Multicomponent green synthesis, spectroscopic and structural investigation of multi-substituted imidazoles. Part 1
-
Shaaban K. Mohamed
 ,
Jim Simpson
,
Jim Simpson

Abstract
Ten 1,2,4,5-tetra-substituted imidazole derivatives have been synthesized with a 2-hydroxyethy substituent at the 1-nitrogen atom and potentially electron releasing hydroxy-, methoxy-, dimethylamino- or nitro substituents in various positions on the benzene ring located on the 2-carbon atom. The prototypical derivative with an unsubstituted phenyl ring at the 2-position is also reported. The compounds are obtained in excellent yields (average 86%) via a four-component cyclocondensation reaction of benzil, ethanolamine, and the appropriate aromatic carbaldehyde together with ammonium acetate. The reaction uses a novel ionic liquid catalyst, DEAHS (diethyl ammonium hydrogen sulfate), under solvent-free conditions and a green synthetic protocol. The key advantages of this process are high yield, shorter reaction times and ease of work-up. Furthermore, the products can be purified by a non-chromatographic method and the catalyst is re-usable. All of these newly synthesized compounds have been characterized from spectral data; the X-ray structures of three representative molecules are also detailed.
1 Introduction
Imidazoles are an important class of heterocycles being the core fragment of numerous natural products and biological systems [1]. In general, compounds incorporating imidazole moieties play important roles in biochemical processes and display significant pharmacological properties [2]. Various substituted imidazole derivatives have also been found to have important biological functions acting as anti-helminthic, analgesic, antibacterial, antifungal, antiviral, tuberculostatic, cytostatic, and anti-inflammatory agents [3].
Multicomponent reactions (MCRs) have excited a great deal of interest among chemists and pharmacists due to their outstanding status in modern organic synthesis and medicinal chemistry. MCRs are one-pot processes bringing together three or more reaction components with high atom economy and good selectivity [4, 5]. Developing new MCRs [6] and improving known multi-component reactions are areas of considerable current interest and MCR processes leading to imidazole derivatives are no exception. The presence of imidazole rings in so many biologically important natural products and pharmacologically active compounds has spawned a diverse array of synthetic approaches to the production of these heterocycles [7], the majority being classical methods [8–10]. In particular, tetrasubstituted imidazoles and their derivatives have been synthesized by several methods [11–14]. In addition, such syntheses are usually carried out in polar organic solvents such as ethanol, methanol, acetic acid, DMF and DMSO and involve complex isolation requirements, side products and tedious recovery procedures. Despite intensive efforts to improve these syntheses, only a handful of general methods exist for the construction of tetrasubstituted imidazoles. These include reactions catalyzed by silica gel or Zeolite HY [15], silica gel-NaHSO4 [16], molecular iodine [17], K5CoW12O40·3H2O [18], hetero-polyacids [19] and HClO4-SiO2 [20]. BF3, as a strong Lewis acid, has also been used in both small and large scale reactions as an acid catalyst [21]. All of these processes generate waste containing both catalyst and solvent, which must be recovered, treated and disposed of. The toxicity and volatile nature of many organic solvents, particularly chlorinated hydrocarbons, which are widely used in huge amounts for organic reactions, have posed a serious threat to the environment [22]. Thus, the design of solvent-free catalytic reactions has received tremendous attention in recent times in the area of green synthesis [23, 24].
Ionic liquid (IL) technology offers a new and environmentally benign approach toward modern synthetic chemistry. Ionic liquids have interesting advantages such as extremely low vapor pressure, excellent thermal stability, reusability and the ability to dissolve many organic and inorganic substrates [25]. Ionic liquids have been successfully employed as solvents and catalysts for a variety of reactions [26–29] which promise widespread applications in both small and large scale organic syntheses. In a continuation of our efforts to develop Lewis and Brønsted acid catalyzed synthetic methodologies [30–33] and further to our study of the synthesis of bio-active molecules [34–36] we report herein a simple and environmentally friendly MCR technique for the synthesis of potentially bio-active tetrasubstituted imidazole compounds (Scheme 1) in high yields using the novel Brønsted acidic ionic liquid, diethyl ammonium hydrogen sulfate (DEAHS) as a catalyst for the first time.

Synthesis of 2a–2j, R= H, 2a, 2-OH, 2b, 4-OH, 2c, 3-OCH3, 2d, 4-OCH3, 2e, 2,4-OCH3, 2f, 3,4-OCH3, 2g, 2,5-OCH3, 2h, 4-(CH3)2N, 2i and4-NO2, 2j.
Because of the pharmaceutical potential of these products, the structures of three representative derivatives have been determined by X-ray crystallography and are reported here. Structures of 2,4,5-triphenyl-substituted imidazole compounds are reasonably common with 214 hits in the Cambridge Structural Database (CSD) [37], including the archetypal lophine (2,4,5-triphenyl-1H-imidazole) [38]. This number drops to 180 if coordination complexes of the imidazole derivatives are excluded. However, only five structures of 2,4,5-triphenyl-substituted imidazole derivative with a alcohol substituents on the imidazole sp3-N atom have been reported. One of these [39] is compound 2e in this paper, with details of the spectroscopic identification of this molecule included here for completeness and comparison with the other, similar derivatives. Two other related compounds have 2-hydroxypropyl substituents [40, 41], with 3-phenylpropan-1-ol substituents on the sp3-N atom in the two other similar compounds [42, 43]. Interestingly, only ten other structures in the database have alkyl chains of two or more C atoms on the 1-nitrogen atom of the imidazole [44–52], with simple aliphatic chains predominating and with no other alcohol derivatives.
2 Results and discussion
2.1 Synthetic procedures
In order to determine the most appropriate reaction conditions and to evaluate the catalytic efficiency of the ionic liquid diethyl ammonium hydrogen sulfate (DEAHS) [53], an initial model study was carried out on the synthesis of 1,2,4,5-tetrasubstituted imidazoles. Among the tested solvents were methanol, ethanol, DMF and DMSO; a solvent-free system was also investigated. Condensation of benzil, benzaldehyde, ethanolamine, and ammonium acetate to form 2-(2,4,5-triphenyl-1H-imidazol-1-yl)ethanol (2a) was found to be significantly more facile under solvent-free conditions and proceeded to give the highest yield in a relatively very short time, Table 1.
Synthesis of 2-(2,4,5-triphenyl-1H-imidazol-1-yl)ethanol (2a) using diethylammonium hydrogen sulfate (DEAHS) catalyst (0.3 equiv) in different solvents.
| Solvent | T (°C) | Time | Yield (%) |
|---|---|---|---|
| Methanol | 67 | 3 h | 40 |
| Ethanol | 78 | 3 h | 45 |
| Acetonitrile | 67 | 3 h | 35 |
| DMF | 100 | 2 h | 55 |
| DMSO | 100 | 2 h | 60 |
| Solvent-free conditions | 100 | 20 min | 98 |
Additionally, we sought to evaluate the generality of this process for the synthesis of a broad range of derivatives with a variety of substituents on the phenyl ring at the 2-position of the imidazole ring, Scheme 1. Thus, reactions of benzil (10 mmol), with various aromatic aldehydes (10 mmol) bearing electron releasing groups (such as hydroxyl, mono- and di-methoxy, or dimethyl amino substituents), ethanolamine (11 mmol) and ammonium acetate (10 mmol), were carried out in the presence of diethylammonium hydrogen sulfate (0.5 g, 3 mmol) as catalyst (Table 1). The yields of all the products so obtained were very good to excellent. Furthermore, the reactions proceeded without the formation of any side products, such as 2,4,5-trisubstituted imidazoles, which are normally observed as an impediment to efficient synthesis under the influence of other strong acids [19, 20]. An earlier report of the preparation of (2a) used a similar preparative route, but with l-proline triflate as the catalyst [54].
The yield and reaction time for the formation of 2a has been recorded using different catalysts as listed in Table 2 and the results represented graphically in Fig. 1. While it is obvious that reaction temperatures will have a significant effect on reaction times, the data in Table 1 clearly confirm that DEAHS is indeed a highly efficient catalyst in terms of both yield and reaction time when compared to other type of catalysts that have been used.
Comparison of the efficiency (yield and reaction time) of various catalysts with diethyl ammonium hydrogen sulfate in the synthesis of the 2-(2,4,5-triphenyl-1H-imidazol-1-yl)ethanol, 2a.
| No | Catalyst | Conditions | Time (min) | Yield (%) | Refs. |
|---|---|---|---|---|---|
| CI | InCl3·3H2O | MeOH, r. t. | 492 | 76 | [12] |
| CII | KH2PO4 | Reflux in EtOH | 60 | 89 | [55] |
| CIII | Yb(OPf)3 | C10F18, 80°C | 360 | 80 | [1] |
| CIV | Zr(acac)4 | Reflux in EtOH | 120 | 95 | [56] |
| CV | l-proline | Methanol, 60°C | 540 | 87 | [57] |
| CVI | [Hbim]BF4 | 100°C | 60 | 94 | [58] |
| CVII | NiCl2·6H2O-Al2O3 | Reflux in EtOH | 90 | 89 | [59] |
| CVIII | Et2NH2+ HSO4– (deahs) | 100°C | 20 | 98 | this work |

Dependence of the yield and time of reaction on the catalyst type.
Hydrogen bond formation between the carbonyl group of the starting aldehyde and the catalyst DEAHS makes the carbonyl very susceptible to nucleophilic attack by an ammonia molecule generated from the ammonium acetate starting material. The resulting intermediate readily loses a water molecule to produce the imine I (Scheme 2). Similarly the catalyst promotes an additional condensation reaction of one of the carbonyl groups from benzil with the aminoethanol reactant to furnish the imine II. DEAHS also catalyzes nucleophilic attack by the imine I on the second carbonyl group in imine II followed by protonation to form the intermediate cation III. This in turn undergoes an intramolecular cyclization to give the unstable cyclic cation IV which eliminates water and then loses a proton to yield the target products 2a–2j. All products were fully characterized by spectroscopic methods such as IR, 1H NMR, 13C NMR and the structures of 2a, 2c and 2j were determined by X-ray crystallography (see below).

General mechanism for the multi-component synthesis.
2.2 Spectroscopic properties
The IR spectra of all of the compounds showed definitive broad bands in the frequency range 3568–3144 cm–1 for the OH stretching vibration of the ethanolic OH groups of all nine compounds. The phenolic OH stretches for 2b and 2c could not be distinguished separately from these vibrations. In addition, C–H stretching modes were found in the range 3084–3053 cm–1 while the C=N stretching mode of the imidazole unit was observed in a relatively narrow range 1601–1603 cm–1 for 2a–2h with the corresponding vibration for the dimethylamino substituted derivative 2i appearing at 1610 cm–1 while that for the 4-nitro derivative 2j appeared at 1595 cm–1. The stretching vibrations of the nitro group of 2j were observed at 1537 cm–1 (asymmetrical) and 1338 cm–1 (symmetrical). Clearly the nature of the substituent on the 2-benzene ring has a noticeable effect on the vibrational spectra.
2.3 Molecular and crystal structures of 2a, 2c and 2j
The molecular structures of the three compounds investigated by X-ray crystallography are shown in Fig. 2 (i–iii). The broad features of the three structures are sufficiently similar to be discussed together. Each molecule comprises a central imidazole ring substituted at N1 with a CH2CH2OH unit which generally lies almost orthogonal to the imidazole ring, Table 3. The imidazole ring also carries an aromatic ring on each C atom with a substituted benzene ring on the C2 carbon atom for all but 2a and simple phenyl substituents on C4 and C5. The relative inclinations of these benzene rings to the central imidazole ring are also detailed in Table 3 which clearly shows that the benzene and imidazole rings are not co-planar in any of the molecules.

The molecular structures of (i) 2a; (ii) 2c; (iii) the two unique molecules in the asymmetric unit of 2j (crystallographic atom numbering and displacement ellipsoids drawn at the 50% probability level; H atoms as spheres with arbitrary radii).
Dihedral angles (deg) between the imidazole and benzene ringsa for 2a, 2c and 2j.
| Compound | 1–2 | 1–3 | 1–4 |
|---|---|---|---|
| 2a | 53.76(19) | 21.02(15) | 71.31(19) |
| 2c | 72.21(7) | 30.70(11) | 73.82(8) |
| 2j (molecule 1) | 42.82(4) | 22.91(3) | 70.19(4) |
| 2j (molecule 2) | 56.10(4) | 17.46(2) | 89.88(4) |
aPlanes are numbered as follows: 1: N1,C2,N3,C4,C5; 2: C21…C26, 3: C41…C46; 4: C51…C56. For 2j, atom numbers defining the planes have additional leading 1 and 2 characters for molecules 1 and 2, respectively [Fig. 2 (iii)].
The C2 benzene ring substituents differentiate the three molecules, starting with the prototypical compound 2a with a simple phenyl ring in the 2-position. 4-Hydroxy substitution is found in the C2 benzene ring of 2b, while 2j has a nitro substituent at the 4-position of the C21…C26 benzene rings of each of the two unique molecules. These are distinguished in the atom numbering by leading 1 and 2 characters; the two molecules differ principally in the inclination of the benzene rings at C2, C4 and C5 relative to the imidazole ring, (vide supra, Table 3). Also for 2j, the planes of the nitro groups are inclined at 12.52(11)° for molecule 1 and 4.3(3)° for molecule 2 to the planes of the benzene ring to which they are bound.
3 Conclusion
We have developed a general procedure for the synthesis of tetrasubstituted imidazoles in high yield by a simple, one-pot, four-component reaction of benzil, aldehydes, ethanolamine and ammonium acetate utilizing diethylammonium hydrogen sulphate (DEAHS) as a new ionic liquid and catalyst. The reactions proceed in the absence of any solvent or additives. This protocol offers a broad scope for access to a wide spectrum of diversely substituted 1,2,4,5-tetrasubstituted imidazoles. The absence of toxic organic solvents from the entire process, multiple reusability of the catalyst, good product purity, simplicity in operation, and no requirement for chromatographic purification, shorter reaction times and high yields of products make this procedure greener, more efficient and importantly cost effective. In view of their pharmaceutical potential the molecular structures of three representative compounds were also determined by X-ray diffraction.
4 Experimental section
All reagents were purchased from Aldrich and Merck supplier companies, and used without further purification. Melting points were determined by the open capillary method using a Gallenkamp melting point apparatus and are uncorrected. CHN microanalyses were determined using a Vario elemental analyzer (Shimadzu, Japan) at the Organic Microanalysis Unit, Faculty of Science, Cairo University, Cairo, Egypt. A SHIMADZU FT-IR-8400s spectrometer was used to record IR spectra as KBr pellets. NMR spectra were recorded on a Bruker (300 and 400 MHz) Ultra Shield NMR spectrometers with [D6]DMSO as the solvent. Chemical shifts δ are given in ppm. The progress of the reactions and purity of the products were monitored by TLC.
4.1 Synthesis of diethyl ammonium hydrogen sulfate (DEAHS)
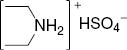
Diethylamine (22.2 g, 0.3 mol) was added into a 250 mL three-necked flask with a magnetic stirrer. Then 29.4 g (0.3 mol) concentrated sulfuric acid (98%) was slowly added dropwise to the flask at room temperature then heated to 80°C for 12 h. The product was washed with diethyl ether three times to remove traces of non-ionic material. The residue was dried under vacuum using a rotary evaporator to obtain the clear viscous product, diethyl ammonium hydrogen sulfate. Yield 98%, pH 1.6 [53].
4.2 General method for the synthesis of 1,2,4,5-tetrasubstituted imidazoles
Benzil (10 mmol), an aldehyde (1a–1j) (10 mmol), ammonium acetate (10 mmol) and ethanolamine (11 mmol) were added to diethylammonium hydrogen sulfate (0.5 g, 3 mmol) in an oil bath at room temperature. The resulting mixtures were heated to 100°C for the time reported in Table 4. Reaction progress was monitored by TLC until completion after 15–35 min. The reaction mixtures were washed with water and the resulting solid products were purified in all cases by recrystallization from ethanol.
Reaction time, yield and physical properties of compounds 2a–2j.
| No. | R | Mol. Formula (Mr in parentheses) | M. p. (°C) | Time (min) | Yield (%) |
|---|---|---|---|---|---|
| 2a | H | C23H20N2O (340.4) | 184–185 | 20 | 98 |
| 2b | 2-OH | C23H20N2O2 (356.4) | 108–110 | 25 | 92 |
| 2c | 4-OH | C23H20N2O2 (356.4) | 287–289 | 15 | 92 |
| 2d | 3-OCH3 | C24H22N2O2 (370.4) | 160–162 | 35 | 82 |
| 2e | 4-OCH3- | C24H22N2O2 (370.4) | 187–188 | 30 | 95 |
| 2f | 2,4-CH3O- | C25H25N2O3 (401.5) | 175–177 | 30 | 88 |
| 2g | 3,4-CH3O- | C25H25N2O3 (401.5) | 212–214 | 35 | 92 |
| 2h | 2,5-OCH3- | C25H25N2O3 (401.5) | 198–200 | 35 | 92 |
| 2i | 4-(CH3)2N- | C25H25N3O (383.5) | 207–209 | 35 | 93 |
| 2j | 4-NO2- | C23H19N3O3 (385.4) | 190–192 | 15 | 85 |
4.3 Spectroscopic properties of 2a–2j
4.3.1 2-(2,4,5-Triphenyl-1H-imidazol-1-yl)ethanol (2a)
M. p. 184–185 oC. – FTIR (KBr): 3265 (OH), 3061 (C–H), 2964 (C–H), 1601 (C=N) 1502 (C=C), 1456, 1181, 1096, 834, 720, 696 cm−1. – 1H NMR (300 MHz, [D6]DMSO, TMS): δ=3.25 (t, 2H, CH2N), 4.07 (t, 2H, CH2O), 5.07 (s, 1H, OH), 7.00–7.00 (m, J=8.8, 2.0 Hz, 15H, Ar-H). – 13C NMR (300 MHz, [D6]DMSO): δ=46.9, 59.8, 126.6, 128.5, 129.0, 129.2, 129.3, 129.6, 130.3, 131.4, 131.6, 135.1, 137.1, 147.7. – Analysis for C23H20N2O (340.4): C 81.16, H 5.92, N 8.23; found C 81.20, H 6.00, N 8.25%.
4.3.2 2-(1-(2-hydroxyethyl)-4,5-diphenyl-1H-imidazol-2-yl)phenol (2b)
M. p. 287–289 oC. – FTIR (KBr): 3518 (OH), 3055 (C–H), 2985 (C–H), 2888 (C–H), 1603 (C=N) 1503 (C=C), 1452, 1169, 1067, 841, 707, 694 cm–1. – 1H NMR (300 MHz, [D6]DMSO, TMS): δ=3.2 (t, 2H, CH2N), 3.95 (t, 2H, CH2O), 4.95 (s br., 1H, CH2OH), 6.8–7.8 (m, J=8.8, 2.0 Hz, 14H, Ar-H), 10 (s br., 1H, C6H4OH). – 13C NMR (300 MHz, [D6]DMSO): δ=46.8, 59.7, 115.8, 122.3, 126.4, 126.6, 128.5, 129.2, 129.5, 129.7, 131.0, 131.5, 131.7, 135.3, 136.6, 147.9, 158.4. – Analysis for C23H20N2O2 (356.4): C 77.51, H 5.66, N 7.89; found C 77.48, H 5.70, N 7.90%.
4.3.3 4-(1-(2-Hydroxyethyl)-4,5-diphenyl-1H-imidazol-2-yl)phenol (2c)
M. p. 287–289 oC. – FTIR (KBr): 3518 (OH), 3055 (C–H), 2985 (C–H), 2888 (C–H), 1603 (C=N) 1503 (C=C), 1452, 1169, 1067, 841, 707, 694 cm–1. – 1H NMR (300 MHz, [D6]DMSO, TMS): δ=3.2 (t, 2H, CH2N), 3.95 (t, 2H, CH2O), 4.95 (s br., 1H, CH2OH), 6.8–7.8 (m, J=8.8, 2.0 Hz, 14H, Ar-H), 10 (s br., 1H, C6H4OH). – 13C NMR (300 MHz, [D6]DMSO): δ=46.8, 59.7, 115.8, 122.3, 126.4, 126.6, 128.5, 129.2, 129.5, 129.7, 131.0, 131.5, 131.7, 135.3, 136.6, 147.9, 158.4. – Analysis for C23H20N2O2 (356.4): C 77.51, H 5.66, N 7.89; found C 77.48, H 5.63, N 7.90%.
4.3.4 2-(2-(3-Methoxyphenyl)-4,5-diphenyl-1H-imidazol-1-yl)ethanol (2d)
M. p. 160–162 °C. – FTIR (KBr): 3334 (OH), 3064 (C–H), 2964 (C–H), 2836 (C–H), 1603 (C=N), 1502 (C=C), 1462, 1262, 1156, 1055, 873, 734, 698 cm–1. – 1H NMR (300 MHz, [D6]DMSO, TMS): δ=3.247 (t, 2H, CH2N), 3.824 (s, 3H, OCH3), 4.005 (t, 2H, CH2O), 4.89 (s br, 1H, CH2OH), 7.109–7.542 (m, 14H, Ar-H). – 13C NMR (300 MHz, [D6]DMSO): δ=46.5, 55.2, 59.3, 114.5, 121.3, 126.1, 126.1, 128.0, 128.9, 129.1, 129.6, 129.8, 131.0, 131.1, 132.5, 146.9, 159.2. – Analysis for C24H22N2O2 (370.5): C 74.56, H 5.99, N 7.60; found C 74.60, H 6.01, N 7.58%.
4.3.5 2-(2-(4-Methoxy)-4,5-diphenyl-1H-imidazol-1-yl)ethanol (2e)
M. p. 187–188 oC. – FTIR (KBr): 3159 (OH), 3058 (C–H), 2965 (C–H), 2836 (C–H), 1603 (C=N), 1466 (C=C),1179, 1081, 834, 721, 695 cm−1. – 1H NMR (300 MHz, [D6]DMSO, TMS): δ=3.25 (t, 2H, CH2N), 4.0 (t, 2H, CH2O), 5.07 (s, 1H, OH), 7.03–7.7 (m, J=8.8, 2.0 Hz, 14H, Ar-H). – 13C NMR (300 MHz, [D6]DMSO): δ=46.8, 55.6, 59.8, 114.4, 123.9, 126.5, 126.6, 128.5, 129,3, 129.5, 129.9, 131.0, 131.4, 131.6, 135.2, 136.8, 147.6. – Analysis for C24H22N2O2 (370.5): C 74.56, H 5.99, N 7.60; found C 74.65, H 6.02, N 7.57%.
4.3.6 2-(2-(2,4-Dimethoxyphenyl)-4,5-diphenyl-1H-imidazol-1-yl)ethanol (2f)
M. p. 175–177 °C. – FTIR (KBr): 3153 (OH), 3060 (C–H), 2980 (C–H), 2837 (C–H), 1614 (C=N) 1579 (C=C), 1454, 1258, 1153, 1072, 837, 720, 701 cm–1. – 1H NMR (300 MHz, [D6]DMSO, TMS): δ=3.73 (s, 6H, 2CH3O), 3.78 (t, 2H, CH2N), 4.00 (t, 2H, CH2O), 4.97 (s br, 1H, CH2OH), 7.02–7.60 (m, 13H, Ar-H). – 13C NMR (300 MHz, [D6]DMSO): δ=46.8, 55.7, 55.7, 59.7, 111.8, 113.3, 122.1, 123.8, 126.4, 126.7, 128.3, 129.1, 129.4, 129.9, 131.5, 131.5, 134.9, 136.7, 147.9, 148.9, 149.8. – Analysis for C25H25N2O3 (401.5): C 74.79, H 6.28, N 6.98; found C 74.77, H 6.30, N 7.02%.
4.3.7 2-(2-(3,4-Dimethoxyphenyl)-4,5-diphenyl-1H-imidazol-1-yl)ethanol (2g)
M. p. 212–214 °C. – FTIR (KBr): 3568, 3100 (OH), 3060 (C–H), 2994 (C–H), 2885, 1601 (C=N) 1585 (C=C), 1481, 1239, 1168, 1057, 872, 725, 701 cm–1. – 1H NMR (300 MHz, [D6]DMSO, TMS): δ=3.75 (s, 6H, 2(CH3O), 3.78 (t, 2H, CH2N), 4.20 (t, 2H, CH2O), 4.91 (s br, 1H, CH2OH), 7.01–7.81 (m, 13H, Ar-H). – 13C NMR (300 MHz, [D6]DMSO): δ=46.8, 55.7, 55.7, 59.7, 111.8, 113.3, 122.1, 123.8, 126.4, 126.7, 128.3, 129.1, 129.4, 129.9, 131.4, 131.5, 134.9, 136.7, 147.9, 148.9, 149.8. – Analysis for C25H25N2O3 (401.5): C 74.79, H 6.28, N 6.98; found C 74.82, H 6.25, N 6.96%.
4.3.8 2-(2-(2,5-dimethoxyphenyl)-4,5-diphenyl-1H-imidazol-1-yl)ethanol (2h)
M. p. 198–200 °C. – FTIR (KBr): 3266 (OH), 3058 (C–H), 2949 (C–H), 2837 (C–H), 1603 (C=N) 1524 (C=C), 1460, 1230, 1181, 1056, 803, 727, 698 cm–1. – 1H NMR (300 MHz, [D6]DMSO, TMS): δ=3.125 (t, 2H, CH2N), 3.737 (t, 2H, CH2O), 3.766 (s, 6H, 2CH3O), 4.716 (s br, 1H, CH2OH), 7.043–7.538 (m, 13H, Ar-H) 13C NMR (300 MHz, [D6]DMSO): δ=46.7, 56.1, 56.4, 59.8, 113.2, 116.4, 118.0, 121.5, 126.5, 126.5, 128.5, 129.3, 129.7, 131.4, 131.6, 135.4, 136.8, 145.0, 151.6, 153.5. – Analysis for C25H25N2O3 (401.5): C 74.79, H 6.28, N 6.98; found C 74.75, H 6.25, N 7.00%.
4.3.9 2-(2-(4-(Dimethylamino)phenyl)-4,5-diphenyl-1H-imidazol-1-yl) ethanol (2i)
M. p. 207–209 oC. – FTIR (KBr): 3320, 3125 (OH), 3064 (C–H), 2970 (C–H), 2890 (C–H), 1670 (C=N) 1579 (C=C), 1450, 1245, 1174, 1098, 874, 724, 694 cm–1. – 1H NMR (300 MHz, [D6]DMSO, TMS): δ=3.95 (s, 6H, (CH3)2N), 3.2 (t, 2H, CH2N), 3.95 (t, 2H, CH2O), 4.91 (s, 1H, CH2OH), 6.78–7.65 (m, 14H, Ar-H), 10 (s br, 1H, C6H4OH). – 13C NMR (300 MHz, [D6]DMSO): δ=40.1, 46.7, 59.8, 112.2, 118.9, 126.5, 128.4, 129.2, 129.5, 130.2, 131.5, 131.8, 135.4, 136.5, 148.2, 150.8. – Analysis for C25H25N3O (383.5): C 78.30, H 6.57, N 10.96; found C 78.32, H 6.60, N 11.00%.
4.3.10 2-(2-(4-Nitrophenyl)-4,5-diphenyl-1H-imidazol-1-yl)ethanol (2j)
M. p. 190–192 oC. – FTIR (KBr): 3144 (OH), 3084 (C–H), 2961 (C–H), 2849 (C–H), 1595 (C=N) 1524 (C=C), 1479, 1286, 1128, 1060, 875, 734, 692 cm–1. – 1H NMR (300 MHz, [D6]DMSO, TMS): δ=3.27 (t, 2H, CH2N), 4.08 (t, 2H, CH2O), 5.00 (s br, 1H, CH2OH), 7.00–8.52 (m, 14H, Ar-H). – 13C NMR (300 MHz, [D6]DMSO): δ=47.5, 59.9, 124.2, 126.7, 126.9, 128.6, 129.6, 130.3, 131.1, 131.4, 134.7, 137.9, 148.1, 145.6, 147.5. – Analysis for C23H19N3O3 (385.4): C 71.68, H 4.97, N 10.90; found C 71.71, H 5.00, N 10.88%.
4.4 X-ray structure determinations
Crystallographic data for 2a, 2c, and 2j are detailed in Table 5. Diffraction data for 2a were collected at Baku Estate University on a Bruker APEXII CCD diffractometer using graphite-monochromatized MoKα radiation (λ=0.71073 Å). Data for 2c and 2j were obtained at the University of Otago on an Agilent SuperNova (Dual, Cu at zero, Atlas) diffractometer using a mirror monochromator and CuKα radiation (λ=1.54184 Å). The Bruker data collection was controlled by Apex2 [60] software with cell refinement and data reduction performed using Saint [60]. Multi-scan absorption corrections were applied using Sadabs [61]. For 2c and 2j these processes were all controlled by CrysAlisPro [62]. The structures were all solved with Shelxs [63] and refined by full-matrix least-squares on F2 using Shelxl-2014 [64] and Titan2000 [65]. All non-hydrogen atoms were assigned anisotropic displacement parameters. The H atoms of the hydroxyl groups for all three molecules were located in difference Fourier maps and their coordinates refined with atomic displacement parameters set to 1.5×Ueq(O). All other H atoms were positioned geometrically and refined using a riding model with d(C–H)=0.95 Å for aromatic and 0.99 Å for CH2 with Uiso=1.2×Ueq(C) and 0.98 Å, Uiso=1.5×Ueq(C) for CH3 atoms. All molecular plots and packing diagrams were drawn using Mercury [66]. Other calculations were performed using Platon [67] and tabular material was produced using WinGX [68].
Crystal data and numbers pertinent to data collection and structure refinement of 2a, 2c, and 2j.
| 2a | 2c | 2j | |
|---|---|---|---|
| Empirical formula | C23H20N2O | C23H20N2O2 | C23H19N3O3 |
| Mr | 40.41 | 356.41 | 385.41 |
| T, K | 296(2) | 100(2) | 100(2) |
| Wavelength; λ, Å | MoKα; 0.71073 | CuKα; 1.54184 | CuKα; 1.54184 |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic |
| Space group | P21/n | Pca21 | P21/c |
| a, Å | 8.926(2) | 39.6826(8) | 15.36052(11) |
| b, Å | 14.538(4) | 5.6950(12) | 8.61192(6) |
| c, Å | 14.401(4) | 7.8390(19) | 28.6080(2) |
| β, deg | 104.701(4) | 90 | 97.1582(7) |
| V, A3 | 1807.7(8) | 1771.6(6) | 3754.88(5) |
| Z | 4 | 4 | 8 |
| Dcalcd, g cm–3 | 1.251 | 1.336 | 1.364 |
| μ, mm–1 | 0.077 | 0.685 | 0.748 |
| F(000), e | 720 | 752 | 1616 |
| Crystal size, mm3 | 0.18 × 0.14 × 0.12 | 0.21 × 0.11 × 0.05 | 0.25 × 0.16 × 0.14 |
| θ range, deg | 2.03–25.49 | 4.46–76.26 | 3.11–76.80 |
| Refl. total/unique/Rint | 15263/3349/0.053 | 8180/2554/0.031 | 39758/7843/0.032 |
| Refl. obs.with I>2 σ(I) | 2543 | 2442 | 7048 |
| Transmission max/min | 0.9908/0.9862 | 1.0000/0.9368 | 1.00000/0.73475 |
| Data/restraints/ref. param. | 3349/0/238 | 2554/1/250 | 7843/0/529 |
| Goodness-of-fit | 1.188 | 1.086 | 1.043 |
| Final R1/wR2 [I>2 σ(I)] | 0.0997/0.2534 | 0.0347/0.0843 | 0.0347/0.0887 |
| Final R1/wR2 (all data) | 0.1204/0.2637 | 0.0375/0.0856 | 0.0392/0.0925 |
| Δρfin (max/min), e Å–3 | 0.37/–0.28 | 0.21/–0.21 | 0.26/–0.26 |
| CCDC reference number | 1024500 | 1024501 | 1024505 |
CCDC 1024500 (2a) 1024501 (2b) and 1024505 (2j) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre viawww.ccdc.cam.ac.uk/data_request/cif.
Acknowledgments
Authors are gratefully thankful to Ministry of Higher Education in Egypt for the financial support of this project in collaboration with National Academy of Sciences of Azerbaijan under their academic exchange protocol. Authors also would like to express their gratitude to the analytical service team (Helen Sutton, Paul Warren and Saeed Gulzar) at Manchester Metropolitan University for providing the spectral results. We also thank the University of Otago for purchase of the Agilent diffractometer and the Chemistry Department, University of Otago, for support of the work of JS.
References
[1] H. R. Shaterian, M. Ranjbar, J. Mol. Liq. 2011, 160, 40.10.1016/j.molliq.2011.02.012Search in Google Scholar
[2] R. Breslow, Acc. Chem. Res. 1995, 28, 146.10.1021/ar00051a008Search in Google Scholar
[3] K. Shalini, N. Kumar, P. K. Sharma, Biointerface Res. Appl. Chem. 2011, 5, 184.Search in Google Scholar
[4] D. M. d’Souza, T. J. J. Mueller, Chem. Soc. Rev. 2007, 36, 1095.10.1039/B608235CSearch in Google Scholar
[5] A. Dömling, Chem. Rev. 2006, 106, 17.10.1021/cr0505728Search in Google Scholar
[6] L. Weber, K. Illgen, M. Almstetter, Synlett1999, 3, 366.10.1055/s-1999-2612Search in Google Scholar
[7] J. Sisko, J. Org. Chem. 1998, 63, 4529.Search in Google Scholar
[8] F. Pozharskii, A. T. Soldatenkov, A. R. Katritzky, Heterocycles in Life and Society, Wiley, Gainesville, Florida 1997.Search in Google Scholar
[9] H. V. D. Bossche, G. Willemsens, W. Cools, P. Marichal, W. Lauwers, Biochem. Soc. Trans. 1983, 11, 665.10.1042/bst0110665Search in Google Scholar
[10] K. Sivakumar, A. Kathirvel, A. Lalitha, Tetrahedron Lett. 2010, 51, 3018.10.1016/j.tetlet.2010.04.013Search in Google Scholar
[11] Y. Xu, Y. Z. Liu, L. Rui, L. Liu, Q. X. Guo, Heterocycles2004, 63, 87.10.3987/COM-03-9896Search in Google Scholar
[12] S. D. Sharma, P. Hazarika, D. Konwar, Tetrahedron Lett. 2008, 49, 2216.Search in Google Scholar
[13] R. Karimi, Z. Almohammadi, J. Azizian, A. A. Mohammadi, M. R. Mohammadizadeh, Catal. Commun. 2006, 7, 728.Search in Google Scholar
[14] M. Atta, Z. H. Abd El Wahab, Z. A. El Shafey, W. I. Zidan, Z. F. Akl, J. Dispersion Sci. Technol. 2010, 31, 1415.10.1080/01932690903269560Search in Google Scholar
[15] S. Balalaei, A. Arabanian, Green Chem. 2000, 2, 274.10.1039/b006201oSearch in Google Scholar
[16] A. R. Karimi, Z. Alimohammadi, J. Azizian, A. A. Mohammadi, M. R. Mohmmadizadeh, Catal. Commun. 2006, 7, 728.10.1016/j.catcom.2006.04.004Search in Google Scholar
[17] M. Kidwai, P. Mothsra, V. Bansal, R. K. Somvanshi, S. A. Ethayathulla, S. Dey, T. P. Singh, J. Mol. Catal. A: Chem. 2007, 265, 177.10.1016/j.molcata.2006.10.009Search in Google Scholar
[18] L. Nagarapu, L. S. Apuri, S. J. Kantevari, J. Mol. Catal. A: Chem. 2007, 266, 104.Search in Google Scholar
[19] M. M. Heravi, F. Derikv, F. F. Bamoharram, J. Mol. Catal. A: Chem. 2007, 263, 112.Search in Google Scholar
[20] S. Kantevari, S. V. N. Vuppalapati, D. O. Biradar, L. Nagarapu, J. Mol. Catal. A: Chem. 2007, 266, 109.10.1016/j.molcata.2006.10.048Search in Google Scholar
[21] B. Sadeghi, B. B. F. Mirjalili, M. M. Hashemi, Tetrahedron Lett. 2008, 49, 2575.10.1016/j.tetlet.2008.02.100Search in Google Scholar
[22] W. M. Nelson in Green Chemistry, (Eds.: P. T. Anastas, T. C. Williamson), Oxford University Press, Oxford 1998, pp. 150.Search in Google Scholar
[23] K. Tanaka, F. Toda, Chem. Rev. 2000, 100, 1025.10.1021/cr940089pSearch in Google Scholar
[24] A. Mohammadi, H. Keshvari, R. Sandaroos, B. Maleki, H. Rouhi, H. Moradi, Z. Sepehr, S. Damavandi, Appl. Catal. A: General2012, 429, 73.10.1016/j.apcata.2012.04.011Search in Google Scholar
[25] M. Armand, F. Endres, D. R. MacFarlane, H. Ohno, B. Scrosati, Nature Material. 2009, 8, 621.10.1038/nmat2448Search in Google Scholar
[26] R. Rogers, K. Seddon, S. Volkov, Green Industrial Application of Ionic Liquids, Environmental Chemistry, Nato Science Series, Vol. 92, Kluwer Academic Publishers, Dordrecht, 2002, p. 295.10.1007/978-94-010-0127-4Search in Google Scholar
[27] M. Freemantle, Introduction to Ionic Liquids, Royal Society of Chemistry, Cambridge 2009.10.1039/9781839168604Search in Google Scholar
[28] P. Wasserscheid, T. Welton, Ionic Liquids in Synthesis, Wiley-VCH, Weinheim, 2008.10.1002/9783527621194Search in Google Scholar
[29] R. A. Sheldon, I. W. C. E. Arends, U. Hanefeld, Green Chemistry and Catalysis, Wiley-VCH, Weinheim, 2007.10.1002/9783527611003Search in Google Scholar
[30] H. R. Shaterian, H. Yarahmadi, M. Ghashang, Tetrahedron2008, 64, 1263.10.1016/j.tet.2007.11.070Search in Google Scholar
[31] H. R. Shaterian, M. Honarmand, A. R. Oveisi, Monatsh. Chem. 2010, 141, 557.10.1007/s00706-010-0302-8Search in Google Scholar
[32] H. R. Shaterian, A. R. Oveisi, Chin. J. Chem. 2009, 27, 2418.10.1002/cjoc.200990327Search in Google Scholar
[33] H. R. Shaterian, A. Hossienian, M. Ghashang, Turk. J. Chem. 2009, 2, 233.Search in Google Scholar
[34] S. K. Mohamed, M. A. A. El-Remaily, A. M. Soliman, H. Abdel-Ghany, Eur. J. Med. Chem. 2012, 47, 138.Search in Google Scholar
[35] S. K. Mohamed, A. A. Abdelhamid, A. M. Maharramov, A. N. Khalilov, A. V. Gurbanov, M. A. Allahverdiyev, J. Chem. Pharm. Res. 2012, 4, 955.Search in Google Scholar
[36] S. H. H. Younes, S. K. Mohamed, M. R. Albayati, Arch. Pharm. (Weinheim, Ger.)2013, 346, 727.Search in Google Scholar
[37] Version 5.36 (November 2014) with two updates. See also: C. R. Groom, F. H. Allen, Angew. Chem. Int. Ed. 2014, 53, 662.10.1002/anie.201306438Search in Google Scholar PubMed
[38] D. Yanover, M. Kaftory, Acta Crystallogr. 2009, E65, o711.10.1107/S1600536809006552Search in Google Scholar
[39] S. K. Mohamed, M. Akkurt, A. A. Marzouk, V. M. Abbasov, A. V. Gurbanov, Acta Crystallogr. 2013, E69, o474.10.1107/S1600536813004285Search in Google Scholar
[40] J. P. Jasinski, S. K. Mohammed, M. Akkurt, A. A. Abdelhamid, M. R. Albayati, Acta Crystallogr. 2015, E71, o77.Search in Google Scholar
[41] M. Akkurt, J. P. Jasinski, S. K. Mohammed, A. A. Marzouk, M. R. Albayati, Acta Crystallogr. 2015, E71, o299.Search in Google Scholar
[42] Y. Xiao, L. Yang, K. He, J. Yuan, P. Mao, Acta Crystallogr. 2012, E68, o264.Search in Google Scholar
[43] Y. Li, P. Mao, Y. Xiao, L. Yang, L. Qu, Acta Crystallogr. 2014, E70, o621.Search in Google Scholar
[44] M. Akkurt, S. K. Mohamed, K. Singh, A. A. Marzouk, A. A. Abdelhamid, Acta Crystallogr. 2013, E69, o846.10.1107/S1600536813011446Search in Google Scholar
[45] Y.-N. Yan, W.-L. Pan, H.-C. Song, Dyes Pigm. 2010, 86, 249.10.1016/j.dyepig.2010.01.011Search in Google Scholar
[46] J. Simpson, S. K. Mohamed, A. A. Marzouk, A. H. Talybov, A. A. Abdelhamid, Acta Crystallogr. 2013, E69, o5.Search in Google Scholar
[47] Y.-N. Yan, D.-Y. Lin, W.-L. Pan, X.-L. Li, Y.-Q. Wan, Y.-L. Mai, H.-C. Song, Spectrochim. Acta, Part A2009, 74, 233.Search in Google Scholar
[48] C. Kison, T. Opatz, Chem. Eur. J. 2009, 15, 843.10.1002/chem.200802175Search in Google Scholar
[49] S. K. Mohamed, M. Akkurt, K. Singh, A. A. Marzouk, A. A. Abdelhamid, Acta Crystallogr. 2013, E69, o1243.10.1107/S1600536813018229Search in Google Scholar
[50] S. K. Mohamed, M. Akkurt, A. A. Marzouk, K. Singh, M. R. Albayati, Acta Crystallogr. 2013, E69, o1833.10.1107/S1600536813031759Search in Google Scholar
[51] S. K. Mohamed, M. Akkurt, K. Singh, A. A. Marzouk, M. R. Albayati, Acta Crystallogr. 2013, E69, o1417.10.1107/S1600536813021983Search in Google Scholar
[52] T. Peppel, M. Köckerling, Z. Naturforsch. 2013, 68b, 245.10.5560/znb.2013-2333Search in Google Scholar
[53] T. Vasantha, P. Attri, P. Venkatesu, R. S. Devi, J. Phys. Chem. B,2012, 116, 11968.10.1021/jp308443fSearch in Google Scholar PubMed
[54] J. Li, S. Lin, J. Dai, W. Su, J. Chem. Res. 2010, 34, 196.Search in Google Scholar
[55] R. S. Joshi, P. G. Mandhane, M. U. Shaikh, R. P. Kale, C. H. Gill, Chin. Chem. Lett. 2010, 21, 429.10.1016/j.cclet.2009.11.012Search in Google Scholar
[56] R. Khosropour, Ultrason. Sonochem. 2008, 15, 659.10.1016/j.ultsonch.2007.12.005Search in Google Scholar
[57] S. Samai, G. C. Nandi, M. S. Singh, Tetrahedron2009, 65, 10155.10.1016/j.tet.2009.10.019Search in Google Scholar
[58] S. S. Qasim, N. Shaikh, S. A. Syed, Int. J. Appl. Biol. Pharma. Tech. 2011, 2, 12.Search in Google Scholar
[59] S. A. Siddiqui, U. C. Narkhede, S. S. Palimkar, T. Daniel, R. J. Lahoti, K. V. Srinivasan, Tetrahedron2005, 61, 3539.10.1016/j.tet.2005.01.116Search in Google Scholar
[60] Apex2, Saint, Area Detector Control and Integration Software, Bruker Analytical X-ray Instruments Inc., Madison, Wisconsin (USA) 2011.Search in Google Scholar
[61] G. M. Sheldrick, Sadabs, Program for Empirical Absorption Correction of Area Detector Data, University of Göttingen, Göttingen (Germany) 2002.Search in Google Scholar
[62] CrysAlisPro Software System, Intelligent Data Collection and Processing Software for Small Molecule and Protein Crystallography, Agilent Technologies Ltd., Yarnton, Oxfordshire (U. K.) 2013.Search in Google Scholar
[63] G. M. Sheldrick, Acta Crystallogr. 2008, A64, 112.10.1107/S0108767307043930Search in Google Scholar
[64] G. M. Sheldrick, Acta Crystallogr. 2015, C71, 3.Search in Google Scholar
[65] K. A. Hunter, J. Simpson, Titan2000, University of Otago, Otago (New Zealand) 1999.Search in Google Scholar
[66] C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek, P. A. Wood, J. Appl. Crystallogr. 2008, 41, 466.10.1107/S0021889807067908Search in Google Scholar
[67] A. L. Spek, Acta Crystallogr. 2009, D65, 148.10.1107/S090744490804362XSearch in Google Scholar
[68] L. J. Farrugia, J. Appl. Crystallogr. 2012, 45, 849.10.1107/S0021889812029111Search in Google Scholar
©2015 by De Gruyter
Articles in the same Issue
- Frontmatter
- In this Issue
- Gd4(BO2)O5F – a gadolinium borate fluoride oxide comprising a linear BO2 moiety
- Hydrolysis of 8-(pinacolboranyl)quinoline: where is the 8-quinolylboronic acid?
- Synthesis of some novel 6′-(4-chlorophenyl)-3,4′-bipyridine-3′-carbonitriles: assessment of their antimicrobial and cytotoxic activity
- Synthesis of some 6-alkylureido-4-aryl-2(1H)-pyridones: further transformations and pharmacological activity
- Multicomponent green synthesis, spectroscopic and structural investigation of multi-substituted imidazoles. Part 1
- Sonochemical synthesis of 5-substituted 1H-tetrazoles catalyzed by ZrP2O7 nanoparticles and regioselective conversion into new 2,5-disubstituted tetrazoles
- Two new taxane-glycosides from the needles of Taxus canadensis
- Cytotoxic 24-nor-ursane-type triterpenoids from the twigs of Mostuea hirsuta
- 4,15-Diamino[2.2]paracyclophane as a useful precursor for the synthesis of novel pseudo-geminal [2.2]paracyclophane compounds
Articles in the same Issue
- Frontmatter
- In this Issue
- Gd4(BO2)O5F – a gadolinium borate fluoride oxide comprising a linear BO2 moiety
- Hydrolysis of 8-(pinacolboranyl)quinoline: where is the 8-quinolylboronic acid?
- Synthesis of some novel 6′-(4-chlorophenyl)-3,4′-bipyridine-3′-carbonitriles: assessment of their antimicrobial and cytotoxic activity
- Synthesis of some 6-alkylureido-4-aryl-2(1H)-pyridones: further transformations and pharmacological activity
- Multicomponent green synthesis, spectroscopic and structural investigation of multi-substituted imidazoles. Part 1
- Sonochemical synthesis of 5-substituted 1H-tetrazoles catalyzed by ZrP2O7 nanoparticles and regioselective conversion into new 2,5-disubstituted tetrazoles
- Two new taxane-glycosides from the needles of Taxus canadensis
- Cytotoxic 24-nor-ursane-type triterpenoids from the twigs of Mostuea hirsuta
- 4,15-Diamino[2.2]paracyclophane as a useful precursor for the synthesis of novel pseudo-geminal [2.2]paracyclophane compounds