Enhanced oral bioavailability of nisoldipine-piperine-loaded poly-lactic-co-glycolic acid nanoparticles
-
Permender Rathee


Abstract
Background:
Piperine helps in the improvement of bioavailability through pharmacokinetic interaction by modulating metabolism when administered with other drugs. Nisoldipine is a substrate for cytochrome P4503A4 enzymes. The study was undertaken to assess the influence of piperine on the pharmacokinetics and pharmacodynamics of nisoldipine nanoparticles in rats.
Methods:
Optimization studies of nanoparticles were performed using Taguchi L9 orthogonal array, and the nanoparticles were formulated by the precipitation method. The influence of piperine and nanoparticles was evaluated by means of in vivo kinetic and dynamic studies by oral administration in rats.
Results:
The entrapment efficiency, drug loading, ζ potential, and average particle size of optimized nisoldipine-piperine nanoparticles was 89.77±1.06%, 13.6±0.56%, −26.5 mV, and 132±7.21 nm, respectively. The in vitro release in 0.1 n HCl and 6.8 pH phosphate buffer was 96.9±0.48% and 98.3±0.26%, respectively. Pharmacokinetic studies showed a 4.9-fold increase in oral bioavailability and a >28.376±1.32% reduction in systemic blood pressure by using nanoparticles as compared to control (nisoldipine suspension) in Wistar rats.
Conclusion:
The results revealed that piperine being an inhibitor of cytochrome P4503A4 enzymes enhanced the bioavailability of nisoldipine by 4.9-fold in nanoparticles.
1 Introduction
Poly-lactic-co-glycolic acid (PLGA)-based nanocarriers have been extensively explored as drug delivery systems. PLGA is considered to be appropriate for most administration routes [1]. It is approved by the Food and Drug Administration and the European Medicines Agency for application in drug targeting [2], [3]. PLGA, due to its adaptive physical properties, gives flexibility to formulate and accomplish an anticipated dosage form by modifying the molecular weight and lactide/glycolide ratio. Moreover, the metabolism and the kinetics of the active ingredient can be regulated [4], [5], [6].
Enhancement of drug bioavailability is always strived for. One of the approaches for enhancing bioavailability is to co-administer drugs with a bioenhancer. Bioenhancers are defined as compounds that themselves are not therapeutic agents but potentiate the therapeutic effect of the co-administered drugs [7]. A number of natural compounds and herbal extracts have the ability to boost the bioavailability by inhibiting metabolism and/or improving absorption [8]. Piperine, obtained from Piper nigrum, has been reported to be an excellent bioenhancer [9]. Piperine improves the bioavailability of co-administered drugs by modulating metabolism. It is reported to downregulate or inhibit phase II enzymes like cytochrome P450 isoforms, UDP-glucuronyltransferase, hepatic arylhydrocarbon hydroxylase, and the glucuronidation process in the liver [10], [11], [12]. Shoba et al. [13] in 1998 showed a remarkable 2000% increase in curcumin bioavailability by piperine.
Nisoldipine is a second-generation long-acting calcium channel blocker. The vascular selectivity of nisoldipine is 10 times more than that of felodipine, isradipine, and nicardipine, and 100 times more than that of amlodipine and nifedipine [14]. The absolute bioavailability of nisoldipine is about 5% due to high presystemic metabolism in the gut wall and intestine [15]. Cytochrome P4503A4 enzymes are supposed to play a foremost role in the metabolism of nisoldipine [16].
A variety of experimental design methods, like the Taguchi and response surface methodologies, have been successfully used for optimization of process parameters [17]. The main purpose of the design of experiments is to lessen the experimental runs required for optimization. The Taguchi design is based upon fractional factorial as provided by a standard orthogonal array. The Taguchi method uses array designs to take into account noise factors (outer) and design factors (inner), which estimate the effect of factors on the response mean and variation. An orthogonal array means the design is well adjusted so that factor levels are weighted equally and each factor can be evaluated autonomously of all the other factors. This allows assessment of the effect of one factor without the interference of effects of other factors [18]. This helps in the reduction of time and cost associated with the experiment when fractionated designs are used [19], [20].
The basic study objective of the current study was to determine the pharmacokinetic and pharmacodynamic changes and bioavailability of nisoldipine using nisoldipine-piperine nanoparticles after oral administration in rats. To test this hypothesis, the nisoldipine-piperine PLGA nanoparticles were formulated and evaluated for in vivo pharmacokinetic and dynamic changes using rats.
2 Materials and methods
2.1 Materials
Orchid Pharma (Chennai, India) provided the nisoldipine and PLGA as gift samples. Piperine, methylprednisolone acetate, was procured from Sigma-Aldrich. PVP-K30, Tween 80, sodium lauryl sulfate (SLS), polyvinyl alcohol (PVA), and acetone were procured from Ranbaxy (India). All other chemicals and reagents used in the study were of AR grade.
2.2 Methods
2.2.1 Experimental design and analysis
The Taguchi design method was used to optimize various parameters for preparing nisoldipine and piperine-loaded nanoparticles. The various variables involved in preparing nisoldipine and piperine-loaded nanoparticles were categorized as dependent and independent variables. In this study, three factors namely polymer concentration (coded as A), piperine concentration (coded as B), and ratio of solvent (coded as C) were considered as variable factors affecting the process performance in terms of encapsulation efficiency (Y1) and nanoparticle size (Y2). The three levels of each of the dependent variables are given in Table 1. Nine experiments were performed to find out the factors and their optimized level ranges having a prominent effect on the proficiency of formulation based on Taguchi’s L9 orthogonal array (Table 2). The run involved the corresponding combination of levels to which the factors in the experiment was set. All nine experiments were performed in triplicate to minimize experimental errors.
Dependent variables and their respective levels used in the experiments.
| Variable | Levels | ||
|---|---|---|---|
| 1 | 2 | 3 | |
| Polymer (%) (A) | 50 mg | 100 mg | 150 mg |
| Piperine (%) (B) | 5 mg | 10 mg | 20 mg |
| Solvent (ml) (C) | 25 ml | 50 ml | 75 ml |
Taguchi’s L9 orthogonal array of experiment for preparing piperine-loaded nisoldipine nanoparticles.
| Experiment number | Polymer (%) | Piperine (%) | Solvent (ml) |
|---|---|---|---|
| 1 | 1 | 1 | 1 |
| 2 | 1 | 2 | 2 |
| 3 | 1 | 3 | 3 |
| 4 | 2 | 1 | 1 |
| 5 | 2 | 2 | 2 |
| 6 | 2 | 3 | 3 |
| 7 | 3 | 1 | 1 |
| 8 | 3 | 2 | 2 |
| 9 | 3 | 3 | 3 |
The responses of signal-to-noise (S/N) ratio effect plots and analysis of variance (ANOVA) of each response, individually as well as simultaneously, were determined to assess significant optimized levels of factors (Table 3). The optimum conditions were determined to yield an improved performance with the minimum possible influence of the noise factor. The first step was to select the factor/level combination to maximize the response. The second step was to find the condition for attaining optimal desirability. All calculations and statistical analysis of the results were carried out by ANOVA to determine the factors having a statistically significant effect on the response parameters (Table 4).
The responses of various factors and output levels of observed vs. predicted levels on nine formulations.
| Experiment number | EE (%, mean) | PS (Nm, mean) | S/N ratio | Fitness (%) | Levels | Predicted “better to best” level | ||
|---|---|---|---|---|---|---|---|---|
| Actual | Predicted | Coded | Fitted | |||||
| 1 | 87.77 | 135.2 | −25.25 | −24.82 | 89.77 | A1B1C1 | Nominal is best | |
| 2 | 88.14 | 132.7 | −27.09 | −25.69 | −0.004 | A1B2C2 | ||
| 3 | 88.47 | 129.5 | −27.15 | −24.98 | 89.80 | A1B3C3 | ||
| 4 | 88.1 | 131.3 | −27.19 | −24.73 | 89.90 | A2B1C2 | ||
| 5 | 88.33 | 129.4 | −27.16 | −24.53 | 89.77 | A2B2C3 | ||
| 6 | 89.5 | 134.5 | −26.59 | −26.18 | −0.004 | A2B3C1 | ||
| 7 | 89.23 | 128.9 | −26.87 | −26.76 | −0.0005 | A3B1C3 | ||
| 8 | 89.77 | 132.01 | −26.52 | −25.81 | 89.77 | A3B2C1 | ||
| 9 | 89.67 | 130.1 | −26.19 | −25.27 | 89.70 | A3B3C2 | ||
| Mean value | 88.78 | 131.69 | −26.6 | −25.4 | – | |||
The responses interpreted were fitted value of observed vs. predicted S/N ratios and the optimized level formula was A2B3C1.
Analysis of variance responses.
| Source | A | B | C | Residual error | Total |
|---|---|---|---|---|---|
| Combined effect on response Y1 and Y2 (EE in% and particle size in nm); nominal is best | |||||
| Degree of freedom | 2.000 | 2.000 | 2.000 | 2.000 | 8.00 |
| Seq SS | 1.755 | 0.244 | 6.139 | 0.0539 | 8.19 |
| F-value | 32.59 | 4.540 | 114.0 | ||
| p-Value | 0.030 | 0.181 | 0.009 | ||
| Rank of levels | 2.000 | 3.000 | 1.000 | A2B3C1 | |
| Individual effect on response Y1; larger is better | |||||
| Degree of freedom | 2 | 2 | 2 | 2 | 8 |
| Seq SS | 0.028 | 0.003 | 0.00004 | 0.0005 | 0.0324 |
| F-value | 0.460 | 2.410 | 4.730 | ||
| p-Value | 0.407 | 0.294 | 0.174 | ||
| Rank of levels | 1.000 | 2.000 | 3.000 | A1B2C3 | |
| Individual effect on response Y2; smaller is better | |||||
| Degree of freedom | 2 | 2 | 2 | 2 | 8 |
| Seq SS | 0.0143 | 0.005 | 0.225 | 0.002 | 0.246 |
| F-value | 8.02 | 2.55 | 126.51 | ||
| p-Value | 0.111 | 0.282 | 0.008 | ||
| Rank of levels | 2.0000 | 3.0000 | 1.000 | A2B3C1 | |
2.2.2 Fabrication of nisoldipine and piperine-loaded nanoparticles
PLGA-based nanoparticles were prepared using a modified precipitation process [21]. Briefly, 100 mg PLGA (50:50, Mw=40,000–75,000) polymer, 10 mg nisoldipine, and 20 mg piperine were dissolved in 25 ml acetone at room temperature to make a clear solution. Then, this solution was added dropwise with continuous magnetic stirring in 25 ml PVA (1%; Mw=30,000–70,000) for 30 min at room temperature. The organic solvent was evaporated at 26°C first by stirring (2029 g using rotary evaporation; Rotavapor R-124, Buchi, Switzerland) at atmospheric pressure for 6 h and then at reduced pressure for 2 h. Thereafter, nanoparticle suspension was centrifuged (REMI, Mumbai, India) for 25 min at 10,956 g at 4°C to remove the polymer aggregates and washed twice with deionized water to remove acetone and PVA residues, and were freeze dried at −40°C (Eclipse 400; RS Biotech, UK) and stored in a vacuum desiccator at 4°C for further use. The percentage yield was >78%.
2.2.3 Preparation of nisoldipine suspension (control)
The nisoldipine suspension was prepared according to the method of Sandhya et al. [22] with modifications, by dissolving 10 mg nisoldipine in 3.0 ml of ethanol. Mixture 1 containing 30 mg PVP-K30, 0.3 ml Tween 80, and 1.0 ml of 0.01% SLS in 30 ml water was prepared and kept on a magnetic stirrer for uniform mixing. The drug solution was slowly added dropwise to mixture 1 and magnetic stirring was continued for 30 min. Then, the solution was kept in a sonicator for 30 min. The suspension was formed and preserved for further use.
2.2.4 Entrapment efficiency and loading capacity
The entrapment efficiency of the prepared nanoparticles was determined by an indirect method [23]. The concentration of nisoldipine in the supernatant after isolation of nanoparticles was determined spectrophotometrically at 238 nm. The percent entrapment efficiency (EE%) was calculated by the following equation:
where Ntotal is the total amount of nisoldipine and Nsupernatant is the free nisoldipine in the supernatant.
For calculating drug loading, the nanoparticles were dissolved in a mixture of acetone and ethanol (10:1) and centrifuged at 1677 g for 15 min. The precipitate was then extracted with acetone. The acetone was evaporated and percent drug loading [24] was calculated by the following equation:
2.2.5 Characterization of nanoparticles
The characterization of nanoparticles was achieved using transmission electron microscopy (TEM). Photon correlation spectroscopy (NanoZS90; Malvern Instruments, Worcestershire, UK) was used to measure the particle size, polydispersity index, and zeta potential (ZP) of the prepared formulation. All samples were diluted with ethanol to achieve sufficient concentration before measurement. All parameters were analyzed in triplicate and data are represented as mean±standard deviation (SD). Thermal [differential scanning calorimetry (DSC)] and spectroscopic [Fourier transform infrared (FTIR)] analyses were used to assess the chemical interaction of the drug with excipients.
2.2.6 Dissolution and release studies
In vitro release of nisoldipine from nanoparticles was studied in 0.1 n HCl (pH 1.2) and in phosphate buffer (pH 6.8) using a type II paddle apparatus (DS 8000; Lab India Ltd., Mumbai, India) at 37±0.5°C and 60 rpm for comparing the release pattern at the two different pHs. The nisoldipine release profile is influenced by pH; therefore, the two different pHs were selected to comparatively evaluate the effect on the release profile of nanoparticles, as some researchers claimed it to be independent of pH. Aliquots of 5 ml were withdrawn from the apparatus at specific intervals. Nisoldipine concentration was measured in the withdrawn aliquots. The dissolution medium volume was kept constant by adding the same amount of fresh medium. Drug dissolution and cumulative drug release was determined for nisoldipine-piperine nanoparticles and conventional nisoldipine formulation at 2, 4, 6, 8, 12, 18, 24, 36, 48, and 60 h.
2.2.7 Antihypertensive and pharmacokinetic studies
The study was carried out in Wistar rats of either sex weighing 150–200 g. The animals were maintained under standard laboratory conditions of temperature (22±2°C) and 12 h light/dark cycle. All animals were kept in quarantine for 1 week prior to experiments. Rats were fed with standard laboratory chow and water ad libitum. All experimental procedures were reviewed and approved by the Institutional Animal Ethical Committee of Chandigarh College of Pharmacy, Landran (IAEC/FEB16/018). The animals were fasted overnight before the experiment. Before experimentation, the animals were trained to stay in the restrainer. This ensured that the rats were calm and unaggressive during blood pressure (BP) measurements.
Hypertension was induced by subcutaneous injection of methyl prednisolone acetate (20 mg/kg/week) for 2 weeks [25]. The rats were then randomly divided into three groups with six animals in each group. The groups were methyl prednisolone+saline (methyl prednisolone group), methyl prednisolone+nisoldipine (nisoldipine group), and methyl prednisolone+nisoldipine-piperine nanoparticles (nanoparticle group). In addition, a normal control group treated with saline alone was also included. Drug administration was started 30 min after the last methylprednisolone injection. Nisoldipine was administered orally once in a dose of 0.9 mg/kg and nanoparticles were also administered orally once in a dose of 0.45 mg/kg in solution form by using a cannula attached to a syringe into the mouth of animals. The dose of nisoldipine used was selected based upon the approved clinical dose in humans [26]. This dose was then converted to the equivalent rat dose on the basis of body surface area formula. A non-invasive tail cuff method was used to measure systolic BP [27]. BP in all groups was measured up to 120 h after drug administration starting from 0 h. At each time point, three readings were taken for each animal and the mean was calculated.
The pharmacokinetic profiles of nisoldipine nanoparticles and conventional nisoldipine formulation were also evaluated after oral administration of drugs. Parameters such as peak serum concentration (Cmax), time for peak serum concentration (tmax), area under the curve (AUC, 0–∞), half-life (t1/2), and mean residence time (MRT) were calculated. The blood samples were withdrawn from the tail vein at 0, 1, 2, 3, 4, 6, 8, 10, 12 and 24 h post-dose. About 0.1 ml of blood sample was withdrawn in Eppendorf tubes and centrifuged at 3000 rpm for 30 min. The serum was transferred to another Eppendorf tube and stored at −20°C until further analyzed by high-performance liquid chromatography (HPLC) using a C-18 analytical column (250 mm×4.6 mm and 5 μm in diameter; Merck, Mumbai, India). The mobile phase consisted of acetonitrile-water (78:22) at a flow rate of 1.1 ml/min and detection at λ max of 237 nm. The injection volume was 10 μl [28]. A stock solution was prepared by dissolving accurately weighed 10 mg of nisoldipine in 10 ml volumetric flask to obtain a 1 mg/ml solution using HPLC-grade methanol. Kinetica software (version 5.0; Innaphase Corporation, Philadelphia, PA, USA) was used to calculate the pharmacokinetic parameters. The values are expressed as mean±SD. The data from BP experiments and pharmacokinetic data are presented as mean±SD. The data were analyzed by unpaired Student’s t-test. A value of p≤0.05 was considered statistically significant.
2.2.8 Accelerated stability studies
Nanoformulation stability analysis was done according to International Council on Harmonization [29] guidelines at temperature of 25±2°C and relative humidity of 5%. The nanoparticle formulation samples were collected at prearranged intervals (30, 60, and 90 days) and tested for particle size, shape, appearance, and DSC analysis. Drug content stability and drug entrapment of nanoparticles were also determined by storing the nanoparticles at 4.0±1°C in a refrigerator and 40±2°C in a stability testing chamber for 3 months.
3 Results and discussion
The Taguchi L9 orthogonal array optimized design was used to prepare nanoparticles of PLGA loaded with nisoldipine and piperine by the precipitation technique, to increase the bioavailability and to obtain a sustained release of drug. Nanoparticles were prepared to study the effect of polymer concentration, amount of bioenhancer, and ratio of solvent on the response variables of nanoparticles. Nine formulations were developed and the calculated values of response variables are shown in Tables 3 and 4. The main effect plots were analyzed to define optimal levels of factors independently and also simultaneously using ANOVA.
The experimental vs. predicted S/N ratio was obtained, and the obtained effect of each factor alone as well as at combined level on responses were plotted (Figure 1A–C). The main responses of S/N ratio plots confirmed the significant factor levels and were used further in the development and characterization of formulation.

Taguchi analysis of significant factors individually: (A) main effect plots on entrapment efficiency (Y1); (B) particle size (Y2); (C) combined effects on Y1 and Y2.
It was concluded that mean particle size (nm) increased with the increase in polymer-drug ratio, whereas it decreased with the increase in piperine concentration. Drug entrapment efficiency (% w/w) was increased with the increase in polymer-drug ratio and piperine concentration, while it remained unaffected by solvent ratio. The optimized formulation had a mean particle size of 132±7.21 nm, drug entrapment efficiency of 89.77±1.06% w/w, drug loading of 13.6±0.56% w/w, and ZP of −26.5 mV. The factors most affecting formulation were polymer ratio and piperine concentration.
3.1 Characterization of nanoparticles
The FTIR spectral analysis showed the characteristic peaks of nisoldipine at 3320 cm−1, N-H stretching at 3001 cm−1, C-H stretching and peak at 1701 cm−1, and esterified carbonyl group stretch. There was also a peak at 1555 cm−1 (aryl nitro group) and at 1230 cm−1 (ether absorption) (Figure 2). Similar peaks were observed in the spectra of nisoldipine nanoparticles without any remarkable change in their positions. Hence, it may be concluded that there was no chemical interaction between the drug and the polymer.

(A) The Fourier transforms spectra of nisoldipine. (B) The Fourier transforms spectra of nanoparticles.
3.2 DSC
If the drug was present in a molecular dispersion or solid solution state in the polymeric nanoparticles, then no detectable endotherm was observed. In the absence of any interaction, the thermogram of a formulation will show patterns corresponding to those of the individual components. Pure nisoldipine shows an endothermic melting peak at 150°C indicating its crystalline nature, piperine shows a peak at 128°C, while PLGA shows an endothermic peak at 59°C corresponding to its glass transition temperature. The thermogram of nanoparticles showed broadening of the characteristic endothermic peak corresponding to nisoldipine and piperine, while the PLGA peak shifted to 54°C, suggesting a decrease in its glass transition temperature. This deviation may be attributed to physical interactions such as hydrogen bonding between the carbonyl group of PLGA and the NH group of nisoldipine. Hence, considering the fact that the drug peak was broadened and the polymer peak was shifted in the drug-loaded nanoparticles, it could be due to physical interaction or H-bonding between the carbonyl groups of PLGA and the NH groups of nisoldipine (Figure 3).

Comparison of DSC of ingredients and formulation.
3.3 Morphological considerations
TEM was used for morphological analysis of nanoparticles. Discrete spherical structures were seen without aggregation (Figure 4). It was observed that the particle sizes were uniform with a narrow size distribution range (average 132±1.21 nm). Additionally, the surface charge of optimized nanoparticles was obtained as −26.5 mV (Zetasizer Nano ZS90). The nanoparticles were dispersed in dilutions of PBS at pH 6.8 (ionic strength 1.5×10−5 m) at 25°C.
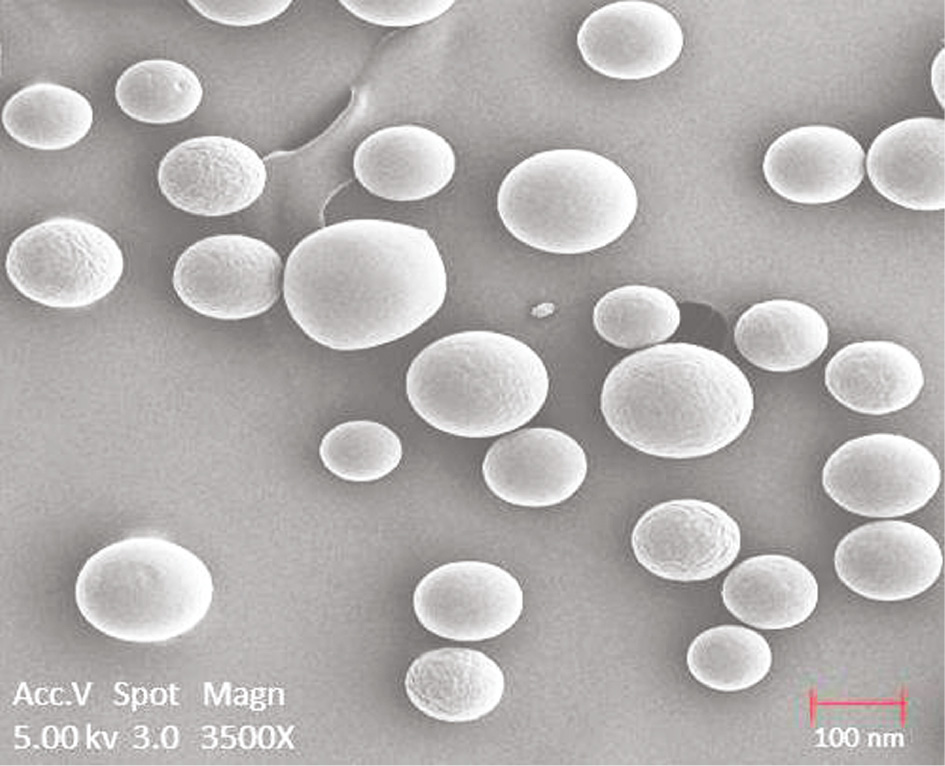
TEM analysis of nanoparticles.
3.4 Drug release studies
The in vitro release behavior of conventional nisoldipine formulation was rapid, consistent, and completed within 12 h (92.5±2.17% for pH 6.8 and 90.4±1.77% for pH 1.2). In 0.1 n HCl (pH 1.2), the cumulative percentage of release from the nanoformulation was 96.9±0.48% over a period of 60 h (Figure 5). In phosphate buffer (pH 6.8), the cumulative percentage of release was 98.3±0.26% in 60 h (Figure 5). No difference was observed in drug release from nanoparticles in phosphate buffer (pH 6.8) and 0.1 n HCl. Thus, the release and solubility of nisoldipine from nanoparticles were independent of pH. Nanoparticles showed a biphasic release behavior of drug. Biphasic release consisted of an initial burst effect up to 6 h followed by a sustained release phase up to 60 h. The initial burst effect was characterized by approximately 32.13±2.3% drug release within 6 h. The initial burst phase was attributed to the dissipation and diffusion of drug adhered to the surface of polymeric nanoparticles or entrapped poorly in polymer matrix. The explosion effect was favorable and helped in realizing the therapeutic concentration of drug in comparatively lesser time [30].
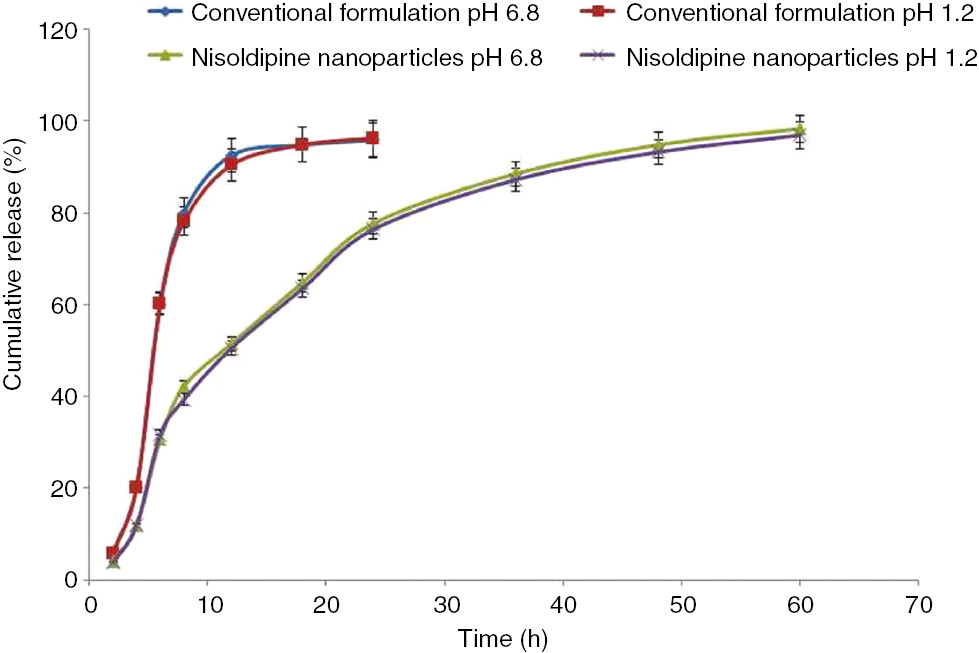
Percentage of cumulative release of nisoldipine from conventional and nanoparticles.
3.5 Release kinetics and stability
The in vitro release data indicated first-order and Higuchi-order release kinetics of nanoparticles, signifying the role of the diffusion principle in drug release. The n-value (0.86–0.33) obtained for the Peppas release model proposed the “non-Fickian diffusion transport” mechanism supported diffusion and relaxation mechanism of controlled drug release (Table 5). The formulation was stable, as no noticeable alteration was noted in terms of particle size and drug content in the tested storage conditions.
Value of regression obtained from different kinetic models.
| Formulation | R2 | ||||
|---|---|---|---|---|---|
| Zero order | First order | Higuchi model | Korsmeyer-Peppas model | n-Value | |
| Nanoparticles | 0.8267 | 0.9317 | 0.9369 | 0.9976 | 0.86–0.33 |
3.6 Antihypertensive study
The antihypertensive effect of nisoldipine-piperine nanoparticles was studied and compared to conventional nisoldipine formulation. Systolic BP was measured and results are given in Table 6. In the normal control group, normal systolic BP was observed. Oral administration of the conventional formulation significantly controlled the hypertension initially, with the maximum effect at 2 h; however, afterwards, the BP gradually increased and stabilized. In contrast, the oral administration of nisoldipine-piperine nanoparticles resulted in a steady decrease of BP. The maximum effect was observed at 36 h, and this effect was maintained up to 72 h. In the hypertensive control group, the systolic BP was significantly higher compared to the normal control group. The 22.30% reduction in BP by nisoldipine nanoparticles as compared to 16.24% reduction by conventional formulation clearly indicated that the nisoldipine nanoparticle formulation gradually released the drug over a period of 72 h. As the administration of nisoldipine-piperine nanoparticles led to sustained and continued drug release, it was proficient in overcoming the limitations of oral administration of conventional nisoldipine formulation [31], [32].
Antihypertensive effect of optimized nisoldipine-piperine nanoparticles and conventional formulation.
| Time (h) | Group 1 Normal control | Group 2 Hypertensive control | Group 3 Nisoldipine conventional | Group 4 Nisoldipine-Piperine nanoparticles |
|---|---|---|---|---|
| Initial | 100.23±0.57 | 165.31±0.38 | 164.12±0.51 | 167.13±0.32 |
| 1 | 110.11±0.42 | 164.91±0.24 | 150.82±0.26 | 158.11±0.35 |
| 2 | 105.54±0.31 | 164.43±0.34 | 110.36±0.22 | 153.17±0.27 |
| 4 | 98.61±0.28 | 164.11±0.38 | 112.61±0.37 | 148.24±0.21 |
| 8 | 110.01±0.33 | 163.73±0.61 | 114.58±0.39 | 142.68±0.41 |
| 12 | 106.82±0.42 | 163.47±0.29 | 116.57±0.42 | 136.41±0.45 |
| 18 | 107.14±0.44 | 161.92±0.28 | 119.67±0.23 | 128.54±0.34 |
| 24 | 112.03±0.29 | 160.18±0.35 | 123.29±0.37 | 119.13±0.29 |
| 36 | 103.71±0.36 | 159.55±0.41 | 128.37±0.25 | 104.19±0.33a |
| 48 | 101.96±0.41 | 158.37±0.43 | 132.61±0.34 | 105.72±0.42a |
| 60 | 103.05±0.52 | 158.12±0.48 | 137.34±0.55 | 106.79±0.22a |
| 72 | 105.11±0.31 | 157.15±0.33 | 139.17±0.61 | 106.97±0.28a |
| 84 | 109.44±0.27 | 156.37±0.27 | 143.01±0.38 | 106.55±0.31a |
| 96 | 100.76±0.45 | 148.44±0.55 | 145.27±0.21 | 108.31±0.36a |
| 108 | 99.61±0.39 | 142.91±0.41 | 147.64±0.39 | 108.81±0.51a |
| 120 | 102.48±0.24 | 142.56±0.43 | 149.38±0.24 | 109.62±0.47a |
Data are presented as mean±SD, n=6. ap-Value <0.05 compared to conventional formulation.
3.7 Pharmacokinetic study
The appropriate pharmacokinetic factors were calculated for the prepared nanoformulation and compared with conventional nisoldipine formulation (Table 7). The Cmax value (22.30±0.56 μg/ml) for nisoldipine-piperine nanoparticles was statistically significant at p<0.0001 with respect to conventional formulation (12.10±0.29 μg/ml). The AUC (0–∞), which symbolizes the magnitude of absorption, was also significantly (p<0.05) higher for nanoparticles (255.54±5.92 μg/ml/h) compared to conventional formulation (52.09±3.76 μg/ml/h). The tmax, t1/2, and MRT values were also higher for nisoldipine-piperine nanoparticles as compared to conventional formulation, resulting in a 4.9-fold improvement in oral bioavailability. The reported literature suggests the role of particle size reduction in improvement of bioavailability by 1.55–2.22 times. Due to the nano size of the formulation, the effective surface increased consequently, increasing the contact time of the nanoparticles and oral bioavailability by 2.17 [33]. The designing of the nisoldipine-loaded self-nanoemulsifying drug delivery system increased the bioavailability by 2.22 times [34]. Similarly, an increase of 1.55-fold bioavailability of nisoldipine was reported in a hydrogel formulation [35]. An additional increase in bioavailability of the prepared nanoparticles was due to the CYP3A4 enzyme inhibitory activity of piperine, which reduces the first-pass metabolism of the drug. Hence, it could be concluded that the enhanced oral bioavailability of nisoldipine-piperine nanoparticles was due to the contribution of both of these factors.
Pharmacokinetic profile of nisoldipine-piperine nanoparticles and nisoldipine conventional formulation.
| Parameters studied | Nisoldipine conventional | Nisoldipine-Piperine nanoparticles |
|---|---|---|
| Cmax | 12.1±0.29 | 22.3±0.56a |
| tmax | 2.0 | 6.0a |
| AUC0−∞ | 52.09±3.76 | 255.54±5.92a |
| t1/2 | 6.25 | 17.74a |
| MRT | 4.63 | 14.19a |
Data are presented as mean±SD. ap-Value <0.05 when compared to conventional formulation. All values are means of three readings.
4 Conclusion
Nanotechnology-based delivery systems for antihypertensive agents are a promising approach in resolving some constraints of antihypertensives. Targeted nanoparticles can effectively take antihypertensives to their site of action, and chronotherapeutics along with nanotechnology can effectively control high BP by not only modifying the release pattern of the drug but also by increasing the bioavailability of the drug. The nisoldipine-piperine-loaded nanoparticles demonstrated good encapsulation efficiency and sustained release behavior. The results obtained from the study are encouraging, as drug-piperine nanoparticulates resulted in a 4.9-fold increase in the bioavailability by nanonization and inhibition of CYP3A4 enzymes. Hence, nisoldipine doses may require special consideration if used along with piperine-containing preparations to avoid complications resulting from drug-drug interactions, leading to alterations of bioavailability.
Acknowledgments
The authors are thankful to Orchid Pharma, Chennai, for providing the model drug gift sample. The research facility provided by IKG Punjab Technical University, Kapurthala, and Chandigarh College of Pharmacy, Landran, India, is gratefully acknowledged.
Conflict of interest statement: The authors declare that they have no conflict of interest.
Funding: No funding was obtained for this study.
References
[1] Makadia HK, Siegel SJ. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers (Basel) 2011, 3, 1377–1397.10.3390/polym3031377Search in Google Scholar PubMed
[2] Jain RA. The manufacturing techniques of various drug loaded biodegradable poly (lactideco-glycolide) (PLGA) devices. Biomaterials 2000, 21, 2475–2490.10.1016/S0142-9612(00)00115-0Search in Google Scholar
[3] Allison SD. Effect of structural relaxation on the preparation and drug release behavior of poly (lactic-co-glycolic) acid microparticle drug delivery systems. J. Pharm. Sci. 2008, 97, 2022–2035.10.1002/jps.21124Search in Google Scholar PubMed
[4] Mundargi R, Babu V, Rangaswamy V, Patel P, Aminabhavi T. Nano/micro technologies for delivering macromolecular therapeutics using poly(d,l-lactide-co-glycolide) and its derivatives. J. Control. Rel. 2008, 125, 193–209.10.1016/j.jconrel.2007.09.013Search in Google Scholar PubMed
[5] Mohamed F, van der Walle CF. Engineering biodegradable polyester particles with specific drug targeting and drug release properties. J. Pharm. Sci. 2008, 97, 71–87.10.1002/jps.21082Search in Google Scholar PubMed
[6] Rathee P, Kamboj A, Sidhu S. Nano-carrier systems: an overview of poly-art and peptide conjugates. Curr. Nanomed. 2016, 6, 50–62.10.2174/1877912306999160122160351Search in Google Scholar
[7] British Pharmacopoeia. The Department of Health Social Services and Public Safety: Great Britain, MHRA: London 2007.Search in Google Scholar
[8] Tatiraju VD, Varsha B, Priya J, Karambelkar J, Jadhav MV, Kadam V. Natural bioenhancers: an overview. J. Pharmacog. Phytochem. 2013, 2, 55–60.Search in Google Scholar
[9] Acharya SG, Momin AH, Gajjar AV. Review of piperine as a bio-enhancer. Am. J. PharmTech Res. 2012, 2, 32–44.Search in Google Scholar
[10] Han HK. The effects of black pepper on the intestinal absorption and hepatic metabolism of drugs. Exp. Opin. Drug Metab. Toxicol. 2011, 7, 721–729.10.1517/17425255.2011.570332Search in Google Scholar PubMed
[11] Bhardwaj RK, Glaeser H, Becquemont L, Klotz U, Gupta SK, Fromm MF. Piperine, a major constituent of black pepper, inhibits human P glycoprotein and CYP3A4. J. Pharmacol. Exp. Ther. 2002, 302, 645–650.10.1124/jpet.102.034728Search in Google Scholar PubMed
[12] Volak LP, Ghirmai S, Cashman JR, Court MH. Curcuminoids inhibit multiple human cytochromes P450 (CYP), UDP-glucuronosyltransferase (UGT), and sulfotransferase (SULT) enzymes, while piperine is a relatively selective CYP3A4 inhibitor. Drug Metab. Dispos. 2008, 36, 1594–1605.10.1124/dmd.108.020552Search in Google Scholar PubMed PubMed Central
[13] Shoba G, Joy D, Joseph T, Majeed M, Rajendran R, Srinivas PS. Influence of piperine on the pharmacokinetics of curcumin in animals and human volunteers. Planta Med. 1998, 64, 353–356.10.1055/s-2006-957450Search in Google Scholar PubMed
[14] Godfraind T, Salomone S, Dessy C, Verhelst B, Dion R, Schoevaerts JC. Selectivity scale of calcium antagonists in the human cardiovascular system based on in vitro studies. J. Cardiovasc. Pharmacol. 1992, 20, S34–S41.Search in Google Scholar
[15] Takanaga H, Ohnishi A, Murakami H, Matsuoa H, Higuchi S, Urae A, Irie S, Furuie H. Relationship between time after intake of grapefruit juice and the effect on pharmacokinetics and pharmacodynamics of nisoldipine in healthy subjects. Clin. Pharmacol. Ther. 2000, 67, 201–214.10.1067/mcp.2000.104215Search in Google Scholar PubMed
[16] Yuan L, Jia P, Sun Y, Zhao C, Zhi X, Sheng N, Zhang L. Study of in vitro metabolism of m-nisoldipine in human liver microsomes and recombinant cytochrome P450 enzymes by liquid chromatography-mass spectrometry. J. Pharm. Biomed. Anal. 2014, 97, 65–71.10.1016/j.jpba.2014.03.030Search in Google Scholar PubMed
[17] Nazzal S, Khan M. Response surface methodology for the optimization of ubiquinone self-nanoemulsified drug delivery system. AAPS Pharm. SciTech. 2002, 3, 23–31.10.1208/pt030103Search in Google Scholar
[18] Taguchi G. System of Experimental Design. UNIPUB, Kraus International Publications: New York, 1987.Search in Google Scholar
[19] Glen S. Taguchi Methods. Peace, Addison Wesley Publishing Company, Inc.: New York, 1992, pp. 338–363.Search in Google Scholar
[20] Roy RK. Design of Experiments Using the Taguchi Approach: 16 Steps to Product and Process Improvement. John Wiley & Sons: New York, 2001.Search in Google Scholar
[21] Muhlen A, Schwarz C, Mehnert W. Solid lipid nanoparticles (SLN) for controlled drug delivery-drug release and release mechanism. Eur. J. Pharm. Biopharm. 1998, 45, 149–155.10.1016/S0939-6411(97)00150-1Search in Google Scholar PubMed
[22] Sandhya J, Pavani A, Reddy RR. Formulation and evaluation of nanosuspension of Nisoldipine. Int. J. Pharm. Sci. Rev. Res. 2014, 24, 177–181.Search in Google Scholar
[23] Bazylinska U, Lewinska A, Lamcha L, Wilk KA. Polymeric nanocapsules and nanospheres for encapsulation and long sustained release of hydrophobic cyanine-type photosensitizer. Colloids Surf. A: Physicochem. Eng. Aspects 2014, 442, 42–49.10.1016/j.colsurfa.2013.02.023Search in Google Scholar
[24] Papadimitriou S, Bikiaris D. Novel self-assembled core–shell nanoparticles based on crystalline amorphous moieties of aliphatic copolyesters for efficient controlled drug release. J. Control. Rel. 2009, 138, 177–184.10.1016/j.jconrel.2009.05.013Search in Google Scholar PubMed
[25] Krakoff LR, Selvadurai R, Sytter E. Effect of methylprednisolone upon arterial pressure and the renin angiotensin system in the rat. Am. J. Physiol. 1975, 228, 613–617.10.1152/ajplegacy.1975.228.2.613Search in Google Scholar PubMed
[26] US Food and Drug Administration, fda.gov/scripts/cder/daf/index.cfm/ApplNo:020356. http://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=020356. Accessed on 5 May, 2017.Search in Google Scholar
[27] Daugherty A, Rateri D, Hong L, Balakrishnan A. Measuring blood pressure in mice using volume pressure recording, a tail-cuff method. J. Visual. Exp. 2009, 27, 1–2.10.3791/1291Search in Google Scholar PubMed PubMed Central
[28] Swetha A, Buchi NN. Development and validation of RP-HPLC-PDA method for the estimation of nisoldipine in bulk and formulations. Ind. J. Pharm. Sci. 2012, 4, 88–91.Search in Google Scholar
[29] ICH Guideline Q1A (R2). Stability Testing of New Drug Substances and Products. 2003, ICH: Geneva, Switzerland, pp. 1–18.Search in Google Scholar
[30] Fu Y, Kao WJ. Drug release kinetics and transport mechanisms of non-degradable and degradable polymeric delivery systems. Exp. Opin. Drug Del. 2010, 7, 429–444.10.1517/17425241003602259Search in Google Scholar PubMed PubMed Central
[31] Langtry HD, Spencer CM. Nisoldipine coat-core: a review of its pharmacodynamic and pharmacokinetic properties and clinical efficacy in the management of ischaemic heart disease. Drugs 1997, 53, 867–884.10.2165/00003495-199753050-00013Search in Google Scholar PubMed
[32] Friedel HA, Sorkin EM. Nisoldipine: a preliminary review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy in the treatment of angina pectoris, hypertension and related cardiovascular disorders. Drugs 1988, 36, 682–731.10.2165/00003495-198836060-00003Search in Google Scholar PubMed
[33] Dudhipala N, Veerabrahma K. Pharmacokinetic and pharmacodynamic studies of nisoldipine-loaded solid lipid nanoparticles developed by central composite design. Drug Dev. Ind. Pharm. 2015, 41, 1968–1977.10.3109/03639045.2015.1024685Search in Google Scholar PubMed
[34] Krishnamoorthy B, Habibur Rahman SM, Tamil Selvan N, Prasad RH, Rajkumar M, Siva Selvakumar M, Vamshikrishna K, Gregory M, Vijayaraghavan C. Design, formulation, in vitro, in vivo, and pharmacokinetic evaluation of nisoldipine-loaded self-nanoemulsifying drug delivery system. J. Nanopart. Res. 2015, 17, 34.10.1007/s11051-014-2818-zSearch in Google Scholar
[35] Xiao Y, Lin Z, Liu J, Zhang W, Wang L, Yu P. A transdermal microemulsion-based hydrogel of nisoldipine: preparation, in vitro characterization and in vivo pharmacokinetic evaluation. Asian J. Pharm. Sci. 2012, 7, 316–328.Search in Google Scholar
©2017 Walter de Gruyter GmbH, Berlin/Boston
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Articles in the same Issue
- Frontmatter
- In this issue
- Communication
- Fabrication and simulation of 1540-nm transmission by 532-nm excitation in photonic crystal of Er:ZnO film
- Self-assembly and enhanced visible-light-driven photocatalytic activity of reduced graphene oxide-Bi2WO6 photocatalysts
- Reviews
- Enhanced oral bioavailability of nisoldipine-piperine-loaded poly-lactic-co-glycolic acid nanoparticles
- Coating strategies for atomic layer deposition
- Magnetically recoverable nano-catalysts in sulfoxidation reactions
- Selective modification of inner surface of halloysite nanotubes: a review
- Analyzing polymeric nanofibrous scaffold performances in diabetic animal models for translational chronic wound healing research
- Photoluminescence from colloidal silicon nanoparticles: significant effect of surface
- Current trends in changing the channel in MOSFETs by III–V semiconducting nanostructures
Articles in the same Issue
- Frontmatter
- In this issue
- Communication
- Fabrication and simulation of 1540-nm transmission by 532-nm excitation in photonic crystal of Er:ZnO film
- Self-assembly and enhanced visible-light-driven photocatalytic activity of reduced graphene oxide-Bi2WO6 photocatalysts
- Reviews
- Enhanced oral bioavailability of nisoldipine-piperine-loaded poly-lactic-co-glycolic acid nanoparticles
- Coating strategies for atomic layer deposition
- Magnetically recoverable nano-catalysts in sulfoxidation reactions
- Selective modification of inner surface of halloysite nanotubes: a review
- Analyzing polymeric nanofibrous scaffold performances in diabetic animal models for translational chronic wound healing research
- Photoluminescence from colloidal silicon nanoparticles: significant effect of surface
- Current trends in changing the channel in MOSFETs by III–V semiconducting nanostructures