetic acid (0.11 mL, 2.0 mmol), and ethanol (2 mL) were added under nitrogen and the solution was heated to 100°C for 24 h. After cooling to room temperature the solvents were removed in vacuo and the residue was dissolved in ethyl acetate (30 mL) and extracted with a saturated aqueous NaHCO3 solution (30 mL). The aqueous layer was extracted with ethyl acetate (20 mL) and the combined organic layers were washed with saturated aqueous NaHCO3 solution (20 mL) and brine (20 mL) and dried with anhydrous magnesium sulfate. The crude product was adsorbed on Celite® and purified by column chromatography on silica gel to give the pure pyridines 4.
Experimental details of the consecutive four-component cBBR synthesis of 3-ethoxycarbonyl-2-methylpyridines 4.
| Entry | Acid chloride 1 | Alkyne 2 | 3-Ethoxycarbonyl-2-methylpyridine 4a |
|---|---|---|---|
| 1 | 110 μL (1.0 mmol) of 1a | 118 μL (1.0 mmol) of 2a | 146 mg (48%) of 4a |
| 2 | 110 μL (1.0 mmol) of 1a | 144 μL (1.0 mmol) of 2cb | 52 mg (21%) of 4i |
| 3 | 138 μL (1.0 mmol) of 1b | 144 μL (1.0 mmol) of 2cb | 84 mg (31%) of 4j |
aIsolated yield after flash chromatography; bcompound 2c is (trimethylsilyl)acetylene.
3.4 Ethyl 2-methyl-4-butyl-6-(thiophen-2-yl)pyridine-3-carboxylate (4a)
After chromatography on silica gel (n-hexane-ethyl acetate 50:1) and according to GP1 149 mg (49%) and according to GP2 146 mg (48%) of compound 4a was obtained as a yellow oil. – 1H NMR (CDCl3, 600 MHz): δ = 0.94 (t, 3J = 7.4 Hz, 3 H), 1.35–1.42 (m, 5 H), 1.61 (tt, 3J = 7.8, 6.5 Hz, 2 H), 2.57 (s, 3 H), 2.71–2.58 (m, 2 H), 4.41 (q, 3J = 7.1 Hz, 2 H), 7.10 (dd, 3J = 4.9, 3.8 Hz, 1 H), 7.32 (s, 1 H), 7.39 (dd, 3J = 5.2, 0.8 Hz, 1 H), 7.57–7.65 (m, 1 H). – 13C NMR (CDCl3, 150 MHz): δ = 13.8 (CH3), 14.19 (CH3), 22.6 (CH2), 23.0 (CH3), 32.6 (CH2), 33.2 (CH2), 61.3 (CH2), 116.6 (CH), 125.0 (CH), 127.4 (Cquat), 127.9 (CH), 128.0 (CH) 144.4 (Cquat), 150.1 (Cquat), 152.1 (Cquat), 155.3 (Cquat), 168.8 (Cquat). – MS ((+)-EI): m/z (%) = 303 (100) [M]+, 274 (3) [M–C2H5]+, 258 (72) [M–C2H5O]+, 246 (13) [M–C4H9]+, 230 (19) [M–C3H5O2]+, 189 (83), 173 (11), 159 (12), 98 (24). – IR: ν̃ (cm−1) = 2957 (w), 2930 (w), 1719 (s), 1584 (s), 1553 (m), 1454 (m), 1267 (s), 1231 (m), 1198 (s), 1105 (s), 1080 (s), 1069 (m), 851 (s), 702 (s). – Anal. for C17H21NO2S (303.4): calcd. C 67.29, H 6.98, N 4.62; found C 67.37, H 6.86, N 4.36.
3.5 Ethyl 2-methyl-4-butyl-6-(4-methoxyphenyl)pyridine-3-carboxylate (4b)
According to GP1 and after flash chromatography on silica gel (n-hexane-ethyl acetate 30:1) 79 mg (24%) of compound 4b was obtained as a yellow oil. – 1H NMR (CDCl3, 300 MHz): δ = 0.94 (t, 3J = 7.3 Hz, 3 H), 1.33–1.47 (m, 5 H), 1.55–1.69 (m, 2 H), 2.61 (s, 3 H), 2.63–2.73 (m, 2 H), 3.86 (s, 3 H), 4.42 (q, 3J = 7.2 Hz, 2 H), 6.85–7.10 (m, 2 H), 7.35 (s, 1 H), 7.91–7.98 (m, 2 H). – 13C NMR (CDCl3, 75 MHz): δ = 13.8 (CH3), 14.2 (CH3), 22.6 (CH2), 23.2 (CH3), 32.7 (CH2), 33.3 (CH2), 55.4 (CH3), 61.1 (CH2), 114.1 (CH), 117.8 (CH), 127.0 (Cquat), 128.4 (CH), 150.1 (Cquat), 155.0 (Cquat), 156.8 (Cquat), 160.6 (Cquat), 164.2 (Cquat), 169.1 (Cquat). – HRMS ((+)-ESI): m/z = 328.190 (calcd. 328.190 for [C20H25NO3–H]+). – MS ((+)-EI): m/z (%) = 327 (100) [M]+, 298 (7) [M–C2H5]+, 282 (35) [M–C2H5O]+, 270 (8) [M–C4H9]+, 254 (9) [M–C3H5O2]+, 213 (32), 135 (14). – IR: ν̃ (cm−1) = 2957 (m), 2932 (m), 2870 (w), 2837 (w), 1721 (s), 1607 (m), 1580 (s), 1553 (m), 1512 (s), 1456 (m), 1443 (m), 1422 (m), 1381 (m), 1366 (m), 1267 (s), 1250 (s), 1211 (s), 1173 (s), 1144 (m), 1121 (m), 1109 (m), 1094 (m), 1072 (s), 1032 (m), 932 (m), 833 (s), 810 (m), 787 (m), 770 (m), 745 (m), 729 (m), 700 (m), 665 (m), 637 (m), 623 (m).
3.6 Ethyl 2-methyl-4-butyl-6-(4-chlorophenyl)pyridine-3-carboxylate (4c)
According to GP1 and after flash chromatography on silica gel (n-hexane-ethyl acetate 50:1) 123 mg (37%) of compound 4c was obtained as an orange oil. – 1H NMR (CDCl3, 600 MHz): δ = 0.94 (t, 3J = 7.4 Hz, 3 H), 1.40 (dt, 3J = 7.3, 10.5 Hz, 5 H), 1.57–1.67 (m, 2 H), 2.61 (s, 3 H), 2.63–2.70 (m, 2 H), 4.43 (q, 3J = 7.1 Hz, 2 H), 7.37 (s, 1 H), 7.42 (d, 3J = 8.5 Hz, 2 H), 7.93 (d, 3J = 8.5 Hz, 2 H). – 13C NMR (CDCl3, 151 MHz): δ = 13.8 (CH3), 14.2 (CH3), 22.6 (CH2), 23.2 (CH3), 32.7 (CH2), 33.3 (CH2), 61.4 (CH2), 118.2 (CH), 127.9 (Cquat), 128.4 (CH), 128.8 (CH), 135.2 (Cquat), 137.5 (Cquat), 150.1 (Cquat), 155.3 (Cquat), 155.9 (Cquat), 169.0 (Cquat). – MS ((+)-EI): m/z (%) = 331 (100) [35Cl-M]+, 303 (4) [35Cl-M–C2H4]+, 286 (74) [35Cl-M–C2H5O]+, 274 (21) [35Cl-M–C4H9]+, 258 (38) [35Cl-M–C3H5O2]+, 245 (22), 217 (47), 139 (34). – IR: ν̃ (cm−1) = 3061 (w), 2957 (w), 2932 (w), 2872 (w), 2862 (w), 1721 (s), 1585 (s), 1576 (m), 1553 (m), 1493 (m), 1454 (m), 1381 (m), 1366 (m), 1265 (s), 1211 (s), 1177 (m), 1119 (s), 1103 (s), 1091 (s), 1070 (s), 1013 (s), 862 (m), 833 (s), 777 (m), 745 (m), 719 (m), 662 (m), 633 (m), 611 (m). – Anal. for C19H22ClNO2 (331.8): calcd. C 68.77, H 6.68, N 4.22; found C 68.73, H 6.77, N 4.13.
3.7 Ethyl 2-methyl-4-butyl-6-(4-nitrophenyl)pyridine-3-carboxylate (4d)
According to GP1 and after flash chromatography on silica gel (n-hexane-ethyl acetate 30:1) 99 mg (29%) of compound 4d was obtained as an orange oil. – 1H NMR (CDCl3, 600 MHz): δ = 0.94 (t, 3J = 7.4 Hz, 3 H), 1.37–1.43 (m, 5 H), 1.63 (tt, 3J = 7.8, 6.6 Hz, 2 H), 2.63 (s, 3 H), 2.66–2.71 (m, 2 H), 4.45 (q, 3J = 7.2 Hz, 2 H), 7.48 (s, 1 H), 8.14–8.19 (m, 2 H), 8.28–8.31 (m, 2 H). – 13C NMR (CDCl3, 150 MHz): δ = 13.8 (CH3), 14.2 (CH3), 22.6 (CH2), 23.1 (CH3), 32.7 (CH2), 33.2 (CH2), 61.6 (CH2), 119.2 (CH), 123.9 (CH), 127.9 (CH), 129.0 (Cquat), 144.80 (Cquat), 148.20 (Cquat), 150.50 (Cquat), 154.30 (Cquat), 155.70 (Cquat), 168.60 (Cquat). – MS ((+)-EI): m/z (%) = 342 (53) [M]+, 313 (29) [M–C2H5]+, 297 (100) [M–C2H5O]+, 285 (29) [M–C4H9]+, 269 (37) [M–C3H5O2]+, 228 (46), 221 (11), 181 (12). – IR: ν̃ (cm−1) = 2959 (m), 2932 (m), 2872 (w), 1722 (s), 1584 (m), 1558 (m), 1518 (s), 1456 (m), 1418 (m), 1383 (m), 1339 (s), 1317 (m), 1267 (s), 1213 (m), 1173 (m), 1121 (m), 1107 (m), 1096 (m), 1070 (m), 1013 (m), 851 (s), 789 (m), 762 (m), 696 (m), 658 (m). – Anal. for C19H22N2O4 (342.4): calcd. C 66.65, H 6.48, N 8.18; found C 66.84, H 6.47, N 8.17.
3.8 Ethyl 2-methyl-4-phenyl-6-(thiophen-2-yl)pyridine-3-carboxylate (4e)
According to GP1 and after flash chromatography on silica gel (n-hexane-dichloromethane 2:1) 64 mg (20%) of compound 4e was obtained as a yellow oil. – 1H NMR (CDCl3, 300 MHz): δ = 0.99 (t, 3J = 7.2 Hz, 3 H), 2.67 (s, 3 H), 4.10 (q, 3J = 7.1 Hz, 2 H), 7.11 (dd, 3J = 5.0, 3.7 Hz, 1 H), 7.38–7.46 (m, 6 H), 7.48 (d, J = 0.6 Hz, 1 H), 7.64 (dd, 3J = 3.7, 1.2 Hz, 1 H). – 13C NMR (CDCl3, 75 MHz): δ = 13.6 (CH3), 23.0 (CH3), 61.3 (CH2), 116.7 (CH), 125.4 (CH), 126.5 (Cquat), 127.8 (CH), 128.1 (CH), 128.3 (CH), 128.5 (CH), 128.5 (CH), 138.8 (Cquat), 144.1 (Cquat), 149.1 (Cquat), 152.3 (Cquat), 156.0 (Cquat), 168.7 (Cquat). – MS ((+)-EI): m/z (%) = 323 (100) [M]+, 295 (8) [M–C2H4]+, 278 (99) [M–C2H5O]+, 250 (3) [M–C3H5O2]+, 209 (33), 84 (37), 56 (71). – IR: ν̃ (cm−1) = 3103 (w), 3059 (w), 2980 (w), 2930 (w), 2853 (w), 1717 (s), 1580 (s), 1541 (s), 1495 (m), 1445 (m), 1412 (m), 1387 (m), 1364 (m), 1265 (s), 1240 (s), 1221 (m), 1171 (m), 1138 (m), 1078 (s), 1038 (m), 1013 (m), 874 (m), 847 (m), 802 (m), 780 (m), 750 (m), 698 (s), 671 (m), 638 (m), 621 (m). – Anal. calcd. for C19H17NO2S (323.4): C 70.56, H 5.30, N 4.33, S 9.91; found C 70.31, H 5.46, N 4.04, S 9.68.
3.9 Ethyl 2-methyl-4-phenyl-6-(4-tolyl)pyridine-3-carboxylate (4f)
According to GP1 and after flash chromatography on silica gel (n-hexane-ethyl acetate 30:1) 81 mg (24%) of compound 4f was obtained as a yellow oil. – 1H NMR (CDCl3, 300 MHz): δ = 1.01 (t, 3J = 7.1 Hz, 3 H), 2.41 (s, 3 H), 2.73 (s, 3 H), 4.12 (q, 3J = 7.1 Hz, 2 H), 7.27–7.31 (m, 2 H), 7.41–7.45 (m, 5 H), 7.55 (s, 1 H), 7.91–7.98 (m, 2 H). – 13C NMR (CDCl3, 75 MHz): δ = 13.6 (CH3), 21.3 (CH3), 23.0 (CH3), 61.3 (CH2), 118.3 (CH), 126.5 (Cquat), 127.1 (CH), 127.9 (CH), 128.5 (CH), 128.5 (CH), 129.5 (CH), 135.7 (Cquat), 139.0 (Cquat), 139.5 (Cquat), 149.1 (Cquat), 155.6 (Cquat), 157.3 (Cquat), 168.9 (Cquat). – MS ((+)-EI): m/z (%) = 331 (87) [M]+, 303 (15) [M–C2H4]+, 302 (62) [M–C2H5]+, 286 (100) [M–C2H5O]+, 258 (8) [M–C3H5O2]+, 237 (19), 216 (31), 213 (18), 202 (25), 119 (21), 77 (10) [C6H5]+. – IR: ν̃ (cm−1) = 3057 (w), 3030 (w), 2978 (w), 2963 (w), 2903 (w), 2860 (w), 2359 (w), 2324 (w), 1719 (s), 1684 (m), 1584 (s), 1543 (s), 1516 (m), 1497 (m), 1445 (m), 1364 (m), 1343 (w), 1264 (s), 1238 (m), 1184 (m), 1140 (m), 1096 (s), 1072 (s), 1018 (m), 881 (m), 856 (m), 822 (s), 768 (s), 750 (m), 719 (m), 700 (s), 664 (m), 642 (m). – Anal. for C22H21NO2 (331.4): calcd. C 79.73, H 6.39, N 4.23; found C 79.55, H 6.45, N 3.99.
3.10 Ethyl 2-methyl-4-phenyl-6-(4-chlorophenyl)pyridine-3-carboxylate (4g)
According to GP1 and after flash chromatography on silica gel (n-hexane-ethyl acetate 50:1) 133 mg (38%) of compound 4g was obtained as an orange oil. – 1H NMR (CDCl3, 300 MHz): δ = 1.01 (t, 3J = 7.1 Hz, 3 H), 2.72 (s, 3 H), 4.13 (q, 3J = 7.1 Hz, 2 H), 7.38–7.48 (m, 7 H), 7.54 (s, 1 H), 7.96–8.02 (m, 2 H). – 13C NMR (CDCl3, 75 MHz): δ = 13.6 (CH3), 23.1 (CH3), 61.3 (CH2), 118.2 (CH), 126.9 (Cquat), 127.1 (CH), 127.8 (CH), 128.4 (CH), 128.5 (CH), 128.9 (CH), 135.4 (Cquat), 137.1 (Cquat), 138.7 (Cquat), 149.0 (Cquat), 155.8 (Cquat), 155.9 (Cquat), 168.7 (Cquat). – MS ((+)-EI): m/z (%) = 351 (73) [35Cl-M]+, 323 (12) [35Cl-M–C2H4]+, 322 (35) [35Cl-M–C2H5]+, 306 (100) [35Cl-M–C2H5O]+, 278 (7) [35Cl-M–C3H5O2]+, 257 (34), 237 (31), 202 (37), 141 (14). – IR: ν̃ (cm−1) = 3802 (w), 3690 (w), 3649 (w), 3059 (w), 2978 (m), 2922 (m), 2901 (m), 2747 (w), 2374 (w), 2311 (w), 1721 (s), 1584 (s), 1572 (m), 1543 (m), 1491 (m), 1445 (m), 1406 (m), 1381 (m), 1364 (m), 1263 (s), 1240 (m), 1227 (m), 1179 (m), 1140 (m), 1098 (s), 1072 (s), 1013 (s), 833 (s), 768 (s), 750 (m), 698 (s). – Anal. for C21H18ClNO2 (351.8): calcd. C 71.69, H 5.16, N 3.98; found C 71.71, H 5.26, N 3.69.
3.11 Ethyl 2-methyl-4-phenyl-6-(2-fluorophenyl)pyridine-3-carboxylate (4h)
According to GP1 and after flash chromatography on silica gel (n-hexane-ethyl acetate 30:1) and 112 mg (33%) of compound 4h was obtained as a red oil. – 1H NMR (CDCl3, 300 MHz): δ = 0.93 (t, 3J = 7.1 Hz, 3 H), 2.64 (s, 3 H), 4.05 (q, 3J = 7.1 Hz, 2 H), 7.06 (ddd, 3JH-F = 11.3 Hz, 3J = 8.2, 1.3 Hz, 1 H), 7.15–7.22 (m, 1 H), 7.25–7.38 (m, 6 H), 7.58 (d, 5JH-F = 1.8 Hz, 1 H), 7.94 (td, 3J = 7.8, 1.9 Hz, 1 H). – 13C NMR (CDCl3, 75 MHz): δ = 13.6 (CH3), 23.0 (CH3), 61.4 (CH2), 116.2 (d, 2JC-F = 23.0 Hz, CH), 122.4 (d, 3JC-F = 9.1 Hz, CH), 124.5 (CH), 124.6 (CH), 126.8 (d, 2JC-F = 11.6 Hz, Cquat), 127.1 (Cquat), 127.9 (CH), 128.5 (CH), 130.7 (d, 3JC-F = 8.5 Hz, CH), 131.2 (d, 4JC-F = 3.0 Hz, CH), 138.6 (Cquat), 148.5 (Cquat), 153.2 (Cquat), 155.6 (Cquat), 160.5 (d, 1JC-F = 250.3 Hz, Cquat), 168.7 (Cquat). – MS ((+)-EI): m/z (%) = 335 (75) [M]+, 320 (3) [M–CH3]+, 306 (60) [M–C2H4]+, 290 (100) [M–C2H5O]+, 262 (7) [M–C3H5O2]+, 242 (16), 220 (33). – IR: ν̃ (cm−1) = 3304 (w), 3061 (w), 2980 (w), 2932 (w), 2864 (w), 1721 (s), 1632 (m), 1614 (m), 1584 (s), 1545 (s), 1491 (s), 1447 (m), 1383 (m), 1364 (m), 1265 (s), 1221 (m), 1211 (m), 1173 (m), 1142 (m), 1111 (m), 1092 (s), 1072 (s), 1032 (m), 1013 (m), 889 (m), 856 (m), 833 (m), 789 (m), 752 (s), 731 (m), 689 (s), 669 (m), 652 (m), 683 (m), 621 (m). – Anal. for C21H18FNO2 (335.4): calcd. C 75.21, H 5.41, N 4.18; found C 75.47, H 5.55, N 3.92.
3.12 Ethyl 2-methyl-6-(thiophen-2-yl)pyridine-3-carboxylate (4i)
According to GP1 and after flash chromatography on silica gel (n-hexane-ethyl acetate 30:1) and 88 mg (36%) and according to GP2 52 mg (21%) of compound 4i was obtained as a pale yellow solid, m. p. 58°C (lit. [73]: 58°C). – 1H NMR (CDCl3, 300 MHz): δ = 1.41 (t, 3J = 7.1 Hz, 3 H), 2.88 (s, 3 H), 4.38 (q, 3J = 7.1 Hz, 2 H), 7.13 (dd, 3J = 5.1, 3.7 Hz, 1 H), 7.45 (dd, 3J = 5.1, 1.1 Hz, 1 H), 7.52 (d, 3J = 8.2 Hz, 1 H), 7.71 (dd, 3J = 3.7, 1.1 Hz, 1 H), 8.21 (d, 3J = 8.3 Hz, 1 H). – 13C NMR (CDCl3, 75 MHz): δ = 14.3 (CH3), 24.9 (CH3), 61.1 (CH2), 115.7 (CH), 123.2 (Cquat), 126.3 (CH), 128.3 (CH), 129.1 (CH), 139.4 (CH), 143.7 (Cquat), 154.0 (Cquat), 160.2 (Cquat), 166.2 (Cquat). – MS ((+)-EI): m/z (%) = 247 (100) [M]+, 219 (20) [M–C2H4]+, 202 (79) [M–C2H5O]+, 174 (11) [M–C3H5O2]+, 147 (13), 140 (11). – IR: ν̃ (cm−1) = 2955 (w), 2922 (m), 2853 (w), 1709 (s), 1582 (s), 1558 (m), 1460 (m), 1425 (m), 1261 (s), 1252 (s), 1236 (s), 1169 (s), 1090 (s), 1078 (s), 1020 (m), 847 (s), 802 (m), 783 (s), 731 (m), 692 (s).
3.13 Ethyl 2-methyl-6-(4-methoxyphenyl)pyridine-3-carboxylate (4j)
After flash chromatography on silica gel (n-hexane-ethyl acetate 20:1) and according to GP1 52 mg (19%) and according to GP2 84 mg (31%) of compound 4j was obtained as a colorless solid, m. p. 66°C (lit. [74]: 68°C). – 1H NMR (CDCl3, 300 MHz): δ = 1.41 (t, 3J = 7.1 Hz, 3 H), 2.91 (s, 3 H), 3.87 (s, 3 H), 4.39 (q, 3J = 7.1 Hz, 2 H), 7.00 (d, 3J = 8.9 Hz, 2 H), 7.56 (d, 3J = 8.3 Hz, 1 H), 8.04 (d, 3J = 8.9 Hz, 2 H), 8.24 (d, 3J = 8.2 Hz, 1 H). – 13C NMR (CDCl3, 75 MHz): δ = 14.3 (CH3), 25.2 (CH3), 55.4 (CH3), 61.1 (CH2), 114.2 (CH), 116.6 (CH), 122.9 (Cquat), 128.8 (CH), 130.9 (CH), 139.4 (Cquat), 158.6 (Cquat), 159.8 (Cquat), 161.1 (Cquat), 166.6 (Cquat). – MS ((+)-EI): m/z (%) = 271 (100) [M]+, 243 (21) [M–C2H4]+, 226 (46) [M–C2H5O]+, 198 (4) [M–C2H5O2]+. – IR: ν̃ (cm−1) = 2980 (w), 2961 (w), 2934 (w), 2835 (w), 1715 (s), 1580 (s), 1564 (m), 1512 (m), 1452 (m), 1288 (s), 1207 (w), 1182 (s), 1125 (m), 1107 (m), 1092 (s), 1074 (s), 1036 (s), 1022 (m), 982 (m), 826 (s), 783 (s), 733 (m), 608 (s).
3.14 Ethyl 2-methyl-6-(4-chlorophenyl)pyridine-3-carboxylate (4k)
According to GP1 and after flash chromatography on silica gel (n-hexane-ethyl acetate 50:1) 130 mg (47%) of compound 4k was obtained as an orange solid, m. p. 42–43°C (lit. [75]: 47–48°C). – 1H NMR (CDCl3, 300 MHz): δ = 1.42 (t, 3J = 7.2 Hz, 3 H), 2.90 (s, 3 H), 4.40 (d, 3J = 7.1 Hz, 2 H), 7.44 (d, 3J = 8.6 Hz, 2 H), 7.59 (d, 3J = 8.2 Hz, 1 H), 8.01 (d, 3J = 8.6 Hz, 2 H), 8.25 (d, 3J = 8.2 Hz, 1 H). – 13C NMR (CDCl3, 75 MHz): δ = 14.3 (CH3), 25.2 (CH3), 61.2 (CH2), 117.0 (CH), 123.9 (Cquat), 128.5 (CH), 129.0 (CH), 135.8 (Cquat), 136.9 (Cquat), 139.4 (CH), 157.7 (Cquat), 160.0 (Cquat), 166.5 (Cquat). – MS ((+)-EI): m/z (%) = 275 (100) [35Cl-M]+, 260 (4) [35Cl-M–CH3]+, 247 (24) [35Cl-M–C2H4]+, 230 (81) [35Cl-M–C2H5O]+, 202 (13) [35Cl-M–C3H5O2]+, 167 (27). – IR: ν̃ (cm−1) = 3414 (w), 3098 (w), 3071 (w), 2982 (w), 2899 (w), 2870 (w), 2849 (w), 1717 (s), 1584 (s), 1557 (m), 1493 (m), 1454 (m), 1435 (m), 1410 (m), 1389 (m), 1364 (m), 1263 (s), 1246 (s), 1194 (m), 1180 (m), 1155 (m), 1092 (s), 1070 (s), 1011 (s), 982 (m), 972 (m), 907 (m), 866 (m), 833 (m), 822 (m), 783 (s), 739 (m), 679 (m), 606 (m).
3.15 Ethyl 2-methyl-6-(4-nitrophenyl)pyridine-3-carboxylate (4l)
According to GP1 and after flash chromatography on silica gel (n-hexane-ethyl acetate 20:1) and 150 mg (52%) of compound 4l was obtained as a yellow solid, m. p. 141°C. – 1H NMR (CDCl3, 300 MHz): δ = 1.43 (t, 3J = 7.1 Hz, 3 H), 2.93 (s, 3 H), 4.42 (q, 3J = 7.1 Hz, 2 H), 7.71 (d, 3J = 8.2 Hz, 1 H), 8.24 (d, 3J = 9.0 Hz, 2 H), 8.28–8.37 (m, 3 H). – 13C NMR (CDCl3, 75 MHz): δ = 14.3 (CH3), 25.1 (CH3), 61.4 (CH2), 118.1 (CH), 124.0 (CH), 125.1 (Cquat), 128.1 (CH), 139.6 (CH), 144.1 (Cquat), 148.5 (Cquat), 156.2 (Cquat), 160.3 (Cquat), 166.2 (Cquat). – MS ((+)-EI): m/z (%) = 286 (100) [M]+, 271 (8) [M–CH3]+, 258 (40) [M–C2H4]+, 241 (96) [M–C2H5O]+, 213 (19) [M–C3H5O2]+, 166 (32), 140 (19). – IR: ν̃ (cm−1) = 2934 (w), 2980 (w), 2932 (w), 2897 (w), 2847 (w), 1717 (s), 1580 (m), 1564 (m), 1516 (s), 1435 (m), 1377 (m), 1366 (m), 1337 (s), 1323 (m), 1206 (m), 1187 (m), 1159 (m), 1107 (m), 1088 (s), 1070 (m), 1024 (m), 1009 (m), 982 (m), 872 (m), 847 (s), 792 (m), 750 (s), 725 (m), 689 (m), 675 (m). Anal. for C15H14N2O4 (286.3): calcd. C 62.93, H 4.93, N 9.79; found C 62.95, H 4.94, N 9.54.
3.16 Ethyl 2-methyl-6-(4-tolyl)pyridine-3-carboxylate (4m)
According to GP1 and after flash chromatography on silica gel (n-hexane-ethyl acetate 40:1) 77 mg (30%) of compound 4m was obtained as a yellow solid, m. p. 50°C (lit. [60]: 54°C). – 1H NMR (CDCl3, 300 MHz): δ = 1.42 (t, 3J = 7.1 Hz, 3 H), 2.41 (s, 3 H), 2.92 (s, 3 H), 4.39 (d, 3J = 7.1 Hz, 2 H), 7.27–7.31 (m, 2 H), 7.60 (d, 3J = 8.2 Hz, 1 H), 7.93–8.00 (m, 2 H), 8.25 (d, 3J = 8.2 Hz, 1 H). – 13C NMR (CDCl3, 75 MHz): δ = 14.3 (CH3), 21.3 (CH3), 25.2 (CH3), 61.1 (CH2), 117.0 (CH), 123.3 (Cquat), 127.2 (CH), 129.5 (CH), 135.6 (Cquat), 139.3 (CH), 139.9 (Cquat), 159.0 (Cquat), 159.8 (Cquat), 166.6 (Cquat). – MS ((+)-EI): m/z (%) = 255 (100) [M]+, 240 (6) [M–CH3]+, 227 (34) [M–C2H4]+, 210 (89) [M–C2H5O]+, 182 (28) [M–C3H5O2]+, 167 (30), 155 (15), 91 (11) [C7H7]+. – IR: ν̃ (cm−1) = 3028 (w), 2980 (w), 2930 (w), 2864 (w), 2733 (w), 1707 (s), 1672 (m), 1578 (s), 1557 (m), 1510 (m), 1481 (m), 1454 (m), 1435 (m), 1412 (m), 1391 (m), 1379 (m), 1362 (m), 1306 (m), 1265 (s), 1246 (s), 1206 (m), 1184 (s), 1155 (m), 1117 (m), 1090 (s), 1072 (s), 1018 (m), 984 (m), 966 (m), 943 (m), 907 (m), 860 (m), 820 (m), 781 (s), 735 (m), 692 (m), 610 (m).
3.17 Ethyl 2-methyl-6-(2-fluorophenyl)pyridine-3-carboxylate (4n)
According to GP1 and after flash chromatography on silica gel (n-hexane-ethyl acetate 50:1) and 142 mg (55%) of compound 4n was obtained as a yellow oil. – 1H NMR (CDCl3, 300 MHz): δ = 1.42 (t, 3J = 7.2 Hz, 3 H), 2.92 (s, 3 H), 4.40 (q, 3J = 7.1 Hz, 2 H), 7.16 (ddd, 3JH-F = 11.4 Hz, 3J = 8.2, 1.2 Hz, 1 H), 7.28 (td, 3J = 7.5, 1.2 Hz, 1 H), 7.34–7.46 (m, 1 H), 7.71 (dd, 3J = 8.2 Hz, 5JH-F = 2.3 Hz, 1 H), 8.08 (td, 3J = 7.8, 1.9 Hz, 1 H), 8.27 (d, 3J = 8.2 Hz, 1 H). – 13C NMR (CDCl3, 75 MHz): δ = 14.3 (CH3), 25.0 (CH3), 61.2 (CH2), 116.2 (d, 2JC F = 23.0 Hz, CH), 121.5 (d, 3JC-F = 10.0 Hz, CH), 124.1 (Cquat), 124.6 (CH), 126.5 (d, 2JC-F = 11.3 Hz, Cquat), 131.1 (d, 3JC-F = 8.7 Hz, CH), 131.3 (d, 4JC-F = 2.7 Hz, CH), 139.0 (CH), 155.0 (d, 4JC-F = 2.2 Hz, Cquat), 159.8 (Cquat), 160.8 (d, 1JC-F = 249 Hz, Cquat), 166.5 (Cquat). – MS ((+)-EI): m/z (%) = 259 (100) [M]+, 244 (6) [M–CH3]+, 231 (22) [M–C2H4]+, 214 (90) [M–C2H5O]+, 186 (29) [M–C3H5O2]+, 159 (18), 133 (12). – IR: ν̃ (cm−1) = 2980 (w), 2932 (w), 2905 (w), 2874 (w), 1719 (s), 1585 (s), 1560 (m), 1489 (m), 1371 (m), 1524 (s), 1153 (m), 1086 (s), 1072 (s), 1032 (m), 1018 (m), 806 (m), 791 (m), 756 (s), 743 (m), 721 (m), 698 (m), 669 (m), 640 (m). – Anal. for C15H14FNO2 (259.3): calcd. C 69.49, H 5.44, N 5.40; found C 69.47, H 5.41, N 5.40.
3.18 Ethyl 2-methyl-6-phenylpyridine-3- carboxylate (4o)
According to GP1 and after flash chromatography on silica gel (n-hexane-ethyl acetate 40:1) and 67 mg (28%) of compound 4o was obtained as an orange oil. – 1H NMR (CDCl3, 300 MHz): δ = 1.42 (t, 3J = 7.2 Hz, 3 H), 2.93 (s, 3 H), 4.40 (q, 3J = 7.1 Hz, 2 H), 7.38–7.57 (m, 3 H), 7.63 (d, 3J = 8.2 Hz, 1 H), 8.01–8.14 (m, 2 H), 8.27 (d, J = 8.2 Hz, 1 H). – 13C NMR (CDCl3, 75 MHz): δ = 14.3 (CH3), 25.2 (CH3), 61.1 (CH2), 117.4 (CH), 123.7 (Cquat), 127.3 (CH), 128.8 (CH), 129.6 (CH), 138.4 (Cquat), 139.3 (CH), 159.0 (Cquat), 159.9 (Cquat), 166.6 (Cquat). – MS ((+)-EI): m/z (%) = 241 (91) [M]+, 226 (7) [M–CH3]+, 213 (26) [M–C2H4]+, 196 (100) [M–C2H5O]+, 168 (44) [M–C3H5O2]+, 141 (27), 115 (18), 84 (11). – IR: ν̃ (cm−1) = 3063 (w), 2980 (w), 2932 (w), 2905 (w), 2872 (w), 1717 (s), 1582 (s), 1558 (m), 1447 (m), 1435 (m), 1381 (m), 1371 (m), 1261 (s), 1246 (s), 1206 (m), 1184 (m), 1152 (m), 1144 (m), 1090 (s), 1074 (s), 1018 (m), 1001 (m), 847 (m), 754 (s), 731 (m), 708 (m), 691 (s), 650 (m), 611 (m).
Supporting information
1H, 13C, and 13-DEPT-135 NMR spectra of 3-ethoxycarbonyl-2-methylpyridines 4 are given as Supporting Information available online (DOI: 10.1515/znb-2016-0046).
Acknowledgments
The authors cordially thank the Fonds der Chemischen Industrie for financial support.
References
[1] F. Brody, P. R. Ruby, R. A. Barnes, Pyridine and its Derivatives, in The Chemistry of Heterocyclic Compounds, Vol. 14, part I (Ed. E. Klingsberg), John Wiley & Sons, Inc., New York, 1960.Search in Google Scholar
[2] R. P. Kreher, in Houben-Weyl, Methoden der organischen Chemie, Vol. E7b, (Ed.: D. Spitzner), Thieme Verlag, Stuttgart, 1992.Search in Google Scholar
[3] J. Jarusiewicz, K. S. Yoo, K. W. Jung, Synlett2009, 482.10.1055/s-0028-1087528Search in Google Scholar
[4] K. Shibatomi, T. Muto, Y. Sumikawa, A. Narayama, S. Iwasa, Synlett2009, 241.10.1055/s-0028-1087675Search in Google Scholar
[5] S. Lin, X. Lu, Org. Lett.2010, 12, 2536.10.1021/ol100767uSearch in Google Scholar
[6] R. Murugan, E. F. V. Scriven, Aldrichimica Acta2003, 36, 21.Search in Google Scholar
[7] G. C. Fu, Acc. Chem. Res.2004, 37, 542.10.1021/ar030051bSearch in Google Scholar
[8] N. De Rycke, F. Couty, O. R. P. David, Chem. Eur. J.2011, 17, 12852.10.1002/chem.201101755Search in Google Scholar
[9] H. Hofmeier, U. S. Schubert, Chem. Soc. Rev.2004, 33, 373.10.1039/B400653BSearch in Google Scholar
[10] A. Mishra, R. Gupta, Dalton Trans.2014, 43, 7668.10.1039/C4DT00277FSearch in Google Scholar
[11] J.-M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, VCH, Weinheim, 1995.10.1002/3527607439Search in Google Scholar
[12] U. S. Schubert, C. Eschbaumer, Angew. Chem. Int. Ed.2002, 41, 2892.10.1002/1521-3773(20020816)41:16<2892::AID-ANIE2892>3.0.CO;2-6Search in Google Scholar
[13] H. Hofmeier, U. S. Schubert, Chem. Commun.2005, 19, 2423.10.1039/b419171dSearch in Google Scholar
[14] T. Šmejkal, B. Breit, Angew. Chem. Int. Ed.2008, 47, 311.10.1002/anie.200703192Search in Google Scholar PubMed
[15] N.-G. Kang, M. Changez, J.-S. Lee, Macromolecules2007, 40, 8553.10.1021/ma071349aSearch in Google Scholar
[16] C. Kaes, A. Katz, M. W. Hosseini, Chem. Rev.2000,100, 3553.10.1021/cr990376zSearch in Google Scholar PubMed
[17] R. Makiura, S. Motoyama, Y. Umemura, H. Yamanaka, O. Sakata, H. Kitagawa, Nat. Mater.2010, 9, 565.10.1038/nmat2769Search in Google Scholar PubMed
[18] D. Wang, H. Wang, H. Li, ACS Appl. Mater. Interfaces2013, 5, 6268.10.1021/am401318aSearch in Google Scholar PubMed
[19] V. Duprez, M. Biancardo, H. Spanggaard, F. C. Krebs, Macromolecules2005,38, 10436.10.1021/ma051274fSearch in Google Scholar
[20] C. Fan, C. Ye, X. Wang, Z. Chen, Y. Zhou, Z. Liang, X. Tao, Macromolecules2015, 48, 6465.10.1021/acs.macromol.5b00493Search in Google Scholar
[21] E. Tekin, E. Holder, V. Marin, B.-J. de Gans, U. S. Schubert, Macromol. Rapid Commun.2005, 26, 293.10.1002/marc.200400507Search in Google Scholar
[22] L. Yang, V.-A. Mihali, D. Brandell, M. Strømme, M. Sjödin, J. Phys. Chem. C2014, 118, 25956.10.1021/jp509606cSearch in Google Scholar
[23] R. B. Lacerda, C. K. F. de Lima, L. L. da Silva, N. C. Romeiro, A. L. P. Miranda, E. J. Barreiro, C. A. M. Fraga, Bioorg. Med. Chem.2009, 17, 74.10.1016/j.bmc.2008.11.018Search in Google Scholar PubMed
[24] C. D. Duffy, P. Maderna, C. McCarthy, C. E. Loscher, C. Godson, P. J. Guiry, ChemMedChem2010, 5, 517.10.1002/cmdc.200900533Search in Google Scholar
[25] G. M. Buckley, N. Cooper, R. J. Davenport, H. J. Dyke, F. P. Galleway, L. Gowers, A. F. Haughan, H. J. Kendall, C. Lowe, J. G. Montana, J. Oxford, J. C. Peake, C. L. Picken, M. D. Richard, V. Sabin, A. Sharpe, J. B. H. Warneck, Bioorg. Med. Chem. Lett.2002, 12, 509.10.1016/S0960-894X(01)00786-7Search in Google Scholar
[26] B. Vacher, B. Bonnaud, P. Funes, N. Jubault, W. Koek, M.-B. Assié, C. Cosi, M. Kleven, J. Med. Chem.1999, 42, 1648.10.1021/jm9806906Search in Google Scholar PubMed
[27] D. O’Hagan, Nat. Prod. Rep.2000, 17, 435.10.1039/a707613dSearch in Google Scholar PubMed
[28] Z. J. Song, M. Zhao, R. Desmond, P. Devine, D. M. Tschaen, R. Tillyer, L. Frey, R. Heid, F. Xu, B. Foster, J. Li, R. Reamer, R. Volante, U. H. Dolling, P. J. Reider, S. Okada, Y. Kato, E. Mano, J. Org. Chem.1999, 64, 9658.10.1021/jo991292tSearch in Google Scholar
[29] Y. Abe, H. Kayakiri, S. Satoh, T. Inoue, Y. Sawada, N. Inamura, M. Asano, I. Aramori, C. Hatori, H. Sawai, T. Oku, H. Tanaka, J. Med. Chem.1998, 41, 4062.10.1021/jm980300fSearch in Google Scholar PubMed
[30] M. Horiuch, C. Murakami, N. Fukamiya, D. Yu, T.-H. Chen, K. F. Bastow, D.-C. Zhang, Y. Takaishi, Y. Imakura, K.-H. Lee, J. Nat. Prod.2006, 69, 1271.10.1021/np060124aSearch in Google Scholar PubMed
[31] S. Follot, J.-C. Debouzy, D. Crouzier, C. Enguehard-Gueiffier, A. Gueiffier, F. Nachon, B. Lefebvre, F. Fauvelle, Eur. J. Med. Chem.2009, 44, 3509.10.1016/j.ejmech.2008.12.026Search in Google Scholar PubMed
[32] C.-M. Lu, Y.-L. Chen, H.-L. Chen, C.-A. Chen, P.-J. Lu, C.-N. Yang, C.-C. Tzeng, Bioorg. Med. Chem.2010, 18, 1948.10.1016/j.bmc.2010.01.033Search in Google Scholar PubMed
[33] T. F. Spande, H. M. Garaffo, M. W. Edwards, H. J. C. Yeh, L. Pannell, J. W. Daly, J. Am. Chem. Soc.1992, 114, 3475.10.1021/ja00035a048Search in Google Scholar
[34] B. Badio, J. W. Daly, Mol. Pharm.1994, 45, 563.Search in Google Scholar
[35] J. W. Daly, T. F. Spande, H. M. Garaffo, J. Nat. Prod.2005, 68, 1556.10.1021/np0580560Search in Google Scholar PubMed
[36] T. Kaneshiro, G. Zweig, Appl. Microbiol.1965, 13, 939.10.1128/am.13.6.939-944.1965Search in Google Scholar PubMed PubMed Central
[37] G. Matolcsy, Pesticide Chemistry, Elsevier Scientific, Amsterdam, Oxford, 1988, pp. 427.Search in Google Scholar
[38] H. Wu, R. Zhang, J. Liu, Y. Guo, E. Ma, Chemosphere2011, 83, 599.10.1016/j.chemosphere.2010.12.004Search in Google Scholar PubMed
[39] T. Kaneshiro, G. Zweig, Appl. Microbiol.1965, 13, 939.10.1128/am.13.6.939-944.1965Search in Google Scholar
[40] É. Lukevits, Chem. Heterocycl. Comp.1995, 31, 723.10.1007/BF01414383Search in Google Scholar
[41] C. Allais, J.-M. Grassot, J. Rodriguez, T. Constantieux, Chem. Rev.2014, 114, 10829.10.1021/cr500099bSearch in Google Scholar PubMed
[42] J. A. Bull, J. J. Mousseau, G. Pelletier, A. B. Charette, Chem. Rev.2012, 112, 2642.10.1021/cr200251dSearch in Google Scholar PubMed
[43] D. Spitzner, Sci. Synth.2005, 15, 11.Search in Google Scholar
[44] G. D. Henry, Tetrahedron2004, 60, 6043.10.1016/j.tet.2004.04.043Search in Google Scholar
[45] A. E. Tschitschibabin, J. Prakt. Chem.1924, 107, 122.10.1002/prac.19241070110Search in Google Scholar
[46] A. Hantzsch, Ber. Dtsch. Chem. Ges.1881, 14, 1637.10.1002/cber.18810140214Search in Google Scholar
[47] R. Lavilla, J. Chem. Soc., Perkin Trans. 12002, 1141.10.1039/b101371hSearch in Google Scholar
[48] M. Baumann, I. R. Baxendale, Beilstein J. Org. Chem.2013, 9, 2265.10.3762/bjoc.9.265Search in Google Scholar
[49] O. G. Dediu, N. A. M. Yehia, T. J. J. Müller, Z. Naturforsch.2004, 59b, 443.10.1515/znb-2004-0413Search in Google Scholar
[50] O. G. Dediu, N. A. M. Yehia, T. Oeser, K. Polborn, T. J. J. Müller, Eur. J. Org. Chem.2005, 1834.10.1002/ejoc.200400828Search in Google Scholar
[51] N. A. M. Yehia, K. Polborn, T. J. J. Müller, Tetrahedron Lett.2002, 43, 6907.10.1016/S0040-4039(02)01615-5Search in Google Scholar
[52] O. G. Schramm (née Dediu), T. Oeser, T. J. J. Müller, J. Org. Chem.2006, 71, 3494.10.1021/jo0602726Search in Google Scholar
[53] T. J. J. Müller, M. Ansorge, D. Aktah, Angew. Chem. Int. Ed.2000, 39, 1253.10.1002/(SICI)1521-3773(20000403)39:7<1253::AID-ANIE1253>3.0.CO;2-XSearch in Google Scholar
[54] R. U. Braun, M. Ansorge, T. J. J. Müller, Chem. Eur.J.2006, 12, 9081.10.1002/chem.200600530Search in Google Scholar
[55] O. G. Schramm (née Dediu), T. J. J. Müller, Adv. Synth. Cat.2006, 348, 2565.10.1002/adsc.200600280Search in Google Scholar
[56] T. J. J. Müller, Synthesis2012, 44, 159.10.1055/s-0031-1289636Search in Google Scholar
[57] F. Bohlmann, D. Rahtz, Chem. Ber.1957, 90, 2265.10.1002/cber.19570901021Search in Google Scholar
[58] C. J. Moody, M. C. Bagley, Synlett1998, 361.10.1055/s-1998-1670Search in Google Scholar
[59] C. J. Moody, M. C. Bagley, Chem. Commun.1998, 2049.10.1039/a805762aSearch in Google Scholar
[60] M. C. Bagley, K. E. Bashford, C. L. Hesketh, C. J. Moody, J. Am. Chem. Soc.2000, 122, 3301.10.1021/ja994247bSearch in Google Scholar
[61] M. C. Bagley, J. W. Dale, J. Bower, Synlett2001, 1149.10.1055/s-2001-15140Search in Google Scholar
[62] M. C. Bagley, J. W. Dale, D. D. Hughes, M. Ohnesorge, N. G. Phillips, J. Bower, Synlett2001, 1523.10.1055/s-2001-17447Search in Google Scholar
[63] M. C. Bagley, C. Brace, J. W. Dale, M. Ohnesorge, N. G. Phillips, X. Xiong, J. Bower, J. Chem. Soc., Perkin Trans. 12002, 1663.10.1039/b203397fSearch in Google Scholar
[64] M. C. Bagley, R. Lunn, X. Xiong, Tetrahedron Lett.2002, 43, 8331.10.1016/S0040-4039(02)01975-5Search in Google Scholar
[65] M. C. Bagley, J. W. Dale, J. Bower, Chem. Commun.2002, 1682.10.1039/b203900aSearch in Google Scholar PubMed
[66] X. Xiong, M. C. Bagley, K. Chapaneri, Tetrahedron Lett.2004, 45, 6121.10.1016/j.tetlet.2004.06.061Search in Google Scholar
[67] B. Willy, T. J. J. Müller ARKIVOC2008, Part I, 195.10.3998/ark.5550190.0009.107Search in Google Scholar
[68] B. Willy, T. J. J. Müller, Curr. Org. Chem.2009, 13, 1777.10.2174/138527209789630479Search in Google Scholar
[69] T. J. J. Müller, Top. Heterocycl. Chem.2010, 25, 25.10.1007/7081_2010_43Search in Google Scholar
[70] T. J. J. Müller, K. Deilhof, in Multicomponent Reactions in Organic Synthesis, (Eds.: J. Zhu, Q. Wang, M.-X. Wang), Wiley-VCH Verlag, Weinheim, 2015, pp. 333.10.1002/9783527678174.ch12Search in Google Scholar
[71] A. S. Karpov, T. J. J. Müller, Org. Lett.2003, 5, 3451.10.1021/ol035212qSearch in Google Scholar PubMed
[72] D. M. D’Souza, T. J. J. Müller, Nat. Protoc.2008, 3, 1660.10.1038/nprot.2008.152Search in Google Scholar PubMed
[73] S. Kantevari, S. R. Patpi, D. Addla, S. R. Putapatri, B. Sridhar, P. Yogeeswari, D. Sriram, ACS Comb. Sci.2011, 13, 427.10.1021/co2000604Search in Google Scholar PubMed
[74] M. C. Bagley, K. Chapaneri, J. W. Dale, X. Xiong, J. Bower, J. Org. Chem.2005, 70, 1389.10.1021/jo048106qSearch in Google Scholar PubMed
[75] M. C. Bagley, J. W. Dale, M. Ohnesorge, X. Xiong, J. Bower, ACS. Comb. Sci.2003, 5, 41.Search in Google Scholar
Supplemental Material:
The online version of this article (DOI: 10.1515/znb-2016-0046) offers supplementary material, available to authorized users.
©2016 by De Gruyter
Articles in the same Issue
- Frontmatter
- In this Issue
- Preface
- Chemistry of the iminium and imine functional groups
- Transformations of β-aryl-N-Cbz-α,β-didehydro-α-amino esters with hydrazine hydrate
- A short synthesis of pyridines from deprotonated α-aminonitriles by an alkylation/RCM sequence
- Palladium complexes of anionic N-heterocyclic carbenes derived from sydnones in catalysis
- Synthesis and characterisation of long wavelength-absorbing donor/acceptor-substituted methine dyes
- Selective di- and monochlorination of pyridazine-annelated bis(imidazolium) salts
- Synthesis and investigation of new cyclic haloamidinium salts
- A simple synthesis of dimethyl 2-[(Z)-3-amino-1-oxo-1-(substituted)but-2-en-2-yl]fumarates: potential intermediates in the synthesis of polysubstituted five- and six-membered heterocycles
- 2-(1,2,3-Triazol-4-yl)-imidazoline, -oxazoline, -thiazoline and -tetrahydropyrimidine as ligands in copper(II) and nickel(II) complexes
- Derivatives of the triaminoguanidinium ion, 4. O-Sulfonylation of N,N′,N″-tris(hydroxybenzylidenamino)guanidinium ions
- Consecutive three- and four-component coupling-Bagley-Bohlmann-Rahtz syntheses of tri- and tetrasubstituted pyridines
- Orthoamide und Iminiumsalze, XCI. N,N′,N″-Peralkylierte Guanidiniumsalze – Ionische Flüssigkeiten als Hilfsmittel in der Elektronenmikroskopie
Articles in the same Issue
- Frontmatter
- In this Issue
- Preface
- Chemistry of the iminium and imine functional groups
- Transformations of β-aryl-N-Cbz-α,β-didehydro-α-amino esters with hydrazine hydrate
- A short synthesis of pyridines from deprotonated α-aminonitriles by an alkylation/RCM sequence
- Palladium complexes of anionic N-heterocyclic carbenes derived from sydnones in catalysis
- Synthesis and characterisation of long wavelength-absorbing donor/acceptor-substituted methine dyes
- Selective di- and monochlorination of pyridazine-annelated bis(imidazolium) salts
- Synthesis and investigation of new cyclic haloamidinium salts
- A simple synthesis of dimethyl 2-[(Z)-3-amino-1-oxo-1-(substituted)but-2-en-2-yl]fumarates: potential intermediates in the synthesis of polysubstituted five- and six-membered heterocycles
- 2-(1,2,3-Triazol-4-yl)-imidazoline, -oxazoline, -thiazoline and -tetrahydropyrimidine as ligands in copper(II) and nickel(II) complexes
- Derivatives of the triaminoguanidinium ion, 4. O-Sulfonylation of N,N′,N″-tris(hydroxybenzylidenamino)guanidinium ions
- Consecutive three- and four-component coupling-Bagley-Bohlmann-Rahtz syntheses of tri- and tetrasubstituted pyridines
- Orthoamide und Iminiumsalze, XCI. N,N′,N″-Peralkylierte Guanidiniumsalze – Ionische Flüssigkeiten als Hilfsmittel in der Elektronenmikroskopie