KRAS-driven pancreatic ductal adenocarcinoma cell lines harbour putative cancer stem cells
-
Yuan Han Teh



Abstract
Objectives
Pancreatic cancer stem cells (CSCs) are known culprits of recurrent pancreatic ductal adenocarcinoma (PDAC). The disputable presence of CSCs in continuous cell lines has led to an extensive application of patient-derived CSC culture for experimentation. However, obtaining patient biopsies remains a challenge for many laboratories. Here, this study aimed to investigate the presence of CSCs in KRAS-driven PDAC cell lines.
Methods
Cell viability assays evaluated the cytotoxicity of gemcitabine and vismodegib in PDAC cell lines. Flow cytometry was used to analyse the expression of pancreatic CSC surface markers. Tumoursphere-forming ability was investigated by culturing cells in serum-free and non-adherent conditions in vitro, while the tumourigenicity of PDAC cells was assessed in immunocompromised mice. Molecular events were delineated by Western blotting, and SOX2-expressing cells in tumour xenografts were detected by immunohistochemistry.
Results
PANC-1 and Capan-2 cell lines showed exceptional chemoresistance, with Capan-2 containing a notable number of putative CSCs that co-expressed CD44, CD24, and CD133 (3.7 % of the total population) and sustained the cytotoxicity of gemcitabine. The Capan-2 cell line harbours tumoursphere-forming cells, of which self-renewal was inhibited by foetal bovine serum. The presence of these cells is consistent with Capan-2 being highly tumourigenic in immunocompromised mice. MAPK, PI3K-AKT, NF-κB, and WNT pathways were activated, and SOX2 expression was upregulated during the self-renewal of putative CSCs. SOX2-expressing cells were correspondingly present in xenografts of highly tumourigenic Capan-2 cell lines.
Conclusion
The Capan-2 cell line contains a subset of putative CSCs, whose self-renewal is driven by MAPK, PI3K-AKT, NF-κB, and WNT pathways.
Introduction
With a global incidence of 511,000 cases in 2022, pancreatic ductal adenocarcinoma (PDAC) is the seventh most lethal cancer, with 467,000 deaths recorded worldwide [1]. Treatment options for PDAC remain scarce, and it is limited to curative resection, which is followed by adjuvant chemotherapy with gemcitabine or fluorouracil [2]. Although the adjuvant gemcitabine therapy was reported to have significantly improved the overall survival and disease-free survival of patients with resectable PDAC [2], 75 % of these patients would experience a recurrence that will eventually lead to fatality within two years after diagnosis [3]. Understanding the molecular events driving PDAC initiation and progression is critical for developing effective therapies.
PDAC begins with a neoplastic precursor lesion, known as pancreatic intraepithelial neoplasia (PanIN), and its development is described as a progressive accumulation of multiple genetic mutations [3], 4]. Among the key proto-oncogenes, KRAS is mutated at the early stage of PDAC development, and oncogenic KRAS protein has been found in more than 90 % of the PDAC cases [5], suggesting that it is a promising therapeutic target in the prevention and treatment of PDAC. Unfortunately, KRAS mutants, notably KRASG12D and KRASG12V remain undruggable. To date, KRASG12C is the only druggable mutant, and it signifies a long battle against pancreatic cancers [5]. The pivotal role of KRAS in promoting stem cell-like characteristics has also been confirmed in pancreatic cancer cells [6]. Importantly, oncogenic KRAS not only drives tumour initiation but also plays a pivotal role in maintaining cancer stem cell properties in pancreatic tumours.
Emerging evidence suggests that pancreatic cancer stem cells (CSCs), which possess self-renewal and tumour-initiating capabilities, may be responsible for tumour recurrence, metastasis, and resistance to therapy. Several surface markers, including CD44, CD24, epithelial cell adhesion molecule (EpCAM), and CD133, have been used to identify and isolate pancreatic CSCs [7], 8]. Notably, KRAS mutations have been implicated in regulating CSC-associated traits such as enhanced plasticity, metabolic reprogramming, and stemness maintenance [9]. However, the molecular link between mutant KRAS signalling and the CSC phenotype remains to be fully elucidated. Vismodegib is the earliest known anti-pancreatic CSC agent that targets Hedgehog signalling, of which activity against pancreatic CSCs was proven to be promising preclinically [10]. In a non-adherent culture condition with the presence of epidermal growth factor (EGF), basic fibroblast growth factor (bFGF), and other known pro-stemness factors in a serum-free culture medium, the formation of sphere-like multicellular colonies is a hallmark characteristic of clonally propagating CSCs, including pancreatic CSCs [7], 8], 11]. In fact, tumoursphere-forming ability is correlated to the self-renewal ability of CSCs [12]. To better understand CSC properties, various functional assays such as tumoursphere formation and xenotransplantation have been developed.
The concept of CSC was first introduced by Lapidot et al. in human acute myeloid leukaemia (AML) through a novel xenotransplantation assay [13]. Since then, the xenotransplantation assay has become the gold standard assay to qualitatively define CSCs in solid tumours of the breast [14], colon [15], brain [16], and pancreas [7], 8]. In these studies, each tumour was successfully engrafted in immunocompromised mice with as few as 100 CSCs, whereas a multiple ten-fold greater number of non-CSCs is required for tumourigenesis.
The existence of CSCs in the continuous culture of cancer cell lines remains controversial. Many have questioned the authenticity of these microenvironment-sensitive cells after prolonged exposure to foetal bovine serum (FBS) in vitro. Although patient-derived CSC culture is preferred for experimentation in CSC research, the inaccessibility of patients’ biopsies poses a great hurdle to research activities. Here, we investigated the presence of CSCs in KRAS-driven PDAC cell lines, namely PANC-1 (KRASG12D), Capan-2 (KRASG12V), and MIA PaCa-2 (KRASG12C), and the molecular mechanism of self-renewing pancreatic CSCs was delineated. To overcome this challenge, KRAS-driven PDAC cell lines offer an accessible in vitro platform to study pancreatic CSC biology and test potential small molecules against pancreatic CSCs.
Materials and Methods
Cell culture
PDAC cell lines were obtained from the American Type Culture Collection (ATCC, USA). PANC-1 (Cat# CRL-1469, RRID: CVCL_0480, ATCC) and MIA PaCa-2 (Cat# CRL-1420, RRID: CVCL_0428, ATCC) cell lines were maintained in high-glucose (4,500 mg/L) Dulbecco’s Modified Eagle’s Medium (Gibco, USA), while Capan-2 (Cat# HTB-80, RRID: CVCL_0026, ATCC) cells were cultured in McCoy’s 5A medium (Sigma-Aldrich, USA). All culture media contained 10 % fetal bovine serum as a supplement (FBS; Sigma-Aldrich, USA).
Tumoursphere culture
Following dissociation with Accutase (Nacalai Tesque, Japan), PDAC cells were seeded as single cells into ultra-low attachment 6-well plates (Corning, USA) at a concentration of 5 cells/µL, with 2000 µL of CSC culture medium added to each well. The medium consisted of Dulbecco’s Modified Eagle’s Medium: Nutrient Mixture F-12 (Gibco, USA), the medium was fortified with 1 × B27 lacking 20 ng/mL bFGF and 20 ng/mL EGF (both from Nacalai Tesque, Japan), and vitamin A (Gibco, USA). Cultures were maintained for seven days, and on day three, the tumoursphere culture was supplemented with fresh CSC medium at 10 % of the original volume.
Preparation of test compounds
Sterile, biocompatible DMSO (Sigma-Aldrich, USA) was used to dissolve both gemcitabine (Gemita; Fresenius Kabi Oncology, India) and vismodegib (GDC-0449; APExBIO Technology, USA).
Cell viability assay
PDAC cells (2 × 103 per well) were seeded into 96-well plates (TPP, Switzerland) and treated with the respective compounds for a duration of 96 h. Cell viability was then assessed based on their ability to metabolize MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide). Each well was treated with MTT solution (0.4 g/L; Gibco, USA), followed by incubation at 37 °C with 5 % CO2 for 4 h. The resulting insoluble formazan crystals were dissolved in analytical-grade DMSO (Thermo Fisher Scientific, USA), and absorbance at 550 nm was measured using a microplate reader (VERSAmax, Molecular Devices, San Jose, CA, USA). Cell growth (in percent) was calculated by using the following equations [17]:
When AbsDay0<AbsTreatment
When AbsDay0>AbsTreatment
Dose-response curves were generated, and growth inhibition metrics – including GI50, TGI, and LC50 – were calculated. GI50 represents the concentration at which 50 % growth inhibition is observed. TGI indicates the concentration required for complete growth arrest (cytostatic effect), while LC50 corresponds to the concentration that causes cytocidal effect or cell death.
Flow cytometry
PDAC cells were detached and dissociated with Accutase. Single cells (1 × 106/100 µL) were stained with human CD44-FITC (Cat# 130-113-334, RRID: AB_2726110, Miltenyi Biotec), CD24-PE (Cat# 130-095-953, RRID: AB_10828818, Miltenyi Biotec), and CD133-APC (Cat# 130-113-106, RRID: AB_2725935, Miltenyi Biotec) antibodies (Miltenyi Biotec, Germany) by diluting the antibodies at 1:50 ratio in fluorescence-activated cell sorting (FACS) staining buffer: 0.5 % (w/v) BSA (Sigma-Aldrich, USA), 2 mM EDTA (Sigma-Aldrich, USA) in 1 × phosphate-buffered saline (PBS; pH 7.4) at 4 °C in the dark for 10 min. Dead cells were stained with 5 µL (0.25 µg) 7-Aminoactinomycin D (7-AAD; BD Biosciences, USA) at 4 °C in the dark for 10 min. Data were acquired by using the BD FACSCanto™ II flow cytometer (BD Biosciences, USA) and analysed with BD FACSDiva™ software (version 8.0, RRID: SCR_001456, BD Biosciences, USA).
FBS-induced inhibition assay
As Capan-2 is the only cell line showing positive in all tests for pancreatic CSC characteristics, namely resisting gemcitabine but responding to vismodegib as well as expressing CD44, CD24, and CD133 surface markers, Capan-2 cells are subjected to further studies. Capan-2 cells were detached and dissociated with Accutase. A single-cell suspension of each cell line was halved. The first half was processed according to the aforementioned tumoursphere culture protocol, while the second half was cultured in Dulbecco’s Modified Eagle’s Medium: Nutrient Mixture F-12 medium supplemented with 10 % (v/v) FBS. In either culture, cells were seeded onto an ultra-low attachment 96-well plate at a density of 5 cells/µL in 200 µL per well of respective media and incubated for seven days. On the third day, both cultures were replenished with 1/10 volume of their respective fresh culture media. The number of tumourspheres, whose diameter is >60 µm [18], was counted under Primo Vert inverted microscope (Carl Zeiss, Germany).
In vivo tumourigenicity assay
An experiment involving animals was approved by the Institutional Animal Care and Use Committee (IACUC) of Universiti Putra Malaysia (NO. UPM/IACUC/AUP-R044/2017). Study design, experimental procedures, and housing and husbandry adhered to IACUC standards for the protection of animals used for scientific purposes. As required by IACUC guidelines, the use of animals was considered to be inevitable to study the presence of CSCs, which are also known as tumour-initiating cells (TICs). Subcutaneous inoculation of pancreatic cancer cells was opted, as it is the least invasive to minimise pain and distress potentially experienced by the animals. During the study, animals were kept in a clean environment with weekly changes of cages and bedding to support their physical and psychological well-being. Sufficient training was given to researchers, who managed subcutaneous inoculation, measured tumour dimensions, transferred mice to clean cages, and changed bedding. IACUC compliance of this study was inspected by trained staffs working at the animal facility. At the endpoint of the study, mice were euthanised with carbon dioxide based on IACUC-approved protocol. There is no inclusion or exclusion criteria in this study. Simple randomisation was done using online random number generators (https://www.graphpad.com/quickcalcs/randomize1/) that splitted a total of 36 NCr nude mice aged 6–8 months (IMSR Cat# TAC:ncrnu, RRID: IMSR_TAC:ncrnu, Taconic Biosciences, USA) into groups of six with an equal number of male and female mice in each group. PDAC cells were detached and dissociated with Accutase and resuspended in PBS at a density of either 1 × 105/100 µL or 1 × 106/100 µL before they were inoculated into each mouse through subcutaneous administration. The length (the longest dimension) and width (the perpendicular dimension of the length) of each tumour were measured every 10 days and the tumour volume was calculated by using the following formula: Tumour volume (mm3)=(Length × Width2)/2. Tumour with a volume of ≥100 mm3 was counted for tumourigenicity. The mean latency period of tumour engraftment (indicated by steep descents on the curve) was calculated by averaging time points of occurrence, at which palpable tumours reach 100 mm3.
Protein analysis
Cells were lysed with immunoprecipitation (IP) lysis buffer: 25 mM Tris-HCl (pH 7.4), 150 mM sodium chloride, 1 % (v/v) Triton X-100, 1 mM EDTA, 50 mM magnesium chloride, 2 % (v/v) glycerol, 1 × protease inhibitor cocktail (Nacalai Tesque, Japan), and 1 × phosphatase inhibitor cocktail (Nacalai Tesque, Japan). Cell lysates were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblotting. The primary antibodies used include p-CRAF (Cat# 9427, RRID: AB_2067317, Cell Signalling Technology, USA), CRAF (Cat# 12552, RRID: AB_2728706, Cell Signalling Technology), p-ERK1/2 (Cat# 9106, RRID: AB_331768, Cell Signalling Technology), ERK1/2 (Cat# 4696, RRID: AB_390780, Cell Signalling Technology), p-AKT (Cat# 4060, RRID: AB_2315049, Cell Signalling Technology), AKT (Cat# 4685, RRID: AB_2225340, Cell Signalling Technology), p-NF-κB P65 (Cat# 3033, RRID: AB_331284, Cell Signalling Technology), NF-κB P65 (Cat# 8242, RRID: AB_10859369, Cell Signalling Technology), p-GSK3β (Cat# 5558, RRID: AB_10013750, Cell Signalling Technology), GSK3β (Cat# 12456, RRID: AB_2636978, Cell Signalling Technology), SOX2 (Cat# 3579, RRID: AB_2195767, Cell Signalling Technology), OCT4 (Cat# 2750, RRID: AB_823583, Cell Signalling Technology), GAPDH (Cat# 5174, RRID: AB_10622025, Cell Signalling Technology), and β-actin (Cat# 4970, RRID: AB_2223172, Cell Signalling Technology). Whereas primary antibodies of KLF4 (Cat# sc-393462, RRID: AB_3662136) and CMYC (Cat# sc-40, RRID: AB_627268) were purchased from Santa Cruz Biotechnology, USA. The HRP-linked secondary antibodies (Cell Signalling Technology, USA) used include mouse IgG (Cat# 7076, RRID: AB_330924, Cell Signalling Technology) and rabbit IgG (Cat# 7074, RRID: AB_2099233, Cell Signalling Technology). All antibodies were used at the dilutions as recommended by their corresponding manufacturers.
Statistical analysis
The results of continuous variables are expressed as mean ± standard deviation (s.d.). Pairwise multiple comparisons were achieved by performing One-Way ANOVA with Tukey adjustment. The statistical significance of the difference between the two groups was determined by the Independent-Samples t-Test. Kaplan-Meier survival analyses were performed with three pairwise comparison algorithms: log-rank method, Breslow test, and Tarone-Ware. All analyses were performed with IBM SPSS Statistics 20 (RRID: SCR_002865, IBM Corp., USA), except for limiting dilution analysis. In the case of limiting dilution transplantation, tumour-initiating cell (TIC) frequency was analysed by the single-hit Poisson model using the ELDA software, which is provided by Walter and Eliza Hall Institute (http://bioinf.wehi.edu.au/software/elda/index.html) [19]. p-values ≤0.05 denote statistically significant, except for the quantitative evaluation of immunoblots (p-values ≤0.1 denote statistically significant).
Results
PANC-1 and Capan-2 cell lines demonstrate remarkable chemoresistance to gemcitabine
The strength of inhibition exerted by gemcitabine on the growth of KRAS-driven PDAC cell lines was invariable, as indicated by statistically non-significant (p>0.05) differences in the GI50 (Table 1) when comparing Capan-2 cell line (p=0.758) and MIA PaCa-2 cell line (p=0.100) to PANC-1 cell line. Nevertheless, it is notable that PANC-1 (p<0.001) and Capan-2 (p<0.001) cells were remarkably more resistant to gemcitabine than MIA PaCa-2 cells, as indicated by their respective TGI values (Table 1). All cell lines resisted the cytocidal effect of gemcitabine, as suggested by their LC50 values >100 µM. Capan-2 cells demonstrated moderate sensitivity to vismodegib treatment (Table 1). PANC-1 and MIA PaCa-2 cells were found to be significantly less responsive to vismodegib treatment as compared with Capan-2 cells (p=0.005; Table 1). The dose-response growth inhibitory effect of gemcitabine and vismodegib on PDAC cell lines is shown in Figure 1A and B.
Growth inhibitory parameters (GI50, TGI, and LC50) of gemcitabine and vismodegib in human PDAC cell lines.
| PDAC cell lines | Gemcitabine | Vismodegib | ||||
|---|---|---|---|---|---|---|
| GI50a | TGIa | LC50a | GI50a | TGIa | LC50a | |
| PANC-1 | 0.11 ± 0.06 | >100 | >100 | >100 | >100 | >100 |
| Capan-2 | 0.08 ± 0.05 | >100 | >100 | 49.3 ± 3.1 | >100 | >100 |
| MIA PaCa-2 | 0.03 | 0.12 ± 0.03 | >100 | >100 | >100 | >100 |
-
PDAC, pancreatic ductal adenocarcinoma; GI50, the concentration that causes 50 % growth inhibition; TGI, the concentration that causes cytostatic effect; LC50, the concentration that causes cytocidal effect. aGI50, TGI, and LC50 values (µM) are expressed as mean ± standard deviation (s.d.) (n=3).
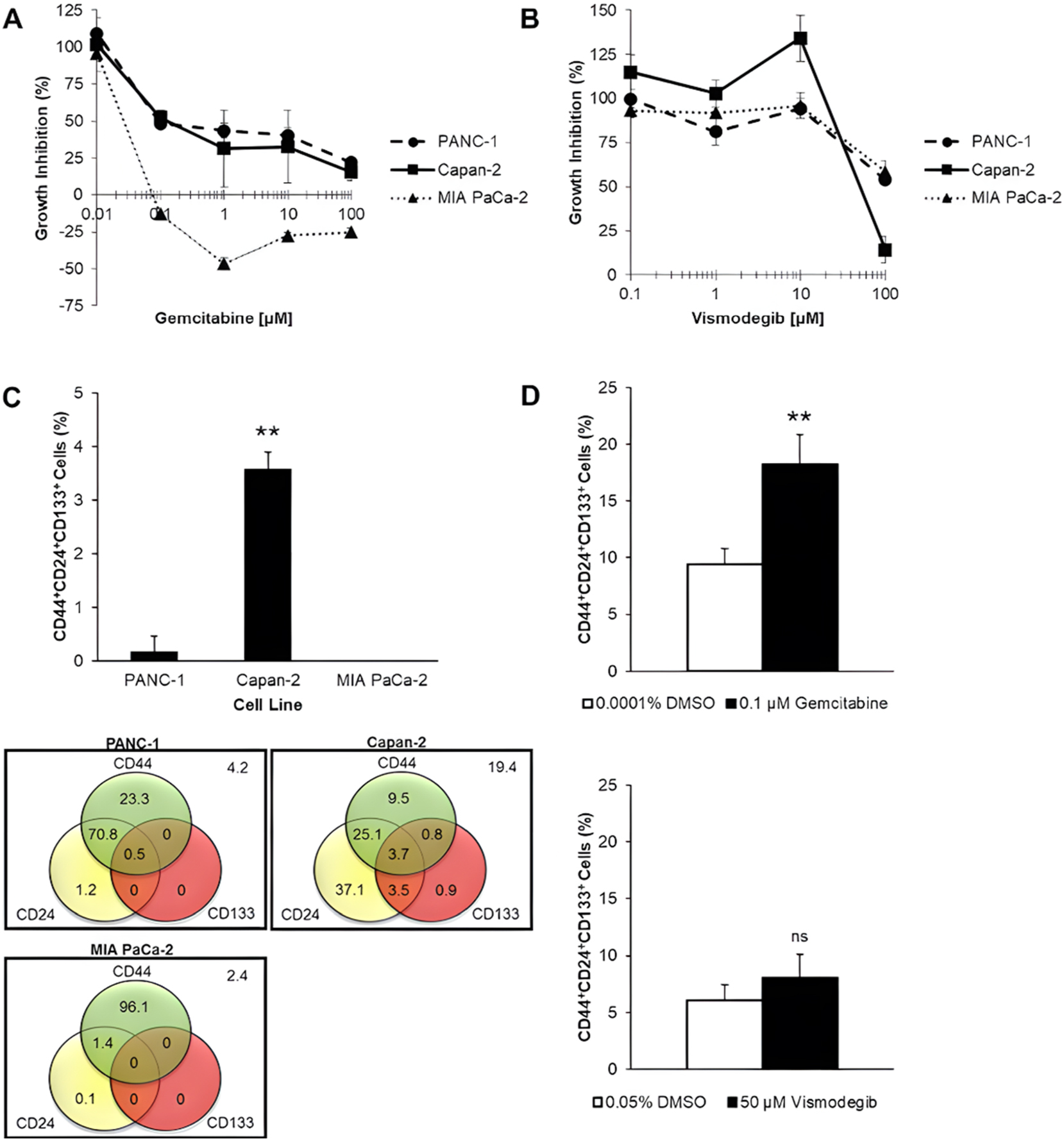
Presence of CSCs in KRAS-driven PDAC cell lines. PDAC cell lines (PANC-1, Capan-2, and MIA PaCa-2) were treated with (A) gemcitabine and (B) vismodegib for 96 h. Data are expressed in mean ± s.d. (n=3 technical replicates). PANC-1 and Capan-2 cells resist gemcitabine, and vismodegib moderately inhibits Capan-2 growth. (C) The composition of the total cell population, in percent, based on pancreatic CSC surface markers (CD44, CD24, and CD133), is displayed in a Venn diagram. The bar chart shows the percentage of CD44+CD24+CD133+ cells in PDAC cell lines. Data are expressed in mean ± s.d. (n=3 independent experiments). Only 3.7 % of Capan-2 cells are CD44+CD24+CD133+. (D) Capan-2 cells were treated with 0.1 µM gemcitabine and 50 µM vismodegib for 96 h. Changes in the CD44+CD24+CD133+ cell subpopulation sizes were detected by flow cytometry. Data are expressed in mean ± s.d. (n=3 independent experiments). Gemcitabine doubled the CD44+CD24+CD133+ cell subpopulation, while vismodegib reduced it. Statistical significance of the differences, which were compared with PANC-1, in the percentage of CD44+CD24+CD133+ cells was analysed by SPSS one-way ANOVA with Tukey adjustment, while the differences between the test compound and its corresponding vehicle control (DMSO) were analysed by SPSS independent-samples t-test (ns, not significant; **p<0.01).
Capan-2 cell line contains CD44+CD24+CD133+ cells
Capan-2 cells exhibited strong resistance to gemcitabine treatment, which implies that the existence of CSCs is probable. It is of great interest to determine whether the intrinsic chemoresistance of Capan-2 cells could be correlated to the presence of CD44+CD24+CD133+ cells, which are considered putative CSCs. Flow cytometric analyses revealed that CD44+CD24+CD133+ cells were detectable in a significantly (p<0.01) larger number in the Capan-2 cell line than in other PDAC cell lines, which accounted for 3.7 % of the total cell line population (Figure 1C). The CD44+CD24+CD133+ cell subpopulation was found in a small quantity of less than 1 % in the PANC-1 cell line (Figure 1C).
CD44+CD24+CD133+ cells of the Capan-2 cell line resist the growth inhibitory effect of gemcitabine
To confirm the gemcitabine-resistant nature of CD44+CD24+CD133+ cells, the effect of gemcitabine on this subpopulation in the Capan-2 cell line was examined. Gemcitabine treatment at a concentration of 0.1 µM is equivalent to its GI50 value in the Capan-2 cell line, significantly (p=0.006) enriching the CD44+CD24+CD133+ cell subpopulation by approximately twofold relative to DMSO vehicle control (Figure 1D). On the other hand, no significant difference was observed in the percentage of CD44+CD24+CD133+ cells when the total population was treated with the GI50 value of vismodegib at 50 µM and the corresponding concentration of DMSO (Figure 1D).
Capan-2 cell line contains cells with tumoursphere-forming ability in vitro
When human PDAC cell lines were cultured in serum-free and non-attached conditions, a proportion of Capan-2 single cells grew into sphere-like colonies with an average diameter of 60 µm, known as tumourspheres, while the other proportion of the single cells remained in single-cell morphology, with their viability being unknown (Figure 2A). Representative microscopic image shows that the tumourspheres were composed of compactly arranged cells within a defined border (Figure 2A). Single cells of PANC-1 and MIA PaCa-2 cell lines, on the other hand, grew larger in number in the form of loosely packed cell aggregates with an irregular morphology, which are generally not considered as tumourspheres (Figure 2A).
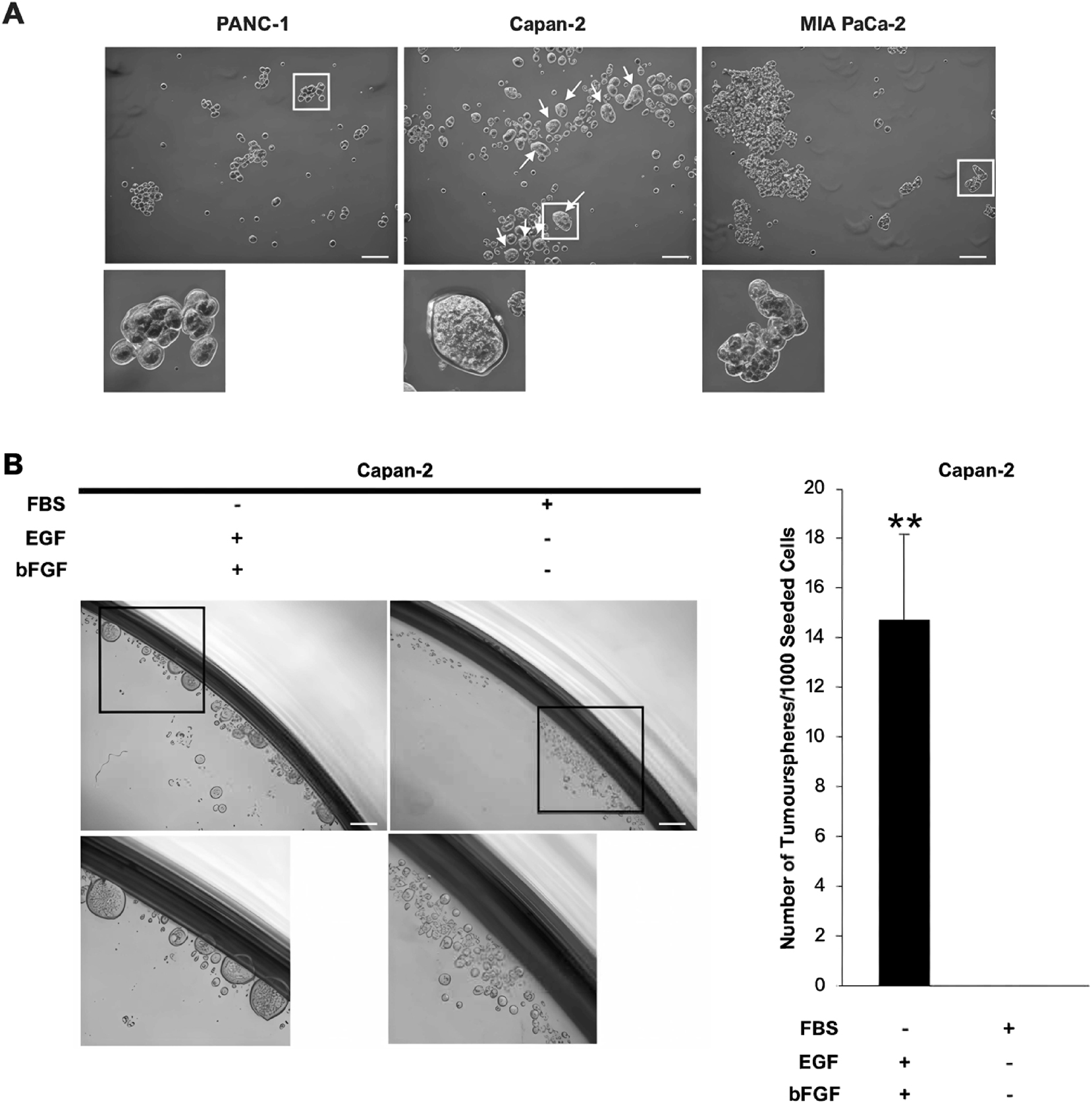
Tumoursphere formation of KRAS-driven PDAC cell lines in vitro. PANC-1, Capan-2, and MIA PaCa-2 cells were cultured in serum-free and non-attached conditions. (A) Representative microscopic images show single cells of Capan-2 propagated into sphere-like colonies (diameter≥60 µm), known as tumourspheres (white arrows). PANC-1 and MIA PaCa-2 cells formed loose aggregates as they grew. Capan-2 cells were also cultured in suspension using either serum-containing or serum-free medium. Scale bar=100 µm. (B) Representative microscopic images of Capan-2 cells in either culture on the day post-seeding. The bar chart shows the number of tumourspheres that arose from 1,000 Capan-2 cells, which is expressed in mean ± s.d. (n=3 independent experiments). 10 % FBS significantly reduced tumoursphere formation and self-renewal of Capan-2 cells. Statistical significance of the difference between serum-containing and serum-free cultures was analysed by SPSS independent-samples t-test (**p<0.01). Scale bar=100 µm. bFGF, basic fibroblast growth factor; EGF, epidermal growth factor; FBS, foetal bovine serum.
It has been well established that CSCs grow into tumourspheres when they are cultured in serum-free and non-adherent conditions, and they differentiate readily in a differentiating agent like FBS [20]. Based on this theory, it was hypothesised that the tumoursphere-forming cells in the Capan-2 cell line would lose their tumoursphere-forming potential in the presence of 10 % FBS if they were CSCs. Indeed, tumoursphere-forming cells of the Capan-2 cell line were unable to form tumourspheres in the presence of 10 % FBS and remained as single cells, albeit these cells were cultured in a non-attached condition that is known to favour the self-renewal of CSCs in the form of propagating into tumourspheres (Figure 2B). As expected, supplementation of culture medium with EGF and bFGF facilitated the formation of tumourspheres in serum-free and non-attached conditions (Figure 2B).
Capan-2 cell line contains cells with tumour-initiating ability in vivo
It is generally accepted that CSCs are tumour-initiating in vivo by nature; therefore, it was hypothesised that tumoursphere-forming cells are potentially TICs. In this study, we found that the in vivo tumourigenicity of human PDAC cell lines coincides with their tumoursphere-forming ability (Figure 3A). In the aforementioned tumoursphere formation assays, the Capan-2 cell line was found harbouring a subset of cells that gave rise to sphere-like colonies or tumourspheres, whereas PANC-1 and MIA PaCa-2 single cells grew into cell aggregates instead of tumourspheres. Being ranked the most tumourigenic in vivo, the Capan-2 cell line was found to harbour significantly higher (p<0.05) mean TIC frequency relative to PANC-1 and MIA PaCa-2 cell lines (Table 2). Noteworthy, there is no significant difference between the mean TIC frequencies of PANC-1 and MIA PaCa-2 cell lines (Table 2).
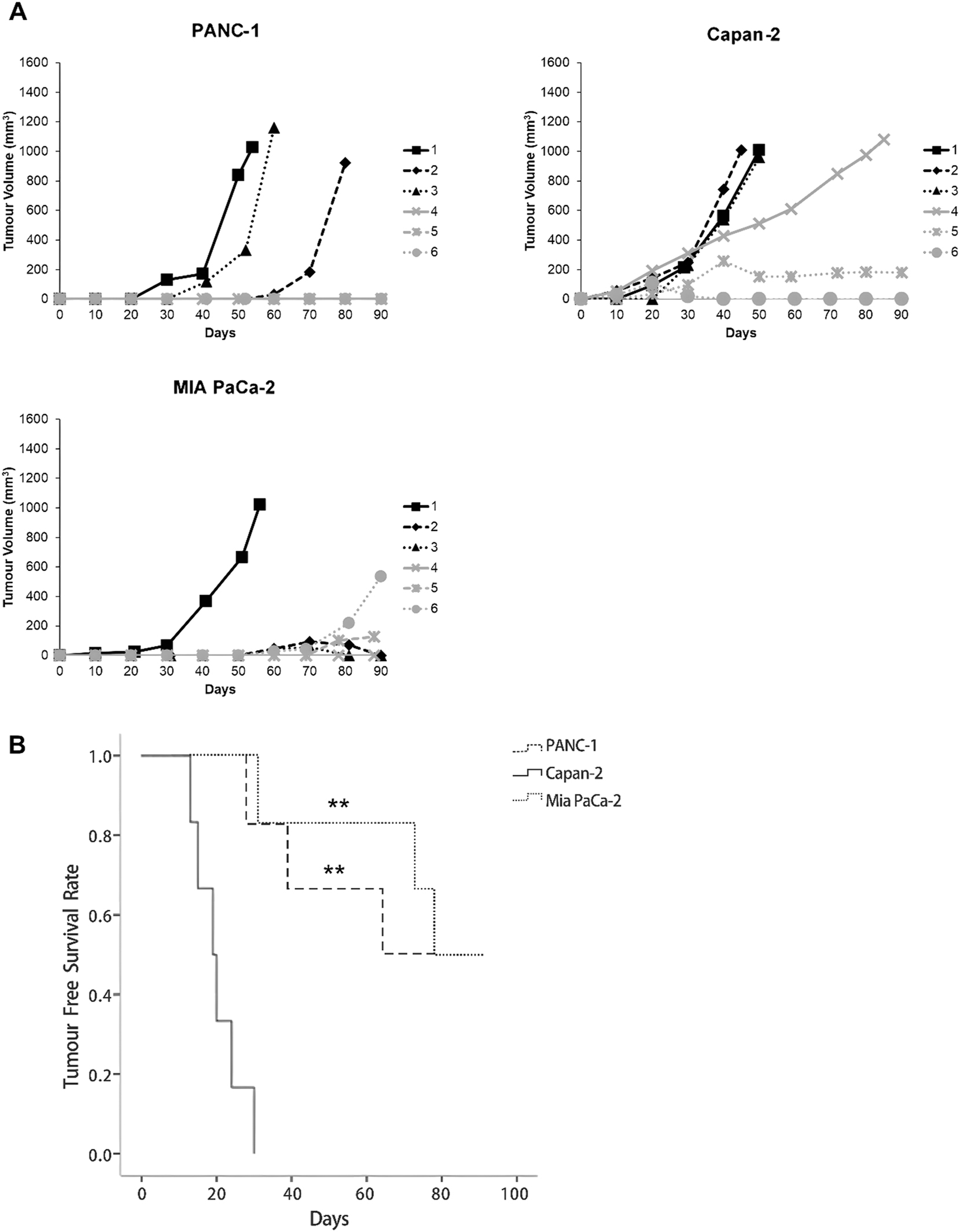
Tumour engraftabilities of KRAS-driven PDAC cell lines in vivo. Tumour xenografts were established in immunocompromised mice via subcutaneous transplantations of 106 cells of each PDAC cell line (PANC-1, Capan-2, and MIA PaCa-2) per animal. (A) Growth curves of tumour xenografts (n=6 per cell line). Capan-2 cells demonstrated a remarkably high rate of tumour engraftment. (B) Tumour free survival curves of immunocompromised mice inoculated with 106 PDAC cells. Capan-2 cells show stronger tumour-initiating ability than PANC-1 and MIA PaCa-2, as shown by earlier tumour onset and shorter tumour-free survival in mice. Statistical significance of the differences relative to Capan-2 was determined by SPSS Kaplan-Meier survival analysis using three pairwise comparison algorithms: log-rank method, Breslow test, and Tarone-Ware (**p<0.01).
Limiting dilution transplantations of human PDAC cells.
| Cell dilution | PANC-1 | Capan-2d | MIA PaCa-2 |
|---|---|---|---|
| 105 cells | 1/6a | 4/6a | 0/6a |
| 106 cells | 3/6a | 6/6a | 3/6a |
| TIC frequencyb,c | 1 in 1,211,340* | 1 in 90,996 | 1 in 1,649,863* |
| (95 % CI)c | (1 in 434,968 – 1 in 3,373,456) |
(1 in 32,544 – 1 in 254,433) |
(1 in 531,912 – 1 in 5,117,476) |
-
TIC, tumour-initiating cell; CI, confidence interval. aThe incident rate of xenografting is expressed in the form of x/y, where x is the number of animals bearing tumour xenografts and y is the number of animals inoculated with human PDAC cells. bTIC frequency is expressed as 1 TIC in a given sample size. cStatistical significance of differences, TIC frequency, and its 95 % CI were calculated via online Extreme Limiting Dilution Analysis (ELDA) software, which is provided by Walter and Eliza Hall Institute (http://bioinf.wehi.edu.au/software/elda/index.html). dStatistical significance of the differences relative to the Capan-2 cell line, which is the most enriched for TICs (*p<0.05).
Kaplan-Meier survival analysis supported the findings of the tumoursphere formation assays. The Capan-2 cell line demonstrated the highest tumour-initiating efficiency. Tumour engraftment was observed in six animals around 20 days after inoculation. Notably, one animal developed a palpable tumour exceeding 100 mm3 by day 19, although this tumour later regressed. In contrast, three animals in each of the PANC-1 and MIA PaCa-2 inoculated groups remained tumour-free throughout the 90-day observation period (Figure 3A). The mean latency period of establishing Capan-2 tumour xenografts at a cell dose of 106 (20 ± 6 days) was significantly shorter than establishing tumour xenografts of PANC-1 (44 ± 18 days) and MIA PaCa-2 (61 ± 26 days) cell lines (p<0.01; Figure 3B). It is noteworthy that immunocompromised mice inoculated with Capan-2 cells demonstrated the lowest tumour-free survival rate. No significant difference in tumourigenic property between PANC-1 and MIA PaCa-2 cell lines was observed following the inoculation of 106 cells into immunocompromised mice (p>0.05; Figure 3B).
Aberrantly activated MAPK, PI3K-AKT, NF-κB, and WNT pathways orchestrate the self-renewal of CSCs
In comparison with the monolayer culture, increased phosphorylations were observed in CRAF (8 ± 1 folds; p=0.011) at Serine-338, ERK1 (4 ± 1 folds; p=0.002) at Threonine-202 and Tyrosine-204, whereas ERK2 (3 ± 1 folds; p=0.043) at Threonine-185 and Tyrosine-187. Notably, despite the reduction in total CRAF expression, its phosphorylation at Serine-338 was markedly increased in tumourspheres, suggesting enhanced upstream signalling or more efficient activation per molecule of CRAF under 3D culture conditions. Phosphorylation of AKT was promoted (4 ± 2 folds; p=0.089) at Serine-473 (Figure 4A). Similarly, phosphorylation of the P65 subunit at Serine-536 was greatly elevated (13 ± 2 folds; p=0.001) in the sphere culture of Capan-2 cells. An increased phosphorylation of glycogen synthase kinase-3 beta (GSK3β) at Serine-9 was clearly observed in Capan-2 tumourspheres (13 ± 9 folds; p=0.087; Figure 4).
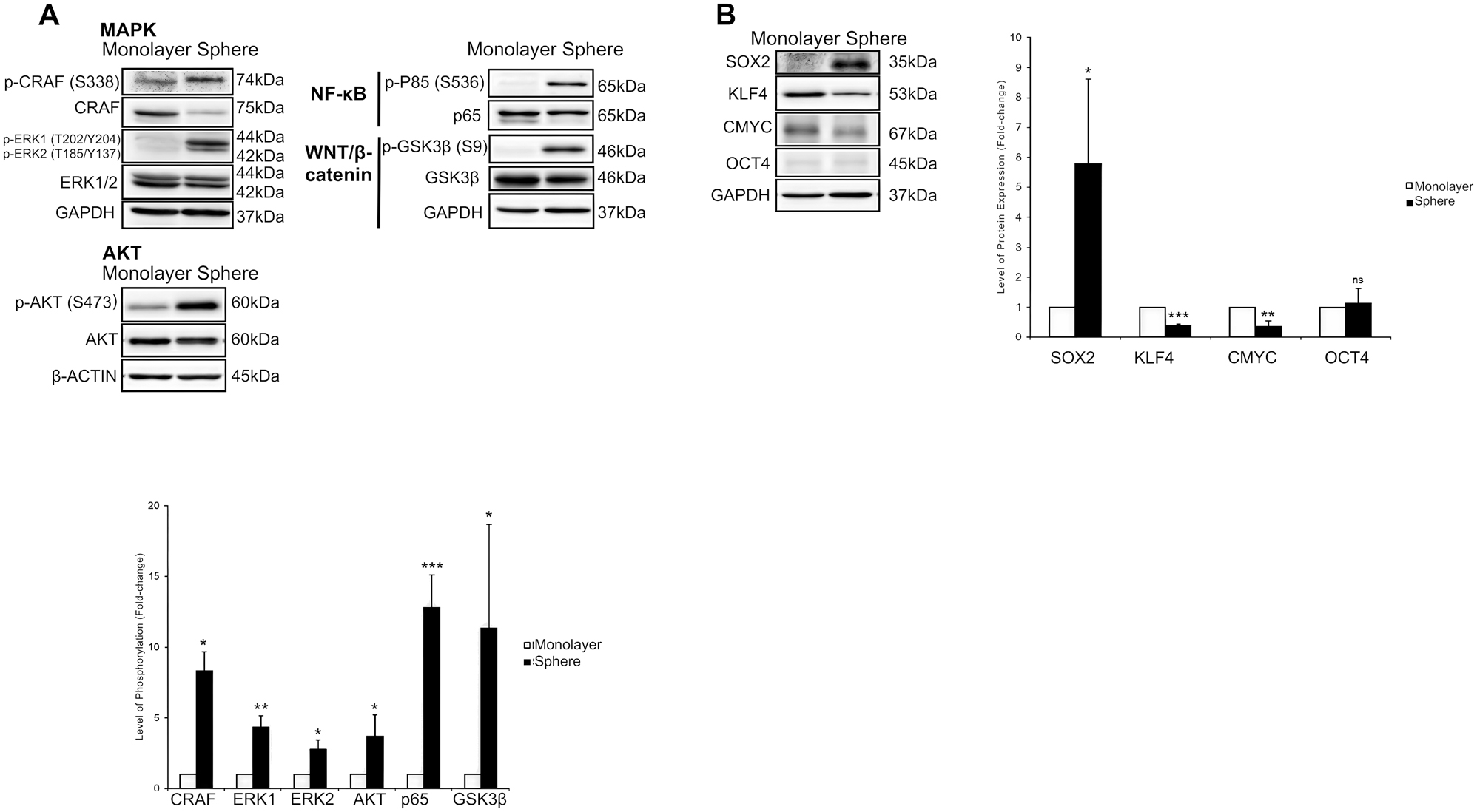
Activations of stemness-related signalling pathways and expressions of embryonic transcription factors in Capan-2 tumourspheres. Capan-2 cells were propagated as a monolayer or tumoursphere (sphere) culture. (A) Immunoblots show strong activation of MAPK, PI3K-AKT, NF-κB, and WNT/β-catenin pathways in Capan-2 tumourspheres. (B) Immunoblots show that only SOX2 expression was elevated among the four embryonic factors in Capan-2 tumourspheres; GAPDH and β-actin served as loading controls. Bar charts show the phosphorylation levels of signalling molecules and the expression levels of embryonic transcription factors. Data are expressed in mean ± s.d. (n=3 independent experiments). Statistical significance of the differences was analysed by SPSS independent-samples t-test (ns, not significant; *p≤0.1; **p≤0.01; ***p≤0.001). CMYC, cellular myelocytomatosis oncogene; ERK1/2, extracellular signal-regulated kinase 1/2; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; GSK3β, glycogen synthase kinase-3 beta; KLF4, kruppel-like factor 4; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor kappa B; OCT4, octamer-binding protein 4; PI3K, phosphoinositide 3-kinase; SOX2, SRY-box transcription factor 2.
Expression of SOX2 embryonic transcription factor is upregulated in putative CSCs of Capan-2 cell line
SRY-box Transcription Factor 2 (SOX2) was the sole embryonic transcription factor with its expression level being upregulated (6 ± 3 folds, p=0.099) in Capan-2 tumourspheres. While Octamer-binding Protein 4 (OCT4) level was comparable between the cultures (p=0.691), Kruppel-like Factor 4 (KLF4; p=0.001) and Cellular Myelocytomatosis Oncogene (CMYC; p=0.004) levels were intriguingly lower in Capan-2 tumourspheres as compared with monolayer culture (Figure 4).
Discussion
CSCs have been implicated as one of the culprits of intrinsic chemoresistance in various cancers [21]. Chemoresistance of PDAC to the gold standard chemotherapeutic drug, gemcitabine, is associated with the presence of pancreatic CSCs [7]. Therefore, establishing a reliable in vitro assay is crucial for discovering effective anti-CSC agents. Remarkable chemoresistance of PANC-1 and Capan-2 cells, as evidenced by TGI values >100 µM, prompted our interest in validating the presence of CSCs in these cell lines based on the co-expression of pancreatic CSC surface markers.
Li et al. reported that pancreatic CSCs express CD44, CD24, and epithelial surface antigen (ESA) surface proteins [22]. In another independent study, Hermann et al. successfully isolated CD133+ pancreatic cancer cells with stem cell-like features, based on which they were called pancreatic CSCs [7]. To the best of our knowledge, the present study is the first to describe a unique subset of CD44+CD24+CD133+ cells in the human PDAC cell line, Capan-2, through the application of flow cytometry. These cells were also found in ambiguously small numbers in the PANC-1 cell line. The presence of CD44+CD24+CD133+ cells in the PANC-1 cell line is questionable, considering that these cells were undetectable in two out of three independent biological replicates.
The distinct existence of CD44+CD24+CD133+ cells in the Capan-2 cell line coincides with the striking chemoresistance of this cell line to gemcitabine, suggesting that resistance was very likely due to the existence of this subpopulation. To investigate whether the viability of this cell subpopulation is affected by gemcitabine treatment, Capan-2 cells were treated with 0.1 µM (GI50) of gemcitabine, and any changes in the size of this subpopulation were observed. It is concluded that CD44+CD24+CD133+ cells were indeed gemcitabine-resistant based on a significant enrichment of these cells demonstrating persistent growth. On the contrary, we deduced that CD44+CD24+CD133+ cells experienced growth inhibition after being treated with vismodegib, based on our observation that the enrichment of the cells, as seen in gemcitabine treatment, was nullified. These observations collectively suggest that CD44+CD24+CD133+ cells are CSCs in the Capan-2 cell line.
Tumourspheres were observed arising from culturing single cells of the Capan-2 cell line at a clonal density in non-adherent and serum-free culture conditions, indicating that CSCs exist in the Capan-2 cell line. On the other hand, PANC-1 and MIA PaCa-2 cells clonally propagated into cell aggregates with irregular outlines, which is suggestive of the absence of CSCs in these cell lines. Tumoursphere-forming cells were previously identified in a human pancreatic cancer cell line, COLO 357 [7], and human pancreatic tumours [11]. Hermann et al. reported that CD133+ CSCs grew into tumourspheres in serum-free and non-adherent conditions, while CD133-non-CSCs died [7]. Simeone showed that CD44+CD24+ESA+ CSCs in patient-derived pancreatic cancer xenografts formed tumourspheres when these cells were cultured clonally in serum-free and non-adherent conditions [11]. Not surprisingly, tumoursphere-forming ability was not observed in CD44-CD24-ESA-non-CSCs. While not all tumoursphere-forming cells are CSCs, a small subset of Capan-2 cells expressing defined pancreatic CSC surface markers and having tumoursphere-forming ability is highly likely the CSC subpopulation of the cell line.
One of the hallmark characteristics of CSCs is that these cells demonstrate remarkable tumourigenic potential in host animals, which is indicative of their pluripotency [23]. With the application of limiting dilution, CSC content can be quantitatively estimated as TIC frequency [19]. In this present study, Capan-2 cells showed noticeably greater potential in tumour engraftment as compared with PANC-1 and MIA PaCa-2 cells, indicating that these cells possess greater tumour-initiating capacity and could be putative CSCs. Moreover, the high tumourigenicity of Capan-2 cells is consistent with their tumoursphere-forming ability, suggesting that tumour-initiating cells in vivo and tumoursphere-forming cells in vitro may represent the same CSC entity in the Capan-2 cell line.
Activated mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K)-AKT pathways are implicated in driving the self-renewal of pancreatic CSCs [24], 25]. In line with previous studies, our results revealed that MAPK and PI3K-AKT pathways were considerably activated in Capan-2 tumourspheres. The present study is the first to show that the P65 subunit was intensively phosphorylated at Serine-536, which is indicative of the activated nuclear factor kappa B (NF-κB) pathway in self-renewing Capan-2 tumourspheres [26]. Previous studies suggested that MAPK and PI3K-AKT pathways regulate the activation of the NF-κB pathway [27], [28], [29]. Therefore, it is not surprising to see a considerably higher level of activated P65 subunits in Capan-2 tumourspheres, in which both MAPK and PI3K-AKT pathways were constitutively active.
MAPK and PI3K-AKT pathways mediate the inhibition of GSK3β kinase activity through its phosphorylation at Serine-9 [30], 31], following which β-catenin is spared from ubiquitin-mediated proteasomal degradation, such that the activation of WNT signalling by β-catenin is possible [32]. On the background of hyperactivated MAPK and PI3K-AKT signalling in Capan-2 tumourspheres, the inhibitory regulation of WNT signalling by GSK3β was likely to be negated by increased phosphorylation at Serine-9, which actively transcribes the direct gene target of β-catenin, CMYC. Nevertheless, CMYC expression was found to be downregulated in tumourspheres, perhaps the functionality of TCF-4 transcription factor and an enhancer element located on the 8q24 chromosome, both of which are known regulators of the CMYC promoter [33], was impaired in tumourspheres. Lower levels of KLF4 and CMYC were found in tumoursphere culture than their monolayer counterparts. The downregulation of KLF4 expression in Capan-2 tumourspheres is in line with a previous study reporting the suppressive effect of KLF4 on tumoursphere-forming ability, chemoresistance, and in vivo tumourigenicity of pancreatic cancer cells [34]. CMYC expression was previously found to be absent in the CSC subset of a CMYC-induced pancreatic cancer model [35]. Furthermore, CMYC expression has an adverse impact on the stemness of pancreatic CSCs [36].
OCT4 expression was minute in both monolayer and sphere cultures of the Capan-2 cell line, which is in line with a previous study reporting an undetectable expression level of OCT4 in SOX2-expressing pancreatic cancer cells [37]. Consistent with this study, SOX2 expression was found to be considerably increased in putative CSCs enriched in Capan-2 tumourspheres. Herreros-Villanueva et al. reported that the SOX2 embryonic transcription factor has an absolute role in imparting pancreatic cancer cells with stem-cell characteristics.
This study has several limitations. Despite the valuable insights provided by this study, several limitations should be acknowledged. First, the findings are based primarily on a single human PDAC cell line, Capan-2, which may not fully represent the heterogeneity observed in primary pancreatic tumours. The tumour microenvironment and stromal interactions, which play crucial roles in CSC maintenance and chemoresistance in vivo, are not fully recapitulated in this in vitro model. Second, although we identified several key signalling pathways associated with the CSC phenotype, mechanistic validation through targeted genetic manipulation was not performed and remains an area for future investigation. Third, the therapeutic evaluation of vismodegib was limited to its effect on CSC marker expression and sphere formation; in vivo validation using patient-derived xenograft models or genetically engineered mouse models would be necessary to confirm its clinical relevance. Finally, the potential off-target effects and toxicity profiles of Hedgehog pathway inhibition were not addressed in this study and warrant further exploration.
Conclusions
Our study indicated that the Capan-2 cell line is a plausible and reliable source of pancreatic CSCs for studying their biology and discovering novel therapeutic agents to eradicate pancreatic cancer recurrence. In addition, this study has provided insight into the molecular mechanism of self-renewing pancreatic CSCs (Figure 5), which serves as an important fundamental in targeting them. KRAS signalling is implicated in tumour initiation. While we believe that constitutively active KRAS mutations maintain stemness in pancreatic cancer stem cells, our study revealed that KRAS signalling is not the sole driver of self-renewal, as not all KRAS-driven pancreatic ductal adenocarcinoma cell lines harbour CD44+CD24+CD133+ pancreatic cancer stem cells.
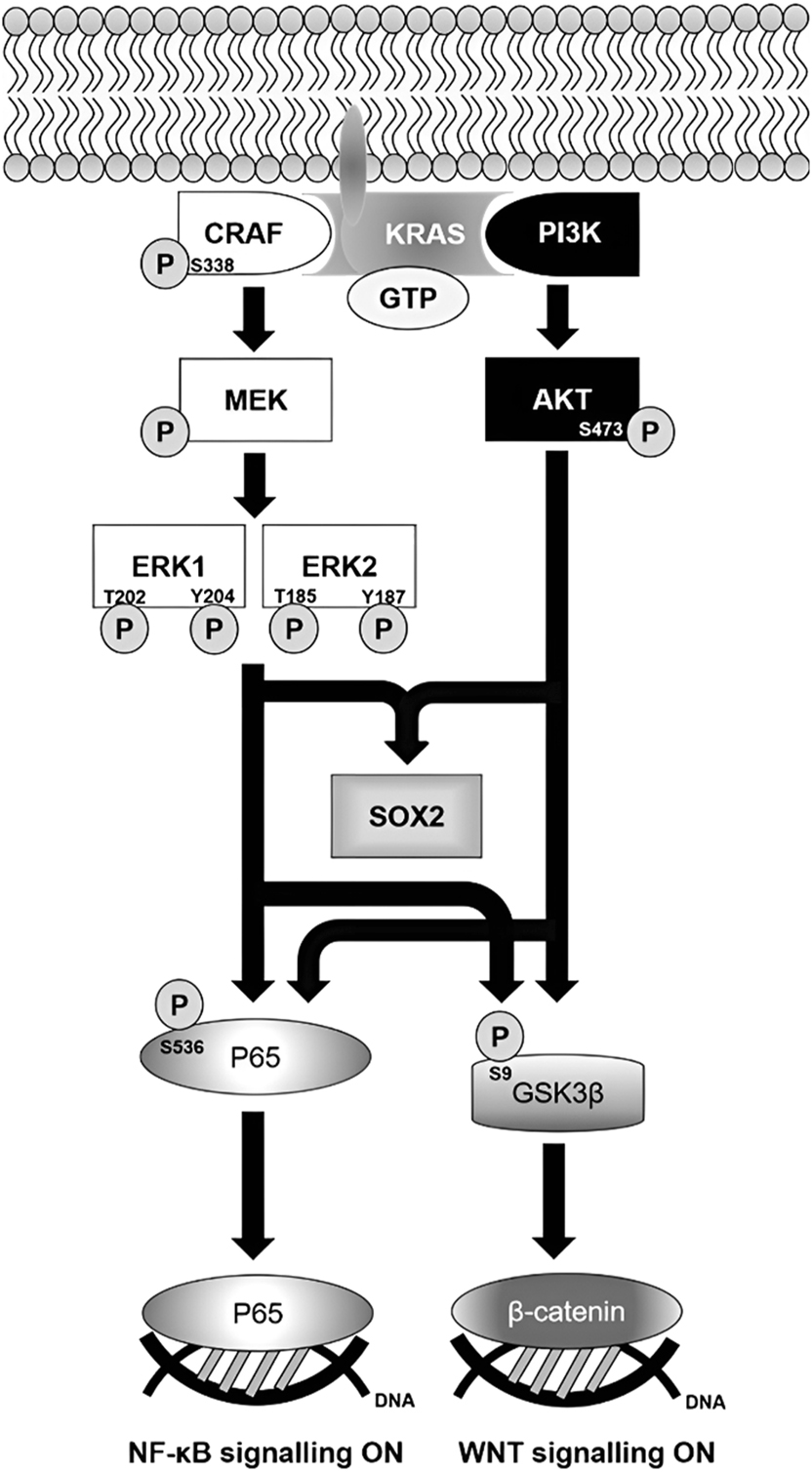
Molecular mechanism of self-renewing Capan-2 tumourspheres. Oncogenic KRAS mediates the activations of MAPK and PI3K-AKT pathways in response to the stimulation of EGF and FGF receptors by their respective growth factors. CRAF is activated by phosphorylation that brings about a cascade of activating phosphorylations of MEK and ERKs, while active PI3K converts phosphatidylinositol (3,4)-bisphosphate (PIP2) into phosphatidylinositol (3,4,5)-triphosphate (PIP3) that facilitates AKT activation. Following the activation of MAPK and PI3K-AKT pathways, downstream NF-κB and WNT pathways are turned on. SOX2 expression is upregulated by dual activation of MAPK and PI3K-AKT pathways. ERK1/2, extracellular signal-regulated kinase 1/2; GSK3β, glycogen synthase kinase-3 beta; GTP, guanosine triphosphate; MEK, mitogen-activated protein kinase kinase; PI3K, phosphoinositide 3-kinase; SOX2, SRY-box transcription factor 2.
Funding source: Ministry of Education, Malaysia through Fundamental Research Grant Scheme Grant
Award Identifier / Grant number: 04-02-13-1324FR
-
Informed consent: Not applicable.
-
Ethics approval: Research involving Animals. The authors confirm that the experiment involving animals was approved by the Institutional Animal Care and Use Committee (IACUC) of Universiti Putra Malaysia (UPM/IACUC/AUP-R044/2017) and they have adhered to IACUC standards for the protection of animals used for scientific purposes.
-
Author contributions: All authors contributed to the study’s conception and design. Johnson Stanslas contributed to the funding of acquisition, study conception, and design as well as supervision. Material preparation, data collection, and analysis were performed by Yuan Han Teh. Rajesh Ramasamy, Kok Lian Ho, and Sreenivasa Rao Sagineedu were involved in supervision and mentorship. The first draft of the manuscript was written by Yuan Han Teh and Jing Rui, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
-
Use of Large Language Models, AI and Machine Learning Tools: None declared.
-
Conflict of interest: The authors have no relevant financial or non-financial interests to disclose.
-
Research funding: This work was supported by the Ministry of Education, Malaysia through Fundamental Research Grant Scheme Grant [04-02-13-1324FR to Johnson Stanslas].
-
Data availability: The authors confirm that the data supporting the findings of this study are available within the article, and the authors declare that this work is part of the doctoral dissertation of Yuan Han Teh at Universiti Putra Malaysia.
References
1. Bray, F, Laversanne, M, Sung, H, Ferlay, J, Siegel, RL, Soerjomataram, I, et al.. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2024;74:229–63. https://doi.org/10.3322/caac.21834.Search in Google Scholar PubMed
2. Halbrook, CJ, Lyssiotis, CA, di Magliano, MP, Maitra, A. Pancreatic cancer: advances and challenges. Cell 2023;186:1729–54. https://doi.org/10.1016/j.cell.2023.02.014.Search in Google Scholar PubMed PubMed Central
3. Gorbudhun, R, Patel, PH, Hopping, E, Doyle, J, Geropoulos, G, Mavroeidis, VK, et al.. Neoadjuvant chemotherapy-chemoradiation for borderline-resectable pancreatic adenocarcinoma: a UK tertiary surgical oncology centre series. Cancers 2022;14:4678. https://doi.org/10.3390/cancers14194678.Search in Google Scholar PubMed PubMed Central
4. Khan, AA, Liu, X, Yan, X, Tahir, M, Ali, S, Huang, H. An overview of genetic mutations and epigenetic signatures in the course of pancreatic cancer progression. Cancer Metastasis Rev 2021;40:245–72. https://doi.org/10.1007/s10555-020-09952-0.Search in Google Scholar PubMed
5. Strickler, JH, Satake, H, George, TJ, Yaeger, R, Hollebecque, A, Garrido-Laguna, I, et al.. Sotorasib in KRAS p.G12C-mutated advanced pancreatic cancer. N Engl J Med 2023;388:33–43. https://doi.org/10.1056/nejmoa2208470.Search in Google Scholar
6. Philip, PA, Azar, I, Xiu, J, Hall, MJ, Hendifar, AE, Lou, E, et al.. Molecular characterization of KRAS wild-type tumors in patients with pancreatic adenocarcinoma. Clin Cancer Res official J Am Assoc Cancer Res 2022;28:2704–14. https://doi.org/10.1158/1078-0432.ccr-21-3581.Search in Google Scholar PubMed PubMed Central
7. Hermann, PC, Huber, SL, Herrler, T, Aicher, A, Ellwart, JW, Guba, M, et al.. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007;1:313–23. https://doi.org/10.1016/j.stem.2007.06.002.Search in Google Scholar PubMed
8. Sumbly, V, Landry, I. Understanding pancreatic cancer stem cells and their role in carcinogenesis: a narrative review. Stem Cel Invest 2022;9:1. https://doi.org/10.21037/sci-2021-067.Search in Google Scholar PubMed PubMed Central
9. Haddadin, L, Sun, X. Stem cells in cancer: from mechanisms to therapeutic strategies. Cells 2025;14:538. https://doi.org/10.3390/cells14070538.Search in Google Scholar PubMed PubMed Central
10. Cortes, JE, Gutzmer, R, Kieran, MW, Solomon, JA. Hedgehog signaling inhibitors in solid and hematological cancers. Cancer Treat Rev 2019;76:41–50. https://doi.org/10.1016/j.ctrv.2019.04.005.Search in Google Scholar PubMed
11. Simeone, DM. Pancreatic cancer stem cells: implications for the treatment of pancreatic cancer. Clin Cancer Res official J Am Assoc Cancer Res 2008;14:5646–8. https://doi.org/10.1158/1078-0432.ccr-08-0584.Search in Google Scholar
12. Zhao, Y, Qin, C, Zhao, B, Wang, Y, Li, Z, Li, T, et al.. Pancreatic cancer stemness: dynamic status in malignant progression. J Exp Clin Cancer Res 2023;42:122. https://doi.org/10.1186/s13046-023-02693-2.Search in Google Scholar PubMed PubMed Central
13. Lapidot, T, Sirard, C, Vormoor, J, Murdoch, B, Hoang, T, Caceres-Cortes, J, et al.. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994;367:645–8. https://doi.org/10.1038/367645a0.Search in Google Scholar PubMed
14. Marangoni, E. Patient-derived xenografts of breast cancer. Adv Exp Med Biol 2025;1464:109–21. https://doi.org/10.1007/978-3-031-70875-6_7.Search in Google Scholar PubMed
15. Zhou, G, Lv, X, Zhong, X, Ying, W, Li, W, Feng, Y, et al.. Suspension culture strategies to enrich colon cancer stem cells. Oncol Lett 2023;25:116. https://doi.org/10.3892/ol.2023.13702.Search in Google Scholar PubMed PubMed Central
16. Alberti, G, Amico, MD, Caruso Bavisotto, C, Rappa, F, Marino Gammazza, A, Bucchieri, F, et al.. Speeding up glioblastoma cancer research: highlighting the zebrafish xenograft model. Int J Mol Sci 2024;25:5394. https://doi.org/10.3390/ijms25105394.Search in Google Scholar PubMed PubMed Central
17. Jada, SR, Matthews, C, Saad, MS, Hamzah, AS, Lajis, NH, Stevens, MF, et al.. Benzylidene derivatives of andrographolide inhibit growth of breast and colon cancer cells in vitro by inducing G(1) arrest and apoptosis. Br J Pharmacol 2008;155:641–54. https://doi.org/10.1038/bjp.2008.368.Search in Google Scholar PubMed PubMed Central
18. Singh, JK, Farnie, G, Bundred, NJ, Simões, BM, Shergill, A, Landberg, G, et al.. Targeting CXCR1/2 significantly reduces breast cancer stem cell activity and increases the efficacy of inhibiting HER2 via HER2-dependent and -independent mechanisms. Clin Cancer Res official J Am Assoc Cancer Res 2013;19:643–56. https://doi.org/10.1158/1078-0432.ccr-12-1063.Search in Google Scholar PubMed PubMed Central
19. Hu, Y, Smyth, GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 2009;347:70–8. https://doi.org/10.1016/j.jim.2009.06.008.Search in Google Scholar PubMed
20. Habanjar, O, Diab-Assaf, M, Caldefie-Chezet, F, Delort, L. 3D cell culture systems: tumor application, advantages, and disadvantages. Int J Mol Sci 2021;22:12200. https://doi.org/10.3390/ijms222212200.Search in Google Scholar PubMed PubMed Central
21. Zhuang, J, Shen, L, Li, M, Sun, J, Hao, J, Li, J, et al.. Cancer-associated fibroblast-derived miR-146a-5p generates a niche that promotes bladder cancer stemness and chemoresistance. Cancer Res 2023;83:1611–27. https://doi.org/10.1158/0008-5472.can-22-2213.Search in Google Scholar
22. Li, C, Heidt, DG, Dalerba, P, Burant, CF, Zhang, L, Adsay, V, et al.. Identification of pancreatic cancer stem cells. Cancer Res 2007;67:1030–7. https://doi.org/10.1158/0008-5472.can-06-2030.Search in Google Scholar
23. Liu, Y, Wang, H. Biomarkers and targeted therapy for cancer stem cells. Trends Pharmacol Sci 2024;45:56–66. https://doi.org/10.1016/j.tips.2023.11.006.Search in Google Scholar PubMed PubMed Central
24. Ischenko, I, Petrenko, O, Hayman, MJ. Analysis of the tumor-initiating and metastatic capacity of PDX1-positive cells from the adult pancreas. Proc Natl Acad Sci U S A 2014;111:3466–71. https://doi.org/10.1073/pnas.1319911111.Search in Google Scholar PubMed PubMed Central
25. Bubin, R, Uljanovs, R, Strumfa, I. Cancer stem cells in pancreatic ductal adenocarcinoma. Int J Mol Sci 2023;24:7030. https://doi.org/10.3390/ijms24087030.Search in Google Scholar PubMed PubMed Central
26. Sasaki, CY, Barberi, TJ, Ghosh, P, Longo, DL. Phosphorylation of RelA/p65 on serine 536 defines an IκBα-independent NF-κB pathway. J Biol Chem 2005;280:34538–47. https://doi.org/10.1074/jbc.m504943200.Search in Google Scholar
27. Sakurai, H, Chiba, H, Miyoshi, H, Sugita, T, Toriumi, W. IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J Biol Chem 1999;274:30353–6. https://doi.org/10.1074/jbc.274.43.30353.Search in Google Scholar PubMed
28. Bai, D, Ueno, L, Vogt, PK. Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int J Cancer 2009;125:2863–70. https://doi.org/10.1002/ijc.24748.Search in Google Scholar PubMed PubMed Central
29. Guo, Q, Jin, Y, Chen, X, Ye, X, Shen, X, Lin, M, et al.. NF-κB in biology and targeted therapy: new insights and translational implications. Signal Transduction Targeted Ther 2024;9:53. https://doi.org/10.1038/s41392-024-01757-9.Search in Google Scholar PubMed PubMed Central
30. Ding, Q, Xia, W, Liu, JC, Yang, JY, Lee, DF, Xia, J, et al.. Erk associates with and primes GSK-3beta for its inactivation resulting in upregulation of beta-catenin. Mol Cell 2005;19:159–70. https://doi.org/10.1016/j.molcel.2005.06.009.Search in Google Scholar PubMed
31. Jope, RS, Johnson, GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci 2004;29:95–102. https://doi.org/10.1016/j.tibs.2003.12.004.Search in Google Scholar PubMed
32. Zhan, T, Rindtorff, N, Boutros, M. Wnt signaling in cancer. Oncogene 2017;36:1461–73. https://doi.org/10.1038/onc.2016.304.Search in Google Scholar PubMed PubMed Central
33. Sotelo, J, Esposito, D, Duhagon, MA, Banfield, K, Mehalko, J, Liao, H, et al.. Long-range enhancers on 8q24 regulate c-Myc. Proc Natl Acad Sci U S A 2010;107:3001–5. https://doi.org/10.1073/pnas.0906067107.Search in Google Scholar PubMed PubMed Central
34. Yan, Y, Li, Z, Kong, X, Jia, Z, Zuo, X, Gagea, M, et al.. KLF4-mediated suppression of CD44 signaling negatively impacts pancreatic cancer stemness and metastasis. Cancer Res 2016;76:2419–31. https://doi.org/10.1158/0008-5472.can-15-1691.Search in Google Scholar PubMed PubMed Central
35. Lin, WC, Rajbhandari, N, Liu, C, Sakamoto, K, Zhang, Q, Triplett, AA, et al.. Dormant cancer cells contribute to residual disease in a model of reversible pancreatic cancer. Cancer Res 2013;73:1821–30. https://doi.org/10.1158/0008-5472.can-12-2067.Search in Google Scholar
36. Sancho, P, Burgos-Ramos, E, Tavera, A, Bou Kheir, T, Jagust, P, Schoenhals, M, et al.. MYC/PGC-1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab 2015;22:590–605. https://doi.org/10.1016/j.cmet.2015.08.015.Search in Google Scholar PubMed
37. Herreros-Villanueva, M, Zhang, JS, Koenig, A, Abel, EV, Smyrk, TC, Bamlet, WR, et al.. SOX2 promotes dedifferentiation and imparts stem cell-like features to pancreatic cancer cells. Oncogenesis 2013;2:e61. https://doi.org/10.1038/oncsis.2013.23.Search in Google Scholar PubMed PubMed Central
© 2025 the author(s), published by De Gruyter on behalf of Tech Science Press (TSP)
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Frontmatter
- Review Articles
- Insights on prevention and gastric cancer detection: an integrative approach through risk factors, microbiome, molecular markers and machine learning
- Periodontal inflammation as a negative stimulus for oral cancerization: the hidden role of periodontitis in oral cancerization
- From dosimetry to deep learning: personalized risk prediction models for radiation pneumonitis
- A mini-review: the role of glycosylation in acute myeloid leukemia and its potential for treatment
- Research Articles
- Impact of ex vivo cell culturing on ABC transporters’ expression and drug sensitivity in patient-derived non-small cell lung cancer models
- Muscle density before neoadjuvant chemoradiotherapy as a prognostic factor in patients with locally advanced rectal cancer: a dual-center cohort study
- KRAS-driven pancreatic ductal adenocarcinoma cell lines harbour putative cancer stem cells
- Macrophage membrane-coated gelatin/laponite nanoparticles containing doxorubicin for anticancer immunotherapy
- Integrated RNA sequencing reveals tumor microenvironment heterogeneity and immunosuppressive role of M2 macrophages in osteosarcoma
- Development of an adenosine-related RiskScore model to predict the prognosis of patients with ovarian cancer
- The dual role of cytopathology in head and neck squamous cell carcinoma: morphologic diagnosis and HPV testing
- Artesunate overcomes icotinib resistance in non-small cell lung cancer with EGFR-sensitive mutations
- Propofol anesthesia for gastric cancer surgery increased cancer suppressor miR-122-5p expression in plasma extracellular vesicles compared with sevoflurane
Articles in the same Issue
- Frontmatter
- Review Articles
- Insights on prevention and gastric cancer detection: an integrative approach through risk factors, microbiome, molecular markers and machine learning
- Periodontal inflammation as a negative stimulus for oral cancerization: the hidden role of periodontitis in oral cancerization
- From dosimetry to deep learning: personalized risk prediction models for radiation pneumonitis
- A mini-review: the role of glycosylation in acute myeloid leukemia and its potential for treatment
- Research Articles
- Impact of ex vivo cell culturing on ABC transporters’ expression and drug sensitivity in patient-derived non-small cell lung cancer models
- Muscle density before neoadjuvant chemoradiotherapy as a prognostic factor in patients with locally advanced rectal cancer: a dual-center cohort study
- KRAS-driven pancreatic ductal adenocarcinoma cell lines harbour putative cancer stem cells
- Macrophage membrane-coated gelatin/laponite nanoparticles containing doxorubicin for anticancer immunotherapy
- Integrated RNA sequencing reveals tumor microenvironment heterogeneity and immunosuppressive role of M2 macrophages in osteosarcoma
- Development of an adenosine-related RiskScore model to predict the prognosis of patients with ovarian cancer
- The dual role of cytopathology in head and neck squamous cell carcinoma: morphologic diagnosis and HPV testing
- Artesunate overcomes icotinib resistance in non-small cell lung cancer with EGFR-sensitive mutations
- Propofol anesthesia for gastric cancer surgery increased cancer suppressor miR-122-5p expression in plasma extracellular vesicles compared with sevoflurane