
The Reaction and Microscopic Electron Properties from Dynamic Evolutions of Condensed-Phase RDX Under Shock Loading
-
Jiao-Nan Yuan
 and
Dong-Qing Wei
and
Dong-Qing Wei

Abstract
Microscopic electron properties of α-hexahydro-1,3,5-trinitro-1,3,5-triazine (α-RDX) with different shock wave velocities have been investigated based on molecular dynamics together with multi-scale shock technique. The studied shock wave velocities are 8, 9 and 10 km ⋅ s−1. It has been said that the shock sensitivity and reaction initiation of explosives are closely relevant with their microscopic electron properties. The reactions, including the reaction products, which are counted from the trajectory during the simulations are analysed first. The results showed that the number of the products strictly rely on shock wave velocities. The reaction rates and decomposition rates are also studied, which showed the differences between the different shock velocities. The results of electron properties show that α-RDX is a wide-gap insulator in the ground state and the metallisation conditions of shocked RDX are determined, which are lower than under-static high pressure.
1 Introduction
To explore the most fundamental questions that include sensitivity and shock response about energetic materials, the shock decomposition has been widely studied by researchers. Shock wave initiation of explosives is a very complex, multi-scale process, and a series of chemical reactions can happen during simulations. The investigation of microscopic properties and reaction process in energetic materials is necessary to understand the decomposition mechanisms and detonation performance. The thermal [1], [2], [3] and shock decomposition [4], [5], [6] of nitromethane (NM) have been widely studied. The primary decomposition mechanisms of 2,4,6-trinitro-1,3,5-benzenetriamine (TATB) [7], [8], [9] and pentaerythritol tetranitrate (PETN) [10], [11], [12] are also studied under shock loading. Our group has a long history of research on shocked energetic materials. Combining molecular dynamics (MD) simulation with multi-scale shock technique (MSST), the decomposition mechanism of shocked octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX) has been investigated. Ge et al. [13] discovered that the primary decomposition of HMX was due to the homolytic cleavage of N–NO2 bond at 8 km ⋅ s−1, while at the primary stage, the route for shocked HMX decomposition was C–H bond fission with the shock velocity higher than 10 km ⋅ s−1. Their results showed that there is a big difference when the shock along different lattice vector at 11 km ⋅ s−1 [14]. However, the primary reaction mechanisms of HMX under different conditions were obviously different. Zhu et al. [15] presented that the decomposition about HMX is because of the N–O bond break and the ring-opening of the molecule at 6.5 km ⋅ s−1. The reaction of HMX which is along the (1 0 0) surface and includes a vacancy in the molecule is studied by He et al. [16]. They found that the primary decomposition is the C–N bond rupture of the molecular ring. All that said, there still remains a lot of controversy about the primary decomposition of energetic materials. Hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX) as the HMX’s smaller cousin has also been studied in our article [17]. Our previous study indicated that the C–N bond rupture of the ring-opening is the primary decomposition pathway of shocked RDX at 10 km ⋅ s−1, which is approved by the results [18] that the polyradicals undergo ring-opening and the N–N bond split is suppressed. Recently, the ring-opening has also been shown to occur earlier than HONO (transferring of a hydrogen from the CH2 to an adjacent NO2 can form the HONO) elimination and N–NO2 homolysis in liquid-phase and gas-phase RDX decomposition by Patidar et al. [19] based on both the experiment of spectrometry analysis and density functional theory calculation. However, we previously observed the primary decomposition in shocked RDX single crystals at 10 km ⋅ s−1, which is different from the result that the primary decomposition mechanism of HMX under shock wave loading depends on the velocities. In this work, on the one hand, we will expand our studies about RDX to different velocities for comparison. On the other hand, in contrast to the studies of primary decomposition and chemical reaction, very few theoretical results have been reported about the electronic properties of energetic materials under shock loading. The metallisation is also a big part in the decomposition of explosives. Ge et al. [20] predicted that the pressure of metallisation is 130 GPa of shocked HMX. Reed et al. [4] showed that there is a transformation from insulator to semi-metallic state of NM under shock loading. He et al. [21] studied microscopic electron properties of β-HMX under shock loading. Bai et al. [22] have shown the microscopic electron properties of the cyclotrimethylenetrinitramine under shock loading; however, in their discussions, only the pressure was taken into account, while the temperature was neglected. Therefore, it is necessary to further study the electronic properties of RDX under shock loading in which both the pressure and temperature would be considered.
2 Methods and Computational Parameters
The shock decomposition is studied by using MD simulation in combination with MSST in CP2K code [23]. The Navier-Stokes equations [24] are adopted in this method, and the purpose is to constrain the MD system to the same thermodynamic states. The unit cell, through the shock wave [25], follows a Lagrangian point. For a specified shock velocity, the system can meet the mass, momentum and energy conservation laws when passing the shock wave first [26], and then the Hugoniot relation can be presented. The Rayleigh line is used to describe the thermodynamic path from the primary state to the final state. The steady shock waves obey the Hugoniot and Rayleigh relations which are based on the continuum theory to describe the stable plane shock wave.
By adopting the self-consistent charge density functional tight binding (SCC-DFTB) scheme [27], the electronic structure is calculated. It has been confirmed that the SCC-DFTB is more suitable for the calculations of organic molecules and bio-molecular systems [28], [29]. It has also been demonstrated to suit and predict reaction energies [30], [31], [32]. This method can be used to calculate the ground-state and excited-state properties and it has been verified to obtain good results in RDX under pressures [33]; the results by these methods are consistent with the results obtained through the density functional theory plus dispersion by Grimme [34]. The optimisation of cell and geometry were performed by the conjugate gradient and Broyden–Fletcher–Goldfarb–Shanno scheme, respectively. We adopted the most stable phase [35] of RDX in our MD simulations. The optimised primary lattice parameters a (39.661 Å), b (11.495 Å) and c (10.536 Å) of RDX are consistent with the experimental values [35] with the error of 0.29 %, –0.68 %, –1.57 %, respectively. The molecules are 24, amounting to 504 atoms in RDX. The simulations were performed at 8, 9 and 10 km ⋅ s−1, which satisfies the mechanical stability criterion of Vs > V0 [36]. The time of every step was 0.05 fs during the shock compression simulations. The separate molecules and reaction products were counted according to the simulation trajectory.
3 Data Analysis and Discussions
3.1 The Parameters Under Shock Loading
The ratio of the compressed volume to the primary volume, system pressure and the temperature of RDX which depends on the time under shock wave loading along a axis with the simulation time of 3.5 ps is presented in Figure 1. In these figures, one can see that the volumes reduce by 38.48 %, 41.25 % and 43 % at 8, 9 and 10 km ⋅ s−1, respectively. Thermodynamic properties of RDX have a great relationship with shock velocities. The temperatures and pressures have experienced an overdrive state, basically reaching a balance from 0.5 ps to the end of our simulations. When the shock wave velocities change from 8 to 9 km ⋅ s−1, the reaction temperatures are nearly stabilised at 1300 and 1700 K, respectively. However, the temperature reached to 3700 K and it kept a persistent increase at 10 km ⋅ s−1. The pressures maintain a near-constant value of 45, 60 and 80 GPa at 8, 9 and 10 km ⋅ s−1, respectively. The reported detonation pressure of RDX is 35.3 GPa [37]. Therefore, the shocked RDX reaches to detonation critical value under the shock wave velocities from 8 to 10 km ⋅ s−1. But shock wave velocity of 8 km ⋅ s−1 is a little lower than the steady detonation velocity of 8.754 km ⋅ s−1 [37]. The strength of the shock wave is too weak to maintain the detonation chemical reaction to continue and gradually attenuate to a sound wave at 8 km ⋅ s−1. So, the steady detonation cannot happen under this shock velocity. Although the shock wave velocity, temperature and pressure are all arrived at the condition of steady detonation of RDX, a few reactions and intermediates are presented. In addition, the calculated time scale may be too short to observe the full decomposition of RDX at 9 and 10 km ⋅ s−1. According to the previous work, to obtain the full chemical behaviors, longer simulation times are required, but it needs much time and computing resources for the huge system in our study. Therefore, in the next discussions, we will mainly focus on investigating the reaction initiation of shocked α-RDX under shock loading with 8, 9 and 10 km ⋅ s−1.
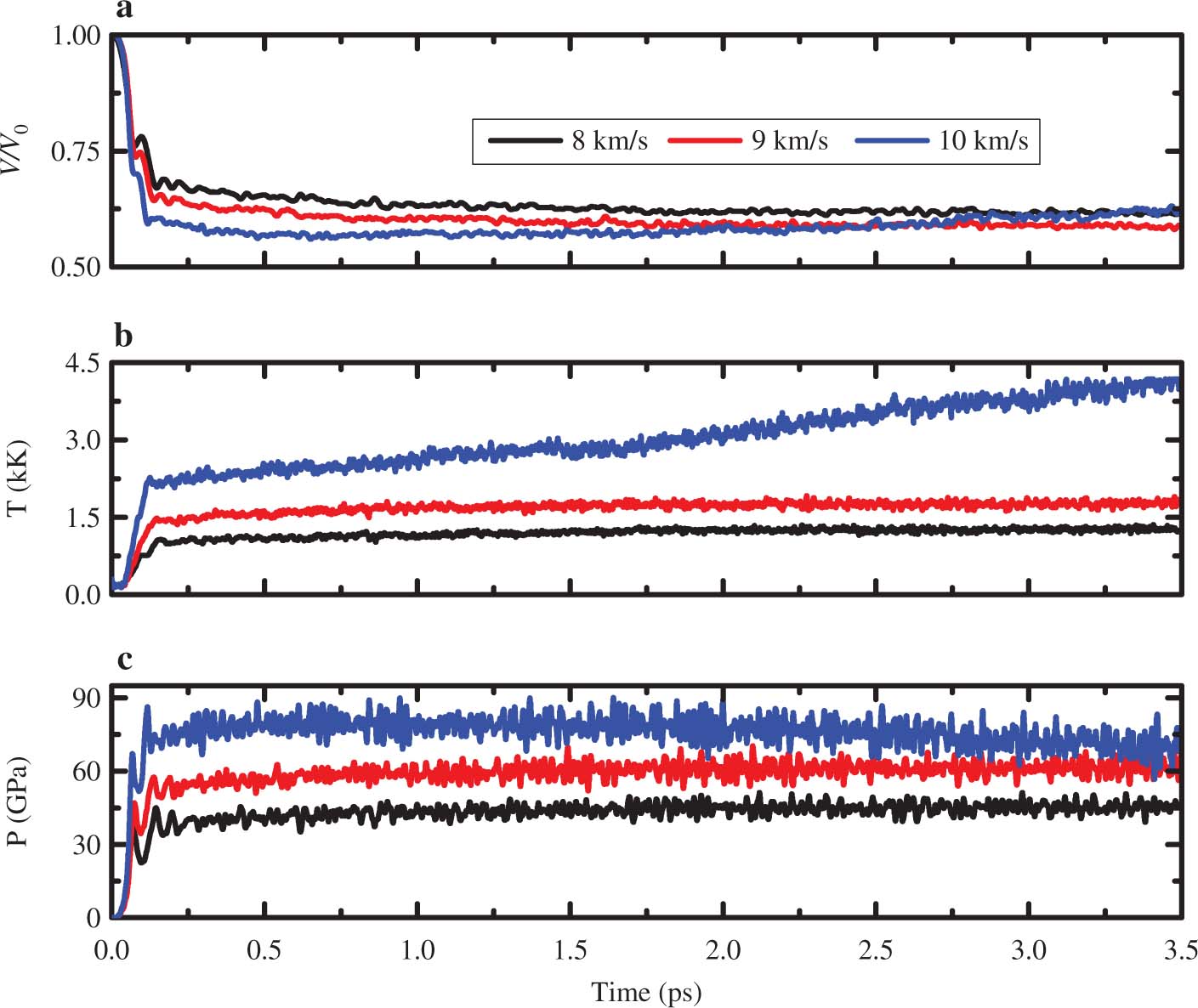
Time dependence of the ratio of the compressed volume to the primary volume (V/V0), average temperature (T), and shock propagation direction pressure (P) calculated for the shock along the lattice vector a at different shock velocities of 8, 9 and 10 km ⋅ s−1 for a time scale of up to 3.5 ps.
3.2 The Reaction Initiation of α-RDX Under Shock Wave Loading
The snapshots of the configurations for four instants under shock loading of 10 km ⋅ s−1 are presented in Figure 2. From the simulation process, we could find that the system undergoes a persistent compressed process along with the molecular rotation. Before the primary decomposition, the system atoms are attracted or repelled with each other. Finally, the bonds between the conjoint atoms are compressed or stretched, which leading to the bonds rupture under shock loading. In our previous study [17], the C–N bond split of the opened ring has been demonstrated as the primary decomposition of shocked RDX at 10 km ⋅ s−1. Our further study showed that the initially produced fragments are all due to the C–N split under different shock wave velocities of 8, 9 and 10 km ⋅ s−1. The other proposed primary decomposition mechanisms are also found. However, from 8 to 10 km ⋅ s−1, until at 115.55, 92.2 and 82.55 fs, respectively, the N–NO2 bond dissociation happens. At 8 km ⋅ s−1, there did not appear HONO elimination, but at 9 and 10 km ⋅ s−1, the HONO was initially formed at 1855.3 and 334 fs, respectively.
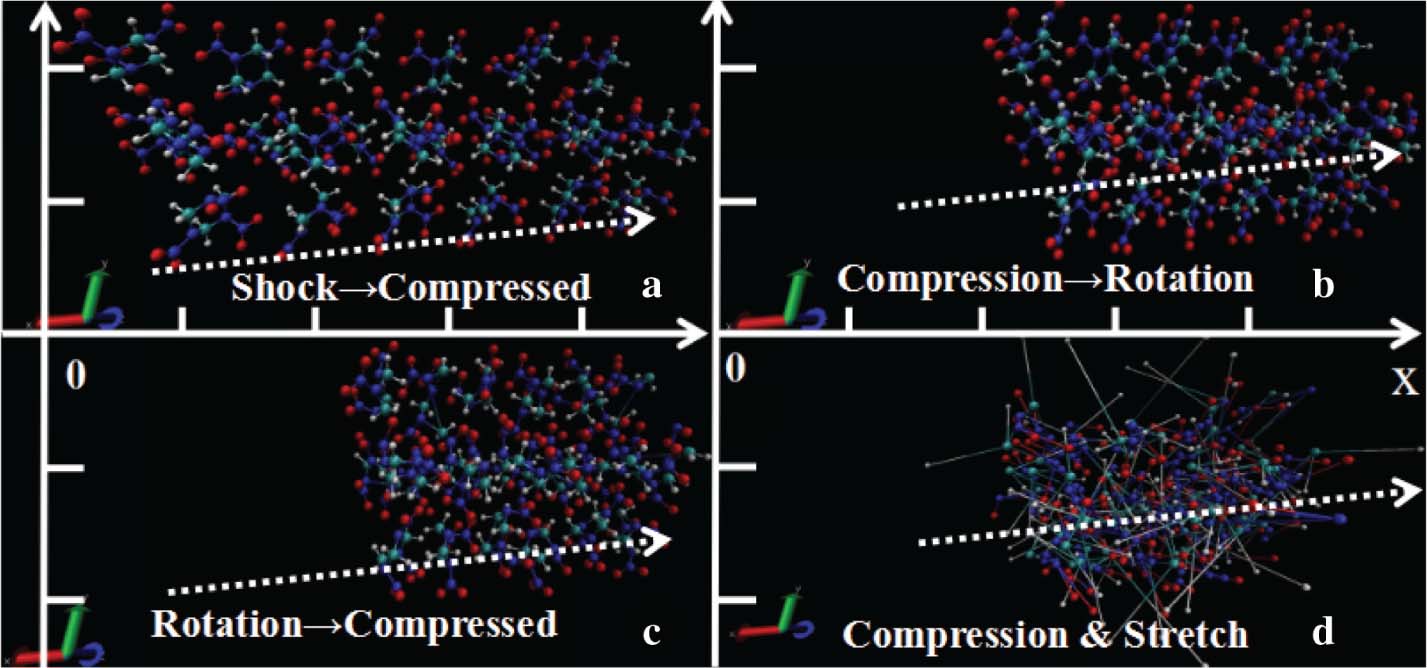
The snapshots of the change in the configurations for four instants under shock compression of 10 km ⋅ s−1 along the lattice vectora. Grey, blue, red, and white spheres stand for C, N, O and H atoms, respectively. (a) Presents a persistent compressed process, (b) shows the rotation during the compression, (c) presents a compressed process while rotating and (d) shows the bonds rupture when compression reaches a certain level.
3.3 The Reaction Rates and Decomposition Products During the Shock Loading
The fitted density numbers of the products, C3H6N6O6 and their reaction rate depending on time under the shock loading of 8, 9 and 10 km ⋅ s−1 are presented in Figure 3. We take derivatives of the density numbers with respect to time to observe the reaction rate. In Figure 3a, we can find that the system is compressed under the primary shock loading and this process lasts to 0.25 ps, the produced density number of the products shows an exponentially increase. In Figure 3b, with the persistent shock loading, the bonds of shocked RDX rupture and the decomposition begins. The opposite rule with the products, before 0.25 ps, the density number of RDX shows an exponentially decrease. There are few decompositions of shocked RDX at 8 and 9 km ⋅ s−1. The shocked RDX shows a full decomposition state and the density numbers of RDX becomes zero from 0.3 ps at 10 km ⋅ s−1. At the same time, the density number of the products shows nearly no change until 0.5 ps. But then the number of the products shows a linear increase to the end of our simulation at 10 km ⋅ s−1, which showed that a large number of secondary reactions happens. From the reaction rate in Figure 3c,d, we can obviously find that both the decomposition rate and produced rate are more fast at the primary shock loading. With the shock velocity increasing, the reaction rate increases. Then the reaction rate tends to become zero at 8 and 9 km ⋅ s−1, but the reaction rate at 10 km ⋅ s−1 is higher than this value, which corresponds to the density numbers of the products increasing with time.
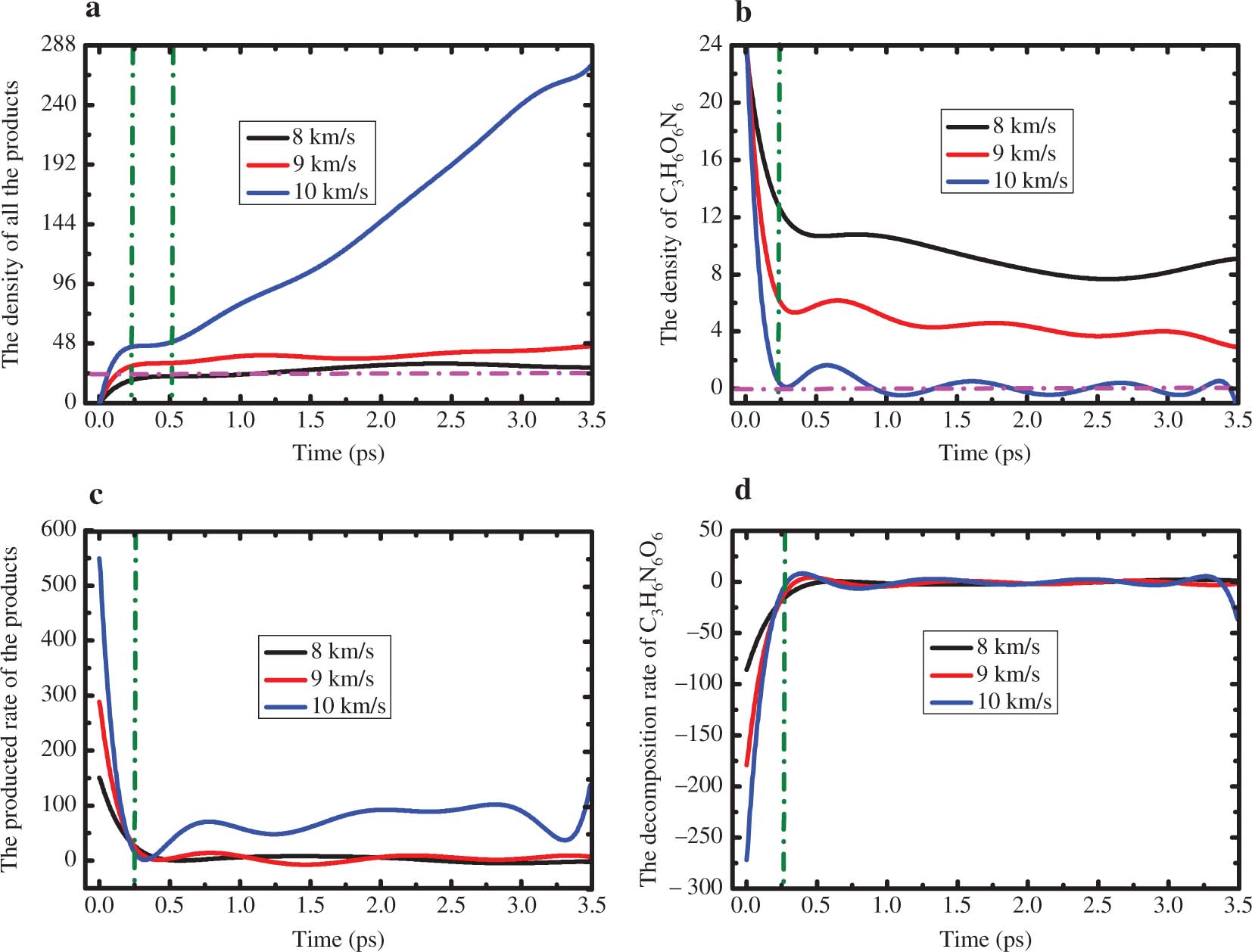
The fitted density numbers of the products (a) and C3H6N6O6 (b) along with the simulation time. The produced rate of products (c) and the decomposition rate of C3H6N6O6 (d) that varied with the simulation time under the shock loading of 8, 9 and 10 km ⋅ s−1 along the lattice vector a.
This difference of the primary decomposition and reaction rate under the shock loading of 8, 9 and 10 km ⋅ s−1 suggests that at higher shock velocities of 9 and 10 km ⋅ s−1, the strong shock loading can lead to faster chemical reaction and the movements of the molecules become more severe than at 8 km ⋅ s−1. Our results show that a few RDX molecules decompose and only NO2 forms below the steady detonation shock velocity (8.754 km ⋅ s−1). This result is consistent with the experimental result [38] by photoelectron spectroscopy analysis and the theoretic study with the nonequilibrium MD simulations [39]. However, newly produced molecules are NO, N2O, H2O at the shock velocity of 9 km ⋅ s−1 and the many kinds of molecules including NO2, NO, N2O, H2O, CO, CO2, H2, N2 at 10 km ⋅ s−1. The frontal analysis shows that at 8 km ⋅ s−1, the temperatures and pressures reach the experimental critical detonation values, but the reaction of the NO2 do not show noticeable change, which confirms no steady detonation formed under this shock velocity. The reaction rates of various products at 9 and 10 km ⋅ s−1 increase quickly, indicating that RDX molecules are easier to decompose and the system arrives at a steady detonation stage. To further understand the decomposition mechanisms for the formation of the gas molecules, the origin of constituent atoms of the key gas molecules was investigated. The investigations showed that most of CO2 and H2O are formed from the intermolecular reaction pathways, while most of CO, NO, NO2 and N2 are formed from the intramolecular reaction pathways. This analysis shows that intramolecular reactions are the dominant pathways for shocked RDX decomposition. Our result showed that N2 is formed by intramolecular reaction pathways. The produced N2 is due to the N–NO2 bonds rupture in a RDX molecule, which is different from the formation of gas products in the TATB molecule [8] and that most of the products are formed via the intermolecular reaction.
3.4 The Microscopic Electron Properties of α-RDX Under Shock Loading
As we all know energy band structure plays a key role in analysing the physical properties of materials. The energy difference that is the highest occupied molecular orbital–lowest unoccupied molecular orbital (HOMO–LUMO) energy gap indicates the insulating behavior of the solids. The HOMO–LUMO energy gap of RDX molecule was 5.269 eV [17] in our previous studies. The energy band structure of RDX in the ground state is presented in Figure 4. The result showed that RDX is a wide-gap insulator with the band gap energy of the system 5.12 eV, which is more consistent with the HOMO–LUMO energy gap by second-order perturbation theory of 5.25 eV [40], and the B3LYP [41] band gap of 5.54 eV for RDX. For more detail, we also studied the band structure of shocked RDX during the simulations at 8, 9 and 10 km ⋅ s−1. We list the band gap energy as a function of the simulations time at 8, 9 and 10 km ⋅ s−1 in Figure 5. The change of the band gap energy under different shock velocities shows the same orderliness. In addition, we can discover that with the simulations continuing, the band gap energy decreases with increasing pressure inside the wave front. From this figure, we can find that at 0.15 ps about 72 GPa and 2100 K, RDX shows obvious metallisation at 10 km ⋅ s−1 that is higher than the experimentally measured steady detonation velocity, which is consistent with the metallisation with HMX under shock loading of 11 km ⋅ s−1 at 0.15 ps about 130 GPa [20]. Until now, there is no available data for comparison of RDX band gap under shock loading on both experimental and theoretical results. The studies indicated that the metallisation pressure is 180 GPa [42] of pure RDX under static pressure conditions. Obviously, our work is studied based on a dynamic simulation, considering the temperature, the metallisation pressure of RDX under shock loading is lower than in the static pressure. At 9 km ⋅ s−1, the temperature and pressure approach a condition of metallisation at 10 km ⋅ s−1, with chemical reactions continuing, the band gap decreases to zero, but then it widens due to the fluctuations of electronic state energies during the simulation, so RDX shows semi-metallisation. However, at 8 km ⋅ s−1 that a little lower than the steady detonation velocity, the pressure and temperature are far away from the condition of metallisation at 10 km ⋅ s−1; it just shows the transformation from insulator to semiconductor.
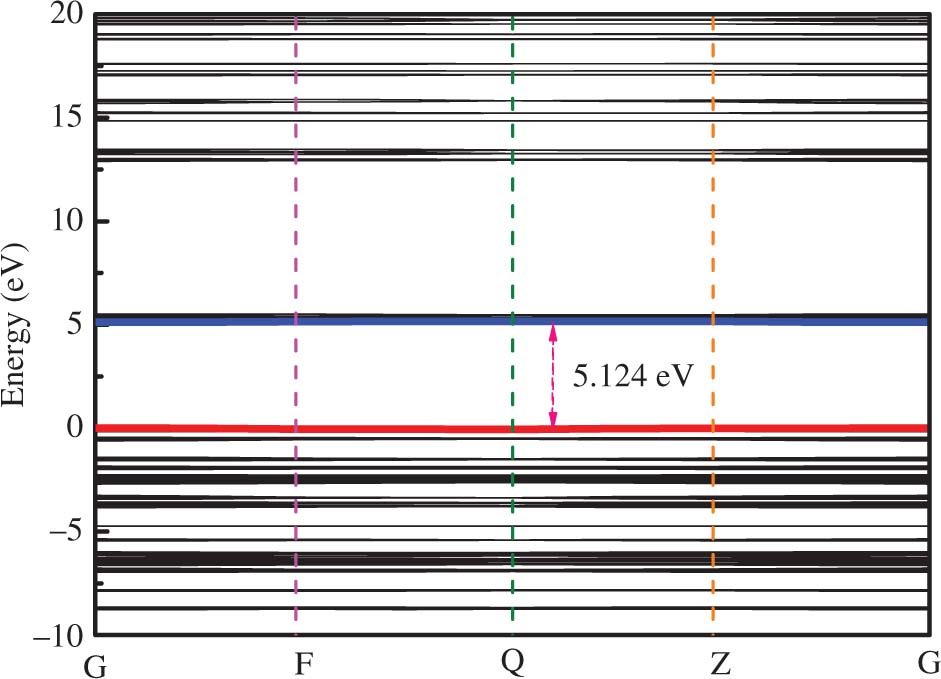
The calculated energy band structures of hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX) at ground states.
![Figure 5: The band gap energy depends on the simulations time under the shock velocities of 8, 9 and 10 km ⋅ s−1, respectively: (a) by Kuklj and Kunz using second-order perturbation theory [40], (b) by Perger using B3LYP [41], (c) present using density functional tight binding.](/document/doi/10.1515/zna-2019-0379/asset/graphic/j_zna-2019-0379_fig_005.jpg)
4 Conclusions
Based on the MD simulations with MSST, our results showed that the C–N bond split of the opened ring is the primary decomposition of shocked RDX. At 8, 9 and 10 km ⋅ s−1, the N–NO2 bond dissociates at 115.55, 92.2 and 82.55 fs, respectively. The HONO is formed just under the shock wave velocities of 9 and 10 km ⋅ s−1 at 1855.3 and 334 fs, respectively. When the velocities are lower than the steady detonation shock velocity, NO2 is the only product, while when the shock velocities are higher than the steady detonation velocity, the products are H2O, NO, N2O, NO2, CO, CO2, H2, N2. The results suggest that intramolecular reaction pathways are dominant to produce these gas molecules for shocked RDX decomposition. From analysing the energy band structure of RDX in the ground states, the results show that RDX is a wide-gap insulator and the band gap of the system is 5.124 eV. However, studies about the microscopic electron properties showed the metallisation of shocked RDX at 0.15 ps was about 72 GPa and 2100 K. This metallisation pressure under the shock loading is lower than metallisation pressure under the static high pressure, which showed that the shock loading reduces the band gap and accelerates the rate of metallisation.
Acknowledgements
This work was supported by the NSAF Joint Fund Jointly set up by the National Natural Science Foundation of China and the Chinese Academy of Engineering Physics under Grant Nos. U1430117 and U1230201; the Science Challenge Project under Grant No. TZ2016001; the National Natural Science Foundation of China for support under Grant No. 11174201 and 11572160; the National Key Laboratory Fund for Shock Wave and Detonation Physics Research of the China Academy of Engineering Physics under Grant No. 2016-LSD-Z-06; the Science and Technology Development Foundation of China Academy of Engineering Physics under Grant Nos.2012A0201007 and 2013B0101002; the Doctoral Fund of Henan University of Technology under Grant No. 2018BS053; the State Key Laboratory of Explosion Science and Technology, Beijing Institute of Technology under the contract KFJJ15-23M.
References
[1] A. Matsugi and H. Shiina, J. Phys. Chem. A 121, 4218 (2017).10.1021/acs.jpca.7b03715Search in Google Scholar PubMed
[2] J. Chang, P. Lian, D. Q. Wei, X. R. Chen, Q. M. Zhang, et al., Phys. Rev. Lett. 105, 188302 (2010).10.1103/PhysRevLett.105.188302Search in Google Scholar PubMed
[3] K. Xu, D. Q. Wei, X. R. Chen, and G. F. Ji, J. Mol. Model. 20, 2438 (2014).10.1007/s00894-014-2438-7Search in Google Scholar PubMed
[4] E. J. Reed, M. R. Manaa, L. E. Fried, K. R. Glaesemann, and J. D. Joannopoulos, Nat. Phys. 4, 72 (2008).10.1038/nphys806Search in Google Scholar
[5] Y. P. Petrov, Y. K. Karasevich, and S. V. Turetskii, J. Phys. Chem. B 4, 566 (2010).10.1134/S1990793110040068Search in Google Scholar
[6] O. Mathieu, B. Giri, A. R. Agard, T. N. Adams, J. D. Mertens, et al., Fuel 182, 597 (2016).10.1016/j.fuel.2016.05.060Search in Google Scholar
[7] Z. H. He, J. Chen, and Q. Wu, J. Phys. Chem. C 121, 8227 (2017).10.1021/acs.jpcc.6b10354Search in Google Scholar
[8] S. C. Tiwari, K. I. Nomura, R. K. Kalia, A. Nakano, and P. Vashishta, J. Phys. Chem. C 121, 16029 (2017).10.1021/acs.jpcc.7b05253Search in Google Scholar
[9] J. A. Carter, J. M. Zaug, A. J. Nelson, M. R. Armstrong, and M. R. Manaa, J. Phys. Chem. A 116, 4851 (2012).10.1021/jp301771ySearch in Google Scholar PubMed
[10] Z. A. Dreger and Y. M. Gupta, J. Phys. Chem. A 117, 5306 (2013).10.1021/jp404283aSearch in Google Scholar PubMed
[11] C. J. Wu, F. H. Ree, and C. S. Yoo, Propell. Explos. Pyrot. 5, 29 (2004).Search in Google Scholar
[12] T. R. Shan, R. R. Wixom, A. E. Mattsson, and A. P. Thompson, J. Phys. Chem. B 117, 928 (2013).10.1021/jp310473hSearch in Google Scholar PubMed
[13] N. N. Ge, Y. K. Wei, G. F. Ji, X. R. Chen, F. Zhao, et al., J. Phys. Chem. B 116, 13696 (2012).10.1021/jp309120tSearch in Google Scholar PubMed
[14] N. N. Ge, Y. K. Wei, Z. F. Song, X. R. Chen, G. F. Ji, et al., J. Phys. Chem. B 118, 8691 (2014).10.1021/jp502432gSearch in Google Scholar PubMed
[15] W. H. Zhu, H. Huang, H. J. Huang, and H. M. Xiao, J. Chem. Phys. 136, 044516 (2012).10.1063/1.3679384Search in Google Scholar PubMed
[16] Z. H. He, J. Chen, G. F. Ji, L. M. Liu, W. J. Zhu, et al., J. Phys. Chem. B 119, 10673 (2015).10.1021/acs.jpcb.5b05081Search in Google Scholar PubMed
[17] J. N. Yuan, Y. K. Wei, X. Q. Zhang, X. R. Chen, G. F. Ji, et al., Appl. Phys. 122, 135901 (2017).10.1063/1.5005804Search in Google Scholar
[18] K. L. Joshi and S. Chaudhuri, Phys. Chem. Chem. Phys. 17, 18790 (2015).10.1039/C5CP00950BSearch in Google Scholar
[19] L. Patidar and S. T. Thynell, Combust. Flame. 178, 7 (2017).10.1016/j.combustflame.2016.12.024Search in Google Scholar
[20] N. N. Ge, Y. K. Wei, F. Zhao, X. R. Chen, and G. F. Ji, J. Mol. Model. 20, 2350 (2014).10.1007/s00894-014-2350-1Search in Google Scholar PubMed
[21] Z. H. He, J. Chen, Q. Wu, and G. F. Ji, Chem. Phys. Lett. 687, 200 (2017).10.1016/j.cplett.2017.08.055Search in Google Scholar
[22] Z. Q. Bai, J. Chang, G. F. Ji, and N. N. Ge, Can. J. Chem. 97, 245, (2018).10.1139/cjc-2018-0256Search in Google Scholar
[23] J. Hutter, M. Iannuzzi, F. Schiffmann, and J. VandeVondele, Wiley Interdiscip. Rev. Comput. Mol. Sci. 4, 15 (2014).10.1002/wcms.1159Search in Google Scholar
[24] E. J. Reed, L. E. Fried, and J. D. Joannopoulos, Phys. Rev. Lett. 90, 235503 (2003).10.1103/PhysRevLett.90.235503Search in Google Scholar PubMed
[25] J. D. Kress, S. R. Bickham, L. A. Collins, B. L. Holian, and S. Goedecker, Phys. Rev. Lett. 83, 3896 (1999).10.1103/PhysRevLett.83.3896Search in Google Scholar
[26] N. Goldman, E. J. Reed, I. F. Kuo, L. E. Fried, C. J. Mundy, J. Chem. Phys. 130, 124517 (2009).10.1063/1.3089426Search in Google Scholar PubMed
[27] M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, et al., Phys. Rev. B 58, 7260 (1998).10.1103/PhysRevB.58.7260Search in Google Scholar
[28] M. Elstner, P. Hobza, T. Frauenheim, S. Suhai, and E. Kaxiras, J. Chem. Phys. 114, 5149 (2001).10.1063/1.1329889Search in Google Scholar
[29] Q. Cui, M. Elstner, E. Kaxiras, T. Frauenheim, and M. Karplus, J. Phys. Chem. B 105, 569 (2001).10.1021/jp0029109Search in Google Scholar
[30] M. R. Manaa, L. E. Fried, C. F. Melius, M. Elstner, and T. Frauenheim, J. Phys. Chem. A 106, 9024 (2002).10.1021/jp025668+Search in Google Scholar
[31] D. Margetis, E. Kaxiras, M. Elstner, T. Frauenhei, and M. R. Manaa, J. Chem. Phys. 117, 788 (2002).10.1063/1.1466830Search in Google Scholar
[32] E. J. Reed, J. D. Joannopoulos, and L. E. Fried, Phys. Rev. B 62, 16500 (2000).10.1103/PhysRevB.62.16500Search in Google Scholar
[33] J. N. Yuan, G. F. Ji, X. R. Chen, D. Q. Wei, F. Zhao, et al., Chem. Phys. Lett. 644, 250 (2016).10.1016/j.cplett.2015.11.062Search in Google Scholar
[34] S. Grimme, J. Comput. Chem. 27, 1787 (2006).10.1002/jcc.20495Search in Google Scholar
[35] C. S. Choi and E. Prince, Acta Crystallogr. B 28, 2857 (1972).10.1107/S0567740872007046Search in Google Scholar
[36] G. E. Duvall, in: Proceedings of the International School of Physics, Physics of High Energy Density (Eds. P. Caldirola, H. Knoepfel), Academic Press, New York 1971, p. 7.Search in Google Scholar
[37] C. L. Mader, Numerical Modeling of Explosives and Propellants, 2nd ed., CRC Press, Boca Raton, FL 1998.Search in Google Scholar
[38] F. J. Owens and J. Sharma, J. Appl. Phys. 51, 1494 (1980).10.1063/1.327798Search in Google Scholar
[39] A. Strachan, A. C. T. van Duin, D. Chakraborty, S. Dasgupta, and W. A. Goddard III, Phys. Rev. Lett. 91, 098301 (2003).10.1103/PhysRevLett.91.098301Search in Google Scholar
[40] M. M. Kuklja and A. B. Kunz, J. Appl. Phys. 89, 4962 (2001).10.1063/1.1359171Search in Google Scholar
[41] W. F. Perger, Chem. Phys. Lett. 368, 319 (2003).10.1016/S0009-2614(02)01879-1Search in Google Scholar
[42] M. M. Kuklja and A. B. Kunz, J. Appl. Phys. 86, 4428 (1999).10.1063/1.371381Search in Google Scholar
© 2020 Walter de Gruyter GmbH, Berlin/Boston
Articles in the same Issue
- Frontmatter
- Atomic, Molecular & Chemical Physics
- The Reaction and Microscopic Electron Properties from Dynamic Evolutions of Condensed-Phase RDX Under Shock Loading
- Trace Hydrogen Sulphide Gas Sensor Based on Cu/rGO Membrane-Coated Photonic Crystal Fibre Michelson Interferometer
- Dynamical Systems & Nonlinear Phenomena
- A General Viscous Model for Some Aspects of Tropical Cyclonic Winds
- Nonlinear Pull-in Instability of Rectangular Nanoplates Based on the Positive and Negative Second-Order Strain Gradient Theories with Various Edge Supports
- Hydrodynamics
- On the Heat Flow Through a Porous Tube Filled with Incompressible Viscous Fluid
- Effect of Magnetic Field on the Unsteady Boundary Layer Flows Induced by an Impulsive Motion of a Plane Surface
- Solid State Physics & Materials Science
- Facile Combustion Synthesis of (Y,Pr)2O3 Red Phosphor: Study of Luminescence Dependence on Dopant Concentration and Enhancement by the Effect of Co-dopant
- Size-Dependent Ultrasonic and Thermophysical Properties of Indium Phosphide Nanowires
Articles in the same Issue
- Frontmatter
- Atomic, Molecular & Chemical Physics
- The Reaction and Microscopic Electron Properties from Dynamic Evolutions of Condensed-Phase RDX Under Shock Loading
- Trace Hydrogen Sulphide Gas Sensor Based on Cu/rGO Membrane-Coated Photonic Crystal Fibre Michelson Interferometer
- Dynamical Systems & Nonlinear Phenomena
- A General Viscous Model for Some Aspects of Tropical Cyclonic Winds
- Nonlinear Pull-in Instability of Rectangular Nanoplates Based on the Positive and Negative Second-Order Strain Gradient Theories with Various Edge Supports
- Hydrodynamics
- On the Heat Flow Through a Porous Tube Filled with Incompressible Viscous Fluid
- Effect of Magnetic Field on the Unsteady Boundary Layer Flows Induced by an Impulsive Motion of a Plane Surface
- Solid State Physics & Materials Science
- Facile Combustion Synthesis of (Y,Pr)2O3 Red Phosphor: Study of Luminescence Dependence on Dopant Concentration and Enhancement by the Effect of Co-dopant
- Size-Dependent Ultrasonic and Thermophysical Properties of Indium Phosphide Nanowires