Treatment for mitochondrial diseases
-
Tongling Liufu


Abstract
Mitochondrial diseases are predominantly caused by mutations of mitochondrial or nuclear DNA, resulting in multisystem defects. Current treatments are largely supportive, and the disorders progress relentlessly. Nutritional supplements, pharmacological agents and physical therapies have been used in different clinical trials, but the efficacy of these interventions need to be further evaluated. Several recent reviews discussed some of the interventions but ignored bias in those trials. This review was conducted to discover new studies and grade the original studies for potential bias with revised Cochrane Collaboration guidelines. We focused on seven published studies and three unpublished studies; eight of these studies showed improvement in outcome measurements. In particular, two of the interventions have been tested in studies with strict design, which we believe deserve further clinical trials with a large sample. Additionally, allotopic expression of the ND4 subunit seemed to be an effective new treatment for patients with Leber hereditary optic neuropathy.
Introduction
Mitochondrial diseases are caused by the impairment of mitochondria due to mutations of mitochondrial DNA (mtDNA) or nuclear DNA (nDNA). Mitochondria function directly in energy production and nutrient metabolism (Pfanner et al. 2019). The structure of mitochondria, which consists of proteins translated by mtDNA (encoding 13 proteins) and nDNA (encoding ∼1,500 proteins), is the foundation of mitochondrial functions (Stewart and Chinnery 2015). In a broad sense, mitochondrial disease is related to a series of diseases accompanied by dysfunction of mitochondria (Schapira 2006). However, other types of diseases, except diseases related to malfunction of mitochondrial protein coding genes, have multifactorial etiologies and mitochondrial deficiency is only one of the causes. In this review, we are only concerned with primary mitochondrial diseases for which the mitochondrial dysfunction is the main cause of the disease.
The prevalence of mitochondrial diseases is estimated to be 11.5:100,000 (Chinnery 2014). Childhood-onset (<16 years of age) mitochondrial diseases are estimated to range from five to 15 cases per 100,000 individuals, mainly caused by mutations in nDNA. In adults (>16 years of age), the prevalence of mitochondrial diseases caused by mutations in mtDNA and nDNA is estimated at 9.6 and 2.9 cases per 100,000 individuals, respectively, in north east England (Gorman et al. 2016). A recent systematic review of the natural history of mitochondrial disorders reported that 59% of disorders had an onset before 18 months and 81% before 18 years (Keshavan and Rahman 2018).
There are currently no effective treatments for mitochondrial disease; most of the measures are supportive (Chinnery 2014). Current recommended treatments fall into three groups: pharmacological agents, dietary supplementation with vitamins and cofactors and exercise therapy (Hirano et al. 2018). Drugs and nutritional supplements used in patients with mitochondrial diseases are supposed to resist oxidation, improve lactic acidosis, correct secondary biochemical deficiencies, transfer electrons and modulate endocrine function (Avula et al. 2014; Parikh et al. 2015). Improvements following dietary modification and exercise therapy have also been documented in individual cases or open-label trials. Recently, new methods and agents have been introduced in clinical trials for patients diagnosed with mitochondrial diseases, and several reviews aimed at the safety and efficiency of these treatments have been published. However, these reviews did not evaluate the design of each trial, nor the reliability of the results. Therefore, our objectives for this systematic review were to analyze recently published studies (during the period of 2010–2020) with revised Cochrane Collaboration guidelines (Higgins et al. 2019), provide detailed information of the design procedures and potential bias of the results in each trial and comment on the direction of future treatment for mitochondrial diseases.
Methods
Search methods for identification of studies
We searched the Cochrane Library (6 March 2020), clinicaltrials.gov (6 March 2020), MEDLINE (5 March 2020) using the terms ‘mitochondrial disease’ and ‘clinical trial’; ‘completed studies’ and ‘mitochondrial disease’; ‘disorders’, ‘mitochondrial disorders’ and ‘mitochondria’ and ‘mitochondrial myopathy’, ‘disorders of mitochondrial function’ and ‘mitochondrial disease’ (Supplementary material). The details of criteria for included studies are shown in Table 1. We primarily focused on randomized clinical trials, and we also gathered information from case reports and open-label trials and incorporated these in the discussion (Table 2).
Criteria for considering studies for this review.
| Different parts of study design | Criteria |
|---|---|
| Types of studies | Randomized controlled trials (crossover trials). |
| Types of participants | Participants of males and females at any age confirmed deficiencies of respiratory complex I, II, III, IV or V, or combinations of these, or had defined mtDNA or nDNA mutations affecting subunits or assembly of these complexes that were associated with known clinical/pathological features of mitochondrial disease. |
| Types of interventions | Interventions of any pharmacological agent, dietary supplement, exercise therapy or other treatment. |
| Types of outcome measures | The outcomes included biochemical markers of the disease, motor function, muscle strength, special sensory (vision, auditory), stroke-like episodes, endocrine deficiency, cardiac disorders, renal disorders, cognition, efficacy of oxygen consumption, quality of life, adverse effect or other measures defining the safety or efficacy of the interventions. |
mtDNA, mitochondrial DNA; nDNA, nuclear DNA.
List of 24 clinical trials.
| Interventions | Mechanism | Diseases | Primary outcomes | Trial number | References |
|---|---|---|---|---|---|
| Aerobic training | Mitochondrial biogenesis | MM | ROS production | NA | Siciliano et al. 2012 |
| Bezafibrate | Pan-PPAR agonist | MM | Safety and tolerability | NCT02398201 | Steele et al. 2020 |
| CoQ10 (included) | Electron carrier and antioxidant | MD | GMFM 88/PedsQoL | NCT00432744 | Kerr et al. 2017 |
| CoQ10 | Electron carrier and antioxidant | MD | Motor function and quality of life | NA | Stacpoole et al. 2012 |
| Elamipretide (MTP-131) (included) | Protect cardiolipin | MM | 6MWT | NCT02367014 | Karaa et al. 2018 |
| Energetic intervention (included) | Mitochondrial biogenesis | NMD | COPM performance | NCT02208687 | Veenhuizen et al. 2019 |
| EPI-743 | Structurally related to CoQ10 | FA | Visual acuity | NCT01728064 | Zesiewicz et al. 2018 |
| EPI-743 | Structurally related to CoQ10 | Leigh syndrome | NPMDS | NCT01370447 | Martinelli et al. 2012 |
| GS010 (included) | Allotopic expression of ND4 | LHON | Visual acuity | NCT02652767 | Nancy 2020 |
| Idebenone | Electron carrier and antioxidant | LHON | Visual acuity | NA | Zhao et al. 2020 |
| Idebenone (included) | Electron carrier and antioxidant | LHON | Visual acuity | NCT00747487 | Klopstock et al. 2011 |
| Idebenone (included) | Electron carrier and antioxidant | MELAS | Cerebral lactate concentration | NCT00887562 | Hirano 2016 |
| KH176 (included) | Antioxidant, redox modulator | m.3243A > G | Gait parameters | NCT02909400 | Janssen et al. 2019 |
| l-Arginine | NO precursor | MELAS | Aerobic capacity, muscle metabolism | NCT01603446 | Rodan et al. 2015 |
| l-Arginine | NO precursor | MELAS | Oral l-arginine: MELAS | JMACTR-IIA00023 JMACTR-IIA00025 | Koga et al. 2018 |
| stroke scale; | |||||
| intravenous l-arginine: | |||||
| improvement rates of headache and nausea/vomiting at 2 h | |||||
| after completion of the initial intravenous | |||||
| administration | |||||
| l-Arginine/l-citrulline | NO precursor | MELAS | NO synthesis rates | NA | El-Hattab et al. 2012 |
| l-Carnitine (included) | Fat oxidation | MM (CPEO) | Exercise tolerance | NA | Gimenes et al. 2015 |
| Omaveloxolone (included) | Potently activated NRF2 | MM | Maximal exercise test | NCT02255422 | Madsen et al. 2020 |
| rAAV2-ND4 | Allotopic expression of ND4 | LHON | Visual acuity | NCT01267422 | Wan et al. 2016 |
| scAAV2(Y444,500,730F)-P1ND4v2 | Allotopic expression of ND4 | LHON | Visual acuity | NA | Guy et al. 2017 |
| Sodium nitrate | NO donor | MM | Oxygen cost of submaximal exercise | NTR3321 | Nabben et al. 2017 |
| Taurine | tRNALeu(UUR) taurine modification | MELAS | Complete prevention of stroke-like episodes | UMIN000011908 | Ohsawa et al. 2019 |
| Tetracycline (included) | Antiapoptotic, antiinflammation, antioxidation | PEO | Eye movements and ptosis | 2007-005274-31 | Mancuso et al. 2011 |
| WBOS | Cysteine donor | PEO | Oxidative stress biomarkers | NA | Mancuso et al. 2010 |
COPM, Canadian occupational performance measure; CoQ10, Coenzyme Q10; CPEO, chronic progressive external ophthalmoplegia; FA, Friedreich ataxia; GMFM 88, McMaster gross motor function measure, version 88; GS010, a recombinant adenoassociated viral vector serotype 2 containing the wild-type ND4 gene (rAAV2/2-ND4); LHON, Leber hereditary optic neuropathy; MD, mitochondrial disease; MELAS, mitochondrial encephalomyopathy, lactic acidosis and stroke-like episode; MM, mitochondrial myopathy; NA, not available; NADH, reduced form of nicotinamide adenine dinucleotide; ND4, NADH dehydrogenase subunit 4; NMD, neuromuscular disease; NO, nitric oxide; NPMDS, Newcastle Pediatric Mitochondrial Disease Scale; NRF2, nuclear factor erythroid 2–related factor 2; PedsQoL, pediatric quality of life scale; P1ND4v2, nuclear encoded ND4 with the appended subunit c of ATP synthase targeting sequences; PPAR, peroxisomal proliferator activated receptor; PEO, progressive external ophthalmoplegia; rAAV2-ND4, recombinant adeno-associated virus 2 containing the wild-type ND4 gene; ROS, reactive oxygen species; scAAV2, self-complementary adenoassociated viral vector; 6MWT, 6-min walk test; WBOS, whey-based oral supplementation (containing 2.5% cysteine).
Data collection and analysis
We first checked approximately 725 abstracts and unpublished studies, then identified 21 potentially eligible abstracts and four unpublished studies for further investigation. In total, seven published studies and three unpublished studies fulfilled the inclusion criteria and were included in the review (Figure 1). The results of those studies were reported after 2011 and thus have not been discussed in other systematic reviews. Second, we extracted details including basic information (review date, registered trial number, citations, contact information), study design (methods, participants, interventions, outcomes), significant results and graded risk of bias according to the Cochrane Handbook for Systematic Reviews of Interventions (Higgins et al. 2019). Seven types of bias were evaluated, including random sequence generation (selection bias), allocation concealment (selection bias), incomplete outcome data (attrition bias), selective reporting (reporting bias), blinding of participants and personnel (performance bias), blinding of outcome assessment (detection bias) and other bias (such as patient distribution between groups, compliance, etc.). For each part, the risk of bias was graded as ‘low risk’, ‘high risk’ or ‘unclear risk’. Third, we compared the available data type, intervention and duration among the included studies to determine whether a meta-analysis was suitable for this study. If possible, we planned to apply Cochrane statistical software Review Manager 5 (RevMan) ([Computer program], Version 5.3, Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014) software using a fixed-effect model to merge the data.
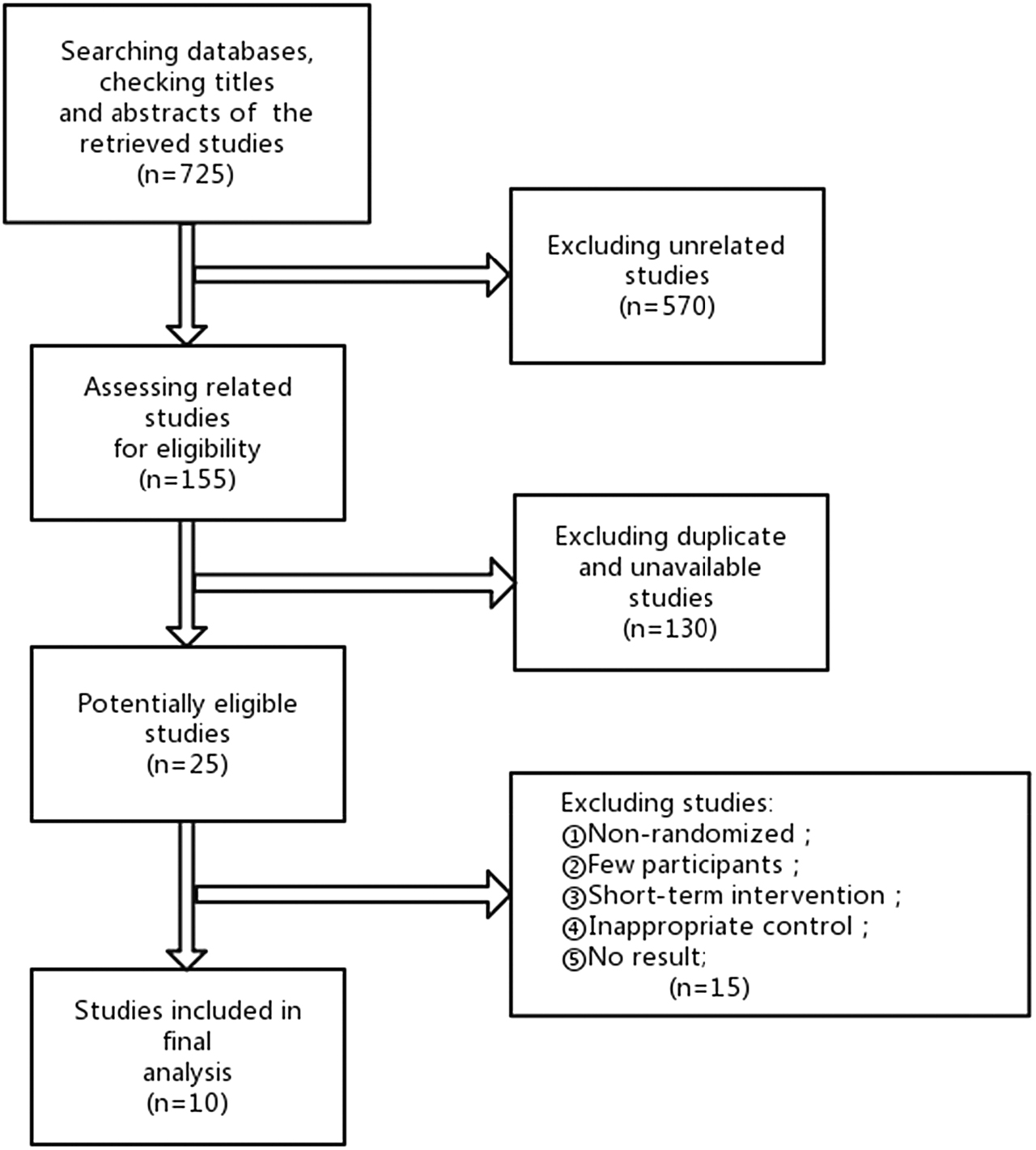
The process of study identification, screening and evaluation of the eligibility of included studies.
Results
Description of studies
Seven published studies and three unpublished studies were further analyzed using revised Cochrane Collaboration guidelines (Figure 1). Most of the studies had unclear risks of bias in random sequence generation. Other risks of bias included incomplete outcome data (attrition bias) and blinding of outcome assessment (detection bias) (Figure 2). Among the different interventions, six studies focused on the clinical effect of drugs, two on dietary supplements, one on energetic intervention and the remaining one on genetic therapy (Table 2; Figure 3). Eight of these studies showed improvement in outcome measurements with the intervention (Table 3). Given the various types of treatments, limited numbers of participants, heterogeneous genetic background of participants and different outcome measurements, we elected not to perform a meta-analysis (Supplementary material).

Risk of bias summary: judgments about each risk of bias item for each included study.
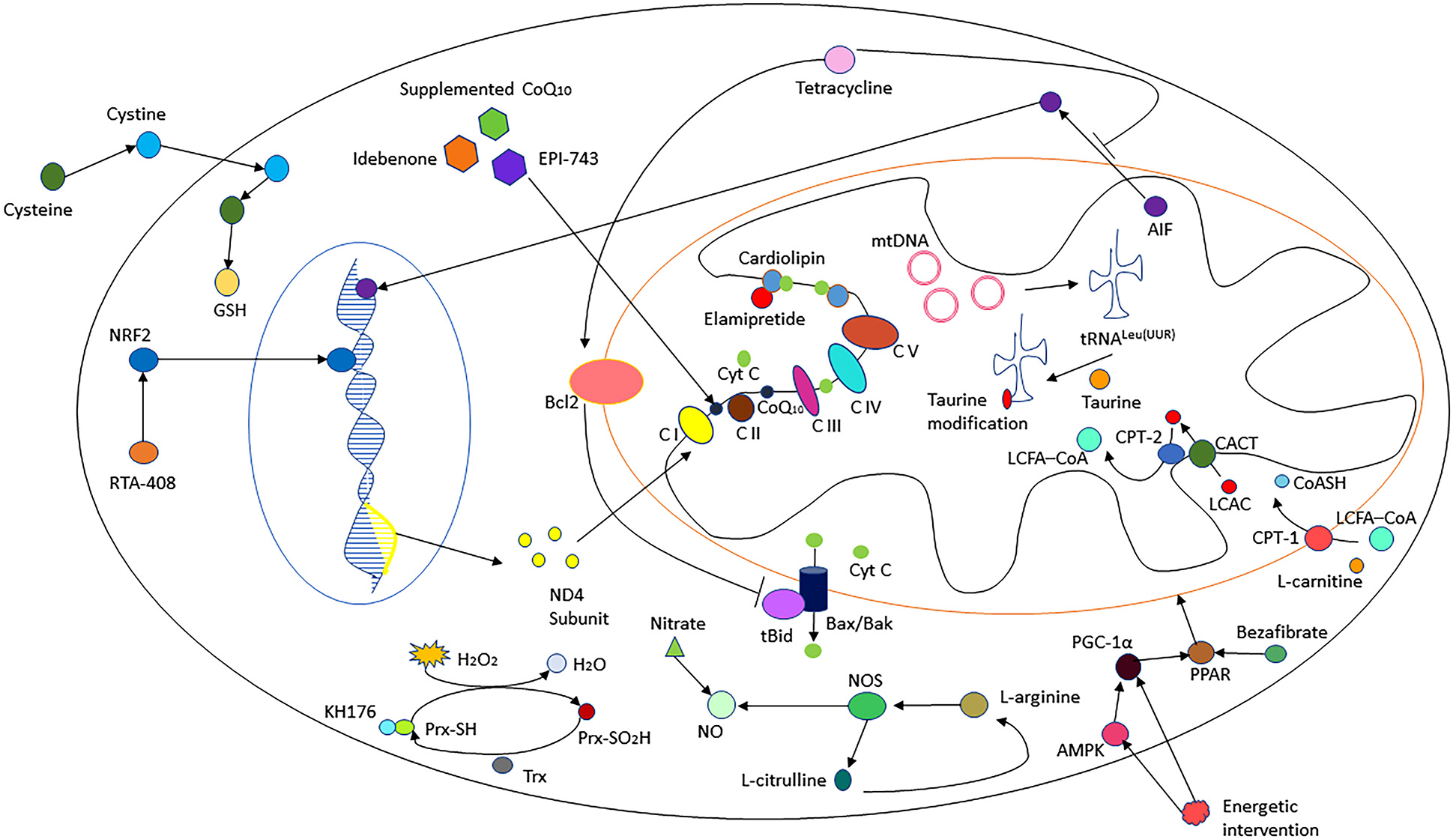
Mechanisms of different therapies. There are 16 kinds of interventions showed in this figure. Tetracycline inhibits the release of the apoptosis-inducing factor (AIF) and cytochrome c (Cyt C) from mitochondria by controlling mitochondrial permeability and upregulating Bcl-2, which prevents cleavage of Bid to truncated Bid (tBid) and antagonizes the death-promoting factors Bax and Bak. CoenzymeQ10, idebenone and EPI-743 might act as electron carriers and antioxidants. Cysteine supplementation might enhance muscle cysteine and glutathione (GSH) availability. Omaveloxolone (RTA-408) prevents nuclear factor erythroid 2–related factor 2 (NRF2) from degradation and induces NRF2 transportation to the nucleus. KH176 might bind to peroxiredoxin (Prx) and promote its role of antioxidation to detoxify hydroperoxides (H2O2) by interacting with the thioredoxin (Trx) systems. Gene therapy like allotopic expression of ND4 subunit may repair the deficient complex I. l-Arginine, l-citrulline and inorganic nitrate induce nitric oxide (NO) synthesis. Energetic intervention and bezafibrate induce mitochondrial biogenesis by activating peroxisomal proliferator activated receptor (PPAR). l-Carnitine is needed for the translocation of fatty acids into the mitochondrial compartment for β-oxidation. Taurine modification of mitochondrial tRNALeu(UUR) is important for mtDNA translation. Elamipretide protects cardiolipin from peroxidation by preventing Cyt C from unfolding and activating. AMPK: AMP-activated protein kinase; C I–IV: complex I–IV; CoA: acyl coenzyme A; CoASH: coenzyme A with a sulfhydryl functional group; CPT1/2: carnitine O-palmitoyltransferase 1/2; LCAC: long-chain acylcarnitines; LCFA: long-chain fatty acids; NOS, nitric oxide synthase; PGC-1α: peroxisomal proliferator activated receptor-γ coactivator-1α mtDNA : mitochondrial DNA.
Eight included studies showed improvement in outcome measurements with the interventions.
| Interventions | Patients | Results |
|---|---|---|
| Coenzyme Q10 | Respiratory complexes deficiency | Favor trend in scores of GMFM 88/PedsQoL |
| Energetic intervention | Neuromuscular disease | Significant increase in the mean COPM performance scores |
| GS010 | LHON | Significant improvement in foveal threshold sensitivities |
| Idebenone | LHON | Significant improvement in the mean visual acuity of all eyes and the tritan color contrast |
| Idebenone | MELAS | Lower fatigue severity scale scores |
| KH176 | m.3243A > G mutation | Significant improvement in mental condition |
| l-Carnitine | Mitochondrial myopathy | Significant increase in exercise tolerance and oxygen consumption |
| Omaveloxolone | Mitochondrial myopathy | Lower increase in blood lactate and lower heart rate during submaximal exercise |
GMFM 88, McMaster gross motor function measure, version 88; PedsQoL, pediatric quality of life scale; COPM, Canadian Occupational Performance Measure; GS010, a recombinant adenoassociated viral vector serotype 2 containing the wild-type ND4 gene (rAAV2/2-ND4); LHON, Leber hereditary optic neuropathy; MELAS, MELAS, mitochondrial encephalomyopathy, lactic acidosis and stroke-like episode.
Effects of interventions
Coenzyme Q10
Coenzyme Q10 (CoQ10) is a lipophilic electron carrier (Schapira 2006). Deficiency of CoQ10 in the inner mitochondrial membrane blocks the flow of electrons and reduces adenosine triphosphate (ATP) synthesis (Horvath et al. 2008). Evidence from different clinical trials shows that patients with primary CoQ10 deficiency and primary mitochondrial diseases had improved clinical symptoms with CoQ10 (Hirano et al. 2018). Studies of CoQ10 analogs, like idebenone and EPI-743, demonstrated more promising effects (Kerr 2013). Kerr et al. (2017) studied the effect of CoQ10 in mitochondrial disease with a randomized, double-blinded, crossover controlled clinical trial. Twenty-four patients with biochemical proof of deficiency of complex I, III or IV or molecular genetic proof of mutations in mtDNA or nDNA mutations in genes known to be associated with dysfunction of the electron transport chain were enrolled in this study. They received CoQ10 (at 10 mg/kg daily up to 400 mg) or placebo for six months, with an intervening washout period of three months. Outcome assessments were performed at the end of each trial period, including McMaster gross motor function measure, version 88 (GMFM 88) scale, and pediatric quality of life scale (PedsQoL). Data were analyzed via the Wilcoxon test, using Kendall’s Tau-B as an estimation parameter. Nine patients did not complete the study for unknown reasons. Generally, three serious adverse events (SAEs) (CoQ10 group: seizure activity; placebo group: lumbar vertebrae fractures, altered mental state) were reported without direct relationships with CoQ10; other adverse events (AEs) included decreased white blood count, elevated liver enzymes, neuromuscular changes and gastrointestinal disorders. There were improvements in scores of GMFM 88 and PedsQoL after treatment.
Idebenone
Idebenone is a synthetic quinone with a shorter and less lipophilic tail than CoQ10, which thus has higher solubility in water compared with CoQ10 and acts as an antioxidant (Hirano et al. 2018). Several studies have assessed the efficacy and safety of idebenone in Friedreich ataxia (FA), mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes (MELASs) and Leber hereditary optic neuropathy (LHON), but their primary endpoints did not reach statistical significance (Avula et al. 2014; Hirano et al. 2018). Klopstock et al. (2011) performed a multicenter double-blind, randomized, placebo-controlled trial of 85 patients with LHON (57 with m.11778G > A mutation, 17 with m.14484T > C mutation, 11 with m.3460G > A mutation), with 900 mg/day of idebenone (300 mg three times a day during meals) or placebo for 24 weeks. Outcomes were assessed at baseline and week 24, including best recovery of visual acuity (VA), change in best VA, change in VA of the best eye, change in VA for both eyes, color contrast sensitivity, retinal nerve fiber layer thickness (RNFLT) and responder analysis. Data were analyzed using the mixed-model repeated measures method.
According to results of the intent-to-treat population, there were two discontinuations related to AEs (one in each group). Two SAEs were reported: infected epidermal cyst and epistaxis. The nature, severity and frequency of AEs were indistinguishable between the treatment and placebo groups. There was no statistically significant difference in best recovery of VA, change in best VA, VA of the best eye, responder analysis, protan color contrast or RNFLT; there were statistically significant differences in the mean VA of all eyes (vs placebo, logMAR −0.100; 95% confidence interval (CI): −0.188,−0.012; p = 0.026) and tritan color contrast (vs placebo, −13.63%; 95% CI: −23.61,−3.66; p = 0.008). However, the authors noted that patients with discordant VA at baseline showed a significant difference in best recovery in VA (p = 0.011), change in best VA (p = 0.003), change in VA of the best eye (p = 0.003) and change in the mean VA of all eyes (p = 0.0001) after idebenone application, and 20% of the eyes of patients unable to read any letters at baseline were able to read at least one full line on the chart at week 24.
Hirano (2016) implemented a double-blind, randomized, placebo-controlled trial among 27 participants with MELASs to determine whether idebenone could relieve the symptoms of these patients. Participants received idebenone (at 900 mg/day or 2,250 mg/day) or placebo for one month. The outcomes of the trial were cerebral lactate levels measured by magnetic resonance spectroscopy, venous lactate concentration and the fatigue severity scale (FSS) score, all of which were recorded after four weeks of treatment. The authors did not provide specific details of the data analysis. Six patients dropped out of the trial without detailed information. There were no SAEs. AEs included symptoms of blood and lymphatic system disorders, cardiac disorders, eye disorders, gastrointestinal disorders, nervous system disorders and renal and urinary disorders. The levels of cerebral lactate concentration and venous lactate concentration increased compared to baseline in the group given idebenone at 2,250 mg/day and decreased in the group given idebenone at 900 mg/day and in the placebo control. Both of the treatment groups had lower FSS scores, while FSS scores in the placebo-controlled group were higher.
KH176
KH176 is a water-soluble derivative of vitamin E and a potent redox-modulating compound that enhances the antioxidant thioredoxin/peroxiredoxin system. A dosage escalating clinical trial with KH176 in healthy volunteers indicated good tolerability and provided a detailed pharmacokinetic profile (Hirano et al. 2018; Koene et al. 2017). Janssen et al. (2019) conducted a double-blind, randomized, placebo-controlled, crossover trial in 19 patients confirmed to have the m.3243A > G mutation with a heteroplasmy percentage of ≥20% in urinary epithelial cells. Patients received oral KH176 at 100 mg twice daily or placebo for 28 days; then, they were given another treatment after a washout period of 28 days. The outcome measurements were determined two days before the start of the intervention and on the last two days of the intervention period and included gait parameters, plasma concentration analysis of KH176, biomarkers, safety measurements, 6-min walk test (6MWT), 6-min mastication test (6MMT), maximal grip strength, the 3-s sit-stand test, accelerometery, spirometry, Newcastle Mitochondrial Disease Adult Scale (NMDAS), checklist individual strength (CIS), SF-36 scale of quality of life (QoL), hospital anxiety and depression scale (HADS), beck depression inventory (BDI), test of attentional performance (AP), sleep quantity and goal attainment scale.
Data were analyzed using the mixed model. No SAEs were reported. The total numbers of treatment-emergent AEs were 35 in the KH176 arm and 51 in the placebo arm. A higher plasma concentration of KH176 was identified as an important potential risk because it was related to cardiac repolarization. The peak plasma concentration of KH176 during treatment ranged from 125 to 351 ng/mL, well below the predefined safety threshold of 1,000 ng/mL. The basic rhythm of total beats per minute was significantly lower during the active treatment period (p = 0.0036). There were no differences in the corrected QT interval. The results showed improvement in AP for alertness in test conditions with and without alarm and a positive trend in the HADS total score. The total score (vs placebo, −2.9; 95% CI: −5.7,−0.13; p = 0.04) and affective subscale (vs placebo, −1.1; 95% CI: −1.7,−0.4; p = 0.004) of BDI was significantly lower after treatment. There were no significant differences in biomarkers, gait parameters, 6MWT, 6MMT, maximal grip strength, the 3-s sit-stand test, accelerometery, spirometry, NMDAS or the CIS scale.
Elamipretide
Elamipretide is a Szeto-Schiller peptide that targets the inner mitochondrial membrane and is associated with cardiolipin. Through this interaction, elamipretide protects cardiolipin from peroxidation and depletion (Szeto 2014). Karaa et al. (2018) carried out a multicenter double-blind, randomized, placebo-controlled trial in 36 participants with primary mitochondrial myopathy (MM) and different genetic backgrounds, including 11 with mtDNA deletion syndrome, eight with mitochondrial tRNA mutations (A3243G, A8344G, T12148C), four with DNA polymerase gamma (POLG)-related disorders, three with LHON, two with Kearns-Sayre syndrome (KSS), five with multisystem mitochondrial disorder, one with Leigh syndrome, one with 3-methylglutaconic acidemia, deafness, encephalopathy, and Leigh-like syndrome (MEGDEL) syndrome and one with MM. During the study, elamipretide (at 0.01, 0.1 and 0.25 mg/kg·h) or placebo were infused intravenously over 2 h for five consecutive days. Apart from the daily symptom questionnaire (DSQ), other outcomes were determined at baseline (day 1), at the end of treatment (day 5) and at the end of the trial (day 7). These outcomes included a 6MWT, cardiopulmonary exercise testing (CPET) parameters, participant-reported symptoms, biomarkers and AEs. Data were analyzed with the analysis of covariance (ANCOVA) model and mixed-model repeated measures method. There were no SAEs, and the most common AE was headache (16.7%), followed by dizziness (8.3%); no differences in AEs were identified between the treatment and placebo groups. In addition, there was no statistically significant difference in distance walked in the 6MWT, CPET parameters, the modified NMDAS symptom scores, DSQ scores, levels of biomarkers (fibroblast growth factor 21 [FGF-21], glutathione, 8-isoprostane and 8-hydroxy-2-deoxyguanosine) after treatment. However, the authors used the ANCOVA model and mixed-model repeated measures method to further analyze the results of 6MWT among different dosage groups and found a dosage-related increase of change in distance (p = 0.014); adjusted change for the highest dosage group was 51.2 vs 3.0 m in the placebo group (p = 0.0297).
L-Carnitine
L-Carnitine is a dietary supplement that functions as a mitochondrial substrate. In normal physiological conditions, l-carnitine is needed for the translocation of fatty acids into the mitochondrial compartment for β-oxidation and has a role in carbohydrate metabolism (Stephens et al. 2007). Gimenes et al. (2015) carried out a double-blind, randomized, placebo-controlled, crossover trial to study the effect of l-carnitine on exercise tolerance in 12 patients with chronic progressive external ophthalmoplegia (CPEO). Patients received l-carnitine at 3 g or placebo in one daily dose (with breakfast or lunch) for eight weeks, with an intervening washout period of four months. Outcomes included body composition, pulmonary function testing, CPET and peripheral muscle testing. After the pretest evaluation, data of the outcomes were obtained after two periods of treatment. Data were analyzed using a generalized linear mixed model. The results showed a significant increase of inspiratory capacity in the l-carnitine group. In the constant work rate exercise, there was a significant increase in the time limit of tolerance (Tlim), oxygen uptake at isotime and Tlim, oxygen uptake per heartbeat at Tlim and a significant decrease in gas exchange ratio at isotime after l-carnitine supplementation. There were no statistically significant differences in body composition, peripheral muscle testing, metabolic, ventilator or cardiovascular variables compared to the placebo group. No AEs were reported.
Omaveloxolone
Omaveloxolone is a synthetic triterpenoid that inhibits KEAP1 and prevents nuclear factor erythroid 2–related factor 2 (NRF2) from degradation. NRF2 is a promising therapeutic target with antiinflammatory and antioxidation effects because it regulates the expression of genes related to inflammation, redox metabolism and proteostasis (Liu et al. 2016). Madsen et al. (2020) tried to prove its effect among 53 patients with MM (18 with CPEO, one with KSS, two with Leigh syndrome, five with MELAS, four with myoclonic epilepsy associated with ragged-red fibers, two with neuropathy, ataxia and retinitis pigmentosa and 21 with unclear disease) with a multicenter double-blind, randomized, placebo-controlled trial. Patients received omaveloxolone at 5, 10, 20, 40, 80 or 160 mg or placebo once daily for 12 weeks. Outcome measurements included change in the maximal exercise test (MET), 6MWT, submaximal exercise test (SET), pharmacodynamic markers and safety measures. These evaluations were conducted after four, eight and 12 weeks of treatment, and a follow-up visit occurred four weeks after treatment termination (week 16). Data were analyzed with the mixed-model repeated measures method. Seven SAEs were reported, three of which were possibly related to the study drug, including tachycardia, fatigue and ventricular tachycardia and atrioventricular dissociation. Four patients in the treatment groups withdrew due to AEs, but the frequency of discontinuation did not significantly differ from the placebo group. Participants at 80 and 160 mg had significantly increased plasma ferritin and γ-glutamyl transferase, alanine transaminase and aspartate transaminase (AST) after four weeks of treatment compared to baseline without increases in bilirubin. Participants at 160 mg had lower increases in blood lactate during the SET (vs baseline, −1.6 ± 0.5 mM; 95% CI: −2.7,−0.6; p = 0.003; vs placebo, −1.4 ± 0.7 mM; 95% CI: −2.8,−0.04; p = 0.04) and a lower heart rate at the end of the SET (vs baseline, −8.7 ± 3.5 bpm; 95% CI: −15.8,−1.6; p = 0.02; vs placebo, −12.0 ± 4.6 bpm; 95% CI: −21.2,−2.7; p = 0.01) at week 12. There were no statistically significant differences in peak workload, maximal oxygen uptake, peak lactate production of MET or 6MWT.
Tetracycline
Tetracyclines, a class of antibiotics, have neuroprotective properties for Huntington disease, Parkinson disease, stroke and multiple sclerosis. Tetracyclines inhibit microglial activation, apoptosis and reactive oxygen species production (Mancuso et al. 2012). Mancuso et al. (2011) carried out a double-blind, randomized, placebo-controlled trial studying tetracycline treatment in 16 patients with progressive external ophthalmoplegia (PEO). Patients received oral tetracycline at 500 mg/day or placebo 14 days a month for three months. Outcomes were quantitative measurements of eye movements and eyelid ptosis, oxidative stress biomarkers (advanced oxidation protein products [AOPPs], ferric reducing antioxidant power [FRAP] and total glutathione [GSH]), lactate concentration, muscle strength, QoL (SF-36), NMDAS and AEs. They were evaluated at baseline and after three months of treatment. Overall, two AEs were observed, including creatine kinase blood elevation (2,432 U/l, normal values < 190) and dermatitis. There was a statistically significant change in GSH levels (vs baseline, +17.8%, p = 0.001). There were no statistically significant differences in FRAP, AOPP, lactate levels, total ocular motility, vertical, horizontal or oblique movements, eyelid ptosis, QoL (SF-36) score, NMDAS score or Medical Research Council scale of muscle strength.
Energetic intervention
Aerobic endurance training may increase mitochondrial mass, muscle mitochondrial enzyme activities and muscle strength. However, there is still a lack of effective individual exercise planning for patients with different degrees of muscle weakness and personal preference of training pattern (Voet et al. 2013). Veenhuizen et al. (2019) carried out a multicenter assessor-blinded, two-arm randomized controlled trial to search for an effective plan of energetic intervention among 53 patients with neuromuscular disease, including five with facioscapulohumeral dystrophy, five with inclusion body myositis, 18 with MM, five with hereditary motor and sensory neuropathy, four with myasthenia gravis, three with myotonic dystrophy type 1, two with hereditary spastic paraplegia, one with chronic idiopathic axonal polyneuropathy, one with congenital myopathy, one with McArdle syndrome, two with congenital fiber type disproportion, one with limb girdle muscular dystrophy, one with autosomal dominant distal and anterior dystrophy, one with Kennedy syndrome, one with hyperkalemic periodic paralysis, two with Duchenne carriers and one with postpolio syndrome.
Patients received energetic intervention combining aerobic exercise training (AET), energy conservation management and relapse prevention or nonprescribed intervention (such as physical therapy, rehabilitation care or no intervention at all) for four months. Outcomes were measured with social participation assessed by the Canadian Occupational Performance Measure (COPM) performance scale, COPM satisfaction scale, 6MWT, CIS fatigue subscale, activity card sort (ACS), HADS, general self-efficacy scale (GSES) and perceived caregiver burden. Outcome data for patients and caregivers were collected before randomization (T0), immediately after the four-month intervention period (T1), at three-month follow-up (T2) and at 11-month follow-up (T3). Data were analyzed using linear models that account for repeated measurements. A total of 76% of patients in the intervention group and 79% of patients in the control group had a caregiver who was able and willing to coparticipate. During the study, one patient (intervention group) was admitted to a hospital for pneumonia and one patient (intervention group) dropped out due to the inability to continue the energetic program. At T1, there was a statistically significant increase in the mean COPM performance scores (vs control, 1.7; 95% CI: 1.0,2.4; p < 0.0001), COPM satisfaction score (vs control, 2.1; 95% CI: 1.2,3.0; p < 0.0001), 6MWT (vs control, 30.3; 95% CI: 12.4,48.2; p = 0.00073) and ACS (vs control, 4.3; 95% CI: −0.02,8.5; p = 0.047) after treatment. Moreover, there was a statistically significant decrease in HADS depression scores (vs control, −2.1; 95% CI: −3.4,−0.71; p = 0.0022). There were no statistically significant differences in the CIS fatigue subscale, GSES, HADS anxiety scores or perceived caregiver burden.
Genetic therapy
The approach of allotopic expression of the ND4 subunit has been widely employed in clinical trials. By delivering a nuclear-encoded version of the ND4 gene into cells, a full-length mitochondrial encoded ND4 subunit would be expressed in the nucleus, translated in the cytoplasm, then imported into the mitochondrion by adding a mitochondrial targeting sequence. A prospective open-label trial studied the effect of nuclear encoded ND4 with the P1 isoform of subunit c of ATP synthase targeting sequences (P1ND4v2), which was inserted into a self-complementary adenoassociated viral vector (scAAV2) (Y444,500,730F) in five patients with LHON. The preliminary results of this study showed improvement in VA of the patients (Feuer et al. 2016).
Recently, Nancy (2020) implemented a double-blind, randomized, sham-controlled trial in 39 patients with LHON. Participants received a single dose of GS010 (rAAV2/2-ND4) in one of their randomly selected eyes, via intravitreal injection containing 9 × 1010 viral genomes (vg) in 90 µL balanced salt solution plus 0.001% Pluronic F68®, while the other eye of participants received a sham injection. Four patients withdrew for unclear reasons. The authors evaluated a series of outcomes including VA, the number of eye responders to treatment, the number of subject responders to treatment, ganglion cell layer (GCL) macular volume, total macular volume, RNFL temporal quadrant thickness, retinal nerve fiber layer (RNFL) papillomacular bundle thickness, foveal threshold sensitivities, visual field (VF), contrast sensitivity and color vision. These data were collected at weeks 48, 72 and 96 after treatment. All-cause mortality risk was 5.13% (two deaths). Five SAEs (alcoholic liver disease, alcohol poisoning, malnutrition, alcohol withdrawal syndrome, renal failure) were reported. Other AEs included symptoms of eye disorders, vascular disorders and nervous system disorders. The study showed significant improvement in foveal threshold sensitivities and a tendency of increase in the number of eye responders and subject responders after treatment. There were no significant differences in early treatment diabetic retinopathy study score at week 48, GCL macular volume, total macular volume, RNFL temporal quadrant thickness, RNFL papillomacular bundle thickness, VF, contrast sensitivity or color vision.
Discussion
General discussion
There is no standard and effective treatment for mitochondrial disease at present due to the lack of high-quality randomized controlled trials, as well as the diversity of study designs and outcome measurements in each trial (Stacpoole 2011). According to the consensus recommendations from the Mitochondrial Medicine Society published in 2015 (Parikh et al. 2015), there are several optimal options for general treatment of mitochondrial disease. l-Arginine and citrulline are recommended for the treatment of MELAS (m.3243A > G)-related stroke; in the acute phase of stroke-like episodes, patients should be treated with l-arginine intravenously, and the role of oral l-arginine and citrulline in preventing strokes should be considered. Vitamins and xenobiotics, like CoQ10, α-lipoic acid and riboflavin, should be offered to patients with mitochondrial disease, though solid evidence of efficacy is sparse. l-Carnitine should be used when there is a documented deficiency, and folinic acid should be considered in patients with mitochondrial disease and central nervous system manifestations. For patients with MM, having endurance exercise and resistance exercise in a supervised, progressive fashion with training that begins at a low intensity and duration is recommended.
However, those reference recommendations are not specific in practical management. Notably, several reviews summarized meaningful results of clinical trials, and some treatments that had not been included in our review should be discussed (Avula et al. 2014; Hirano et al. 2018; Horvath et al. 2008; Pfeffer et al. 2012). In 2006, a long-term study conducted in adults using dichloroacetate at a dosage of 25 mg/kg·day had to be terminated prematurely due to documented peripheral nerve toxicity and no therapeutic benefit. Three studies of treatment with creatine showed conflicting data in locomotor function, indicating the treatment response of creatine may be unsustainable. There was no evidence of clinical response to dimethylglycine. A trial studying the effect of curcumin in patients with LHON was completed, but no results were available. One retrospective study of 24 patients with mitochondrial neurogastrointestinal encephalomyopathy treated with allogeneic hematopoietic stem cell transplant to replace thymidine phosphorylase which is encoded by the TYMP gene showed that only nine (37.5%) were alive at the last follow-up. In terms of exercise training, a systematic review analyzed six randomized controlled design studies on the effect of training in patients with muscle disease. The authors concluded that no clearly defined exercise protocols could be drawn from the research evidence (Voet et al. 2013).
Included and excluded studies
Ten included studies followed randomized, placebo-controlled design, all but one of the studies were double blind. The study using energetic intervention as treatment was unable to keep the information of patients secret from caregivers and therapists. Three of the studies in a range of 12–24 patients were crossover with different washout periods according to the pharmacokinetic profile of each drug, while these studies might ignore the natural clinical courses of individuals, and thus reduce the comparability between the treatment and placebo groups. There were 16 kinds of interventions in both the included studies and excluded studies (14 studies). For most of the studies, participants had a variety of genetic backgrounds, excepting five studies (two included studies) of patients with LHON carrying m.11778G > A, m.14484T > C or m.3460G > A mutation and five studies (one included study) of patients with MELAS carrying m.3243A > G mutation. The primary outcomes of the studies were quite different as listed in Table 2. The major endpoints for patients with MELAS included symptoms and biomarkers related to stroke-like episodes, which are the most common features of patients with MELAS. For patients with LHON, all of the studies used VA as the primary outcome. Ten of the studies (six included) focused on patients with MM, namely patients who experienced muscle weakness and fatigue, and used biochemical, physiologic and questionnaire data indicating motor function and muscle strength as primary outcome measurements. Three studies for children with mitochondrial diseases mainly evaluated motor function and quality of life. The remaining one study measured change in VA of patients with FA. Eight included studies applied the mixed model to analyze repeated measurement data, effectively and comprehensively. Five included studies had low risks for selection bias and reporting bias (Figure 2); in two of them, idebenone in patients with LHON and energetic intervention in patients with neuromyopathy showed significant results of primary outcomes after treatment according to the original data. There were significant results in excluded studies, despite the lack of randomized, placebo-controlled design. The results of those studies provide important information about potential effective indicators and the AEs of each drug; thus, we incorporated these results with the results of included studies in the following discussion to evaluate the effect of each intervention objectively.
LHON causes progressive and mostly irreversible loss of central VA and dyschromatopsia. Among the five studies for patients with LHON, two discussed the effect of idebenone (one retrospective case-controlled, one multicenter double-blind, randomized, placebo-controlled study) (Klopstock et al. 2011; Zhao et al. 2020). The identical result was improvement in VA over time in all eyes, while the multicenter study emphasized that patients with discordant VA at baseline had significant change in all outcomes assessing VA after treatment. In addition, genetic treatment seemed to be effective for patients with the G11778A mutation according to three studies (one double-blind, randomized, sham-controlled, two open-label studies) (Guy et al. 2017; Nancy 2020; Wan et al. 2016) using viral vector carrying the nuclear encoded ND4 with mitochondrial targeting sequences to relieve the visual defect of patients with LHON through intravitreal injection. All three studies showed improvement in VA, while the double-blind study showed a tendency to improve after 72–96 weeks; one open-label trial with 14 participants showed improvement in five participants after one month and the remaining trial showed significant improvement in six of nine patients after nine months of follow-up.
Patients with MELAS have common symptoms including muscle weakness, headaches, seizures and stroke-like episodes (Horvath et al. 2008). The included study that treated patients with MELAS with idebenone (Hirano 2016) showed no significant results in primary outcomes, while patients in treatment groups had an improvement trend in muscle strength. For l-arginine, one nine-year open-label trial (Koga et al. 2018), in which the authors abandoned the initially designed randomized placebo-controlled trial considering the severe features of the disease, demonstrated the effect of oral l-arginine and intravenous l-arginine in preventing the ictuses and progression and fatal outcome of MELAS. In both the two-year clinical trials and the seven-year follow-up, the mortality rate was 17.39%, while the mortality rates of a five-year cohort study (Yatsuga et al. 2012) and an eight-year cohort study (Zhang et al. 2018) were 20.8 and 23.1%, respectively. The other dosage of oral l-arginine (Rodan et al. 2015) showed an improvement in oxygen and ATP consumption. In addition to these studies, one study compared the efficacy between l-arginine and l-citrulline (El-Hattab et al. 2012), and the results indicated that the plasma nitric oxide concentration showed no difference between the two drugs. One new study (Ohsawa et al. 2019) that focused on the effect of taurine was carried out based on the theory that the defect in taurine modification in mutant mitochondrial tRNALeu(UUR) causes a failure in deciphering the cognate codon and leads to mitochondrial dysfunction in patients with MELAS harboring the m.3243A > G or m.3271T > C mutation. Patients enrolled in this study experienced a significant decrease in the frequency of stroke-like episodes.
Skeletal muscle is one of the most affected sites in mitochondrial diseases. The treatments of 10 studies for patients with MM included drugs (elamipretide, omaveloxolone, tetracycline, KH176, bezafibrate), nutritional agents (l-carnitine, inorganic nitrate, cysteine) and exercise training. Patients treated with omaveloxolone (Madsen et al. 2020) had lower increases in blood lactate during submaximal exercise and lower heart rate at the end of submaximal exercise, while the drug may cause tachycardia, fatigue and ventricular tachycardia and atrioventricular dissociation. Treatment with tetracycline (Mancuso et al. 2011) significantly increased the GSH level, while there was no improvement in related clinical syndrome. KH176 (Janssen et al. 2019) affected mental condition of patients with MM and the m.3243A > G mutation. Bezafibrate (Steele et al. 2020) may lead to an exacerbation of the mitochondrial pathology in patients, even though it reduced the number of complex IV-deficient muscle fibers. The result of 12 patients with CPEO treated with l-carnitine (Gimenes et al. 2015) indicated improvement in oxygen consumption. Whey-based oral cysteine supplementation (Mancuso et al. 2010) in patients with PEO showed improvement in oxidative stress biomarkers at rest and after exercise. However, no significant result was observed in treatment with elamipretide (Karaa et al. 2018) or inorganic nitrate (Nabben et al. 2017). A study using energetic intervention (Veenhuizen et al. 2019) showed improvement in social participation, motor function and mental condition after treatment. Another study with 10 weeks of AET (Siciliano et al. 2012) showed a reduction of the increase of lactate during the exercise test and lower mean blood lipoperoxide level.
In three studies (one included) for children with mitochondrial disease, one open-label trial treated Leigh syndrome with EPI-743 (Martinelli et al. 2012), a parabenzoquinone analog resembling CoQ10, which has shown an effect in patients with FA (Zesiewicz et al. 2018). The results showed significant increases in Newcastle Pediatric Mitochondrial Disease Scale scores, GMFM scores and QoL score on the PedsQoL neuromuscular module. The other two studies used CoQ10 (Kerr et al. 2017; Stacpoole et al. 2012) to treat children with mitochondrial disease. According to the results, there was an improvement in scores of GMFM 88 and PedsQoL after treatment.
Conclusion
We evaluated 24 studies in this review and provided a detailed introduction of ten randomized clinical trials. In particular, two of the interventions, idebenone in patients with LHON and energetic intervention in patients with neuromyopathy, followed strict study design and showed significant results, which we believe deserve further clinical trials. Additionally, recombinant adenoassociated viral vector serotype 2 (rAAV2/2) containing the wild-type ND4 gene (rAAV2/2-ND4) seemed to be a new effective treatment for patients with LHON, while the comparable effect between idebenone and this invasive treatment is worthy of further evaluation.
Author contribution: All the authors have accepted responsibility for the entire content of this submitted manuscript and approved submission.
Research funding: None declared.
Conflict of interest statement: The authors declare no conflicts of interest regarding this article.
References
Avula, S., Parikh, S., Demarest, S., Kurz, J., and Gropman, A. (2014). Treatment of mitochondrial disorders. Curr. Treat. Options Neurol. 16: 292, https://doi.org/10.1007/s11940-014-0292-7.Search in Google Scholar PubMed PubMed Central
Chinnery, P.F. (20141993-2020). Mitochondrial disorders overview. In: Adam, M.P., Ardinger, H.H., Pagon, R.A., et al. (Eds.), GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle, pp. 1–21.Search in Google Scholar
El-Hattab, A.W., Hsu, J.W., Emrick, L.T., Wong, L.-J.C., Craigen, W.J., Jahoor, F., and Scaglia, F. (2012). Restoration of impaired nitric oxide production in MELAS syndrome with citrulline and arginine supplementation. Mol. Genet. Metabol. 105: 607–614, https://doi.org/10.1016/j.ymgme.2012.01.016.Search in Google Scholar PubMed PubMed Central
Feuer, W.J., Schiffman, J.C., Davis, J.L., Porciatti, V., Gonzalez, P., Koilkonda, R.D., Yuan, H., Lalwani, A., Lam, B.L., and Guy, J. (2016). Gene therapy for leber hereditary optic neuropathy: initial results. Ophthalmology 123: 558–570, https://doi.org/10.1016/j.ophtha.2015.10.025.Search in Google Scholar PubMed PubMed Central
Gimenes, A.C., Bravo, D.M., Nápolis, L.M., Mello, M.T., Oliveira, A.S.B., Neder, J.A., and Nery, L.E. (2015). Effect of L-carnitine on exercise performance in patients with mitochondrial myopathy. Braz. J. Med. Biol. Res. 48: 354–362, https://doi.org/10.1590/1414-431x20143467.Search in Google Scholar PubMed PubMed Central
Gorman, G.S., Chinnery, P.F., DiMauro, S., Hirano, M., Koga, Y., McFarland, R., Suomalainen, A., Thorburn, D.R., Zeviani, M., and Turnbull, D.M. (2016). Mitochondrial diseases. Nat Rev Dis Primers 2: 16080, https://doi.org/10.1038/nrdp.2016.80.Search in Google Scholar PubMed
Guy, J., Feuer, W.J., Davis, J.L., Porciatti, V., Gonzalez, P.J., Koilkonda, R.D., Yuan, H., Hauswirth, W.W., and Lam, B.L. (2017). Gene therapy for leber hereditary optic neuropathy: low- and medium-dose visual results. Ophthalmology 124: 1621–1634, https://doi.org/10.1016/j.ophtha.2017.05.016.Search in Google Scholar PubMed PubMed Central
Higgins, J.P.T., Thomas, J., Chandler, J., Cumpston, M., Li, T., Page, M.J., and Welch, V.A. (2019). Cochrane handbook for systematic reviews of interventions version 6.0: Cochrane.10.1002/9781119536604Search in Google Scholar
Hirano, M., Emmanuele, V., and Quinzii, C.M. (2018). Emerging therapies for mitochondrial diseases. Essays Biochem 62: 467–481, https://doi.org/10.1042/ebc20170114.Search in Google Scholar
Hirano, M. (2016). Study of idebenone in the treatment of mitochondrial encephalopathy lactic acidosis & stroke-like episodes (MELAS): clinicaltrials.gov.NCT00887562.Search in Google Scholar
Horvath, R., Gorman, G., and Chinnery, P.F. (2008). How can we treat mitochondrial encephalomyopathies? Approaches to therapy. Neurotherapeutics 5: 558–568, https://doi.org/10.1016/j.nurt.2008.07.002.Search in Google Scholar PubMed PubMed Central
Janssen, M.C.H., Koene, S., Laat, d.P., Hemelaar, P., Pickkers, P., Spaans, E., Beukema, R., Beyrath, J., Groothuis, J., Verhaak, C., and Smeitink, J. (2019). The KHENERGY study: safety and efficacy of KH176 in mitochondrial m.3243A>G spectrum disorders. Clin. Pharmacol. Ther. 105: 101–111, https://doi.org/10.1002/cpt.1197.Search in Google Scholar PubMed PubMed Central
Karaa, A., Haas, R., Goldstein, A., Vockley, J., Weaver, W.D., and Cohen, B.H. (2018). Randomized dose-escalation trial of elamipretide in adults with primary mitochondrial myopathy. Neurology 90: e1212–e1221, https://doi.org/10.1212/wnl.0000000000005255.Search in Google Scholar
Kerr, D.S., deGrauw, T.J., and Feigenbaum, A.S. (2017). Phase III trial of coenzyme Q10 in mitochondrial disease: clinicaltrials.gov, NCT00432744.Search in Google Scholar
Kerr, D.S. (2013). Review of clinical trials for mitochondrial disorders: 1997-2012. Neurotherapeutics 10: 307–319, https://doi.org/10.1007/s13311-013-0176-7.Search in Google Scholar PubMed PubMed Central
Keshavan, N., and Rahman, S. (2018). Natural history of mitochondrial disorders: a systematic review. Essays Biochem. 62: 423–442, https://doi.org/10.1042/ebc20170108.Search in Google Scholar PubMed
Klopstock, T., Yu-Wai-Man, P., Dimitriadis, K., Rouleau, J., Heck, S., Bailie, M., Atawan, A., Chattopadhyay, S., Schubert, M., Garip, A., et al. (2011). A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain 134: 2677–2686, https://doi.org/10.1093/brain/awr170.Search in Google Scholar PubMed PubMed Central
Koene, S., Spaans, E., Van, B.L., Van, L.G., Delafontaine, B., Badilini, F., Beyrath, J., and Smeitink, J. (2017). KH176 under development for rare mitochondrial disease: a first in man randomized controlled clinical trial in healthy male volunteers. Orphanet J. Rare Dis. 12: 163, https://doi.org/10.1186/s13023-017-0715-0.Search in Google Scholar PubMed PubMed Central
Koga, Y., Povalko, N., Inoue, E., Nakamura, H., Ishii, A., Suzuki, Y., Yoneda, M., Kanda, F., Kubota, M., Okada, H., et al. (2018). Therapeutic regimen of L-arginine for MELAS: 9-year, prospective, multicenter, clinical research. J. Neurol. 265: 2861–2874, https://doi.org/10.1007/s00415-018-9057-7.Search in Google Scholar PubMed PubMed Central
Liu, X., Ward, K., Xavier, C., Jann, J., Clark, A.F., Pang, I., and Wu, H. (2016). The novel triterpenoid RTA 408 protects human retinal pigment epithelial cells against H2O2-induced cell injury via NF-E2-related factor 2 (Nrf2) activation. Redox Biol 8: 98–109, https://doi.org/10.1016/j.redox.2015.12.005.Search in Google Scholar PubMed PubMed Central
Madsen, K.L., Buch, A.E., Cohen, B.H., Falk, M.J., Goldsberry, A., Goldstein, A., Karaa, A., Koenig, M.K., Muraresku, C.C., Meyer, C., et al. (2020). Safety and efficacy of omaveloxolone in patients with mitochondrial myopathy: MOTOR trial. Neurology 94: e687–e698, https://doi.org/10.1212/wnl.0000000000008861.Search in Google Scholar
Mancuso, M., Orsucci, D., Calsolaro, V., LoGerfo, A., Allegrini, L., Petrozzi, L., Simoncini, C., Rocchi, A., Trivella, F., Murri, L., et al. (2011). Tetracycline treatment in patients with progressive external ophthalmoplegia. Acta Neurol. Scand. 124: 417–423, https://doi.org/10.1111/j.1600-0404.2011.01536.x.Search in Google Scholar PubMed
Mancuso, M., Orsucci, D., Filosto, M., Simoncini, C., and Siciliano, G. (2012). Drugs and mitochondrial diseases: 40 queries and answers. Expet Opin. Pharmacother. 13: 527–543, https://doi.org/10.1517/14656566.2012.657177.Search in Google Scholar PubMed
Mancuso, M., Orsucci, D., Logerfo, A., Rocchi, A., Petrozzi, L., Nesti, C., Galetta, F., Santoro, G., Murri, L., and Siciliano, G. (2010). Oxidative stress biomarkers in mitochondrial myopathies, basally and after cysteine donor supplementation. J. Neurol. 257: 774–781, https://doi.org/10.1007/s00415-009-5409-7.Search in Google Scholar
Martinelli, D., Catteruccia, M., Piemonte, F., Pastore, A., Tozzi, G., Dionisi-Vici, C., Pontrelli, G., Corsetti, T., Livadiotti, S., Kheifets, V., et al. (2012). EPI-743 reverses the progression of the pediatric mitochondrial disease--genetically defined Leigh Syndrome. Mol. Genet. Metabol. 107: 383–388, https://doi.org/10.1016/j.ymgme.2012.09.007.Search in Google Scholar
Nabben, M., Schmitz, J.P.J., Ciapaite, J., leClercq, C.M.P., vanRiel, N.A., Haak, H.R., Nicolay, K., deCoo, I.F.M., Smeets, H., Praet, S.F., et al. (2017). Dietary nitrate does not reduce oxygen cost of exercise or improve muscle mitochondrial function in patients with mitochondrial myopathy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 312: R689–R701, https://doi.org/10.1152/ajpregu.00264.2016.Search in Google Scholar
Nancy, J.N. (2020). Efficacy Study of GS010 for the Treatment of Vision Loss up to 6 Months From Onset in LHON Due to the ND4 Mutation (RESCUE): clinicaltrials.gov, NCT02652767.Search in Google Scholar
Ohsawa, Y., Hagiwara, H., Nishimatsu, S., Hirakawa, A., Kamimura, N., Ohtsubo, H., Fukai, Y., Murakami, T., Koga, Y., Goto, Y., et al. (2019). Taurine supplementation for prevention of stroke-like episodes in MELAS: a multicentre, open-label, 52-week phase III trial. J. Neurol. Neurosurg. Psychiatry 90: 529–536, https://doi.org/10.1136/jnnp-2018-317964.Search in Google Scholar
Parikh, S., Goldstein, A., Koenig, M.K., Scaglia, F., Enns, G.M., Saneto, R., Anselm, I., Cohen, B.H., Falk, M.J., Greene, C., et al. (2015). Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet. Med. 17: 689–701, https://doi.org/10.1038/gim.2014.177.Search in Google Scholar
Pfanner, N., Warscheid, B., and Wiedemann, N. (2019). Mitochondrial proteins: from biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 20: 267–284, https://doi.org/10.1038/s41580-018-0092-0.Search in Google Scholar
Pfeffer, G., Majamaa, K., Turnbull, D.M., Thorburn, D., and Chinnery, P.F. (2012). Treatment for mitochondrial disorders. Cochrane Database Syst. Rev. CD004426. https://doi.org/10.1002/14651858.CD004426.pub3.Search in Google Scholar
Horvath, Rita, Gorman, Grainne, and Chinnery, Patrick F. (2008). How can we treat mitochondrial encephalomyopathies? Approaches to therapy. Neurotherapeutics 5: 558–568, https://doi.org/10.1016/j.nurt.2008.07.002.Search in Google Scholar
Rodan, L.H., Wells, G.D., Banks, L., Thompson, S., Schneiderman, J.E., and Tein, I. (2015). L-Arginine affects Aerobic capacity and muscle metabolism in MELAS (mitochondrial encephalomyopathy, lactic Acidosis and stroke-like episodes) syndrome. PloS One 10: e0127066, https://doi.org/10.1371/journal.pone.0127066.Search in Google Scholar
Schapira, A.H.V. (2006). Mitochondrial disease. Lancet 368: 70–82, https://doi.org/10.1016/s0140-6736(06)68970-8.Search in Google Scholar
Siciliano, G., Simoncini, C., Lo, G.A., Orsucci, D., Ricci, G., and Mancuso, M. (2012). Effects of aerobic training on exercise-related oxidative stress in mitochondrial myopathies. Neuromuscul. Disord. S172–S177. https://doi.org/10.1016/j.nmd.2012.10.005.Search in Google Scholar PubMed PubMed Central
Stacpoole, P.W., deGrauw, T.J., Feigenbaum, A.S., Hoppel, C., Kerr, D.S., McCandless, S.E., Miles, M.V., Robinson, B.H., and Tang, P.H. (2012). Design and implementation of the first randomized controlled trial of coenzyme CoQ₁₀ in children with primary mitochondrial diseases. Mitochondrion 12: 623–629, https://doi.org/10.1016/j.mito.2012.09.005.Search in Google Scholar PubMed PubMed Central
Stacpoole, P.W. (2011). Why are there no proven therapies for genetic mitochondrial diseases?. Mitochondrion 11: 679–685, https://doi.org/10.1016/j.mito.2011.05.002.Search in Google Scholar PubMed PubMed Central
Steele, H., Gomez-Duran, A., Pyle, A., Hopton, S., Newman, J., Stefanetti, R.J., Charman, S.J., Parikh, J.D., He, L., Viscomi, C., et al. (2020). Metabolic effects of bezafibrate in mitochondrial disease. EMBO Mol. Med. 12: e11589, https://doi.org/10.15252/emmm.201911589.Search in Google Scholar PubMed PubMed Central
Stephens, F.B., Constantin-Teodosiu, D., and Greenhaff, P.L. (2007). New insights concerning the role of carnitine in the regulation of fuel metabolism in skeletal muscle. J. Physiol. (Lond.). 581: 431–444, https://doi.org/10.1113/jphysiol.2006.125799.Search in Google Scholar PubMed PubMed Central
Stewart, J.B., and Chinnery, P.F. (2015). The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat. Rev. Genet. 16: 530–542, https://doi.org/10.1038/nrg3966.Search in Google Scholar PubMed
Szeto, H.H. (2014). First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 171: 2029–2050, https://doi.org/10.1111/bph.12461.Search in Google Scholar PubMed PubMed Central
Veenhuizen, Y., Cup, E.H.C., Jonker, M.A., Voet, N.B.M., Keulen, B.J., Maas, D.M., Heeren, A., Groothuis, J.T., Engelen, B.G.M., and Geurts, A.C.H. (2019). Self-management program improves participation in patients with neuromuscular disease: a randomized controlled trial. Neurology 93: e1720–e1731, https://doi.org/10.1212/wnl.0000000000008393.Search in Google Scholar
Voet, N.B.M., vanderKooi, E.L., Riphagen, I.I., Lindeman, E., vanEngelen, B.G.M., and Geurts, A.C.H. (2013). Strength training and aerobic exercise training for muscle disease. Cochrane Database Syst. Rev. 7: CD003907. https://doi.org/10.1080/02713683.2020.1736307.Search in Google Scholar PubMed
Wan, X., Pei, H., Zhao, M., Yang, S., Hu, W., He, H., Ma, S., Zhang, G., Dong, X., Chen, C., et al. (2016). Efficacy and safety of rAAV2-ND4 treatment for leber’s hereditary optic neuropathy. Sci. Rep. 6: 21587, https://doi.org/10.1038/srep21587.Search in Google Scholar PubMed PubMed Central
Yatsuga, S., Povalko, N., Nishioka, J., Katayama, K., Kakimoto, N., Matsuishi, T., Kakuma, T., and Koga, Y. (2012). MELAS: a nationwide prospective cohort study of 96 patients in Japan. Biochim. Biophys. Acta 1820: 619–624, https://doi.org/10.1016/j.bbagen.2011.03.015.Search in Google Scholar PubMed
Zesiewicz, T., Salemi, J.L., Perlman, S., Sullivan, K.L., Shaw, J.D., Huang, Y., Isaacs, C., Gooch, C., Lynch, D.R., and Klein, M.B. (2018). Double-blind, randomized and controlled trial of EPI-743 in Friedreich’s ataxia. Neurodegener. Dis. Manag. 8: 233–242, https://doi.org/10.2217/nmt-2018-0013.Search in Google Scholar PubMed
Zhang, Z., Zhao, D., Zhang, X., Xiong, H., Bao, X., Yuan, Y., and Wang, Z. (2018). Survival analysis of a cohort of Chinese patients with mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) based on clinical features. J. Neurol. Sci. 385: 151–155, https://doi.org/10.1016/j.jns.2017.12.033.Search in Google Scholar PubMed
Zhao, X., Zhang, Y., Lu, L., and Yang, H. (2020). Therapeutic effects of idebenone on leber hereditary optic neuropathy. Curr. Eye Res. 1–9. https://doi.org/10.1002/14651858.CD003907.pub4.Search in Google Scholar PubMed
Supplementary Material
Supplementary data to this article can be found online at https://doi.org/10.1515/revneuro-2020-0034.
© 2020 Tongling Liufu and Zhaoxia Wang, published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Frontmatter
- Circulating microparticles in patients after ischemic stroke: a systematic review and meta-analysis
- Efficacy and safety of endovascular treatment for patients with acute intracranial atherosclerosis–related posterior circulation stroke: a systematic review and meta-analysis
- Neuroprotection of hypoxic/ischemic preconditioning in neonatal brain with hypoxic-ischemic injury
- Treatment for mitochondrial diseases
- The role of melatonin and its analogues in epilepsy
- Gut microbiota on gender bias in autism spectrum disorder
- BDNF and nicotine dependence: associations and potential mechanisms
- Brain metabolic DNA: recent evidence for a mitochondrial connection
- Channels to consciousness: a possible role of gap junctions in consciousness
Articles in the same Issue
- Frontmatter
- Circulating microparticles in patients after ischemic stroke: a systematic review and meta-analysis
- Efficacy and safety of endovascular treatment for patients with acute intracranial atherosclerosis–related posterior circulation stroke: a systematic review and meta-analysis
- Neuroprotection of hypoxic/ischemic preconditioning in neonatal brain with hypoxic-ischemic injury
- Treatment for mitochondrial diseases
- The role of melatonin and its analogues in epilepsy
- Gut microbiota on gender bias in autism spectrum disorder
- BDNF and nicotine dependence: associations and potential mechanisms
- Brain metabolic DNA: recent evidence for a mitochondrial connection
- Channels to consciousness: a possible role of gap junctions in consciousness