Unusual behavior of bimetallic nanoparticles in catalytic processes of hydrogenation and selective oxidation
-
Elena A. Redina
 ,
Olga A. Kirichenko
,
Olga A. Kirichenko


Abstract
Recent results obtained in studying mono- and bimetallic catalysts for selective hydrogenation of unsaturated carbonyl compounds, even unsaturated ones, acetylenic and nitro compounds as well as CO and bio-available alcohols oxidation are reviewed from the standpoint of the strong interaction between the metal nanoparticles, on the one hand, and two metals in the composition of bimetallic nanoparticles, on the other hand. Such interactions were demonstrated to result in partial positive or negative charging of metal nanoparticles, which, in turn, changes their adsorption and catalytic properties, especially with respect to the reactions involving hydrogen. Among the systems studied, Au–Pt, Au–Pd, Au–Cu, Au–Fe, Pt–WO x , Fe–Pd, Fe–Pt, Fe–Cu nanoparticles prepared by the redox procedure are considered to be most perspective in diverse catalytic applications because of the proper combination of the particle size and the electronic state of the metals.
Introduction
Metal nanoparticles deposited on porous carriers are widely studied due to their various practical applications, especially in catalysis. The properties of catalysts with supported metal nanoparticles are determined by the structural, electronic, and other characteristics of the deposited nanoparticles. The high catalytic activity of metal catalysts can be due to such factors as the morphology and size of metal nanoparticles, the interaction of the metal with the carrier, and the method of catalyst preparation [1], [2]. The carrier contributes to the stabilization of small nanoparticles, in some cases with a size smaller than 1 nm, and also governs the morphology of the deposited nanoparticles and the uniformity and width of the particle size distribution. This is true even in cases where the carrier is an inert material that does not interact with the supported metal particles, but due to its porous structure, especially in the case of micropores, contributes to the stabilization of very small particles via topological (geometric) effects.
The choice of optimal conditions for the synthesis of catalysts with supported metal nanoparticles can result in a strong interaction of the metal with the carrier and in the electron transfer from the metal to the carrier with the formation of a partial positive charge on the metal or transfer in the opposite direction, as a result of which the metal acquires a partial negative charge [3], [4], [5], [6]. As a result of such a charge transfer, the properties of metal particles change, including catalytic properties, such as activity and selectivity [7]. For example, for supported gold particles, it is shown that as the particle size decreases, the metal-metal bond length decreases and the d-electron density increases [8]. The relative number of metal atoms on the facets, edges, and vertices of a nanoparticle is uniquely determined by its size [9], [10].
The charge transfer is reliably determined by physicochemical spectroscopic methods [11]. The effect of the charge transfer on catalytic properties is described in the literature for metal nanoparticles containing noble metals (Pt, Pd, Au, Rh, Ir, Ru) [3], [4], [12], [13]. There are three main methods for determining the variation of the charge of metal nanoparticles, two of which are direct methods: (1) X-ray photoelectron spectroscopy (XPS) and (2) X-ray absorption spectroscopy using synchrotron radiation (EXAFS/XANES). The third method – IR spectroscopy using adsorbed molecules-probes, mainly CO is not direct, since an indirect assessment of the charge of metal particles can be obtained from the values of the shifts of carbonyl complexes of metals in relation to complexes of neutral (uncharged particles). Note that all three methods have certain disadvantages, including the possibility of electrostatic charges of the sample (metal particles) under the action of X-rays, and the variation of the electron density or the modification of metal particles due to the adsorption of CO. In some cases, for example, for particles of iridium or rhodium, such an interaction of the metal particles with CO may lead to disintegration of nanoparticles [14], which, however, is not observed in the case of Pt, Pd, Au and some other metals.
X-ray absorption spectroscopy (XAS) is used for studying both the electronic (oxidation) and coordination state of a metal. The Fermi level of the metal is shifted when the particle is charged to the higher energy region if the particle is negatively charged or to the lower energy region if the particle is positively charged. Moreover, the intensity of the so-called white line increases when the particles are positively charged or, conversely, decreases when they are negatively charged.
X-ray photoelectron spectroscopy (XPS) has proven to be a very effective and sensitive method for studying the electronic state of supported metals. The position of the electron binding energy indicates the oxidation state of the metal. However, it should be noted that the displacement of the core levels when changing the particle size should be taken into account. Studies of supported metal nanoparticles with the size smaller than 10 nm by the XPS method indicate a significant shift in the binding energy of the core-level electrons in comparison with the bulk metal. Usually the reason for this shift is the transfer of electron density between the metal and the carrier, however, dimensional and geometric effects can contribute to the value of the shift. In particular, geometric factors such as particle shape or morphology, including deviation from the spherical shape and the formation of flat (disc-shaped) or elongated (filaments, rods) particles can play a significant role in changing the electronic state of supported metal nanoparticles [15].
Note that the carriers can have a fundamentally different nature, both in terms of acid-base properties and electrical properties: insulators, semiconductors, and conductors. In this mini-review, interaction of supported metal nanoparticles with carriers will be considered and the catalytic activity/selectivity patterns will be interpreted from this standpoint.
Au-based metal nanoparticles in selective oxidation
Catalysis on gold is a relatively new but rapidly developing area of both homogeneous and heterogeneous catalysis. Since the discovery by M. Haruta of the unique properties of supported gold nanoparticles in the low-temperature oxidation of CO [16], more than 500 thousand publications have been published devoted to catalysis on gold. As shown in reviews [17], [18], [19], the number of reactions using gold catalysts is constantly expanding. Over the past decade, supported gold nanoparticles have been established as active and selective catalysts for the oxidation and hydrogenation of various organic substances, in particular, hydrocarbons of various structures [20], [21], [22], [23] and oxygen-containing organic compounds, including classes of alcohols and carbohydrates [24], [25], [26], [27], [28], [29], [30].
Today of particular interest are bimetallic systems based on gold nanoparticles [31], [32], [33], [34]. The combination of gold with a second metal (Au–Me) allows one to develop catalytic systems with unique physical and chemical properties not observed for individual monometallic catalysts due to the synergistic effect. It was noted that the nature of the second metal, the Au:Me ratio, the structure of bimetallic catalysts, the size, and, consequently, the shape of nanoparticles, which is, in turn, determined by the method of synthesis of bimetallic catalysts, have a great influence on their activity, selectivity, and stability [35], [36], [37], [38].
One of the ways to obtain a bimetallic Au-containing catalyst is the modification of a support with one metal (M1) and subsequent Au nanoparticle deposition on it. This strategy allowed us to obtain efficient and stable Au catalysts supported on the high-temperature θ-Al2-xFexO3 carrier for the process of selective CO oxidation in the presence of NH3 in the feed for the needs of exhaust gases neutralization both from diesel engine auto transport and industry [39], [40]. The prepared Au/θ-Al2-xFexO3 nanomaterials maintain high catalytic activity even after prolonged thermal treatment at 750 °C. A strong synergetic effect was observed between Au and Fe species; the activity, selectivity and stability of bimetallic catalysts were significantly higher compared to monometallic Au and Fe samples (Fig. 1). Moreover, the preparation method played a crucial role in obtaining high activity in selective CO oxidation in the presence of ammonia. The Au catalysts on the θ-Al2-xFexO3 supports (prepared by calcination of the precursor ‘‘Fe-salt/γ-Al2O3”at 1000 °C) were more active in CO oxidation in comparison with the catalysts on the FeO x /θ-Al2O3 supports prepared by deposition of Fe2O3 on pure θ-Al2O3. ESR studies revealed the presence of both the isolated and exchange-bonded Fe3+ ions in the θ-Al2-xFexO3 supports. The supported FeO x phases and the exchange-bonded Fe3+ ions favored NH3 oxidation, whereas the Fe3+ isolated ions decreased the activity in NH3 oxidation. The preferential oxidation of CO in the rich mixture with ammonia showed the possible purification way of the kiln gas from industrial production of lithium iron phosphate batteries (LFP) through utilization of ammonia and carbon monoxide by means of their processing into the initial components of the mixture for the LFP synthesis, i. e., ammonia can be transformed to (NH4)2HPO4, while CO should be first transformed into CO2 to be used in the synthesis of initial Li2CO3. In addition, the high thermal stability of the prepared nanomaterials makes possible their application as a catalytic layer in the engine exhaust gas-purifying systems equipped with oxidation catalytic section for CO and CH treatment coupled with NH3-SCR methodology.
![Fig. 1:
Temperature dependence of the CO (a) and NH3 (b) conversion in the gas mixture consisting of 1.5 vol.% NH3, 4.5 vol.% CO, 22.5 vol.% O2 and He balance for the samples calcined at 300 °C or at 500 °C [39].](/document/doi/10.1515/pac-2020-0207/asset/graphic/j_pac-2020-0207_fig_001.jpg)
Temperature dependence of the CO (a) and NH3 (b) conversion in the gas mixture consisting of 1.5 vol.% NH3, 4.5 vol.% CO, 22.5 vol.% O2 and He balance for the samples calcined at 300 °C or at 500 °C [39].
The other way of bimetallic catalyst preparation implies the formation of bimetallic nanoparticles on the surface of a support. However, the major problem faced in the preparation of bimetallic supported catalysts is the difficulties to ensure an extended area of contact between the monometallic phases. The most common method of bimetallic particles synthesis is the sol-method with subsequent immobilization of prepared nanoparticles on the support, yet unstable solid solutions are often formed by this way [37], [41]. Another drawback is stabilizing agent removal [42], [43]. Impregnation and co-precipitation methods are considered to be non-selective and lead to formation of the “non-true” bimetallic particles with the presence of isolated monometallic phases. In addition, the structure of the obtained bimetallic particles is hard to control [44]. In our works we showed the simplicity and high efficiency of the redox-methods that are based on the reduction of the ions of the second metal either by the pre-supported first [45], [46] or by a reducing agent adsorbed at the surface of the first metal, for instance, hydrogen [47], [48], [49]. Several catalytic systems were prepared combining gold and other noble metals (Ru, Pd, Pt) or non-noble ones (Cu, Fe). A strong synergetic effect was observed for Au-Ru nanoparticles supported on θ-Al2O3 for the preferential CO oxidation in the presence of ammonia in the treatment of exhaust gases in diesel engines [49]. The crucial role of the method of bimetallic nanoparticles formation was also demonstrated. The conventional method of deposition-precipitation with urea (DPU) lead to big particles of both RuO2 and Au (>40 nm), while the redox-method (RR) allowed obtaining nanoparticles below 7 nm even after calcination of the catalyst at 300 °C. The redox-prepared catalysts showed the highest activity in CO oxidation and the largest window between the temperature of full CO conversion and the temperature at which NH3 oxidation starts to proceed (Fig. 2). Another examples are redox-prepared Au/Rh/TiO2 catalysts. This system was an extremely efficient in dichloromethane degradation processes. The complete DCM conversion was observed at 400 °C over the low-loaded 0.1Au/0.3Rh and 0.1Au catalysts with selectivity of destruction to HCl 87 and 72%, correspondingly, and the CO release 3.2 and 400 ppm, respectively, indicating the strong interaction between Au and Rh in bimetallic nanoparticles [47].
![Fig. 2:
Temperature dependence of the CO (a) and NH3 (b) conversion in the gas mixture consisting of 1.5 vol.% NH3, 4.5 vol.% CO, 22.5 vol.% O2 and He balance [49].](/document/doi/10.1515/pac-2020-0207/asset/graphic/j_pac-2020-0207_fig_002.jpg)
Temperature dependence of the CO (a) and NH3 (b) conversion in the gas mixture consisting of 1.5 vol.% NH3, 4.5 vol.% CO, 22.5 vol.% O2 and He balance [49].
Our special attention in the preparation of bimetallic catalysts is devoted to the systems with an ultra-low amount of the active phase. Applying the redox method and working with diluted precursor solutions, we have obtained low-loaded Au-containing bimetallic catalysts, which showed unprecedently high activity and selectivity in bio-available alcohol oxidation. For the first time, we have developed an ultra-low-loaded Au/CuO x /SiO2 catalytic system, which was able to oxidize ethanol to acetaldehyde in the gas phase with a full conversion and 100% selectivity at 250 °C, while monometallic Au catalyst with the same metal loading was less active and CuO x /SiO2 showed only a 7% of ethanol conversion (Fig. 3). The interaction between Au0 and Cu2+ isolated cations led to the pronounced oxidation activity and selectivity in acetaldehyde formation [45].

Performance of low-loaded Au-Cu bimetallic catalysts in ethanol oxidation. Reaction conditions: EtOH:O2 = 0.3:1; VHSV: 4600 h−1 (left) and STEM image of 0.8%Au/0.8%Cu/SiO2 catalyst (right).
We also observed that Au redox-deposition in the amount of only 0.05% wt onto the 0.5%Pd/TiO2 catalyst can change the route of ethanol conversion, and instead of full complete ethanol oxidation to CO2, acetaldehyde and ethyl acetate can be formed with selectivity 70 and 20%, respectively, due to the formation of bimetallic Au–Pd nanoparticles proved by the DRIFTS-CO method [47].
Catalytic valorization of bio-available glycerol and 1,2-propanediol represents a very challenging problem since such processes open a new era of non-petrochemical synthesis of valuable products. Among them lactic acid is a key molecule for the production of bio-degradable polymer – polylactic acid (PLA). We have succeeded in the preparation of active and stable ultra-low-loaded Au/1%Pt/TiO2 bimetallic catalysts with an Au content of 0.025% wt. This system appeared to be active in selective primary hydroxyl-group oxidation in 1.2-propanediol with the lactic acid yield 87% [48] (Fig. 4). Moreover, the 0.025%Au/1%Pt/TiO2 catalyst is the first example of an active and selective system with the extremely low Au content for the formation of lactic acid from glycerol just in air at 60 °C. The addition of Au greatly enhanced the activity and selectivity of Pt/TiO2 catalysts both in 1,2-propanediol and glycerol oxidation due to the observed strong interaction between the two metals in Au/Pt bimetallic particles and between the bimetallic particles and the support, which was proved by XPS analysis, DRIFTS-CO technique, and temperature programmed reduction method (TPR-H2) [47] (Fig. 4).
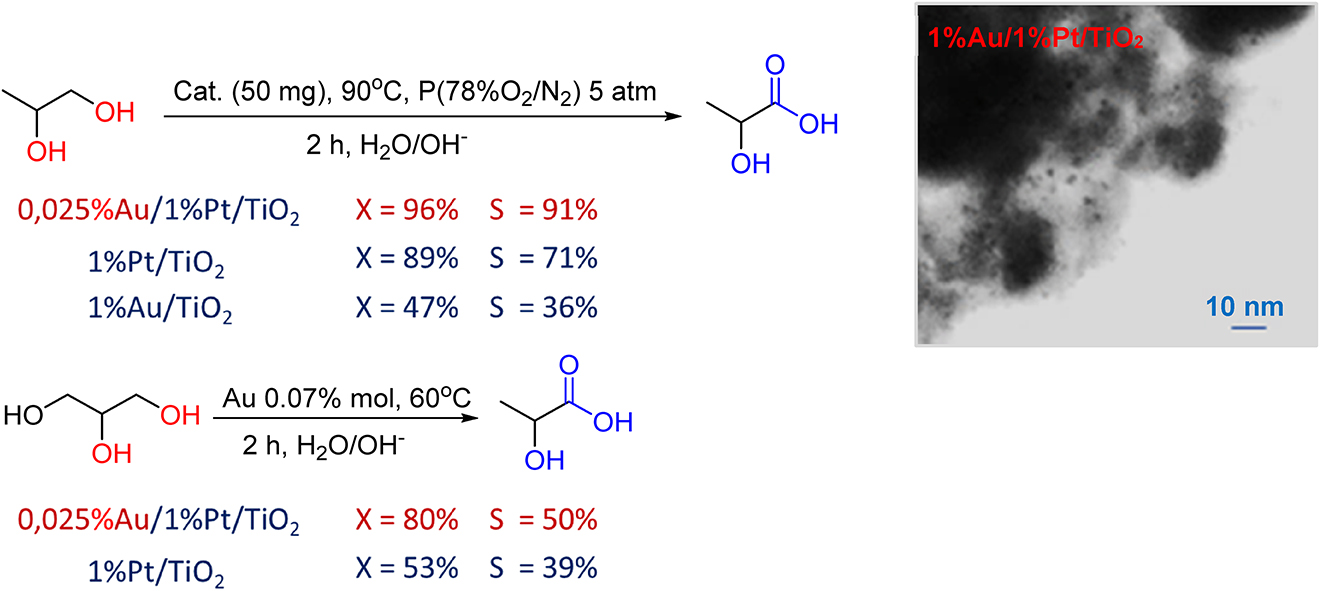
The performance of low-loaded bimetallic and monometallic catalysts in 1,2-propanediol and glycerol oxidation (left) and STEM image of 1%Au/1%Pt/TiO2 catalyst (right).
Hydrogenation processes
Pt-containing bimetallic nanocatalysts in hydrogenation reactions
Pt catalysts are universal systems for both oxidation and reduction reactions. The ability of Pt-group metals to activate hydrogen predetermines their application for hydrogenation of different classes of organic substances such as nitro-compounds, carbonyl compounds, alkenes and alkynes, nitriles, etc. [50], [51], [52], [53], [54], [55]. The activity and selectivity, as well as the form, in which Pt is present in the catalytic system (nanoparticles or clusters), greatly depend on the support. Strong Pt-support interaction in hydrogenation reactions plays a crucial role to obtain high activity and selectivity in hydrogenation reactions. Thus, widely used supports for Pt nanoparticles are reducible metal oxides such as СeO2, TiO2, Fe2O3 [56], [57], [58], [59], [60], [61], [62]. It is known that interaction of hydrogen with supported metal nanoparticles (Pt, Ni, Cu) causes a H2 spillover effect with the formation of active hydrogen species (H*=H• or H+ and H−) [63], [64], [65], [66], [67], [68], [69]. Split-over hydrogen species can cause reduction of the support, and therefore can interact with an adsorbed substrate leading to the hydrogenation product formation [70], [71]. Thus, the lower the energy barrier of H2 activation and transition of H* species to the carrier (the spillover), the milder the conditions required for the reaction to proceed. Decreasing the energy of hydrogen dissociation and inducing the hydrogen spillover effect can be achieved by the synthesis of supported Pt-containing bimetallic nanoparticles [72], [73], [74], [75], formation of ultra-small or single-atom Pt NPs [76], [77], [78], [79], and, finally, by the modification of oxide support [80], [81]. The use of mixed oxide systems or solid solution can greatly enhance the spillover effect due to the defects in the crystalline structure of the obtained carrier, that can appear, for example, in easier reduction of the metal oxide [82]. We have applied this strategy to develop a catalytic system characterized by a low energy barrier for H2 activation [83]. The main attention was paid to the synthesis of a mixed oxide support, and CeO2–ZrO2 system was chosen as it is characterized by the easier reduction of Ce4+ to Ce3+ compared to CeO2 oxide [84], [85]. The preparation technique used led to the highly dispersed mixed nano-oxide CeO2–ZrO2 with the particle size less than 5 nm and specific surface area higher than 100 m2/g. Deposited Pt NPs on the prepared support were stabilized with the size <1 nm. The giant hydrogen adsorption caused by hydrogen spillover effect was observed at temperatures −50 to +25 °C on the prepared 1%Pt/CeO2–ZrO2 catalytic system for the first time (Fig. 5). The unique ability of intense hydrogen activation at sub-ambient and ambient temperatures allowed us to perform selective hydrogenation of carbonyl compounds with different structures at room temperature and atmospheric pressure of H2 [83], [86], [87]. The developed catalytic system was much more active than Pt NPs supported on monometallic CeO2 and ZrO2 supports, as well as Pt supported on non-reducible SiO2, which correlated with the magnitude of the observed low-temperature hydrogen spillover effect. The activity of Pt/CeO2–ZrO2 supported catalysts is greatly affected by the specific surface area of the mixed-oxide support [87].
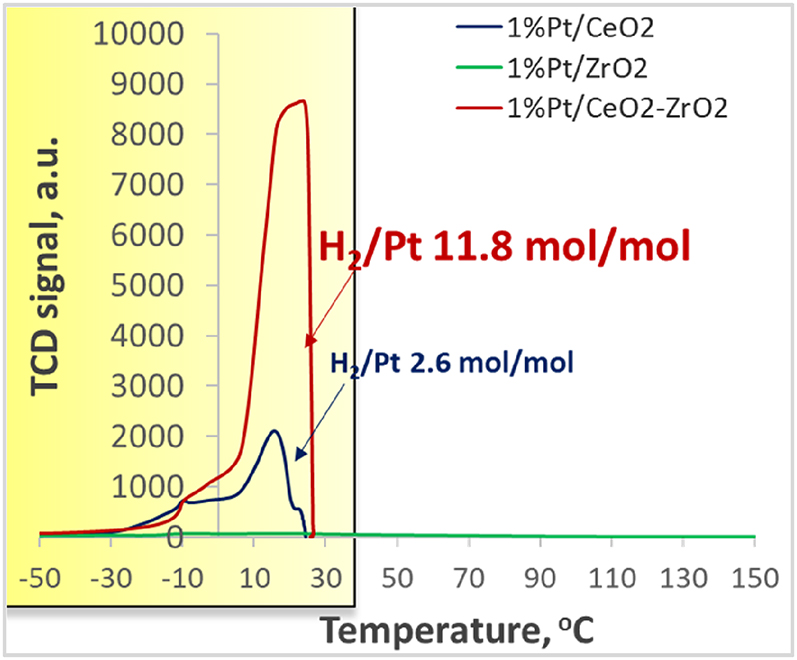
TPR-H2 profiles for 1%Pt/CeO2–ZrO2, 1%Pt/CeO2, and 1%Pt/ZrO2 catalysts.
In some cases, the modification of the support used for the preparation of Pt-containing catalysts with a second metal oxide can also lead to the change in their acidity [88], [89]. This technique is usually applied for the reactions implying the isomerization of a hydrogenated substrate or for hydrodeoxygenation reactions. One of examples of such a process is hydrogenolysis of tetrahydrofurfural alcohol to 1,5-pentanediol [90]. The combination of Pt with reducible oxides (WOx, MoOx) was responsible for the enhanced selectivity, while monometallic catalysts were not selective. Another example is the hydrodeoxygenation of glycerol to a value-added product, 1,3-propanediol, used as a monomer for the synthesis of a new type of thermoplastic polymer PTT [91], [92]. The most efficient system for this reaction appeared to be Pt/WOx/Al2O3 [88], [93], [94]. It was suggested that hydrogen dissociation takes place at the the Pt−WOx interface with spillover to a neighboring oxygen atom and the formation of strong Brønsted acid sites, i. e., protons formed in situ in the reaction are responsible for the high activity and selectivity of this type of catalytic system in glycerol to 1,3-propanediol transformation [93]. On the other hand, WOx provides strong adsorption sites for the primary hydroxy group of glycerol molecule and stabilizes the secondary carbocation formed during the reaction [94]. However, we showed that the activity of these catalysts greatly depends on the method and conditions of their preparation [95]. To obtain strong Pt-WO x interaction, special attention should be paid to the conditions of preparation of W-modified alumina: the complete ammonium tungstate (10 %wt) deposition is observed only after 7 h of the reaction at 80 °C. In addition, to exclude leaching of W-species during glycerol dehydrodeoxigenation reaction, the W-modified alumina support should be calcined at 800 °C [95].
The results presented clearly illustrate that high hydrogenation activity of Pt containing catalysts is mainly due to the strong metal-support interaction and hydrogen spillover effect. The bimetallic catalytic system, when second metal modifies the support, are the most perspective catalysts for hydrogenation reactions.
Fe-based metal nanoparticles in selective hydrogenation processes
Selective hydrogenation of triple C–C bonds to form the desired alkene is a key step in the synthesis of many important compounds of petrochemical industry, as well as for the synthesis of fine and specialty chemicals (i. e., vitamins A and E, linalool) [96], [97], [98], [99]. The conventional heterogeneous catalysts of this process are based on noble metals, in particular Pd [100], [101], [102], [103]. Although these catalytic systems are highly effective, they are quickly deactivated due to the blocking of the active surface by the reaction products and by-products; we can also note the high content of the noble metal in the catalysts and hence their high cost, and, in some cases, the toxicity of the systems used, in particular, of the Lindlar palladium–lead catalyst [104], [105].
An addition of a second metal or metal oxide to Pd-based catalysts is known to increase the activity and selectivity of catalysts [106]. The well-known bimetallic nanoparticlesfor selective alkyne hydrogenation include Pd–In [107], Pd–Ag [108] and Pd–Cu [109], [110]. However, the Fe influence on the catalytic properties of Pd catalysts in these reactions remains scarcely investigated. There is increasing research interest in iron as a catalyst component for the homogeneous and heterogeneous hydrogenation since this transition metal is abundant in nature, cheap, non-toxic, and potentially amenable to magnetic recovery [111]. Despite the large number of reports on the selective hydrogenation on Pd-containing catalysts, the improvements in efficiency and environmental safety of catalytic systems are remaining an active area of research and development.
In a number of our published papers [112], [113], [114], [115], [116], [117], we paid special attention to the study of catalytic properties of bimetallic catalytic systems Fe–M (Pt, Pd, Cu), in particular, to interrelation between the redistribution of electron density between metals in bimetallic catalysts, representing a combination of metallic nanoparticles of a noble metal (Pd, Pt) and Fe or FeOx, and their activity/selectivity in reactions of selective hydrogenation of triple C≡C bonds to C=C bonds under mild conditions. The high catalytic activity of bimetallic Fe–Me systems appears due to many factors, such as the structure, size and dispersion of nanoparticles, the method of synthesis and conditions of thermal treatments and reduction, the metal ratio, as well as the nature of the support.
In 2016 we published [115], [116] the results of catalytic activity of the supported bimetallic Fe–Pd nanoparticles in hydrogenation of phenylacetylene to styrene. The bimetallic of Fe–Pd/SiO2 catalysts were synthesized by incipient wetness impregnation of SiO2 carriers (LS-30 m2/g and HS-300 m2/g) with aqueous solutions of the active metal precursors (NH4)3[Fe(C2O4)3] and [Pd(NH3)4]Cl2 with subsequent thermal treatment (air calcination, reduction in a H2 flow). We investigated the influence of many factors, such as the solvent, the nature of the support, the composition and conditions of thermal treatment on the catalytic properties. It is shown that the use of ethanol as a solvent of phenylacetylene leads to an increase in the rate of the hydrogenation reaction by 3 times, and there is an increase in the selectivity of styrene formation in comparison with the results obtained in the iso-propyl alcohol.
The application of the support with a high specific surface area (HS) makes it possible to obtain the 90% selectivity to styrene at the full conversion of phenylacetylene at 23 °C and 0.1 MPa H2 for 25 min compared to a sample on a low specific surface area carrier, where the maximum selectivity was 82%. The XRD and TEM methods have shown that the average size of supported nanoparticles in the sample on the HS support is 8 nm, compared to the sample supported on a carrier with a low specific surface area (15 nm).
We also showed that variation of thermal treatment conditions (air calcination at different temperatures, reduction in a H2 flow) have a significant effect on the phase composition of the catalyst, the size of the bimetallic particles and the electronic state of metals, which leads to different catalytic properties of the obtained systems. The activity of the calcined bimetallic 8%Fe-3%Pd/SiO2 catalyst exceeds the activity of the reduced monometallic 3%Pd/SiO2 catalyst and is 10 times higher than the activity of an industrial Lindlar catalyst (5% Pd/CaCO3-Pb). The high selectivity of styrene formation 86% is achieved on a bimetallic sample for 20 min compared to a monometallic palladium sample. Reduction of catalysts in the hydrogen flow leads to a decrease in hydrogenation activity, as well as to an increase in the selectivity of styrene formation up to 90%. The XRD results of the calcined 8Fe-3Pd/SiO2 catalyst indicate the formation of Pd0, α-Fe2O3 and Fe3O4 phases. The reduction of the calcined bimetallic samples in a H2 flow at 400 °C provides the formation of Fe0 nanoparticles 17-20 nm in size, whereas the reduction of the dried samples results in the formation of X-ray amorphous Fe phases.
It should be noted that the support material plays an importantrole in the overall catalytic performance for achieving the high conversion and better selectivity towards the desired products [105]. In our work [117], we have shown that the use of γ-Al2O3 (SBET of 240 m2 g–1) as a carrier for Fe–Pd nanoparticles made it possible to increase the initial phenylacetylene hydrogenation rate several times in comparison with nanoparticles supported on SiO2 and Lindlar catalyst. The bimetallic 8Fe-3Pd/γ-Al2O3 catalysts also showed a high catalytic activity in hydrogenation of the acetylene alcohol. The selectivity towards 2-methylbut-3-en-2-ol on this catalyst was 95% at the complete conversion of 2-methylbut-3-yn-2-ol at a high rate of hydrogenation at room temperature and 0.1 MPa H2. The reduced 8Fe-3Pd/Al2O3 samples demonstrated promising magnetic properties and were easily separated from the reaction solution with a permanent magnet. The catalytic activity and the selectivity remained high even after four successive catalytic recycles. In addition, no metal leaching occurred during the cyclic operation after four cycles.
Thus, our stable 8Fe-3Pd/SiO2 and 8Fe-3Pd/Al2O3 catalysts are a good alternative to the traditional toxic Lindlar catalyst (5Pd/CaCO3 inhibited by Pb) in the selective hydrogenation of acetylene alcohols and alkynes under mild conditions.
The particular interest for the process of selective hydrogenation of unsaturated compounds are catalysts with an ultra low content of a noble metal, 0.05–0.1%, which are currently poorly studied.
In our investigation [115], the maximum value of the selectivity of styrene formation of 93% was achieved on the calcined bimetallic Fe–Pd/SiO2 sample with a Pd content of 0.05 wt.% and the atomic ratio of Fe:Pd is 2 times greater than in a reduced catalyst with a higher Pd content of 3 wt.% at room temperature and 0.1 MPa H2.
In 2018 [113], [114] we published for the first time a new method for the preparation of bimetallic FeOx/Pd/SiO2 catalysts synthesized by the redox methods including reduction with H2 at ambient conditions. Redox methods of the deposition of a second metal ensure large contact surfaces in bimetallic particles resulting in stronger synergistic effects between two metals and support [47], [48], [49]. It was shown in [113] that redox methods can also be used to support oxides onto the surface of noble metal nanoparticles via the reduction of soluble oxo-anions to insoluble hydroxides. The goal of our work was to develop the procedure and conditions of redox deposition of FeOx species on the surface of the silica-supported Pd nanoparticles, as well as to study the reducibility of thus synthesized materials and their catalytic activity in the hydrogenation of phenylacetylene with molecular H2. The method proposed is based on the difference in the solubility of Fe3+ and Fe2+ oxalates. First the monometallic 3%Pd/SiO2 catalyst was prepared by the impregnation of the support (SBET = 240 m2/g) with the [Pd(NH3)4]Cl2 solution followed by drying and reduction in hydrogen at 400 °C. Then the 1.5% Fe/3% Pd/SiO2 catalyst was synthesized using the redox method of catalytic reduction with hydrogen with further calcination at 300 °C to decompose the precipitated oxalate to iron oxide. The scheme of the precipitation of iron ions can be presented as:
The TPR curve profile obtained in our work indicates the strong interaction between the deposited iron oxide and palladium, confirming the deposition of iron particles on the surface of palladium nanoparticles. The negative peak with a minimum at 60 °C shows that the surface of palladium nanoparticles in the prepared bimetallic materials is still available for the adsorption of hydrogen to form palladium hydride that is important for catalysis in hydrogenation processes.
The easily reducible FeOx/Pd/SiO2 materials exhibited a multiple increase in the catalytic activity as compared to the starting Pd/SiO2 system in the selective hydrogenation of triple carbon bonds with molecular H2 in hydrogenation of phenylacetylene in the liquid phase at room temperature and 0.1 MPa hydrogen pressure.
In our work [112], we have shown that the strong contact interaction between Pt and Fe and the charge transfer from Fe to Pt in a bimetallic particle, proven by XPS, TPR, and DRIFTS-CO methods, provides both high selectivity (85% at 90% conversion) of the bimetallic catalyst and increased activity in liquid-phase hydrogenation of triple bonds in phenylacetylene to C=C bonds in comparison with monometallic 1%Pt/SiO2 catalyst (5% selectivity at 90% conversion) at 70 °C and 0.5 MPa H2.
Some of the most significant results on the study of the catalytic properties of iron-containing nanoparticles without the addition of noble metals are presented in our papers [118], [119]. We have shown for the first time that deposited iron oxide nanoparticles are active in the hydrogenation of unsaturated compounds under relatively mild reaction conditions (1.3 MPa H2, 80–110 °C). The activity of FexOy nanoparticles obtained by decomposition of the (NH4)3[Fe(C2O4)3] precursor and stabilized on the SiO2 support significantly exceeds that of the unsupported α-Fe2O3 phase (Fig. 6). The catalytic properties of the FexOy/SiO2 monometallic catalysts depend on the conditions of their heat treatment. Thus, an increase in the temperature of calcination of the samples from 250 to 500 °C led to the oxidation of Fe3O4 to Fe2O3 and a twofold increase in the catalytic activity and to a high selectivity of the process (Figs. 6 and 7). A TEM study showed that an increase in the calcination temperature from 250 to 500 °C results in a growth of nanoparticles to reach an average size of 15 nm and aggregation of particles. The hydrogenation of the C≡C bond preferably occurs on larger particles, which are accessible for the reaction. According to XRD and TPR-H2 results, the further reduction of Fe2O3 in a H2 flow led to the formation of nanoparticles of Fe0 and an increase in both activity and selectivity in the studied process compared to the supported Fe2O3 nanoparticles. Thus, a high selectivity of styrene formation (78%) at full phenylacetylene conversion at 110 °C and 1.3 MPa H2 in ethanol was obtained for the sample with metal state of iron.
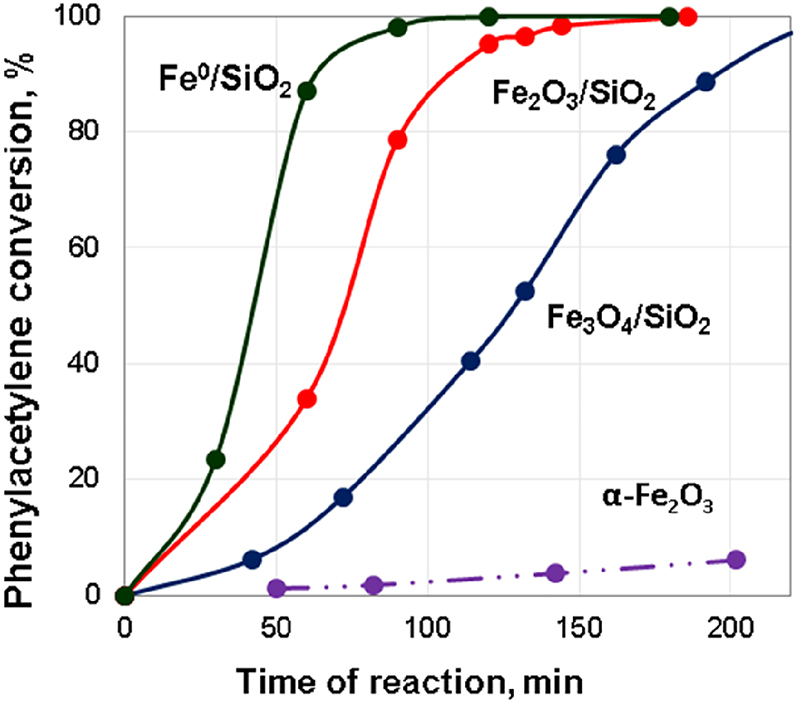
The influence of the phase composition on the activity in phenylacetylene hydrogenation at 110 °C and 1.3 MPa H2 in ethanol.
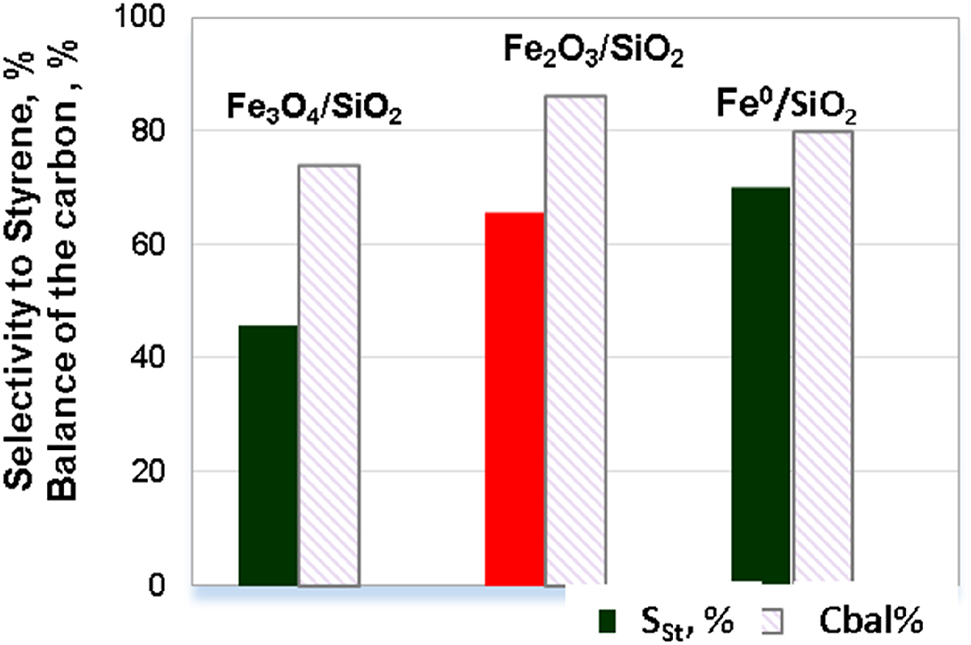
Effect of the phase composition of catalysts on the selectivity (Sst) of styrene formation and carbon balance at a 100% conversion of phenylacetylene.
The paper also showed that modification of the iron oxide phase with small amounts of copper oxides (0.4 wt%) makes it possible to exclude the stage of the reduction of FeOx to Fe0 in hydrogen at high temperatures, while the high activity and 78% selectivity of styrene formation are retained. Based on the TPR-H2 results of bimetallic Fe-Cu/SiO2 catalysts, we can conclude that Fe and Cu oxide nanoparticles have a strong contact interaction already at the calcination stage. An increase in the copper content in the samples promotes hydrogenation and polymerization of styrene at elevated temperatures and hence has an adverse effect on the selectivity of the formation of the desired styrene product.
Thus, based on our previously published works, we can conclude that the highly effective catalysts based on iron or iron oxide nanoparticles are very promising systems that represent an alternative to existing hydrogenation catalysts with a high content of noble metals. Thus, the development of catalytic systems that do not require the use of expensive and dangerous stoichiometric reducing agents, are highly efficient under mild reaction conditions, are affordable and environmentally friendly remains a key task today.
Cu-based metal nanoparticles in hydrogenation of nitrocompounds
Aromatic amines obtained via the catalytic reduction of corresponding nitro compounds are widely used in the synthesis of different compounds for fine organic synthesis, production of dyes, urethanes, and pharmaceutical industry [120]. Nickel-containing catalysts, as well as catalysts based on platinum-group metals are the most commonly used systems for the selective hydrogenation of nitro- and dinitrobenzene to corresponding amines [121]. The main drawback of these catalysts is a high content of the active components and their toxicity. The development of more effective and environmentally safe catalytic systems remains an important goal of catalysis.
For the first time, we proposed supported Fe-Cu nanoparticles for reducing nitro compounds to the corresponding amines [122], [123]. The preparation procedure of the supported bimetallic Fe–Cu/SiO2 catalysts and the conditions of the thermal treatment affect the catalytic activity and selectivity of p-phenylenediamine formation from p-dinitrobenzene. Silica-supported mono- and bimetallic samples were synthesized via (1) co-deposition–precipitation of Cu and Fe precursors (copper nitrate and iron (III) nitrate) predominantly on the outer surface of the silica support using thermal hydrolysis of urea and (2) deposition of Cu and Fe precursors by consecutive incipient wetness impregnation of porous silica with an ammonium trioxalatoferrate solution and, after intermediate drying, with copper (II) nitrate solutions. Commercial silica gel (SBET = 108 m2 g–1) was used as a carrier. Once the metal precursors were supported, the samples were dried at 60–110 °С and then calcined at 300 °С. The calcined samples were reduced in a Н2 flow at 300 °С. The character, temperature, and degree of reduction of the metals in bimetallic samples depend largely on the preparation procedure used. Analysis of the TPR curve shows that Cu and Fe were present mostly in the form of individual oxides in the bimetallic sample obtained via impregnation. The strong contact interaction of Fe and Cu species in the prepared nanoparticles has been revealed by the method of TPR, which exhibited the Cu-enhanced reducibility of Fe3+ to Fe0 and stability to reoxidation for the metallic particles. Variations in the preparation procedures influence the reducibility and the interaction between Cu and Fe components resulting in considerable changes of the catalytic activity and selectivity in hydrogenation of nitro compounds. The catalytic hydrogenation was conducted in the temperature range of 145–180 °C and initial hydrogen pressure of 1.3 MPa. The calcined Fe–Cu/SiO2 catalysts obtained by co-deposition are more active and selective (89%) toward phenylenediamine at complete conversion of nitroaniline than catalysts synthesized by impregnation.
The purposes of the another our work [124], [125] was to elucidate the applicability of chrysocolla-like (Cu2Si2O5(OH)2·xH2O) Cu/SiO2 and Cu–Zn/SiO2 catalysts synthesized by the urea deposition method as catalysts for reduction of nitroaromatic compounds with molecular hydrogen. The catalysts were synthesized similarly to a previously described procedure [35]. Copper hydrosilicates demonstrated high catalytic activity and selectivity towards nitrobenzene hydrogenation to aniline (100%) and p-dinitrobenzene hydrogenation to p-nitroaniline and p-phenylenediamine (94%) under relatively mild conditions (150–170 °C, hydrogen pressure of 1.3 MPa). At the same time, the introduction of Zn additives leads to an increase in the selectivity to p-phenylenediamine to 99%.
Thus, we were able for the first time to synthesize mono- and bimetallic catalysts and to carry out the selective hydrogenation of nitrobenzenes in the absence of noble metals under mild conditions.
Electronic and size effects
The behavior of monometallic and bimetallic metal nanoparticles in catalytic reactions is determined not exclusively by the nature of the metal and the size of the metal nanoparticles. The electronic effects may be equally or even more important, since the electron transfer between the metal and the carrier or between two metals in a bimetallic particle changes, sometimes dramatically, the catalytic properties of the metals. This is especially important in the case of bimetallic nanoparticles based on Fe, when the d electron density depends on the extent of electron transfer usually from Fe to a more electronegative noble metal. However, the examples of such studies in the literature related to catalytic properties of iron-based catalysts applied in hydrogenation reactions are scarce and non-systematic. Nevertheless, some pattern can be revealed on the basis of available data obtained using spectroscopic methods, such as IR spectroscopy of adsorbed carbon monoxide, X-ray photoelectron spectroscopy, and EXAFS/XANES.
Au-Pt catalysts. Strong interaction between Au and Pt components in bimetallic particles was confirmed by XPS [47], [48]. The shift of the Pt 4f line to BE = 72.3 eV, and that of the Au 4f line to BE = 84.3 eV, in comparison with the expected energies for metals [126] (71.1 and 84 eV, respectively) indicated the strong interaction between Au and Pt in the 1Au/1Pt catalysts resulting in the formation of partially positively charged metallic Pt NPs (Ptδ+) and negatively charged metallic Au NPs (Auδ−). A similar shift of Pt BE to the higher values with increasing Au content was observed recently for Au(core)–Pt(shell)/C catalysts [127]. Strong Au–Pt interaction in Au-Pt/CeO2 was also found for platinum moieties in the XPS spectrum [128]. On the contrary, the Pt 4f BE in 0.05Au/1Pt particles was close to that of metallic platinum, and BE in 0.025Au/1Pt particles was somewhat shifted to the higher values [47], [48]. We assume that upon deposition of a very small amount of gold onto the Pt-NPs surface by the redox method, the most active sites of Pt nanoparticles covered by preadsorbed hydrogen are involved in the oxidation–reduction reaction and strong Au–Pt contacts are formed. The XPS study of the Au–Pt catalysts with a low Au loading is difficult since the intensity of Au 4f lines is extremely low. The surface Au:Pt atomic ratio in the low-loaded Au/Pt samples significantly exceeded the ratio in the bulk as a result of the higher dispersion of Au than Pt and coverage of the Pt surface with Au subparticles. For the 1Au/1Pt system, we observed a different effect. The surface Au:Pt ratio was almost twice lower than in the bulk [47], [48]. This can be accounted for by partial coverage of Au NPs with Pt. La Parola et al. [129] reported the decrease in the surface Au/Pt ratio measured by XPS in AuPt/SiO2 catalysts with an increase in the thickness of the Pt shell on the surface of the Au core. The bands at 2068 and 1837 cm−1 due to the linear form of CO on Pt and bridged CO on Pt, respectively, are observed in the IR spectrum of the monometallic Pt catalyst reduced at 200 °C [48], [130]. The DRIFT spectra of the Au/Pt sample reduced at 200 °C contain three bands of metal carbonyls at 2107, 2069, 1852 cm−1 [48]. The band at 2107 cm−1 was assigned to CO adsorbed on metallic Au. It can also be assigned to Auδ−−CO [131], the formation of Au with a partial negative charge in the 1Au/1Pt catalyst being revealed by XPS. It is known that the strength of CO adsorption on metal nanoparticles is size-dependent and small NPs adsorb CO stronger than the large particles, for example for Au NPs [132]. The strength of CO adsorption in the bridged form on Pt NPs in the bimetallic Au/Pt catalyst was weaker than in the monometallic Pt/TiO2 catalyst, since the frequency of bridged CO for the 1Au/1Pt catalyst was higher (1852 cm−1 compared to 1837 cm−1) and thus adsorbed CO was more easily removed by evacuation at 20 °C compared to the monometallic Pt catalyst [48]. This effect can be accounted for by covering of Pt NPs by Au NPs as a result of direct redox deposition of gold. Gold redox deposition on TiO2 supported Pt NPs was also responsible for the noticeable strong metal support interaction. Comparing the ratio between the intensity of the Ti4+–CO and Pt0–CO bands, we found an increase of the Ti4+–CO/Pt0–CO ratio for the Pt/TiO2 sample with an increase of the reduction temperature from 200 to 500 °C. This ratio for the bimetallic Au/Pt catalyst was much lower, and after the reduction at 400 °C, it dramatically decreased. This can be accounted for by the higher rate of the TiO2 reduction with the formation of Ti3+ cations due to the effect of strong Au–Pt and TiO2 interaction in the bimetallic catalyst [48]. The geometrical factor of such strong interaction, i. e., covering of metal NPs with the reducible support, can be responsible for the increase in the Ti4+–CO/Pt–CO ratio with increasing reduction temperature for both samples. Therefore, the results of the DRIFTS study also proved the selective Au deposition on the Pt NPs by the redox method and the effect of strong Au–Pt and TiO2 interaction [48].
In the XPS spectra of TiO2 supported 1Au/1Pt catalysts, the doublet of Au 4f was shifted to the lower binding energies (BE) by 0.4–0.6 eV in comparison to the value for the metallic Au with BE Au 4f7/2 = 84.0 eV [47]. This indicated that Au NPs in Au/Pt/TiO2 catalysts had an effective negative charge. The electronic state of Pt in the bimetallic catalyst Au/Pt/TiO2 was affected by the preparation method, atomic ratio Au:Pt, and applied conditions of redox Au deposition. We assumed that a partial negative charge of Au NPs was due to electron transfer from TiO2 observed in several studies [47], [48], [133]. The electron transfer could occur on the contact perimeter between the bimetallic Au/Pt particles and TiO2. The back donation of the electron density from Pt to the reducible oxide support and/or to gold NPs was observed in [134] for Au/Pt/CeO2 catalysts and in [135] for Au/Pt/C catalysts. To sum up, XPS data provided direct evidence that there was a strong interaction between Au–Pt in the bimetallic Au/Pt particles and between Au/Pt particles and TiO2 in the redox-prepared catalysts. The surface of low-loaded Au/Pt/TiO2 catalysts was enriched with Au and the surface Au:Pt ratio was much higher than that calculated from the chemical composition of the sample indicating high dispersion of Au species on Pt NPs.
Au-Pd/TiO 2 catalysts . Direct selective Au deposition from the HAuCl4 solution on the surface of the titania-supported Pd NPs [47] has been proved by a decrease of the CO adsorption with an increase in the Au:Pd ratio. In preliminary experiments, it was found that CO adsorption under the used conditions is negligible on the surface of the Au0 nanoparticles of the size 1–15 nm. Most likely, the observed decrease in the CO adsorption for the Au/Pd samples is due to the coverage of the Pd surface with Au0 species.
The surface properties of Au/Pd/TiO2 catalysts were studied with the use of DRIFTS-CO examining the low-loaded 0.05Au/0.5Pd/TiO2 catalyst [47] in comparison with the monometallic 0.5Pd/TiO2 sample. Two bands were observed in the DRIFT spectrum of adsorbed CO on the 0.5Pd/TiO2 sample at 2089 and 1955 cm−1 assigned to the linear adsorbed CO on top of Pd (111) facets and 2-fold-bridged carbonyls on Pd (100)In the DRIFT-CO spectra of the 0.05Au/0.5Pd/TiO2 catalyst, there is an additional band at 1936 cm−1 assigned to two-fold-bridged CO on Au/Pd sites. The IR data provide evidence for deposition of Au species on the Pd NPs surface, especially on top of Pd (111) facets. The comparison of the DRIFT-CO spectra of the 0.05Au/0.5Pd/TiO2 catalyst and the Pd/TiO2 sample indicated selective Au deposition on the Pd surface by the redox procedure [47]. The increase in the ratio of intensities of the bands of Auδ+-CO-linear and Pd-CO could be caused by covering of Pd NPs surface with Au species [47].
Au-Cu/SiO 2 catalysts . The state of gold was studied by EXAFS using the Au L3-edge [45]. In the spectrum of the Au foil, the first double peak corresponds to the first coordination shell containing 12 Au atoms at a 2.885 Å real distance from the central Au atom. The analysis of the EXAFS oscillations shows that the nearest neighbors of the central Au atom in the samples are the Au atoms with an average coordination number (CN) about 10.8 at 2.85 Å real distances.
Pd-Fe catalysts . The relationship of electron transfer between two metals in deposited bimetallic Fe-Pd/SiO2 и Fe-Pd/Al2O3 catalysts and catalytic properties in furfural hydrogenation was investigated [136]. The use of an inert carrier like SiO2 makes it possible to ensure sufficient mobility of the metal precursors to intermingle efficiently and thus the formation of a metallic alloy upon reduction. When Al2O3 was used, the signals that characterize the formation of a Fe–Pd alloy by XRD, XPS methods were not detected. The formation of the Fe–Pd alloy in the bimetallic Fe-Pd/SiO2 catalyst has been proven by XRD, XPS methods. For the Pd-Fe/SiO2 catalyst, there is a clear shift about 0.4 eV to higher binding energies relative to the peaks characterizing Pd0 in the monometallic catalyst. Tang et al. observed a positive binding energy shift in Pd-Fe/C samples compared to a Pd/C sample [137]. In addition, Felicissimo et al. observed shifts to higher binding energies up to 0.6 eV when Fe was deposited on top of Pd [138]. The same trend has been reported by other authors, who have consistently observed a positive shift when the Pd–Fe alloy is formed with respect to pure Pd. It can be assumed that the accompanying shifts in binding energies reflect an electron transfer from Fe to Pd, which was predicted by an analysis of the electronegativity of the two metals (Pd: 2.2 and Fe: 1.83) [139]. Alloy formation in these works is confirmed by DFT calculations of the Pd3d core-level shift in the monometallic Pd and bimetallic Pd–Fe alloy bulk structures, which clearly show that alloying Pd with Fe leads to the Pd3d core-level shift of 0.35 eV in the Pd3-Fe1 alloy and 0.52 eV in the Pd2-Fe2 alloy [136]. The TEM studies of samples showed that the average particle size in a bimetallic Fe-Pd/SiO2 sample is relatively smaller (4.1 nm) than in a monometallic Pd/SiO2 catalyst (6.9 nm). Studies of the catalytic properties of synthesized Fe–Pd catalysts have shown that the formation of bimetallic particles with a strong contact interaction of two metals affects not only the activity, but also the reaction route of furfural hydrogenation. In the presence of a monometallic Pd catalyst, the main product of furfural hydrogenation with a conversion of 68% was furan with a yield of 36%, while the yield of the target product – furfuryl alcohol was only 7% and the formation of methylfuran on the monometallic catalyst did not occur. However, on bimetallic 0.5Fe-1Pd/SiO2 and 0.5Fe-1Pd/Al2O3 catalysts, the conversion of furfural was 84 and 89%, respectively. It should be noted that the effect of a strong metal-metal interaction affects the distribution of reaction products. Thus, the use of a bimetallic catalyst 0.5Fe-1Pd/SiO2 allowed to increase the yield of the main reaction products–furfuryl alcohol and methylfuran to 21 and 36%, respectively, in comparison with catalysts 0.5Fe-1Pd/Al2O3, where the yields of the corresponding products were only 6 and 20%. It can be noted that the carbonyl group is more strongly adsorbed on the surface of the Fe–Pd alloy due to the presence of electron-deficient Fe, which accelerates the hydrogenation of furfural to furfuryl alcohol and further deoxygenation to methylfuran, while the decarbonylation reaction predominantly proceeds on a monometallic catalyst.
Thus, both the physical and chemical properties of bimetallic catalysts and their catalytic properties in various oxidation and hydrogenation reactions differ significantly from those of monometallic analogs due to the presence of additional electronic effects in bimetallic systems.
Conclusions
The reactions involving hydrogen and oxygen in the presence of Fe0 and FeO x nanoparticles, mono- and bimetallic Au and Pt nanoparticles supported on reducible carriers with ultra-low amount of noble metals are a promising trend in modern catalysis. A significant breakthrough on this topic was made recently in metal-complex catalysis, while heterogeneous mono- and bimetallic catalytic systems in the reactions of selective hydrogenation of unsaturated, nitro-, and carbonyl compounds as well as selective oxidation are only beginning to be actively investigated. Thus, the development of novel catalytic systems that can provide atom-efficient hydrogenation and oxidation processes with high selectivity under mild reaction conditions meeting the principles of green chemistry remains a challenging task today.
The Fe–and Cu-containing catalysts are a good alternative to traditional catalysts containing a large amount of noble metals, including the toxic Lindlar catalyst (5%Pd/CaCO3 inhibited by Pb complexes), in the selective hydrogenation of unsaturated and nitro- compounds under mild reaction conditions. Low-loaded bimetallic catalysts are promising systems for both oxidation and reduction reactions with unique characteristics that make them “next generation” catalysts. However, such systems call for a new challenge in the investigation of their physio-chemical properties as today there are no instruments and methods to study the catalytic materials with ppm amounts of active components. At the same time, the development of a new type of multi-metallic supports opens up great prospects in the formation of a novel catalytic systems with an easily manageable properties for a variety of applications.
Funding source: Russian Foundation for Basic Research
Award Identifier / Grant number: 19-33-60001
Funding source: Ministry of Education and Science of the Russian Federation
Award Identifier / Grant number: К2-2019-005
Funding source: Council on grants of the President of Russian Federation
Award Identifier / Grant number: MK-3811.2019.3
-
Funding: This work was supported by the Russian Foundation for Basic Research, project no. 19-33-60001 in the part related to the synthesis of the Fe- and Cu- containing nanomaterials and their catalytic study, by the Council on grants of the President of Russian Federation МК-3811.2019.3 in the part of low-loaded Au containing catalysts. The characterization of materials was carried out with a financial support from the Ministry of Education and Science of the Russian Federation in the framework of Increase Competitiveness Program of NUST “MISiS” (grant no. К2-2019-005).
References
[1] T. W. van Deelen, C. Hernández Mejía, K. P. de Jong. Nat. Catal. 2, 955 (2019). https://doi.org/10.1038/s41929-019-0364-x.Search in Google Scholar
[2] E. S. Lokteva, E. V. Golubina. Pure Appl. Chem. 91, 609 (2019). https://doi.org/10.1515/pac-2018-0715.Search in Google Scholar
[3] J. Feng, W. Jiang, C. Yuan, X. Shi, K. Zang, Y. Zhang. Appl. Catal. Gen. 562, 106 (2018). https://doi.org/10.1016/j.apcata.2018.06.002.Search in Google Scholar
[4] Y. Chen, J. Chen. Appl. Surf. Sci. 387, 16 (2016). https://doi.org/10.1016/j.apsusc.2016.06.067.Search in Google Scholar
[5] A. Vicente, G. Lafaye, C. Especel, P. Marécot, C. T. Williams. J. Catal. 283, 133 (2011). https://doi.org/10.1016/j.jcat.2011.07.010.Search in Google Scholar
[6] R. P. Doherty, J.-M. Krafft, C. Méthivier, S. Casale, H. Remita, C. Louis, C. Thomas. J. Catal. 287, 102 (2012). https://doi.org/10.1016/j.jcat.2011.12.011.Search in Google Scholar
[7] P. Wu, Y. Cao, Y. Wang, W. Xing, Z. Zhong, P. Bai, Z. Yan. Appl. Surf. Sci. 457, 580 (2018). https://doi.org/10.1016/j.apsusc.2018.06.300.Search in Google Scholar
[8] J. A. van Bokhoven, J. T. Miller. in AIP Conference Proceedings, p. 582, Stanford, California (USA), 882 (2007). https://doi.org/10.1063/1.2644599.Search in Google Scholar
[9] L. Liu, A. Corma. Chem. Rev. 118, 4981 (2018). https://doi.org/10.1021/acs.chemrev.7b00776.Search in Google Scholar
[10] R. Van Hardeveld, F. Hartog. Surf. Sci. 15, 189 (1969). https://doi.org/10.1016/0039-6028(69)90148-4.Search in Google Scholar
[11] F. Wang, J. Jiang, B. Wang. Catalysts 9, 477 (2019). https://doi.org/10.3390/catal9050477.Search in Google Scholar
[12] A. J. Pamphile-Adrián, P. P. Florez-Rodriguez, M. H. M. Pires, G. Perez, F. B. Passos. Catal. Today 289, 302 (2017). https://doi.org/10.1016/j.cattod.2016.07.026.Search in Google Scholar
[13] J. Wang, C. Yuan, N. Yao, X. Li. Appl. Surf. Sci. 441, 816 (2018). https://doi.org/10.1016/j.apsusc.2018.02.070.Search in Google Scholar
[14] A. Berkó, F. Solymosi. Surf. Sci., 411, L900 (1998). https://doi.org/10.1007/s10562-014-1398-y.Search in Google Scholar
[15] J. Radnik, C. Mohr, P. Claus. Phys. Chem. Chem. Phys. 5, 172 (2003). https://doi.org/10.1039/b207290d.Search in Google Scholar
[16] M. Haruta. Catal. Today 36, 153 (1997). https://doi.org/10.1016/S0920-5861(96)00208-8.Search in Google Scholar
[17] A. S. K. Hashmi, G. J. Hutchings. Angew. Chem. Int. Ed. 45, 7896 (2006). https://doi.org/10.1002/anie.200602454.Search in Google Scholar PubMed
[18] R. Ciriminna, E. Falletta, C. Della Pina, J. H. Teles, M. Pagliaro. Angew. Chem. Int. Ed. 55, 14210 (2016). https://doi.org/10.1002/anie.201604656.Search in Google Scholar PubMed
[19] X. Liu, L. He, Y.-M. Liu, Y. Cao. Acc. Chem. Res. 47, 793 (2014). https://doi.org/10.1021/ar400165j.Search in Google Scholar PubMed
[20] M. E. Ali, Md. M. Rahman, S. B. A. Hamid. AMR 925, 38 (2014). https://doi.org/10.4028/www.scientific.net/AMR.925.38.Search in Google Scholar
[21] L. Torrente-Murciano, B. Solsona, S. Agouram, R. Sanchis, J. M. López, T. García, R. Zanella. Catal. Sci. Technol. 7, 2886 (2017). https://doi.org/10.1039/C7CY00635G.Search in Google Scholar
[22] S. Albonetti, R. Bonelli, R. Delaigle, C. Femoni, E. M. Gaigneaux, V. Morandi, L. Ortolani, C. Tiozzo, S. Zacchini, F. Trifirò. Appl. Catal. Gen. 372, 138 (2010). https://doi.org/10.1016/j.apcata.2009.10.029.Search in Google Scholar
[23] W. Zhan, J. Wang, H. Wang, J. Zhang, X. Liu, P. Zhang, M. Chi, Y. Guo, Y. Guo, G. Lu, S. Sun, S. Dai, H. Zhu. J. Am. Chem. Soc. 139, 8846 (2017). https://doi.org/10.1021/jacs.7b01784.Search in Google Scholar PubMed
[24] S. Guadix-Montero, H. Alshammari, R. Dalebout, E. Nowicka, D. J. Morgan, G. Shaw, Q. He, M. Sankar. Appl. Catal. Gen. 546, 58 (2017). https://doi.org/10.1016/j.apcata.2017.07.045.Search in Google Scholar
[25] E. Falletta, C. Della Pina, M. Rossi, Q. He, C. J. Kiely, G. J. Hutchings. Faraday Discuss. 152, 367 (2011). https://doi.org/10.1039/c1fd00063b.Search in Google Scholar PubMed
[26] P. Kaminski, M. Ziolek, J. A. van Bokhoven. RSC Adv. 7, 7801 (2017). https://doi.org/10.1039/C6RA27671G.Search in Google Scholar
[27] B. Hao, M. J. Gunaratna, M. Zhang, S. Weerasekara, S. N. Seiwald, V. T. Nguyen, A. Meier, D. H. Hua. J. Am. Chem. Soc. 138, 16839 (2016). https://doi.org/10.1021/jacs.6b12113.Search in Google Scholar PubMed PubMed Central
[28] E. Pakrieva, E. Kolobova, G. Mamontov, N. Bogdanchikova, M. H. Farias, L. Pascual, V. Cortés Corberán, S. Martinez Gonzalez, S. A. C. Carabineiro, A. Pestryakov. ChemCatChem 11, 1615 (2019). https://doi.org/10.1002/cctc.201801566.Search in Google Scholar
[29] Y. Kotolevich, O. Martynyuk, S. Martínez-González, H. Tiznado, A. Pestryakov, M. Avalos Borja, V. Cortés Corberán, N. Bogdanchikova. Fuel 236, 589 (2019). https://doi.org/10.1016/j.fuel.2018.09.050.Search in Google Scholar
[30] A. Villa, D. Wang, D. S. Su, L. Prati. Catal. Sci. Technol. 5, 55 (2015). https://doi.org/10.1039/C4CY00976B.Search in Google Scholar
[31] C. Yang, F. Zhang, N. Lei, M. Yang, F. Liu, Z. Miao, Y. Sun, X. Zhao, A. Wang. Chin. J. Catal. 39, 1366 (2018). https://doi.org/10.1016/S1872-2067(18)63103-1.Search in Google Scholar
[32] C. M. Olmos, L. E. Chinchilla, A. Villa, J. J. Delgado, A. B. Hungría, G. Blanco, L. Prati, J. J. Calvino, X. Chen. J. Catal. 375, 44 (2019). https://doi.org/10.1016/j.jcat.2019.05.002.Search in Google Scholar
[33] L. A. Calzada, C. Louis, C. Wan Han, V. Ortalan, R. Zanella. Appl. Catal. B Environ. 264, 118503 (2020). https://doi.org/10.1016/j.apcatb.2019.118503.Search in Google Scholar
[34] L.-M. Luo, W. Zhan, R.-H. Zhang, Q.-Y. Hu, Y.-F. Guo, X.-W. Zhou. J. Catal. 381, 316 (2020). https://doi.org/10.1016/j.jcat.2019.11.010.Search in Google Scholar
[35] B. Katryniok, H. Kimura, E. Skrzyńska, J.-S. Girardon, P. Fongarland, M. Capron, R. Ducoulombier, N. Mimura, S. Paul, F. Dumeignil. Green Chem. 13, 1960 (2011). https://doi.org/10.1039/c1gc15320j.Search in Google Scholar
[36] A. Wang, X. Y. Liu, C.-Y. Mou, T. Zhang. J. Catal. 308, 258 (2013). https://doi.org/10.1016/j.jcat.2013.08.023.Search in Google Scholar
[37] H.-L. Jiang, Q. Xu. J. Mater. Chem. 21, 13705 (2011). https://doi.org/10.1039/c1jm12020d.Search in Google Scholar
[38] L. Prati, A. Villa. Acc. Chem. Res. 47, 855 (2014). https://doi.org/10.1021/ar400170j.Search in Google Scholar PubMed
[39] O. A. Kirichenko, G. I. Kapustin, O. P. Tkachenko, V. D. Nissenbaum, I. V. Mishin, N. A. Davshan, E. A. Redina, L. M. Kustov. Mater. Res. Bull. 80, 139 (2016). https://doi.org/10.1016/j.materresbull.2016.03.042.Search in Google Scholar
[40] O. A. Kirichenko, N. A. Davshan, E. A. Redina, G. I. Kapustin, I. V. Mishin, O. P. Tkachenko, A. V. Kucherov, L. M. Kustov. Chem. Eng. J. 292, 62 (2016). https://doi.org/10.1016/j.cej.2016.01.101.Search in Google Scholar
[41] P. Mäki-Arvela, D. Yu. Murzin. Appl. Catal. Gen. 451, 251 (2013). https://doi.org/10.1016/j.apcata.2012.10.012.Search in Google Scholar
[42] C. L. Bianchi, P. Canton, N. Dimitratos, F. Porta, L. Prati. Catal. Today 102–103, 203 (2005). https://doi.org/10.1016/j.cattod.2005.02.003.Search in Google Scholar
[43] J. A. Lopez-Sanchez, N. Dimitratos, C. Hammond, G. L. Brett, L. Kesavan, S. White, P. Miedziak, R. Tiruvalam, R. L. Jenkins, A. F. Carley, D. Knight, C. J. Kiely, G. J. Hutchings. Nat. Chem. 3, 551 (2011). https://doi.org/10.1038/nchem.1066.Search in Google Scholar PubMed
[44] Y. Nakagawa, K. Tomishige. Catal. Commun. 12, 154 (2010). https://doi.org/10.1016/j.catcom.2010.09.003.Search in Google Scholar
[45] E. A. Redina, A. A. Greish, I. V. Mishin, G. I. Kapustin, O. P. Tkachenko, O. A. Kirichenko, L. M. Kustov. Catal. Today 241, 246 (2015). https://doi.org/10.1016/j.cattod.2013.11.065.Search in Google Scholar
[46] E. A. Redina, K. V. Vikanova, A. A. Shesterkina, L. M. Kustov. Russ. J. Phys. Chem. 92, 2143 (2018). https://doi.org/10.1134/S003602441811033X.Search in Google Scholar
[47] E. A. Redina, O. A. Kirichenko, A. A. Greish, A. V. Kucherov, O. P. Tkachenko, G. I. Kapustin, I. V. Mishin, L. M. Kustov. Catal. Today 246, 216 (2015). https://doi.org/10.1016/j.cattod.2014.12.018.Search in Google Scholar
[48] E. Redina, A. Greish, R. Novikov, A. Strelkova, O. Kirichenko, O. Tkachenko, G. Kapustin, I. Sinev, L. Kustov. Appl. Catal. Gen. 491, 170 (2015). https://doi.org/10.1016/j.apcata.2014.11.039.Search in Google Scholar
[49] O. Kirichenko, E. Redina, N. Davshan, I. Mishin, G. Kapustin, T. Brueva, L. Kustov, W. Li, C. Kim. Appl. Catal. B Environ. 134–135, 123 (2013). https://doi.org/10.1016/j.apcatb.2012.12.039.Search in Google Scholar
[50] L. Ning, S. Liao, C. Dong, M. Zhang, W. Gu, X. Liu. ACS Appl. Mater. Interfaces 12, 7198 (2020). https://doi.org/10.1021/acsami.9b19867.Search in Google Scholar PubMed
[51] N. Singh, U. Sanyal, G. Ruehl, K. A. Stoerzinger, O. Y. Gutiérrez, D. M. Camaioni, J. L. Fulton, J. A. Lercher, C. T. Campbell. J. Catal. 382, 372 (2020). https://doi.org/10.1016/j.jcat.2019.12.034.Search in Google Scholar
[52] C. Sakthivel, L. Keerthana, I. Prabha. Johnson Matthey Technol. Rev. 63, 122 (2019). https://doi.org/10.1595/205651319X15498900266305.Search in Google Scholar
[53] G. Wang, H. Xin, Q. Wang, P. Wu, X. Li. J. Catal. 382, 1 (2020). https://doi.org/10.1016/j.jcat.2019.12.004.Search in Google Scholar
[54] A. Huang, R. Nie, B. Zhang, Y. Pei, M. Chen, R. Behera, J. Yu, X. Luan, N. T. Hunter, M. Ke, W. Huang. Chem. Cat. Chem. 12, 602 (2020). https://doi.org/10.1002/cctc.201901930.Search in Google Scholar
[55] Y. Nishida, C. Chaudhari, H. Imatome, K. Sato, K. Nagaoka. Chem. Lett. 47, 938 (2018). https://doi.org/10.1246/cl.180349.Search in Google Scholar
[56] D. Zhu, L. Long, J. Sun, H. Wan, S. Zheng. Appl. Surf. Sci. 504, 144329 (2020). https://doi.org/10.1016/j.apsusc.2019.144329.Search in Google Scholar
[57] H. Xin, Y. Xue, W. Zhang, P. Wu, X. Li. J. Catal. 380, 254 (2019). https://doi.org/10.1016/j.jcat.2019.09.042.Search in Google Scholar
[58] Y. Ren, Y. Tang, L. Zhang, X. Liu, L. Li, S. Miao, D. Sheng Su, A. Wang, J. Li, T. Zhang. Nat. Commun. 10, 4500 (2019). https://doi.org/10.1038/s41467-019-12459-0.Search in Google Scholar PubMed PubMed Central
[59] J. Zhang, Z. Gao, S. Wang, G. Wang, X. Gao, B. Zhang, S. Xing, S. Zhao, Y. Qin. Nat. Commun. 10, 4166 (2019). https://doi.org/10.1038/s41467-019-11970-8.Search in Google Scholar PubMed PubMed Central
[60] P. Concepción, A. Corma, J. Silvestre-Albero, V. Franco, J. Y. Chane-Ching. J. Am. Chem. Soc. 126, 5523 (2004). https://doi.org/10.1021/ja031768x.Search in Google Scholar PubMed
[61] C. Guo, W. Lu, G. Wei, L. Jiang, Y. Yu, Y. Hu. Appl. Surf. Sci. 457, 1136 (2018). https://doi.org/10.1016/j.apsusc.2018.07.050.Search in Google Scholar
[62] L. Zhang, M. Zhou, A. Wang, T. Zhang. Chem. Rev. 120, 683 (2020). https://doi.org/10.1021/acs.chemrev.9b00230.Search in Google Scholar
[63] Y. Lykhach, T. Staudt, M. Vorokhta, T. Skála, V. Johánek, K. C. Prince, V. Matolín, J. Libuda. J. Catal 285, 6 (2012). https://doi.org/10.1016/j.jcat.2011.09.002.Search in Google Scholar
[64] S. Salasc, V. Perrichon, M. Primet, M. Chevrier, N. Mouaddib-Moral. Studies in Surface Science and Catalysis, Elsevier, 130, 3297, (2000). https://doi.org/10.1016/S0167-2991(00)80531-5.Search in Google Scholar
[65] T. Braunschweig, U. Roland, H. Winkler. Studies in Surface Science and Catalysis, Elsevier, 77, 183 (1993). https://doi.org/10.1016/S0167-2991(08)63172-9.Search in Google Scholar
[66] A. Gutsze, U. Roland. Studies in Surface Science and Catalysis, Elsevier, 112, 417 (1997). https://doi.org/10.1016/S0167-2991(97)80864-6.Search in Google Scholar
[67] D.-L. Hoang, H. Berndt, H. Lieske. Catal Lett 31, 165 (1995). https://doi.org/10.1007/BF00808830.Search in Google Scholar
[68] S. Alayoglu, K. An, G. Melaet, S. Chen, F. Bernardi, L. W. Wang, A. E. Lindeman, N. Musselwhite, J. Guo, Z. Liu, M. A. Marcus, G. A. Somorjai. J. Phys. Chem. C 117, 26608 (2013). https://doi.org/10.1021/jp407280e.Search in Google Scholar
[69] F. Yang, B. Hu, W. Xia, B. Peng, J. Shen, M. Muhler. J. Catal. 365, 55 (2018). https://doi.org/10.1016/j.jcat.2018.06.020.Search in Google Scholar
[70] G. Dutta, U. V. Waghmare, T. Baidya, M. S. Hegde. Chem. Mater 19, 6430 (2007). https://doi.org/10.1021/cm071330m.Search in Google Scholar
[71] W. Karim, C. Spreafico, A. Kleibert, J. Gobrecht, J. VandeVondele, Y. Ekinci, J. A. van Bokhoven. Nature 541, 68 (2017). https://doi.org/10.1038/nature20782.Search in Google Scholar PubMed
[72] J.-H. Guo, X.-D. Li, X.-L. Cheng, H.-Y. Liu, S.-J. Li, G. Chen. Int. J. Hydrogen Energy 43, 19121 (2018). https://doi.org/10.1016/j.ijhydene.2018.08.143.Search in Google Scholar
[73] H. Miyamura, A. Suzuki, T. Yasukawa, S. Kobayashi. J. Am. Chem. Soc. 140, 11325 (2018). https://doi.org/10.1021/jacs.8b06015.Search in Google Scholar PubMed
[74] S. Bai, L. Bu, Q. Shao, X. Zhu, X. Huang. J. Am. Chem. Soc. 140, 8384 (2018). https://doi.org/10.1021/jacs.8b03862.Search in Google Scholar PubMed
[75] K. Ding, D. A. Cullen, L. Zhang, Z. Cao, A. D. Roy, I. N. Ivanov, D. Cao. Science 362, 560 (2018). https://doi.org/10.1126/science.aau4414.Search in Google Scholar PubMed
[76] Y. Wang, H. Arandiyan, J. Scott, K.-F. Aguey-Zinsou, R. Amal. ACS Appl. Energy Mater. 1, 6781 (2018). https://doi.org/10.1021/acsaem.8b00817.Search in Google Scholar
[77] H. Zhao, G. Yu, M. Yuan, J. Yang, D. Xu, Z. Dong. Nanoscale 10, 21466 (2018). https://doi.org/10.1039/C8NR05756G.Search in Google Scholar
[78] Z. Zhang, Y. Li, J. Gu, L. Ding, N. Xue, L. Peng, X. Guo, Y. Zhu, J. Ma, W. Ding. Catal. Sci. Technol 8, 6384 (2018). https://doi.org/10.1039/C8CY01867G.Search in Google Scholar
[79] S. Li, Y. Yang, Y. Wang, H. Liu, J. Tai, J. Zhang, B. Han. Catal. Sci. Technol. 8, 4314 (2018). https://doi.org/10.1039/C8CY00969D.Search in Google Scholar
[80] S. Takenaka, R. Kaji, K. Sugiyama, R. Ida. Appl. Catal. Gen. 566, 52 (2018). https://doi.org/10.1016/j.apcata.2018.08.021.Search in Google Scholar
[81] Y. Long, S. Song, J. Li, L. Wu, Q. Wang, Y. Liu, R. Jin, H. Zhang. ACS Catal. 8, 8506 (2018). https://doi.org/10.1021/acscatal.8b01851.Search in Google Scholar
[82] S. Damyanova, B. Pawelec, K. Arishtirova, M. V. M. Huerta, J. L. G. Fierro. Appl. Catal. Gen. 337, 86 (2008). https://doi.org/10.1016/j.apcata.2007.12.005.Search in Google Scholar
[83] E. A. Redina, K. V. Vikanova, G. I. Kapustin, I. V. Mishin, O. P. Tkachenko, L. M. Kustov. Eur. J. Org. Chem. 2019, 4159 (2019). https://doi.org/10.1002/ejoc.201900215.Search in Google Scholar
[84] N. Kakuta, Y. Kudo, H. Rachi, H. Ohkita, T. Mizushima. Top. Catal. 42–43, 377 (2007). https://doi.org/10.1007/s11244-007-0209-8.Search in Google Scholar
[85] O. A. Kirichenko, G. W. Graham, W. Chun, R. W. McCabe. in Studies in Surface Science and Catalysis, 118, 411, Elsevier (1998). https://doi.org/10.1016/S0167-2991(98)80207-3.Search in Google Scholar
[86] K. V. Vikanova, E. A. Redina, G. I. Kapustin, N. A. Davshan, L. M. Kustov. Russ. J. Phys. Chem. 93, 231 (2019). https://doi.org/10.1134/S003602441901031X.Search in Google Scholar
[87] K. V. Vikanova, E. A. Redina. Russ. J. Phys. Chem. 92, 2355 (2018). https://doi.org/10.1134/S0036024418120464.Search in Google Scholar
[88] S. García-Fernández, I. Gandarias, J. Requies, M. B. Güemez, S. Bennici, A. Auroux, P. L. Arias. J. Catal. 323, 65 (2015). https://doi.org/10.1016/j.jcat.2014.12.028.Search in Google Scholar
[89] Y. Fan, S. Cheng, H. Wang, J. Tian, S. Xie, Y. Pei, M. Qiao, B. Zong. Appl. Catal. B Environ. 217, 331 (2017). https://doi.org/10.1016/j.apcatb.2017.06.011.Search in Google Scholar
[90] C. Wang, J. D. Lee, Y. Ji, T. M. Onn, J. Luo, C. B. Murray, R. J. Gorte. Catal Lett 148, 1047 (2018). https://doi.org/10.1007/s10562-018-2323-6.Search in Google Scholar
[91] W. Liu, A. K. Mohanty, L. T. Drzal, M. Misra, J. V. Kurian, R. W. Miller, N. Strickland. Ind. Eng. Chem. Res. 44, 857 (2005). https://doi.org/10.1021/ie049112f.Search in Google Scholar
[92] C. Sarathchandran, C. Chan, S. R. Karim. in Physical Chemistry of Macromolecules, S. Thomas, (Ed.), 573, Apple Academic Press (2014). https://doi.org/10.1201/b16706-22.Search in Google Scholar
[93] N. Lei, X. Zhao, B. Hou, M. Yang, M. Zhou, F. Liu, A. Wang, T. Zhang. ChemCatChem 11, 3903 (2019). https://doi.org/10.1002/cctc.201900689.Search in Google Scholar
[94] S. García-Fernández, I. Gandarias, J. Requies, F. Soulimani, P. L. Arias, B. M. Weckhuysen. Appl. Catal. B. Environ. 204, 260 (2017). https://doi.org/10.1016/j.apcatb.2016.11.016.Search in Google Scholar
[95] O. Kirichenko, V. Nissenbaum, G. Kapustin, E. Redina, K. Vikanova, N. Davshan, L. Kustov. J. Therm. Anal. Calorim. 138, 2205 (2019). https://doi.org/10.1007/s10973-019-08322-5.Search in Google Scholar
[96] W. Bonrath, J. Medlock, J. Schutz, B. Wustenberg, T. Netscher. in Hydrogenation, I. Karam, (Ed.), InTech (2012). https://doi.org/10.5772/48751.Search in Google Scholar
[97] M. Crespo-Quesada, F. Cárdenas-Lizana, A.-L. Dessimoz, L. Kiwi-Minsker. ACS Catal. 2, 1773 (2012). https://doi.org/10.1021/cs300284r.Search in Google Scholar
[98] S. A. Nikolaev, V. V. Smirnov. Catal. Today 147, S336 (2009). https://doi.org/10.1016/j.cattod.2009.07.032.Search in Google Scholar
[99] E. V. Golubina, E. S. Lokteva, A. V. Erokhin, A. A. Veligzhanin, Ya. V. Zubavichus, V. A. Likholobov, V. V. Lunin. J. Catal. 344, 90 (2016). https://doi.org/10.1016/j.jcat.2016.08.017.Search in Google Scholar
[100] E. A. Karakhanov, A. L. Maximov, E. M. Zakharyan, A. V. Zolotukhina, A. O. Ivanov. Mol. Catal. 440, 107 (2017). https://doi.org/10.1016/j.mcat.2017.07.011.https://doi.org/10.1016/j.mcat.2017.07.011Search in Google Scholar
[101] E. V. Belyaeva, V. I. Isaeva, E. E. Said-Galiev, O. P. Tkachenko, S. V. Savilov, A. V. Egorov, L. M. Kozlova, V. Z. Sharf, L. M. Kustov. Russ. Chem. Bull. 63, 396 (2014). https://doi.org/10.1007/s11172-014-0443-8.Search in Google Scholar
[102] S. Domínguez-Domínguez, Á. Berenguer-Murcia, B. K. Pradhan, Á. Linares-Solano, D. Cazorla-Amorós. J. Phys. Chem. C 112, 3827 (2008). https://doi.org/10.1021/jp710693u.Search in Google Scholar
[103] W. Yu, H. Hou, Z. Xin, S. Niu, Y. Xie, X. Ji, L. Shao. RSC Adv. 7, 15309 (2017). https://doi.org/10.1039/C7RA00123A.Search in Google Scholar
[104] W. Zhang, F. Wang, X. Li, Y. Liu, Y. Liu, J. Ma. Appl. Surf. Sci. 404, 398 (2017). https://doi.org/10.1016/j.apsusc.2017.01.298.Search in Google Scholar
[105] F. P. da Silva, L. M. Rossi. Tetrahedron 70, 3314 (2014). https://doi.org/10.1016/j.tet.2013.10.051.Search in Google Scholar
[106] Z. Wang, L. Yang, R. Zhang, L. Li, Z. Cheng, Z. Zhou. Catal. Today 264, 37 (2016). https://doi.org/10.1016/j.cattod.2015.08.018.Search in Google Scholar
[107] I. S. Mashkovsky, N. S. Smirnova, P. V. Markov, G. N. Baeva, G. O. Bragina, A. V. Bukhtiyarov, I. P. Prosvirin, A. Y. Stakheev. Mendeleev Commun. 28, 603 (2018). https://doi.org/10.1016/j.mencom.2018.11.013.Search in Google Scholar
[108] A. V. Rassolov, P. V. Markov, G. O. Bragina, G. N. Baeva, I. S. Mashkovskii, I. A. Yakushev, M. N. Vargaftik, A. Y. Stakheev. Kinet. Catal. 57, 853 (2016). https://doi.org/10.1134/S0023158416060124.Search in Google Scholar
[109] P. V. Markov, G. O. Bragina, A. V. Rassolov, G. N. Baeva, I. S. Mashkovsky, V. Yu. Murzin, Y. V. Zubavichus, A. Y. Stakheev. Mendeleev Commun. 26, 502 (2016). https://doi.org/10.1016/j.mencom.2016.11.014.Search in Google Scholar
[110] K. C. K. Swamy, A. S. Reddy, K. Sandeep, A. Kalyani. Tetrahedron Lett. 59, 419 (2018). https://doi.org/10.1016/j.tetlet.2017.12.057.Search in Google Scholar
[111] A. V. B. Reddy, Z. Yusop, J. Jaafar, Y. V. M. Reddy, A. B. Aris, Z. A. Majid, J. Talib, G. Madhavi. J. Environ. Chem. Eng. 4, 3537 (2016). https://doi.org/10.1016/j.jece.2016.07.035.Search in Google Scholar
[112] A. A. Shesterkina, O. P. Tkachenko, E. V. Shuvalova, G. I. Kapustin, V. B. Kazansky, L. M. Kustov. Mendeleev Commun. 29, 666 (2019). https://doi.org/10.1016/j.mencom.2019.11.021.Search in Google Scholar
[113] O. Kirichenko, A. Strekalova, G. Kapustin, A. Shesterkina, E. Redina, L. Kustov. J. Therm. Anal. Calorim. 138, 1913 (2019). https://doi.org/10.1007/s10973-019-08786-5.Search in Google Scholar
[114] O. A. Kirichenko, A. A. Strekalova, G. I. Kapustin, A. A. Shesterkina. Russ. J. Phys. Chem. 92, 2396 (2018). https://doi.org/10.1134/S003602441812021X.Search in Google Scholar
[115] A. A. Shesterkina, O. A. Kirichenko, L. M. Kozlova, G. I. Kapustin, I. V. Mishin, A. A. Strelkova, L. M. Kustov. Mendeleev Commun. 26, 228 (2016). https://doi.org/10.1016/j.mencom.2016.05.002.Search in Google Scholar
[116] A. A. Shesterkina, L. M. Kozlova, O. A. Kirichenko, G. I. Kapustin, I. V. Mishin, L. M. Kustov. Russ. Chem. Bull. 65, 432 (2016). https://doi.org/10.1007/s11172-016-1317-z.Search in Google Scholar
[117] A. A. Shesterkina, L. M. Kozlova, I. V. Mishin, O. P. Tkachenko, G. I. Kapustin, V. P. Zakharov, M. S. Vlaskin, A. Z. Zhuk, O. A. Kirichenko, L. M. Kustov. Mendeleev Commun. 29, 339 (2019). https://doi.org/10.1016/j.mencom.2019.05.034.Search in Google Scholar
[118] A. A. Shesterkina, E. V. Shuvalova, E. A. Redina, O. A. Kirichenko, O. P. Tkachenko, I. V. Mishin, L. M. Kustov. Mendeleev Commun. 27, 512 (2017). https://doi.org/10.1016/j.mencom.2017.09.028.Search in Google Scholar
[119] A. A. Shesterkina, E. V. Shuvalova, O. A. Kirichenko, L. M. Kustov. Russ. J. Phys. Chem. 92, 2412 (2018). https://doi.org/10.1134/S0036024418120348.Search in Google Scholar
[120] F. Cárdenas-Lizana, S. Gómez-Quero, C. J. Baddeley, M. A. Keane. Appl. Catal. Gen. 387, 155 (2010). https://doi.org/10.1016/j.apcata.2010.08.019.Search in Google Scholar
[121] M. H. Brijaldo, F. B. Passos, H. A. Rojas, P. Reyes. Catal. Lett. 144, 860 (2014). https://doi.org/10.1007/s10562-014-1218-4.Search in Google Scholar
[122] O. Kirichenko, G. Kapustin, V. Nissenbaum, A. Strelkova, E. Shuvalova, A. Shesterkina, L. Kustov. J. Therm. Anal. Calorim. 134, 233 (2018). https://doi.org/10.1007/s10973-018-7122-1.Search in Google Scholar
[123] A. A. Shesterkina, E. V. Shuvalova, O. A. Kirichenko, A. A. Strelkova, V. D. Nissenbaum, G. I. Kapustin, L. M. Kustov. Russ. J. Phys. Chem. 91, 201 (2017). https://doi.org/10.1134/S0036024417020285.Search in Google Scholar
[124] O. A. Kirichenko, E. V. Shuvalova, E. A. Redina. Russ. Chem. Bull. 68, 2048 (2019). https://doi.org/10.1007/s11172-019-2665-2.Search in Google Scholar
[125] E. V. Shuvalova, O. A. Kirichenko, G. I. Kapustin, L. M. Kustov. Russ. Chem. Bull. 65, 2850 (2016). https://doi.org/10.1007/s11172-016-1667-6.Search in Google Scholar
[126] E. R. Essinger-Hileman, D. DeCicco, J. F. Bondi, R. E. Schaak. J. Mater. Chem. 21, 11599 (2011). https://doi.org/10.1039/c0jm03913f.Search in Google Scholar
[127] S. Yan, S. Zhang. Int. J. Hydrogen Energy 37, 9636 (2012). https://doi.org/10.1016/j.ijhydene.2012.03.058.Search in Google Scholar
[128] R. K. P. Purushothaman, J van Haveren, D. S. van Es, I. Melián-Cabrera, J. D. Meeldijk, H. J. Heeres. Appl. Catal. B Environ. 147, 92 (2014). https://doi.org/10.1016/j.apcatb.2013.07.068.Search in Google Scholar
[129] V. La Parola, M. Kantcheva, M. Milanova, A. M. Venezia. J. Catal. 298, 170 (2013). https://doi.org/10.1016/j.jcat.2012.11.007.Search in Google Scholar
[130] K. I. Hadjiivanov, G. N. Vayssilov. Advances in Catalysis, Elsevier, 47, 307 (2002). https://doi.org/10.1016/S0360-0564(02)47008-3.Search in Google Scholar
[131] Chen, Y., Cai, Z, Yan, D. W., Goodman. J. Am. Chem. Soc. 128, 6341 (2006). https://doi.org/10.1021/ja0557536.Search in Google Scholar
[132] C. Lemire, R. Meyer, Sh. K. Shaikhutdinov, H.-J. Freund. Surf. Sci. 552, 27 (2004). https://doi.org/10.1016/j.susc.2004.01.029.Search in Google Scholar
[133] M. Bonarowska, B. Burda, W. Juszczyk, J. Pielaszek, Z. Kowalczyk, Z. Karpiński. Appl. Catal. B Environ. 35, 13 (2001). https://doi.org/10.1016/S0926-3373(01)00227-2.Search in Google Scholar
[134] X. Hong, Y. Sun, T. Zhu, Z. Liu. Catal. Sci. Technol. 6, 3606 (2016). https://doi.org/10.1039/C5CY01744K.Search in Google Scholar
[135] S. Demirel, K. Lehnert, M. Lucas, P. Claus. Appl. Catal. B Environ. 70, 637 (2007). https://doi.org/10.1016/j.apcatb.2005.11.036.Search in Google Scholar
[136] N. Pino, S. Sitthisa, Q. Tan, T. Souza, D. López, D. E. Resasco. J. Catal. 350, 30 (2017). https://doi.org/10.1016/j.jcat.2017.03.016.Search in Google Scholar
[137] Y. Tang, S. Cao, Y. Chen, T. Lu, Y. Zhou, L. Lu, J. Bao. Appl. Surf. Sci. 256, 4196 (2010). https://doi.org/10.1016/j.apsusc.2010.01.124.Search in Google Scholar
[138] M. P. Felicissimo, O. N. Martyanov, T. Risse, H.-J. Freund. Surf. Sci. 601, 2105 (2007). https://doi.org/10.1016/j.susc.2007.02.023.Search in Google Scholar
[139] J. C. Bertolini, J. L. Rousset, P. Miegge, J. Massardier, B. Tardy. Surf. Sci. 287–288, 346 (1993). https://doi.org/10.1016/0039-6028(93)90800-Y.Search in Google Scholar
© 2020 IUPAC & De Gruyter. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License. For more information, please visit: http://creativecommons.org/licenses/by-nc-nd/4.0/
Articles in the same Issue
- Frontmatter
- In this issue
- Preface
- Research papers from the 21st Mendeleev Congress on General and Applied Chemistry
- Conference papers of the 21st Mendeleev Congress on General and Applied Chemistry
- Unusual behavior of bimetallic nanoparticles in catalytic processes of hydrogenation and selective oxidation
- Soft chemistry of pure silver as unique plasmonic metal of the Periodic Table of Elements
- Catalytic hydrogenation with parahydrogen: a bridge from homogeneous to heterogeneous catalysis
- Azidophenylselenylation of glycals towards 2-azido-2-deoxy-selenoglycosides and their application in oligosaccharide synthesis
- Bis-γ-carbolines as new potential multitarget agents for Alzheimer’s disease
- Octafluorobiphenyl-4,4′-dicarboxylate as a ligand for metal-organic frameworks: progress and perspectives
- Some aspects of the formation and structural features of low nuclearity heterometallic carboxylates
- Particular kinetic patterns of heavy oil feedstock hydroconversion in the presence of dispersed nanosize MoS2
- Concentration profiles around and chemical composition within growing multicomponent bubble in presence of curvature and viscous effects
- Application of gold nanoparticles in the methods of optical molecular absorption spectroscopy: main effecting factors
- Membrane materials for energy production and storage
- New types of the hybrid functional materials based on cage metal complexes for (electro) catalytic hydrogen production
- Conference paper of the 15th Eurasia Conference on Chemical Sciences
- Discovery of bioactive drug candidates from some Turkish medicinal plants-neuroprotective potential of Iris pseudacorus L.
Articles in the same Issue
- Frontmatter
- In this issue
- Preface
- Research papers from the 21st Mendeleev Congress on General and Applied Chemistry
- Conference papers of the 21st Mendeleev Congress on General and Applied Chemistry
- Unusual behavior of bimetallic nanoparticles in catalytic processes of hydrogenation and selective oxidation
- Soft chemistry of pure silver as unique plasmonic metal of the Periodic Table of Elements
- Catalytic hydrogenation with parahydrogen: a bridge from homogeneous to heterogeneous catalysis
- Azidophenylselenylation of glycals towards 2-azido-2-deoxy-selenoglycosides and their application in oligosaccharide synthesis
- Bis-γ-carbolines as new potential multitarget agents for Alzheimer’s disease
- Octafluorobiphenyl-4,4′-dicarboxylate as a ligand for metal-organic frameworks: progress and perspectives
- Some aspects of the formation and structural features of low nuclearity heterometallic carboxylates
- Particular kinetic patterns of heavy oil feedstock hydroconversion in the presence of dispersed nanosize MoS2
- Concentration profiles around and chemical composition within growing multicomponent bubble in presence of curvature and viscous effects
- Application of gold nanoparticles in the methods of optical molecular absorption spectroscopy: main effecting factors
- Membrane materials for energy production and storage
- New types of the hybrid functional materials based on cage metal complexes for (electro) catalytic hydrogen production
- Conference paper of the 15th Eurasia Conference on Chemical Sciences
- Discovery of bioactive drug candidates from some Turkish medicinal plants-neuroprotective potential of Iris pseudacorus L.