Thermodynamic and kinetic insights into B10H14 and B10H14 2−
-
Gerardo Hernández-Juárez

 and
Gabriel Merino
and
Gabriel Merino

Abstract
The dianion [B10H14]2− has attracted renewed attention due to its structural characteristics and a mercury-free synthesis route. Although its arachno structure has been reported, we explored the potential energy surface of B10H14 2− to identify more stable species. Subsequent analyses included bonding evaluation, kinetic studies, and Born-Oppenheimer molecular dynamics (BO-MD) simulations. Calculations indicate that the closo-[B10H10]2−⋯2H2 complex is 21.5 kcal mol−1 more stable than the arachno structure. The complex is stable below 110 K but dissociates into closo-[B10H10]2− and H2 at higher temperatures. The arachno isomer remains kinetically stable because a 30 kcal/mol activation barrier prevents its conversion. BO-MD simulations corroborate this kinetic stability, as the arachno framework is maintained during the trajectories. The effects of the solvent and the counterions were also examined and found to have a negligible influence on the relative energetics.
Introduction
Decaborane (B10H14) is a colorless crystalline boron hydride whose synthesis in the early 20th century marked a milestone in boron chemistry. 1 It is widely used as a precursor for various borohydrides, including the decaborate dianion, [B10H10]2−, 2 , 3 the dodecaborate dianion [B12H12]2−, 4 , 5 and the tetradecahydrodecaborate dianion [B10H14]2−. 6 , 7 , 8 , 9 Its corresponding dianion [B10H14]2− was first prepared in 1958 via the reaction of sodium with B10H14 in ammonia. 10 Subsequent attempts to obtain its alkali salts (e.g., Na2B10H14 and K2B10H14) 11 , 12 encountered low yields and byproduct formation, particularly when using sodium amalgam or KBH4. 6 In 2023, Chen and co-workers 9 reported a mercury-free, high-yield (83 %) synthesis of M2B10H14 (M = Na and K) using alkali metal amidoboranes (MNH2BH3) in tetrahydrofuran at room temperature. This approach avoids reactive sodium metal and provides a safer, more practical route. 6 , 10 , 11 , 12 This improved accessibility and purity of these compounds has renewed interest in their fundamental properties and potential applications.
The structures of B10H14 and [B10H14]2− were elucidated by Harker 13 and Lipscomb, 7 , 14 through X-ray diffraction and later supported by ab initio SCF-MO calculations. 15 According to Wade-Mingos rules, these compounds are classified as nido- and arachno-borohydrides, respectively. 16 , 17 , 18 Both adopt C 2v symmetry and share an incomplete octadecahedral boron skeleton, differing mainly in hydrogen arrangements (Fig. 1). nido-B10H14 contains ten BH units and four B–H–B bridges, whereas arachno-[B10H14]2− has eight BH units, two BH2 groups, and two bridging hydrogens. In principle, adding two electrons to nido-B10H14 induces minor structural changes, 19 breaking two B–H–B bridges to form two BH2 units while retaining C 2v symmetry (Fig. 1).
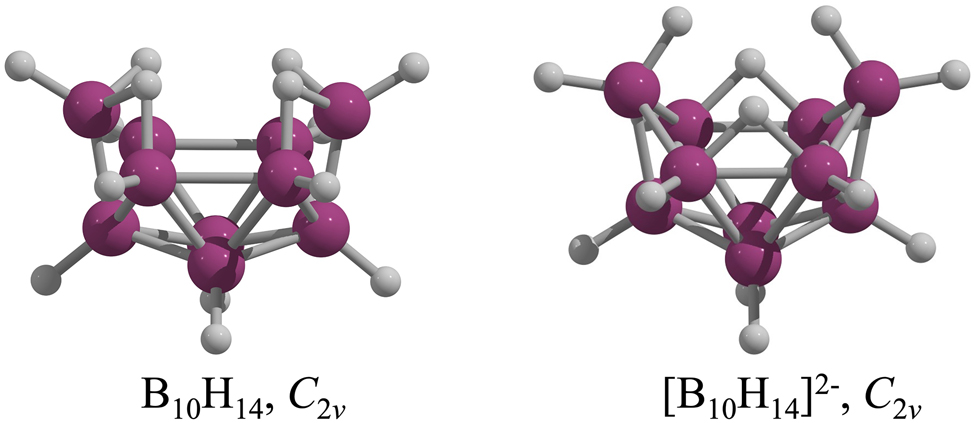
Structures of neutral (left) and dianionic (right) B10H14.
Our recent studies indicate that some boranes deviate from their predicted structures upon electron removal. 20 , 21 For instance, [B12H12]2−, an icosahedron in its anionic form, adopts a structure with ten B–H bonds and one B–H2 bond after two-electron removal. 20 Similarly, [B5H5]2− has a trigonal bipyramidal form with D 3h symmetry, whereas its neutral counterpart, B5H5, forms an asymmetric 3D structure rather than a planar or cage-like arrangement. 21 These findings raise the question of whether the arachno structure represents the global minimum for B10H14 2−. Our systematic exploration shows that it does not; the global minimum corresponds to a complex formed by a closo-[B10H10]2− fragment interacting with two hydrogen molecules. The arachno structure lies approximately 21 kcal mol−1 higher in energy but persists as a kinetically stable local minimum. This behavior contrasts with [B12H12]2−, where the deltahedral cage is the global minimum, and underscores the stability of B10H14 2−, motivating the present study.
Computational details
The potential energy surface (PES) of B10H14 2− was explored using a genetic algorithm implemented in GLOMOS, 22 as described elsewhere. 23 Initial screening was performed at the PBE0/def2-SVP level, 24 , 25 considering both singlet and triplet spin states. Identified structures were reoptimized and fully characterized at the TPSS/aug-cc-pVTZ 26 , 27 level, including Grimme’s D3 dispersion correction. 28 A quadratically convergent SCF procedure (XQC) was used to improve the convergence process. Single-point energies were calculated on TPSS geometries using CCSD(T) 29 method with the aug-cc-pVTZ basis set. Relative Gibbs free energies were calculated at the CCSD(T)/def2-TZVP//TPSS-D3/def2-TZVP level, including entropic and thermal corrections derived from the TPSS-D3/aug-cc-pVTZ level at 298.15 K in the gas phase. Solvent effects of tetrahydrofuran were assessed using the SMD model 30 , 31 at the TPSS-D3/aug-cc-pVTZ level. Conversion mechanisms between arachno-[B10H14]2− and the complex [B10H10]2−⋯2H2 were evaluated using the intrinsic reaction coordinate (IRC) 32 method. All calculations were performed with Gaussian 16. 33
Bonding was analyzed at the TPSS-D3/aug-cc-pVTZ level using the natural bond orbital (NBO) method in NBO 6.0, 34 and the adaptive natural density partitioning (AdNDP) 35 approach implemented in Multiwfn. 36 AdNDP describes bonding through n-center-two-electron (nc-2e) bonds, recovering both classical Lewis concepts (e.g., lone pairs, 2c-2e bonds) and delocalized bonds. Non-covalent interactions (NCI) of the global minimum were analyzed by generating the reduced density gradient (RDG) isosurfaces (s = 0.5 au) with NCIPLOT. 37 , 38 The isosurfaces were colored according to sign(λ 2)ρ in the range −0.5 au < sign(λ 2)ρ < +0.5 au, highlighting strong interactions in blue, weak interactions in green, and repulsions in red.
The dynamic behavior of B10H14 2− was investigated via Born-Oppenheimer Molecular Dynamics (BO-MD) 39 simulations at 1200 K for 30 ps with 1 fs time step, using a Nosé–Hoover chain thermostat for temperature control. 40 , 41 , 42 This temperature provides sufficient kinetic energy to overcome barriers, enabling efficient interconversions of structures within the simulation time. BO-MD computations were conducted with deMon-2k 43 at the PBE0/DZVP level. 44
Results and discussion
The global minimum (1) is a singlet state with C s symmetry, corresponding to a closo-[B10H10]2− bicapped-square antiprism with two H2 molecules oriented toward adjacent triangular faces of the boron skeleton (Fig. 2). The next lowest-energy structure, 2, lies 21.5 kcal mol−1 higher and corresponds to the arachno-[B10H14]2− dianion with C 2v symmetry reported by Chen and coworkers (Fig. 1). 9 Structures 3, 4, and 5 involve a dissociated hydrogen molecule from the borohydride cluster. Specifically, 3 is a closo-[B9H9]2− cluster with an attached BH3 fragment, while 4 and 5 adopt nido-[B10H12]2− framework, with relative Gibbs free energy ranges from 32 and 39 kcal mol−1 above 1. Structure 6 (C s symmetry) resembles 2, containing ten BH units, two BH2 units, and two B–H–B bridges, but with different bridge arrangements, resulting in a ΔG 2-6 of 17.9 kcal mol−1. In 2, the bridges are opposite each other and distant from the BH2 units, whereas in 6, one bridge connects a BH2 unit to an adjacent boron atom (B–H–BH2). Structure 7 is an irregular half-open cage with a relative energy of 52.8 kcal mol−1 above 1, and 8, a butterfly-like half-open cage linked by an HB–BH fragment, is 55.7 kcal mol−1 higher. Finally, 9 is a C 2v half-open cage that binds a hydrogen molecule, with a relative energy of 58.5 kcal mol−1.
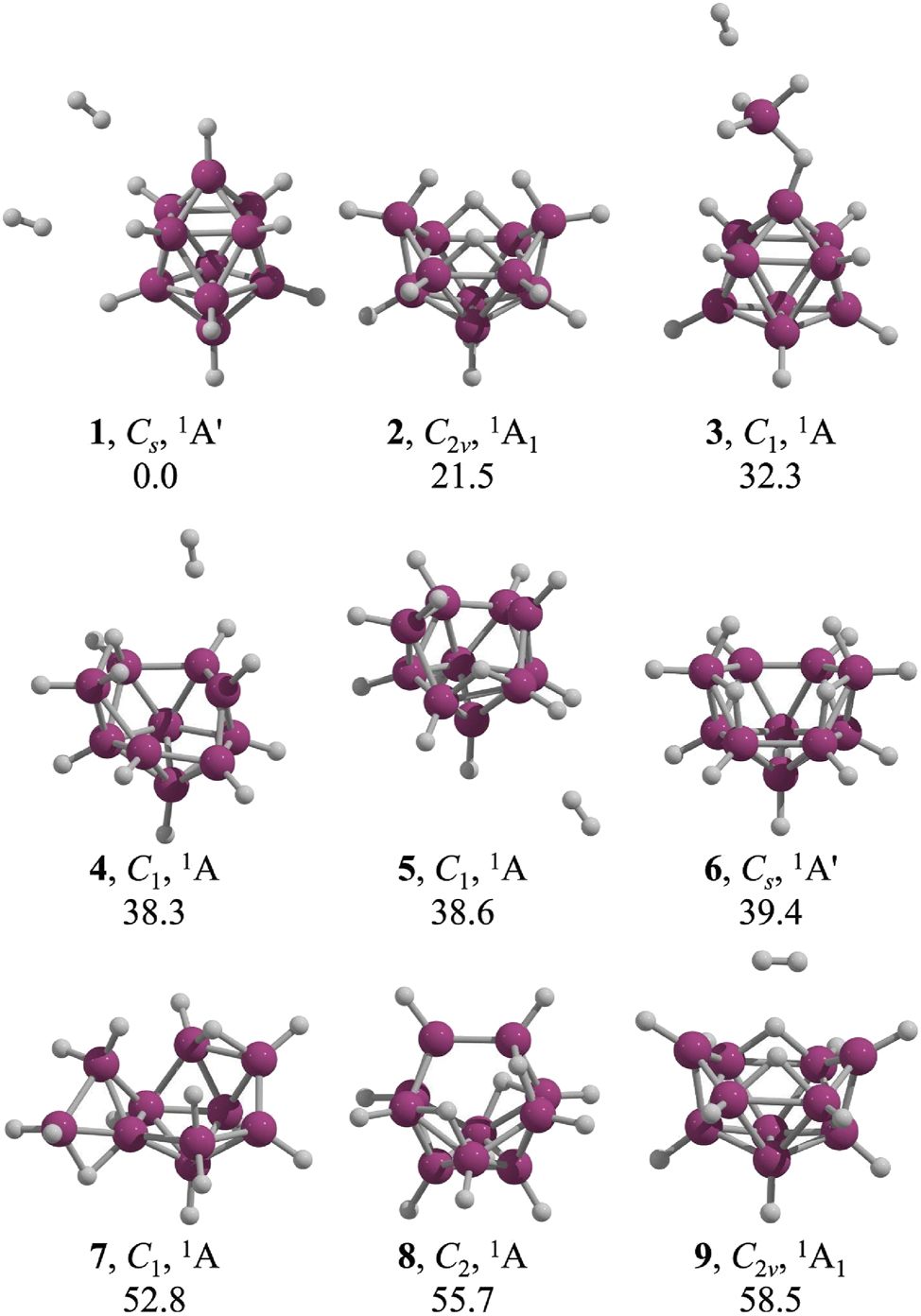
Low-lying local minima of B10H14 2−. Relative Gibbs free energies (kcal mol−1) were computed at the CCSD(T)/aug-cc-pVTZ//TPSS-D3/aug-cc-pVTZ level at 298.15 K. All structures were confirmed as local minima by frequency analysis.
Comparison of the geometric parameters of 2, 1, and nido-B10H14 shows B–H distances of 1.18–1.20 Å (Fig. 3). The B–H–B bridges in nido-B10H14 and 2 have comparable bond lengths (around 1.34 Å). In nido-B10H14, the four boron atoms (B1 to B4) forming the closed cap have B–B bond lengths of 1.79 Å, which shorten by 0.04 Å in 2. A similar trend is found at the open side (B5 to B10). In nido-B10H14, the B5–B10 distance is 1.98 Å, 0.09 Å longer than in 2. In contrast, bonds B5–B6, B6–B7, B8–B9, and B9–B10 are 0.11 Å longer in 2 than in nido-B10H14. In structure 1, B–B lengths are slightly shorter than in nido-B10H14 and 2 forms. The closo-[B10H10]2− fragment in 1 consists of two four-membered rings at the equatorial position and two boron atoms at the apical position. Bond lengths between equatorial boron atoms (1.84 Å) or between the four-membered rings (1.82 Å) are longer than those between apical and equatorial boron atoms (1.71 Å), indicating compaction of the boron framework. Notably, interaction with the hydrogen molecules does not alter the [B10H10]2− framework in 1.
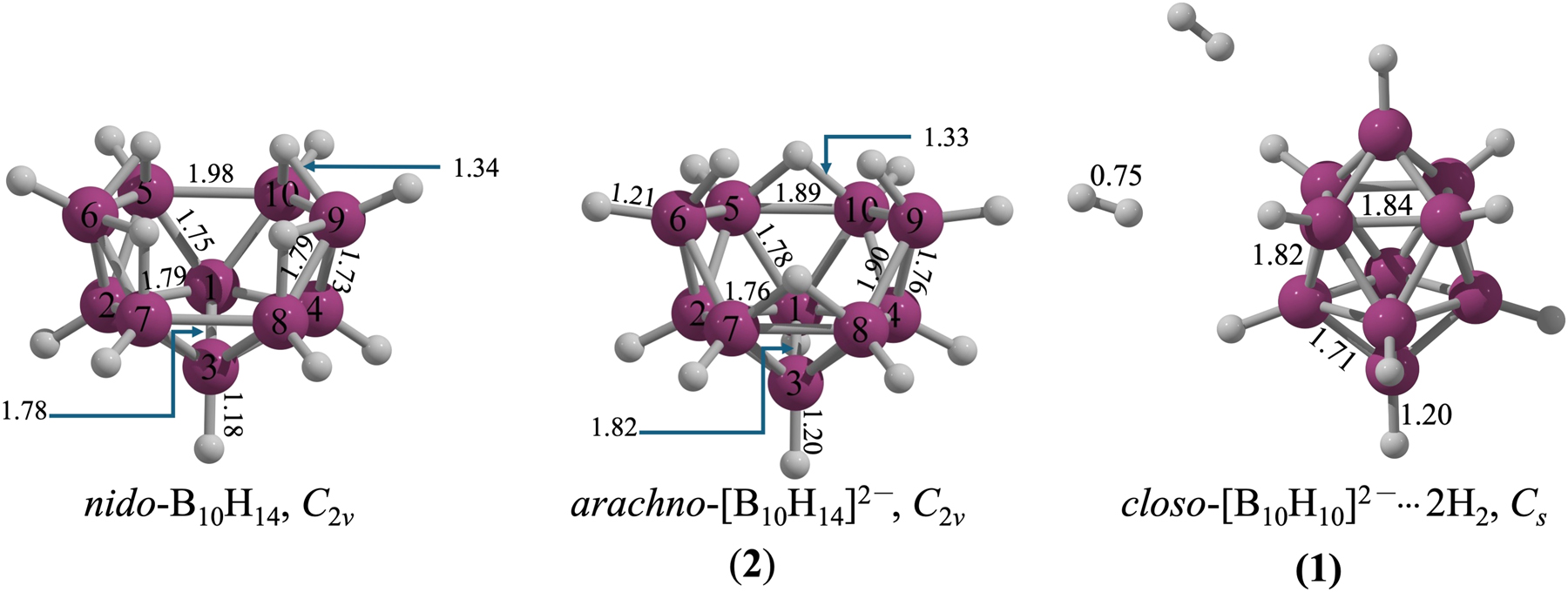
Structural comparison among B10H14, 2, and 1 structures.
To evaluate the thermal stability of complex 1, we computed its Gibbs free energies and its dissociated products, relative to the intact complex, from 0 to 400 K (Fig. 4a) in the gas phase using the Eyringpy software, 45 , 46 at the TPSS-D3/aug-cc-pVTZ level. At 0 K, 1 is more stable than both [B10H10]2−⋯H2 + H2 and [B10H10]2− + 2H2 by 0.8 and 2.0 kcal mol−1, respectively. Above 110 K, 1 dissociates into [B10H10]2− and 2H2. At room temperature, dissociation into [B10H10]2−⋯H2 + H2 is exergonic (ΔG = 4.2 kcal mol−1), and [B10H10]2−⋯H2 further dissociates into [B10H10]2− + H2 with ΔG = 2.5 kcal mol−1. So, these results indicate that 1 is stable only at low temperatures.
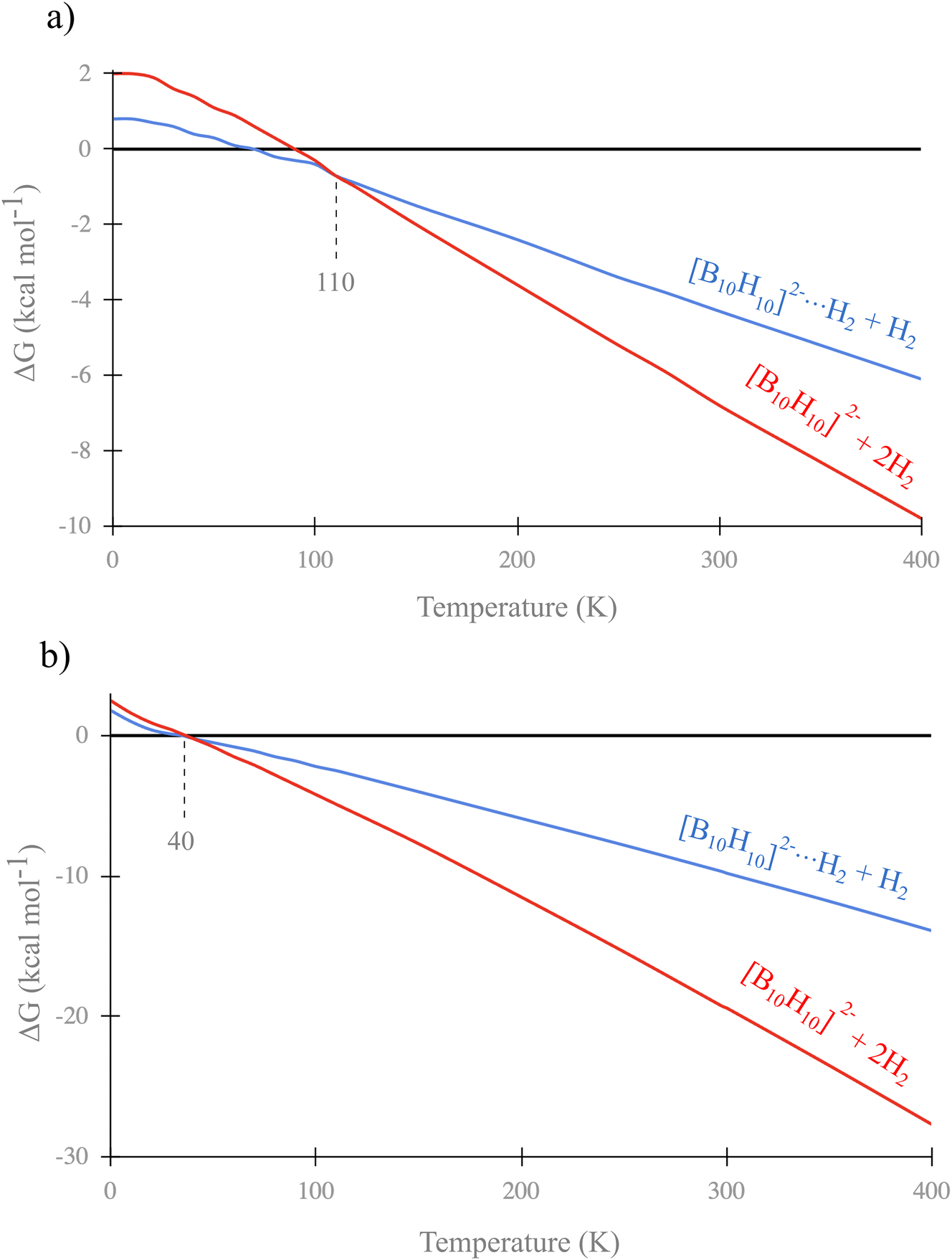
Stability of 1 at different temperatures a) in the gas phase and b) in solution.
In both phases, structure 1 is the global minimum at the DFT level (see Figure S1). Structure 2 lies 20.5 kcal mol−1 above 1 in the gas phase and 24.7 kcal mol−1 in solution, showing that solvent effects slightly influence the relative Gibbs energies. However, the dissociation differs between phases. In solution, 1 begins to dissociate into [B10H10]2−⋯H2 + H2 and [B10H10]2− + 2H2 at 40 K (Fig. 4b), approximately 70 K lower than in the gas phase (T < 110 K). Therefore, 1 is viable in solution only at very low temperatures, dissociating as the temperature increases.
To analyze the bonding, an AdNDP study was performed for nido-B10H14, 2 and 1 (Fig. 5). nido-B10H14 contains ten 2c-2e σ-bonds, whereas 2 and 1 have twelve, two of which in 1 correspond to H2 molecules. nido-B10H14 also shows six delocalized 3c-2e bonds (four in a B2H ring and two in a B3 ring) and six 5c-2e bonds that stabilize the boron skeleton; these are absent in 2 and 1. In contrast, 2 retains only two 3c-2e bonds in a B2H ring, while the closo-[B10H10]2− framework in 1 lacks 3c-2e bonding altogether. The stability of 2 arises from nine 4c-2e bonds distributed over B4 units, while the boron moiety in 1 is stabilized by 11 4c-2e bonds delocalized throughout the boron cage. So, the number of electron pairs in the [B10H10]2− fragment of 1 stabilizes the closo structure in accordance with Wade’s rules for n = 10. 16 Moreover, the NCI analysis of 1 reveals non-covalent interactions not only between the B10H10 framework and the H2 molecules, but also between the two H2 molecules (Figure S2). These non-covalent interactions contribute to the stabilization of 1 at low temperatures.
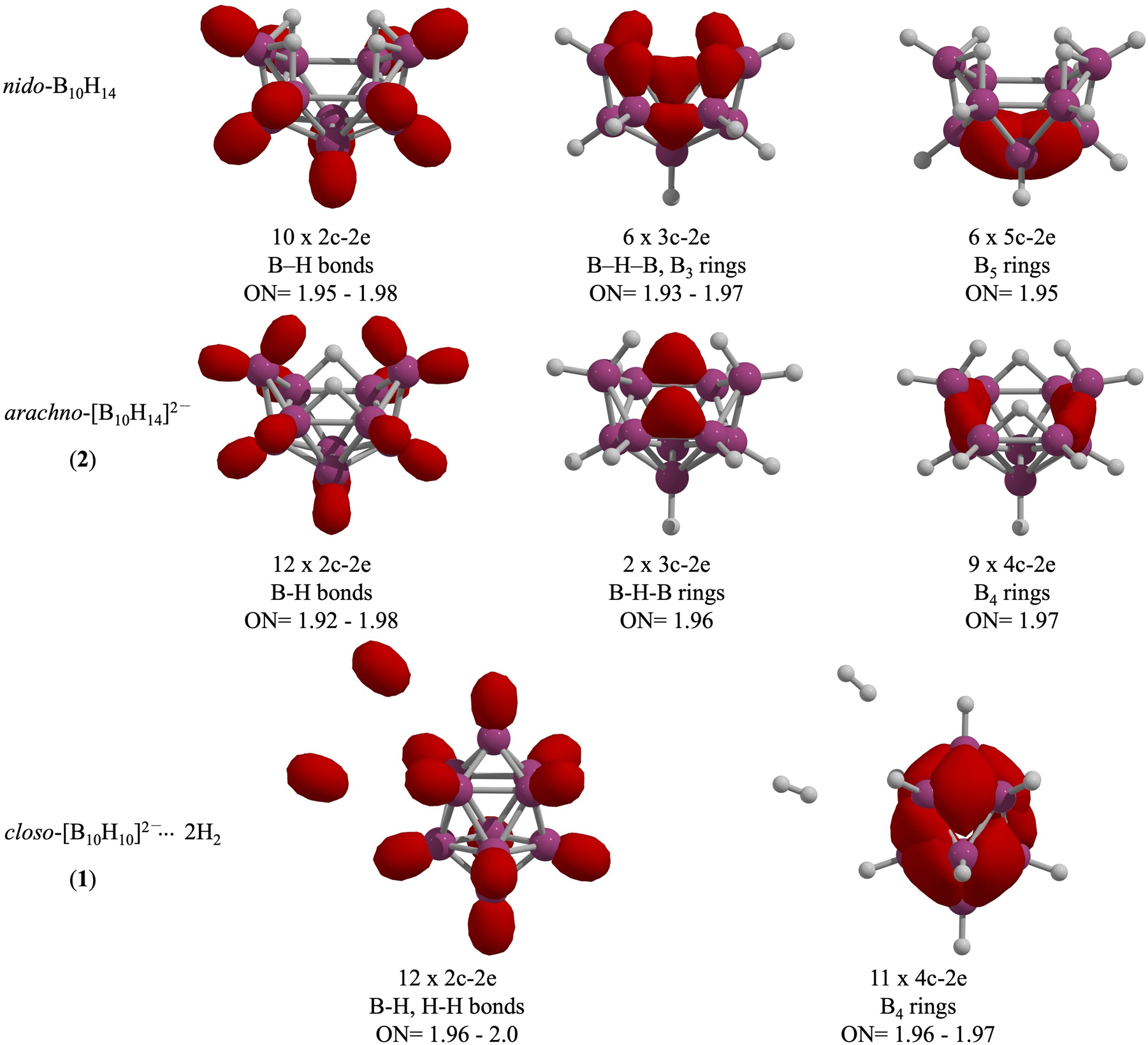
AdNDP analysis for B10H14, 2, and 1. Occupation numbers (ONs) are given in |e|.
The thermodynamic analysis indicates that the bonding in 1 enhances delocalization, contributing to its lower energy. However, the isolation of 2 by Chen et al. shows that it is kinetically viable. Transformation of 2 into 1 requires detachment of two H2 molecules and closure of the boron skeleton to form a closo-B10H10 moiety.
Two pathways for this conversion were explored. The first, involving H2 removal from BH2 units (Fig. 6a), has a high activation barrier (ΔG ⧧ = 65.7 kcal mol−1) leading to 9, and the formation of 9 is highly endergonic (ΔG = 58.5 kcal mol−1), rendering this pathway unfeasible. The second pathway involves stepwise hydrogen elimination (Fig. 6b). Initially, 2 converts to 6 via TS 2-6 with a moderate barrier (ΔG ⧧ = 19.9 kcal mol−1), during which hydrogen atoms rearrange into B–H–B bridges and migrate toward BH2 groups to form B–H–BH2 units. Next, two hydrogen atoms are released as H2 through TS 6-4 (ΔG ⧧ = 22.8 kcal mol−1). Finally, another H2 molecule is eliminated, promoting closure of the boron skeleton in 4 to yield 1 via TS 4-1 (ΔG ⧧ = 31.3 kcal mol−1). The overall formation of [B10H10]2− and two H2 molecules is exergonic (ΔG rxn = −6.7 kcal mol−1), indicating spontaneous dissociation of 1. Although the first hydrogen detachment via TS 6-4 is feasible, the second is hindered by its high barrier, preventing 2 from converting into 1 or dissociated into [B10H10]2− and molecular hydrogen.
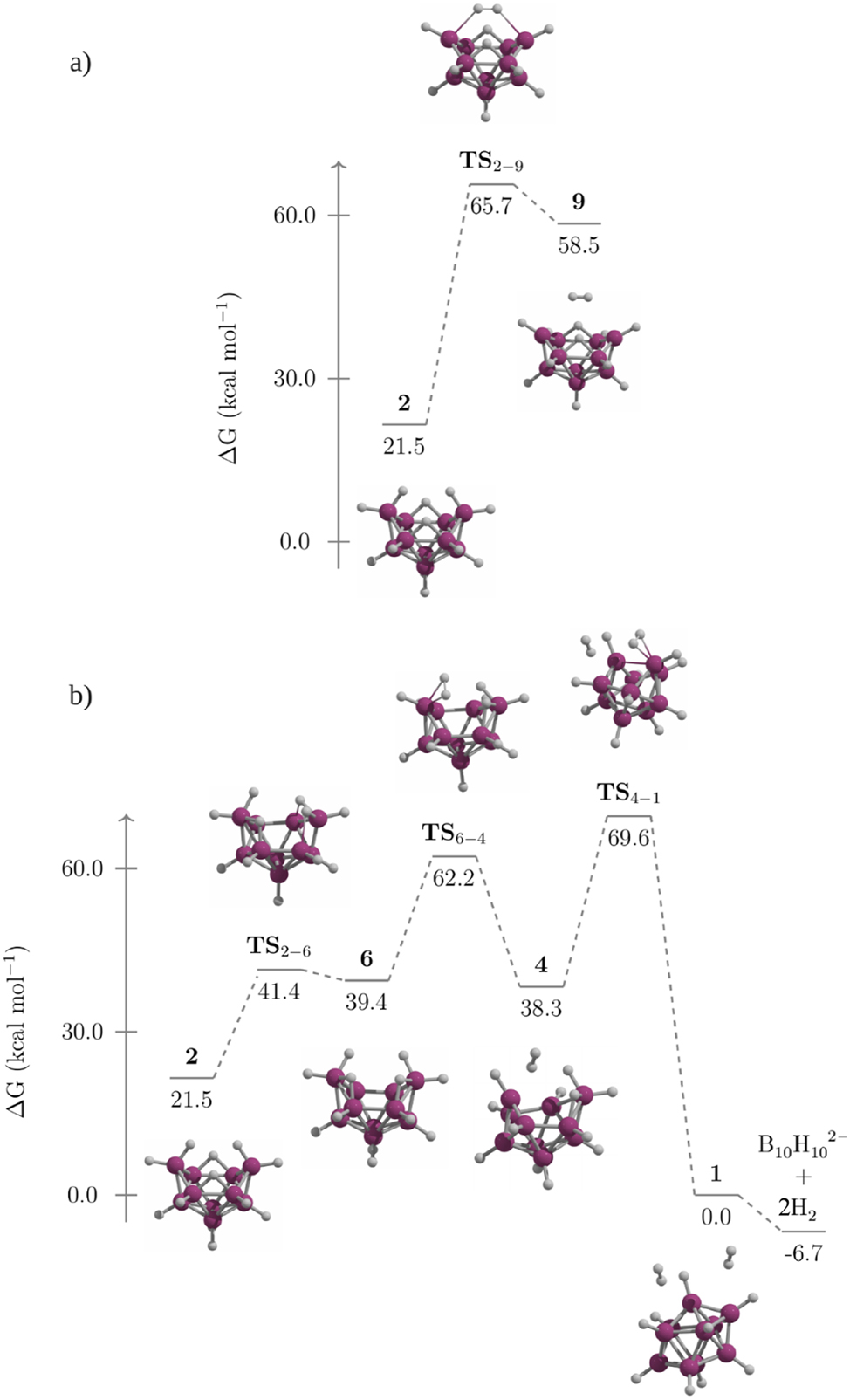
Gibbs energy profile for the conversion of 2 to 1.
Born-Oppenheimer molecular dynamics simulations at 1200 K for 30 ps confirm the kinetic stability of 2, which undergoes only minor distortions while maintaining its arachno structure (see Movie S1 in the ESI). Solvent effects on the barriers are minimal at room temperature, with relative energy variations of only 0.7–3.7 kcal mol−1 (Figure S3).
Finally, to evaluate the effect of counterions, we performed structural optimizations of dianionic isomers 1 and 2 in the presence of Na+ at the TPSS-D3/aug-cc-pVTZ level (Figure S4). The Na2B10H10⋯2H2 structure is 25.5 kcal mol−1 more stable than the Na2B10H14 structure, indicating that the addition of counter-ions increases the relative energy difference between 1 and 2 by 4.0 kcal mol−1 (see Figure S1). Importantly, the Na2B10H10⋯2H2 structure remains energetically more stable than the Na2B10H14 form. Structurally, the B–B and B–H bond distances in both dianionic forms interacting with sodium counter-ions change only slightly, by 0.01–0.04 Å.
Conclusions
This study shows that arachno-[B10H14]2−, although kinetically stable, is not the global minimum. The [B10H10]2−⋯2H2 complex is more stable by approximately 21.5 kcal mol−1. Conversion pathways indicate that the arachno form is stabilized by a high activation barrier (ΔG ⧧ = 31.3 kcal mol−1), preventing its transformation to the [B10H10]2−⋯2H2 complex under standard conditions. Molecular dynamics simulations confirm the kinetic stability of the arachno form, with minimal distortions over time. Our calculations show that the global minimum remains stable at low temperatures (T < 110 K) but dissociates into [B10H10]2−⋯H2 + H2 and closo-[B10H10]2− + 2H2 at high temperatures, suggesting that closo-[B10H10]2− can adsorb two hydrogen molecules under cold conditions. Solvent effects are minor, although they reduce the dissociation temperature of the global minimum to T < 40 K relative to the gas phase. The effects of counterions induce negligible structural and energetic changes. So, the addition of two electrons to neutral B10H14 significantly alters the structure of the global minimum. These results provide insight into borohydride chemistry, particularly regarding the design and stabilization of borane structures.
Funding source: National Agency for Research and Development
Award Identifier / Grant number: FONDECYT project 1221019
Acknowledgments
G. H.-J. and F. M. thank Secihti for their PhD and postdoc fellowships, respectively. AVE thanks the support of the National Agency for Research and Development (ANID) through FONDECYT project 1221019. Authors also acknowledge research support from Clemson University with allotment of computation time on the Palmetto cluster (this material is based on work supported by the National Science Foundation under Grant Nos. MRI# 2024205, MRI# 1725573, and CRI# 2010270). The work in México was supported by Cinvestav.
-
Research ethics: Not applicable.
-
Informed consent: All authors have consented to participate in the study and to the publication of the manuscript.
-
Author contributions: All authors have accepted responsibility for the entire content of this manuscript and approved its submission.
-
Use of Large Language Models, AI and Machine Learning Tools: Improve language.
-
Conflict of interest: The authors state no conflict of interest.
-
Research funding: National Agency for Research and Development (ANID) through FONDECYT project 1221019.
-
Data availability: Not applicable.
References
1. Stock, A.; Pohland, E.; Borwasserstoffe, X. I. I. Zur Kenntnis des B10H14. Ber. Dtsch. Chem. Ges. 1929, 62, 90–99.10.1002/cber.19290620111Search in Google Scholar
2. Sivaev, I. B.; Prikaznov, A. V.; Naoufal, D. Fifty Years of the Closo-Decaborate Anion Chemistry. Collect. Czechoslov. Chem. Commun. 2010, 75, 1149–1199; https://doi.org/10.1135/cccc2010054.Search in Google Scholar
3. Mahfouz, N.; Ghaida, F. A.; El Hajj, Z.; Diab, M.; Floquet, S.; Mehdi, A.; Naoufal, D. Recent Achievements on Functionalization within Closo-Decahydrodecaborate [B10H10]2− Clusters. ChemistrySelect 2022, 7, e202200770; https://doi.org/10.1002/slct.202200770.Search in Google Scholar
4. Sivaev, I. B.; Bregadze, V. I.; Sjöberg, S. Chemistry of Closo-Dodecaborate Anion [B12H12]2−: A Review. Collect. Czechoslov. Chem. Commun. 2002, 67, 679–727.10.1135/cccc20020679Search in Google Scholar
5. Zhao, X.; Yang, Z.; Chen, H.; Wang, Z.; Zhou, X.; Zhang, H. Progress in Three-Dimensional Aromatic-like Closo-Dodecaborate. Coord. Chem. Rev. 2021, 444, 214042; https://doi.org/10.1016/j.ccr.2021.214042.Search in Google Scholar
6. Muetterties, E. L. Chemistry of Boranes. VI. Preparation and Structure of B10H14–. Inorg. Chem. 1963, 2, 647–648; https://doi.org/10.1021/ic50007a061.Search in Google Scholar
7. Kendall, D. S.; Lipscomb, W. N. Crystal Structure of Tetramethyl Ammonium Tetradecahydrodecaborate. Structure of the B10H142– Ion. Inorg. Chem. 1973, 12, 546–551; https://doi.org/10.1021/ic50121a010.Search in Google Scholar
8. McKee, M. L. Deconvoluting the Reaction Path from B10H14 Plus BH4− to B12H122−. Can Theory Make a Contribution? In Boron: The Fifth Element; Hnyk, D.; McKee, M., Eds.; Springer: Cham, 2015; pp. 121–138.10.1007/978-3-319-22282-0_5Search in Google Scholar
9. Chen, X.-M.; Yu, X.-C.; Chi, J.-X.; Jing, Y.; Wang, H.; Zhang, N.; Zhang, C.; Ge, Y.-W.; Chen, X. An Improved Method for the Synthesis and Formation Mechanism of MBH Based on the Reactions of BH with MNHBH (M = Na, K). Dalton Trans. 2023, 52, 17684–17688; https://doi.org/10.1039/d3dt03403h.Search in Google Scholar PubMed
10. Toeniskoetter, R. H.; Schaeffer, G. W.; Evers, E. C.; Hughes, R. E.; Bagley, G. E. In Abstracts of Papers; 134th National Meeting of the American Chemical Society: Chicago, Vol. I11, 1958; p. 23N.Search in Google Scholar
11. Hough, W. V.; Edwards, L. J. In Abstracts of Papers; 133rd National Meeting of the American Chemical Society: San Francisco, Calif., 1958; p. 28L.Search in Google Scholar
12. Rupp, E. B.; Smith, D. E.; Shriver, D. F. Mechanisms of Electrolytic Reduction for decaborane(14), B10H14, in an Aprotic Solvent. I. First Reduction Step. J. Am. Chem. Soc. 1967, 89, 5562–5567; https://doi.org/10.1021/ja00998a010.Search in Google Scholar
13. Kasper, J. S.; Lucht, C. M.; Harker, D. The Crystal Structure of Decaborane, B10H14. Acta Crystallogr. 1950, 3, 436–455; https://doi.org/10.1107/s0365110x50001270.Search in Google Scholar
14. Lipscomb, W. N.; Wiersema, R. J.; Hawthorne, M. F. Structural Ambiguity of the B10H142– Ion. Inorg. Chem. 1972, 11, 651–652; https://doi.org/10.1021/ic50109a052.Search in Google Scholar
15. Guest, M. F.; Hillier, I. H. Electronic Structure of Boron Hydrides. Ab Initio Study of B10H14, B10H142–, and B10H122–. J. Chem. Soc., Faraday Trans. 2 1974, 70, 2004.10.1039/f29747002004Search in Google Scholar
16. Wade, K. The Structural Significance of the Number of Skeletal Bonding Electron-Pairs in Carboranes, the Higher Boranes and Borane Anions, and Various Transition-Metal Carbonyl Cluster Compounds. J. Chem. Soc. Dalton Trans. 1971, 15, 792–793; https://doi.org/10.1039/c29710000792.Search in Google Scholar
17. Mingos, D. M. P. A General Theory for Cluster and Ring Compounds of the Main Group and Transition Elements. Nat. Phys. Sci. 1972, 236, 99–102; https://doi.org/10.1038/physci236099a0.Search in Google Scholar
18. Mingos, D. M. P. Polyhedral Skeletal Electron Pair Approach. Acc. Chem. Res. 1984, 17, 311–319; https://doi.org/10.1021/ar00105a003.Search in Google Scholar
19. McKee, M. L.; Wang, Z.-X.; Schleyer, P. v. R. Ab Initio Study of the Hypercloso Boron Hydrides BnHn and BnHn–. Exceptional Stability of Neutral B13H13. J. Am. Chem. Soc. 2000, 122, 4781–4793; https://doi.org/10.1021/ja994490a.Search in Google Scholar
20. Hernández-Juárez, G.; Vásquez-Espinal, A.; Murillo, F.; Quintal, A.; Ortíz-Chi, F.; Zarate, X.; Barroso, J.; Merino, G. Unveiling the Electronic and Structural Consequences of Removing Two Electrons from B12H122–. Dalton Trans. 2023, 52, 17398–17406; https://doi.org/10.1039/d3dt02652c.Search in Google Scholar PubMed
21. Hernández-Juárez, G.; Barroso, J.; Vásquez-Espinal, A.; Ortíz-Chi, F.; Tiznado, W.; Murillo, F.; Merino, G. Breaking the Plane: B5H5 is a Three-Dimensional Structure. Phys. Chem. Chem. Phys. 2024, 26, 8089–8093; https://doi.org/10.1039/d4cp00029c.Search in Google Scholar PubMed
22. Ortiz-Chi, F.; Merino, G. GLOMOS: Mérida, México, 2020.Search in Google Scholar
23. Grande-Aztatzi, R.; Martínez-Alanis, P. R.; Cabellos, J. L.; Osorio, E.; Martínez, A.; Merino, G. Structural Evolution of Small Gold Clusters Doped by One and Two Boron Atoms. J. Comput. Chem. 2014, 35, 2288–2296; https://doi.org/10.1002/jcc.23748.Search in Google Scholar PubMed
24. Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170; https://doi.org/10.1063/1.478522.Search in Google Scholar
25. Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305; https://doi.org/10.1039/b508541a.Search in Google Scholar PubMed
26. Tao, J.; Perdew, J. P.; Staroverov, V. N.; Scuseria, G. E. Climbing the Density Functional Ladder: Nonempirical Meta-Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401; https://doi.org/10.1103/physrevlett.91.146401.Search in Google Scholar
27. Dunning, T. H.Jr Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023; https://doi.org/10.1063/1.456153.Search in Google Scholar
28. Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104; https://doi.org/10.1063/1.3382344.Search in Google Scholar PubMed
29. Watts, J. D.; Gauss, J.; Bartlett, R. J. Coupled-Cluster Methods with Noniterative Triple Excitations for Restricted Open-Shell Hartree–Fock and Other General Single Determinant Reference Functions. Energies and Analytical Gradients. J. Chem. Phys. 1993, 98, 8718–8733; https://doi.org/10.1063/1.464480.Search in Google Scholar
30. Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396; https://doi.org/10.1021/jp810292n.Search in Google Scholar PubMed
31. Ribeiro, R. F.; Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. Prediction of SAMPL2 Aqueous Solvation Free Energies and Tautomeric Ratios Using the SM8, SM8AD, and SMD Solvation Models. J. Comput. Aided Mol. Des. 2010, 24, 317–333; https://doi.org/10.1007/s10822-010-9333-9.Search in Google Scholar PubMed
32. Fukui, K. The Path of Chemical Reactions – The IRC Approach. Acc. Chem. Res. 1981, 14, 363–368; https://doi.org/10.1021/ar00072a001.Search in Google Scholar
33. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A. V.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery, J. A.Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M. J.; Heyd, J. J.; Brothers, E. N.; Kudin, K. N.; Staroverov, V. N.; Keith, T. A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A. P.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Fox, D. J. Gaussian 16; Gaussian, Inc.: Wallingford CT, 2016.Search in Google Scholar
34. Glendening, E. D.; Landis, C. R.; Weinhold, F. NBO 6.0: Natural Bond Orbital Analysis Program. J. Comput. Chem. 2013, 34, 1429–1437; https://doi.org/10.1002/jcc.23266.Search in Google Scholar PubMed
35. Zubarev, D. Y.; Boldyrev, A. I. Developing Paradigms of Chemical Bonding: Adaptive Natural Density Partitioning. Phys. Chem. Chem. Phys. 2008, 10, 5207–5217; https://doi.org/10.1039/b804083d.Search in Google Scholar PubMed
36. Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592; https://doi.org/10.1002/jcc.22885.Search in Google Scholar PubMed
37. Johnson, E. R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A. J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506; https://doi.org/10.1021/ja100936w.Search in Google Scholar PubMed PubMed Central
38. Contreras-García, J.; Johnson, E. R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D. N.; Yang, W. NCIPLOT: A Program for Plotting Non-Covalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632; https://doi.org/10.1021/ct100641a.Search in Google Scholar PubMed PubMed Central
39. Millam, J. M.; Bakken, V.; Chen, W.; Hase, W. L.; Schlegel, H. B. Ab Initio Classical Trajectories on the Born–Oppenheimer Surface: Hessian-Based Integrators Using Fifth-Order Polynomial and Rational Function Fits. J. Chem. Phys. 1999, 111, 3800–3805; https://doi.org/10.1063/1.480037.Search in Google Scholar
40. Nosé, S. A Unified Formulation of the Constant Temperature Molecular Dynamics Methods. J. Chem. Phys. 1984, 81, 511–519; https://doi.org/10.1063/1.447334.Search in Google Scholar
41. Martyna, G. J.; Klein, M. L.; Tuckerman, M. Nosé–Hoover Chains: The Canonical Ensemble via Continuous Dynamics. J. Chem. Phys. 1992, 97, 2635–2643; https://doi.org/10.1063/1.463940.Search in Google Scholar
42. Hoover, W. G. Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A 1985, 31, 1695; https://doi.org/10.1103/physreva.31.1695.Search in Google Scholar PubMed
43. Koster, A. M.; Geudtner, G.; Alvarez-Ibarra, A.; Calaminici, P.; Casida, M. E.; Carmona-Espindola, J.; Dominguez, V. D.; Flores-Moreno, R.; Gamboa, G. U.; Goursot, A.; Heine, T.; Ipatov, A.; de la Lande, A.; Janetzko, F.; del Campo, J. M.; Mejia-Rodriguez, D.; Reveles, J. U.; Vasquez-Perez, J.; Vela, A.; Zuniga-Gutierrez, B.; Salahub, D. R. deMon2k, Version 5; The deMon Developers: Cinvestav, Mexico City, 2018.Search in Google Scholar
44. Chiodo, S.; Russo, N.; Sicilia, E. Newly Developed Basis Sets for Density Functional Calculations. J. Comput. Chem. 2005, 26, 175–184; https://doi.org/10.1002/jcc.20144.Search in Google Scholar PubMed
45. Dzib, E.; Cabellos, J. L.; Ortíz-Chi, F.; Pan, S.; Galano, A.; Merino, G. Eyringpy: A Program for Computing Rate Constants in the Gas Phase and in Solution. Int. J. Quantum Chem. 2019, 119, e25686.10.1002/qua.25686Search in Google Scholar
46. Quintal, A.; Dzib, E.; Ortíz-Chi, F.; Jaque, P.; Restrepo, A.; Merino, G. Automating the IRC-Analysis within Eyringpy. Int. J. Quantum Chem. 2021, 121, e26684.10.1002/qua.26684Search in Google Scholar
Supplementary Material
This article contains supplementary material (https://doi.org/10.1515/pac-2025-0521).
© 2025 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.