PLA/PLGA nanoparticles for delivery of drugs across the blood-brain barrier
-
Jingyan Li
Jingyan Li received her undergraduate degree in Bioengineering in June 2010 from East China University of Science and Technology. In autumn 2010, she joined the Biological and Agricultural Engineering Department at Louisiana State University, Baton Rouge, LA, USA. Jingyan Li received her Master of Science in Biological and Agricultural Engineering in December 2012. She is an active member of the Phi Kappa Phi Honor Society. She currently resides in China.
Cristina Sabliov, PhD is an Associate Professor in the Biological and Agricultural Engineering Department at Louisiana State University and LSU Agcenter. She came to LSU in 2003 from North Carolina State University where she received a double-major PhD in Food Science and Biological and Agricultural Engineering, and a MS in Chemical Engineering. During her tenure at LSU, Dr. Sabliov quickly developed an expertise in the field of nanotechnology, specifically on polymeric nano particles designed for delivery of bioactive components for improved food quality and human health. Projects pursued in her laboratory range from design and synthesis of multifunctional polymeric nanoparticles for delivery of bioactives of controlled properties (size, surface charge, controlled-release profile and targeting properties) to
in vitro andin vivo evaluation of the nanoparticle functiona lity and biotoxicity under the conditions of use. During the past 10 years, Dr. Sabliov’s research was supported by federal (USDA, NSF, ACS, NOAA) and state (LA-BOR, SBB, RRB, ASCL) agencies. She published extensively in reputable journals such as ACS Nano, Nanomedicine, Journal of Biomaterial Science – Polymer Edition, Nanotechnology, and Journal of Physical Chemistry. Dr. Sabliov is a recognized national and international leader in food nanotechno logy. She organized several Institute of Food Technologists (IFT) and American Institute of Chemical Engineers sessions on the topic of Nano technology and Food, and is currently serving on the IFT Food Nanoscience Advisory Panel.

Abstract
The blood-brain barrier (BBB), which protects the central nervous system (CNS) from unnecessary substances, is a challenging obstacle in the treatment of CNS disease. Many therapeutic agents such as hydrophilic and macromolecular drugs cannot overcome the BBB. One promising solution is the employment of polymeric nanoparticles (NPs) such as poly (lactic-co-glycolic acid) (PLGA) NPs as drug carrier. Over the past few years, significant breakthroughs have been made in developing suitable PLGA and poly (lactic acid) (PLA) NPs for drug delivery across the BBB. Recent advances on PLGA/PLA NPs enhanced neural delivery of drugs are reviewed in this paper. Both in vitro and in vivo studies are included. In these papers, enhanced cellular uptake and therapeutic efficacy of drugs delivered with modified PLGA/PLA NPs compared with free drugs or drugs delivered by unmodified PLGA/PLA NPs were shown; no significant in vitro cytotoxicity was observed for PLGA/PLA NPs. Surface modification of PLGA/PLA NPs by coating with surfactants/polymers or covalently conjugating the NPs with targeting ligands has been confirmed to enhance drug delivery across the BBB. Most unmodified PLGA NPs showed low brain uptake (<1%), which indirectly confirms the safety of PLGA/PLA NPs used for other purposes than treating CNS diseases.
1 Introduction
Treatment of central nervous system (CNS) diseases including Alzheimer’s disease (AD), Parkinson’s disease, Huntington’s disease, schizophrenia, HIV infection of the brain and brain tumors remain limited due to the low transport of drugs across the blood-brain barrier (BBB) [1]. The BBB is composed of special endothelial cells with tight junctions, pericytes, astrocytes and microglial cells [2]; tight junctions (TJs) are formed by a complex network of proteins and linked with the cytoskeleton to restrict the passage of substances from the bloodstream to the brain [3]. The expression of different transporters on the BBB such as transferring receptors, insulin receptors, lipoprotein receptors, ATP-binding cassette (ABC) efflux transporters p-glycoprotein (P-gp) could either help the internalization or prevent the entry of molecules. P-gp is the principal efflux method for extruding drugs form the brain. Only a small amount of water-soluble molecules diffuse paracellularly through the TJ. Small lipid soluble molecules enter transcellularly through the lipid membrane. Cell-mediated transcytosis is another recently identified route for drugs entering the brain through the BBB; for example, some pathogens such as HIV or Cryptococcal could enter the BBB via cell-mediated transcytosis [3, 4]. For other substances, adsorptive transcytosis and specific receptor-mediated are the main transport routes across the BBB [3]. Adsorptive-mediated transcytosis (AMT) is induced by the electrostatic interaction between the negatively charged membrane surface and a positively charged substance. Receptor-mediated transcytosis (RMT) is a selective uptake of a specific substance. The expression of different transporters such as transferring receptors, insulin receptors and lipoprotein receptors could help the internalization of molecules [3].
Transport of many therapeutic agents for CNS disease such as hydrophilic drugs and macromolecular drugs through the impermeable TJ of the BBB is limited [5]. Therefore, invasive and non-invasive methods were employed to enhance brain delivery of drugs. Compared with invasive methods which include changing the permeability of the BBB, or direct intracerebral infusion and implantation of the drug, non-invasive methods such as systemic administration are preferred [1].
For systemically administered drugs, the goal for the drug delivery system is to cross the BBB in sufficient concentrations without a negative impact on the function of the BBB. A number of drug carriers have been developed to improve brain delivery of drugs, and among carriers, nanoparticles (NPs) have been receiving increased attention because of their unique properties [3]. In the size range from 1 to 1000 nm, nanocarriers are able to carry therapeutically significant amounts of drug, which cannot otherwise be transported through the BBB in free form. These delivery systems can be designed to provide a sustained controlled release profile, which reduces negative side effects of drugs to healthy tissue and prolong their retention time in the body. After surface modification, nanocarriers, especially at sizes under 300 nm can provide enhanced brain delivery of loaded drug. Biodegradable polymeric NPs such as poly (lactic-co-glycolic acid) (PLGA) and poly (lactic acid) (PLA) have been well studied for drug delivery as stable drug carriers. Compared with other nanocarriers, such as liposomes, micelles and dendrimers, the advantages of polymeric NPs include improved stability, high loading capacity, sustained drug release, non-immunogenic property, reduced drug toxicity, improved bioavailability, enhanced therapeutic efficacy of the entrapped drug and versatile surface modification [6]. PLGA and PLA are commonly used polymers for nano-delivery because of their biocompatibility, biodegradation and well-studied degradation kinetics; PLGA and PLA have also been approved for pharmaceutical application by the US Food and Drug Administration [6]. Brain delivery can be achieved by well-designed polymeric NPs fabricated from polymers such as PLGA and PLA of different molecular weights, with surfactants and other surface modifications that confer the surface characteristics (i.e., zeta potential and hydrophilicity) desired for improved BBB uptake via adsorptive transcytosis. In addition, ligands known to target BBB surface receptors such as transferring, insulin and lipoprotein receptors can be linked to the surface of the polymeric NPs to initiate receptor-mediated endocytosis and thus cross the BBB [7].
Both in vitro and in vivo studies have been conducted to study cytotoxicity and effectiveness of polymeric NPs designed for delivery of drugs to the CNS. Prior to complex and expensive in vivo studies, in vitro studies on cytotoxicity, cellular uptake, endocytosis mechanism and drug efficiency of the drug delivery system were investigated with different cell lines such as brain endothelial cells (BCECs) and glioma cells. Next, in vivo studies performed on mouse and rats were used to study brain uptake, location of nanocarriers and therapeutic efficacy of the entrapped drug.
Aspects of NP synthesis, general properties, mechanisms of action and potential pitfalls of NPs for drug delivery to the CNS have been reviewed over the past years [1, 8–11]. Recent findings on polymeric NPs as brain delivery carriers are also available in the literature [6, 12].
However, to the authors’ knowledge, no reviews have been specifically and solely dedicated to the effect of NPs properties on delivery of drugs to the brain with PLGA/PLA NPs; this aspect was only partially mentioned or briefly discussed in previous review papers. The goal of this review is to highlight the recent developments on PLGA or PLA NPs designed for neural delivery, with emphasis on NP cytotoxicity, therapeutic efficacy of the entrapped drug and brain uptake of PLGA/PLA NPs as a function of their properties.
In this review, a literature search was conducted on the subject of PLGA/PLA NPs and brain delivery of drugs using PubMed, ScienceDirect and SpringerLink databases. The following keywords were used in the search: polymeric/PLGA nanoparticles, brain delivery, blood-brain barrier, central nervous system and neuron delivery. Only papers relevant to PLGA/PLA NPs focused on systemic administration, with the goal of delivering drugs across the BBB were selected. In total, 29 papers published after 2004 on PLGA/PLA NPs for drug delivery across the BBB were used in this review.
The papers are summarized with an emphasis on NP characteristics (core polymer, PLGA composition and molecular weight, emulsifier, surface modification, loaded drug, size, zeta potential), the in vitro and in vivo models used, methods, and significant findings are compiled in Table 1. The discussion section is divided into two sections: in vitro studies and in vivo studies. For the in vivo studies, the percentage (%) brain uptake over 24 h was calculated as percentage of dose administered per animal, and plotted versus time (h). Potential effects of NP properties such as size, dose and administration on brain uptake of different NPs were compared and discussed.
Summaries of NP characteristics (core polymer, emulsifier, surface modification, loaded drug, fluorescent marker, size and zeta potential), animal study and result.
| Core polymer | Emulsifier | Surface modification | Loaded drug | Fluorescent marker | Size (nm) | Zeta potential (mV) | Experimental details | Results | References |
|---|---|---|---|---|---|---|---|---|---|
| PLGA (50:50, 5–15 kDa) | PVA | Curcumin | 163 | -12.5 | In vivo: male Sprague-Dawley rats, i.v. administration | Significantly increased retention times of curcumin in the cerebral cortex and hippocampus | [13] | ||
| PLGA (50:50, 35–40 kDa) | DMAB | 1% T80 2% T80 3% T80 4% T80 5% T80 | Estradiol | 134.7 138.8 141.8 153.1 157 172.4 | 68.5, pH 4.1 19.2, pH 5.6 7.7 -1.1 -3.6 -6.5 | In vitro: simulated gastric fluid and intestinal fluid In vivo: male Sprague-Dawley rats, oral administration | T-80 coat was stable in simulated gastric fluid and intestinal fluid Orally administered T80-coated nanoparticles reached higher levels after 24 h than uncoated drugs | [14] | |
| PLGA (50:50) | PVA | Doxorubicin | 238.6 | 11.8 | In vivo: tumor-bearing male Wistar rats, tail vein injection | Loperamide loaded PVA-stabilized PLGA nanoparticles coated with poloxamer proved the highest and long-lasting anti-tumor(glioblastoma) effect in rats | [15] | ||
| PVA | F68 | 243.4 | 6 | ||||||
| PVA | T80 | 239.9 | 8.2 | ||||||
| HSA | 401.7 | 9.5 | |||||||
| HSA | F68 | 408.6 | 8.1 | ||||||
| HSA | T80 | 412 | 16.2 | ||||||
| PVA | Loperamide | 177.7 | -11.4 | ||||||
| PVA | F68 | 168.5 | -17.9 | ||||||
| PVA | T80 | 166.9 | -25 | ||||||
| HSA | 288.9 | -11.9 | |||||||
| HSA | F68 | 287.7 | -17.5 | ||||||
| HSA | T80 | 292.4 | -18.9 | ||||||
| PLA (5 kDa) | T80 | FITC FITC | 162.1 194.2 202.6 | -29.5 -13.4 -10.17 | In vivo: mice, tail vein injection | T80 coat was necessary for brain targeting. Free FITC, T80 and blank NPs were not observed in brain tissues | [16] | ||
| PLGA (50:50, 40–75 kDa) | PVA TPGS PVA PVA PVA | F68 T80 F127 | Docetaxel 1 | 227.7 222.3 235.5 250.2 242.5 | -27.5 -41.3 -23.8 -21.3 -22.4 | In vitro: C6 glioma cell line, cytotoxicity studies | The lowest IC50 was achieved after 72 h by the F68-PVA-NPs | [17] | |
| PVA TPGS PVA PVA PVA | F68 T80 F127 | Coumarin-6 | 177 165 196 194 188 | -31.1 -38.5 -20.2 -23.1 -20.6 | In vitro: MDCK cells uptake studies In vivo: male Sprague-Dawley rats, tail vein injection | TPGS coated PLGA NPs showed 1.5-fold higher cellular uptake than PVA-emulsified NPs. F68-coated PLGA NPs displayed the highest uptake compared with T80- and F127-coated NPs. F68-coated PLGA nanoparticles showed highest brain accumulation over those with T80 and F127 on the surface | |||
| PLGA (75:25, 20 kDa) | PVA | T80 F68 CS | Coumarin-6 | 269.3 231.7 252.2 396.2 | -21.2 -20.3 -19.8 6.1 | In vivo: male Wister rats, carotid artery/jugular administration | Carotid artery administration of all NPs showed higher brain distribution than after jugular vein administration. T80-PLGA NP prolonged circulation time in the blood. CS-PLGA NP and T80-PLGA NP proved higher brain distribution than uncoated and F68 coated NPs. CLSM studies demonstrated that only T80-PLGA crossed the BBB and located in the parenchyma | [18] | |
| PLGA (50:50, 40–75 kDa) | TPGS | Loperamide | 173.5 | -24.9 | In vitro: rat brain endothelial cell and C6 co-cultures | P188-coated NPs showed better permeability than P80-coated NPs (1.47-fold); no toxicity | [19] | ||
| PLGA-PEG-PLGA (PEG 2 kDa) | P80 | 152.3 149 | -0.5 1.5 | In vivo: male ICR mice, tail vein injection | P188 NPs showed better effect over P80 NPs followed by PEP NP and blank NPs | ||||
| P188 | 147 | 3.1 | |||||||
| PLGA PLGA-PEG- PLGA | TPGS | P80 P188 | Coumarin-6 | 224.2 196.5 190 200 | -22.4 -0.4 1.2 2.4 | In vivo: male SD rats, tail vein injection | P188-coated NPs distributed more in the brain than P80-coated NPs. Surfactant-coated NPs showed enhanced brain delivery over uncoated NPs; PEGylated NPs showed increased uptake than blank NPs | ||
| PLGA | PEG TPGS P80 F127 | Coumarin-6 | 215 225 248 256 234 | n/r n/r n/r n/r n/r | In vitro: MDCK cells uptake studies; C6 glioma cells cytotoxicity study (entrapped paclitaxel instead of coumarin-6) | The addition of P80 and TPGS during synthesis helped improve cellular uptake more than surface-coated nanoparticles; P80 emulsified NPs showed lowest cell viability | [20] | ||
| PEG TPGS F127 | 239 245 251 | n/r n/r n/r | |||||||
| PLA-b-PEG | n/r | T80 | Amphotericin B (AmB) | 120 120 | n/r n/r | In vivo: Cryptococcal meningitis-bearing mice, intravenous administration | T80 enhanced the stability and entrapment efficiency, and brain targeting | [21] | |
| PLGA (75:25, 10 kDa) | TPGS | TMC TMC | Coenzyme Q10 | Coumarin-6 | 94.7 136.8 99.6 146.7 | -22.3 17.7 -18.3 21 | In vitro: cytotoxicity in SH-SY5Y cells by MTT assay In vivo: CD-1 wild type mice, caudal veins injection In vivo: APP/PS1 double transgenic mice, caudal veins injection | No significant cytotoxic effects Accumulated in the periventricular region of the cortex and the third ventricle, mechanism was AMT; behavior tests, senile plaque and biochemical parameters were confirmed superior to the blank NPs | [22] |
| PLGA (50:50) | TPGS | 250 | n/r | In vitro: PC 12 cell line | No cytotoxicity, the more TPGS on the surface, the more effective therapeutic effects | [23] | |||
| PLGA (50:50, 153 kDa) | PVA | CS CS, anti-Aβ antibody | Coumarin-6 | 267.7 217.3 265 | -7.6 32.0 16.8 | In vitro: polarized MDCK cell monolayers | Selectively targeted the intracellular Ab antigen | [24] | |
| PEG-PLA | Sodium Cholate | Alexa Fluor 488-labeled CBSA BSA | Sulpiride | Rhodamine B | 218 329 308 | -39 -19 n/r | In vivo: male Sprague-Dawley rats, injection in caudal vein | Faster clearance from the plasma and a high biodistribution in brain | [25] |
| PEG-PLA | |||||||||
| (43–44 kDa) | Coumarin-6 | 100 | -16.8 | In vitro: co-culture of brain endothelial cells (BCECs) and astrocytes | Permeability of CBSA-NPs was higher than blank NPs, and decreased while incubated with free CBSA | [26] | |||
| BSA | Coumarin-6 | 97.4 | -16.2 | ||||||
| CBSA | Coumarin-6 | 105 | -12.2 | In vivo: male BALB/c mice, tail vein injection | AMT process while no brain targeting of native BSA | ||||
| PLGA | |||||||||
| (11 kDa) | F68 | Peptide 1 Peptide 2 Peptide 2 Peptide 3 Peptide 4 Peptide 5 | Fluorescein (f)-linked f-linked f-linked Tetramethylrhodamine (TMR)-linked f-linked f-linked f-linked | 212 170 162 211 178 191 185 | -17.1 -6.9 -8.6 -14.6 -15.2 -6.1 -9.9 | In vivo: rat brain perfusion technique; rat femoral vein injection | Not able to cross the BBB, localized into the blood vessels Penetrate into cerebral parenchyma Reach CNS after systemic administration Reach CNS after systemic administration Penetrate into cerebral parenchyma Not able to cross the BBB Penetrate into cerebral parenchyma | [27] | |
| PEG-PLGA (25:75) | n/r | 12-amino acid peptide | Coumarin-6 | 109.3 121.5 | -23.4 -18.3 | In vitro: bEnd.3 cells, a model of the BBB In vivo: male nude mice intravenously infection | Significantly higher bEnd.3 cells uptake and brain delivery Significantly higher brain delivery | [28] | |
| PLA (40 kDa) PLGA | PVA | TAT peptide | 3H-ritonavir 3H-ritonavir | Coumarin-6 | 300 340 n/r | -19.3 2.4 n/r | In vitro: MDCK cells overexpressing (MDCK-MDR1) and non-P-gp-expressing (MDCK-wt) cells In vivo: male mice, tail veil In vivo: male mice, tail veil | TAT-NPs showed 40-fold increasing uptake than free ritonavir, similar with unconjugated NP in MDCK-MDR1 cells 800-fold higher brain ritonavir level than free ritonavir and 7-fold higher than unconjugated NP Located within the parenchyma | [29] |
| (50:50, 7–17 kDa) | F68 | SA-g7 | Loperamide | 180 | -22.8 | In vivo: male albino Wistar-Hannover rats, tail vein injection | Significantly increase the central opioid activity of LOP for 24 h than other nano-delivery system | [30] | |
| g7 | Loperamide | 155 | 15.2 | ||||||
SA-g7 | Rhodamine-123 Rhodamine-123 | 187 197 | -21.7 -15.9 | SA-g7 NPs can cross the BBB, prolonged retention time | |||||
| g7 | Rhodamine-123 | 169 | -18.9 | ||||||
| SA-g7 | TMR-linked | 154 | -24.2 | ||||||
| PLGA (50:50, 7–17 kDa) | PVA | Tet-1 peptide | Curcumin | 150–200 | -30 to -20 | In vitro: cytotoxicity on LAG cells; uptake on GI-1 glioma cells | No significant cytotoxic effects; Tet-1 conjugated NPs showed increased uptake compared with unconjugated NPs | [31] | |
| PEG- PLGA (50:50) | n/r | 2% glutathione | PTX PTX | 168 237.6 | n/r n/r | In vitro: RG2 glioma cells | Higher cytotoxicity in RG2 cells due to increased microtubule stabilization than uncoated NP | [32] | |
| Coumarin-6 | n/r | n/r | In vivo: mice | Higher brain uptake | |||||
| PLGA (50:50, 24–38 kDa) | n/r | Tween 20 | DiI DiI | 79.2 80.9 | -44.2 -21.4 | In vitro: co-culture of BCECs and astrocytes | Cellular endocytosis of Tf-NPs was 20-fold greater than blank NPs; 2-fold greater than BSA-NPs. T20-NPs showed higher toxicity | [33] | |
| BSA | DiI | 88.8 | -33.1 | ||||||
| FITC-Tf | DiI | 90.2 | -32.5 | ||||||
| PLA-TPGS | TPGS | Coumarin-6 | 228.6 | -30 | In vitro: C6 glioma cell line | Increasing uptake; active receptor-mediated Tf/TfR endocytosis of Tf-conjugated NPs | [34] | ||
| Tf | Coumarin-6 | 245.8 | -29 | In vivo: male Sprague-Dawley rats, i.v. | Much higher concentration of coumarin-6 in the brain than non-conjugated NPs | ||||
Tf | Docetaxel Docetaxel | 121.6 137.6 | -36.5 -31.1 | In vitro: C6 cells | Cell viability and IC50 were determined, Tf-TPGS-PLA was most efficient drug delivery | ||||
| PLGA (50:50, 20 kDa) | PVA | Temozolomide | 112.9 | -35.3 | In vitro: eight human cancer cell lines of six different cancer tissues | Tf-PEG-PLGA NPs and PEG-PLGA NPs showed higher cytotoxicity than PLGA NPs | [35] | ||
| PEG-PLGA | 117.2 | -4.2 | |||||||
| Tf | 121 | -4.3 | |||||||
| Tf | Coumarin-6 | n/r | n/r | In vivo: albino rats, i.v. | Tf-PEG-PLGA was located within the brain parenchyma | ||||
| PLGA (85:15) | DODAB and P80 | Tf | Nevirapine | 170–200 | 30–40 | In vitro: HBMECs, cytotoxicity and permeability | Tf and DODAB had effect on cell viability, and permeability of Tf-NPs | [36] | |
| Fluorescein conjugated dextran 7000 | In vitro: HBMECs, uptake | Tf-PLGA NPs showed higher uptake than unconjugated NPs | |||||||
| PEG-PLGA (50:50) | n/r | 92 | n/r | In vitro: RG2 glioma cells | Enhanced cellular uptake compared with unconjugated NPs and free Tempol | [37] | |||
| Tf antibody (OX 26) | Tempol | Coumarin-6 | 105 | n/r | |||||
| PLGA (50:50, 7–17 kDa) | PVA | Glycopeptides NCAM1 antibody CD44 antibody | ZnSO4 | TMR-linked TMR-linked TMR-linked | 190–210 | -0.5 to -10 | In vitro: neurons/glial cultures In vitro: NCAM1 labeled microtubule associated protein positive cells and CD44 labeled glial cells | PLGA NPs were not toxic. Glycopeptides conjugated NPs enhanced endocytosis NCAM-NPs targeted more to neurons stained for MAP2, less to glial cells CD44-NPs targeted to glial cells more, and decreased targeting to neurons | [38] |
| PEG-PLGA | Sodium cholate | Lactoferrin | 90 | -24 | In vitro: bEnd.3 cells, a model of the BBB | Significantly increased uptake in bEnd.3 cells; clathrin-mediated endocytosis | [39] | ||
| Coumarin-6 | 95 | n/r | In vivo: KM mice, injection in caudal vein | 2.49 times of coumarin-6 accumulation in the brain when delivered with NPs | |||||
| Urocortin (UCN) | 120 | -14 | In vivo: 6-OHDA rats model of PD, injection in caudal vein | Enhanced delivery and therapeutic effect of UCN loaded in Lf-NPs was demonstrated | |||||
| In vivo: BALB/c mice, injection in caudal vein | Transient acute dose-related inflammatory reactions in liver, spleen and kidney | ||||||||
| PLGA | DDAB | Poly-(-γ glutamic acid) (γ-PGA) (6 kDa) | Saquinavir (SQV) | 188 | -28 | In vitro: a monolayer of HBMECs with human astrocyte (HA) | A higher grafting efficiency and a lower molecular weight of g-PGA increased the permeability of SQV cross the BBB, enhanced the endocytosis and expression of ornithine decarboxylase (ODC) | [40] | |
| PEG-PLGA (50:50, 15 kDa) | Sodium cholate | AS1411(AP) | PTX | Coumarin-6 | n/r 121 | n/r -23 | In vitro: C6 glioma cells In vitro: C6 glioma cells | Improved cellular association Enhanced cytotoxicity of AP conjugated NP, IC50 was determined | [41] |
| AP | PTX | 156 | -32.9 | In vivo: rats bearing glioma xenografts and rats bearing intracranial C6 gliomas | Target brain tumor, improved drug efficacy and prolonged the lifespan of rats bearing gliomas |
Abbreviations: n/r, not reported; FITC, fluorescein isothiocyanate; CLSM, confocal laser scanning microscopy; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; DiI, 3′,3′,3′,3′-tetramethylindo-carboxycyanate perchlorate; DMAB, didodecyl dimethyl ammonium bromide; PD, Parkinson’s disease; SD rat, Sprague-Dawley rat; wt, wild type; for other abbreviations please see the text.
Among 29 papers cited in this review, 9 papers performed in vitro studies [20, 23, 24, 31, 33, 37, 38, 40], 8 papers focused on in vivo studies [13, 15, 16, 18, 21, 25, 27, 30], and 12 papers did both in vitro and in vivo studies [14, 17, 19, 22, 26, 28, 29, 32, 34, 35, 39, 41].
2 In vitro studies
In vitro studies were usually conducted in brain endothelial cell lines (bovine, human and rat), Madin-Darby canine kidney (MDCK) cells and glioma cells. In vitro studies were performed to investigate cytotoxicity, uptake efficiency, endocytosis mechanism and drug efficiency with different cell lines. All aspects are discussed in the following sections.
2.1 Cytotoxicity
The uptake efficiency, internalization mechanisms and cytotoxicity of NPs have been studied with different cell lines. In addition to naked NPs, PLGA/PLA NPs were coated to change the surface property or linked with targeting ligands to trigger AMT or RMT to the CNS [6]. The cytotoxicity studies of trimethylated chitosan (TMC)-d-α-tocopheryl polyethylene glycol 1000 succinate (TPGS)-PLGA NPs (136 nm) with SH-SY5Y cells [22], TPGS-PLGA NPs (250 nm) with PC 12 cell line [23], TGN-polyethylene glycol (PEG)-PLGA NPs (119 nm) with bEnd.3 cells [28], Tet-1 PLGA NPs (150–200 nm) with LAG cell line (mouse fibroblast-like connective tissue) [31] and lactoferrin (Lf)-PEG-PLGA (120 nm) with bEnd.3 cells [39] demonstrated no significant cytotoxicity of all formations of NPs at a dose range of 0.075–8000 μg/ml. One paper claimed that mild cytotoxicity was observed for dioctadecyldimethylammonium bromide (DODAB) stabilized (Tf)-PLGA NPs (170–200 nm) as indicated by a decrease in the viability of human brain microvascular endothelial cells (HBMECs) to 70–75%. The low cytotoxicity was believed to be caused by the inferring electrical repulsion between NPs and the membrane of HBMECs which induced possible inflammation. However, the increasing of Tf amount could improve viability and inhibit the secretion of tumor necrosis factor-α (TNF-α) [36]. The cytotoxic effect of different antitumor drugs after their entrapment in modified NPs to glioma cells was investigated in the literature and will be discussed in the therapeutic efficacy section of this paper.
2.2 Uptake efficiency
Cellular uptake was investigated by fluorescent or confocal microscopy studies, showing enhanced internalization of surface-modified NPs compared with unmodified NPs. Different cells including MDCK cells [17, 20, 24, 29], BCECs [19, 26, 33, 40], bEnd.3 cells [28, 39], and glioma cells [20, 34, 37, 41] were used as in vitro cell models treated at doses ranging from 10 to 15,000 μg/ml. Small size, positively charged and less hydrophilic NPs were associated with enhanced cellular uptake [17]. After coating with TMC, a cationic ligand, zeta potential of NPs increased from -18 to 21 mV, and the active transport of NPs via AMT was confirmed [22]. TPGS and surfactants such as Tween 80/polysorbate 80 (T80) and poloxamer 188 (F68) coated on the surface NPs have been proven to have an improved cellular uptake due to changes of NP surface properties such as surface charge and surface hydrophilicity [17]. MDCK cellular uptake of TPGS-PLGA NPs (222 nm) was 1.5-fold higher than poly(vinyl alcohol) (PVA)-emulsified NPs 4 h after incubation [17]. The permeation percentages of loperamide entrapped in F68- and T80-PLGA NPs (21% and 14.5%) were significantly higher compared with that of loperamide loaded in PLGA NPs (4.5%) and free loperamide (0.4%) [19].
Targeted ligands conjugated on the surface of the NPs could provide a means for receptor-mediated endocytosis as opposed to non-specific endocytosis. Cellular uptake of Tf-PLGA NPs (90 nm) was found to be 20-fold and 2-fold greater than unmodified PLGA NPs and bovine serum albumin (BSA)-PLGA NPs (88 nm), respectively, 1 h after incubation with BCECs. In one study, anti-NCAM1 antibody conjugated PLGA NPs (190–210 nm) distributed more in neurons labeled with NCAM1, whereas anti-CD44 antibody conjugated PLGA NPs (190–210 nm) targeted more glial cells labeled with CD44 [38].
The endocytosis process was proven by changing the temperature-, concentration- and time-dependent uptake and co-incubating cells with free targeting ligand and NaN3, which stops energy-dependent processes indicating an active endocytosis process [33, 39]. Furthermore, the uptake mechanisms of different ligand-conjugated NPs were studied with cells preincubated with inhibitors. By using chlorpromazine which inhibits clathrin-dependent and caveolae-dependent pathways, colchicines which inhibit the macropinocytosis pathway, and Brefeldin A (BFA) which inhibits the Golgi apparatus and lysosome-related endocytosis process, transport of Lf-TPGS-PLA NPs (120 nm) across the membrane was demonstrated to be clathrin-mediated endocytosis [39]. The mechanism of uptake of Tf-PLGA NPs (90 nm) was confirmed as caveolae-dependent by co-incubating cells with filipin, a caveolae inhibitor, and other different types of inhibitors. Co-incubated blank NPs with filipin did not show inhibition of NP cellular uptake which indicated that endocytosis of blank NPs might be adsorptive-mediated endocytosis [33].
2.3 Therapeutic efficacy
A number of in vitro experiments have demonstrated that surface functionalized NPs enhanced the therapeutic efficacy of antitumor drugs against the glioma cells such as RG2 and C6 glioma cells when compared with that of free drugs in solution and unmodified NPs with entrapped drugs [17, 20, 28, 32, 37, 41]. Besides cell viability, IC50, the concentration of the drug required to kill 50% of the incubated cells over a fixed time period, was employed to quantify and evaluate the cytotoxicity of NPs loaded with chemotherapeutic drugs, and their therapeutic efficiency. The lowest IC50 (1.63 μg/ml) for surfactant coated NPs with entrapped docetaxel was achieved with the F68-PLGA NPs (196 nm) after 24 h in C6 glioma cell lines, which was 94.30% more efficient than docetaxel [17]. AP-PEG-PLGA with entrapped paclitaxel NPs (156 nm) showed 2–4 times lower IC50 than unmodified NPs and free drug [41]. Glutathione-coated PEG-PLGA NPs (237 nm) with entrapped paclitaxel showed significantly higher therapeutic efficiency than uncoated NPs and paclitaxel solution. Tubulin immunofluorescent and Western blotting studies also confirmed the enhanced therapeutic efficacy of paclitaxel entrapped in glutathione-coated NPs [32].
3 In vivo studies
In vivo studies were conducted in rats or mouse after administration (oral, carotid artery or intravenous) of the NPs. Several experiments including brain uptake quantification, confocal laser scanning microscopy, behavior studies, therapeutic efficacy of drugs and in vivo toxicity were carried out. The percentage NP uptake was calculated in the brain and expressed as percentage of dose administered per animal, and data was plotted over a 24-h time span [13, 14, 17, 18, 21, 26, 29, 30, 34, 39]. In all cases, the brain weights of rat and mice used to convert % dose/g reported by some authors to % dose were 2 g and 400 mg, respectively [42]. NP types were divided into three categories: unmodified PLGA/PLA NPs, surface-coated NPs and ligand-conjugated NPs (Figure 1). Brain uptake of unmodified PLGA/PLA NPs, which were the control group in most cases, was studied to confirm the safety of PLGA/PLA NPs not specifically designed for shuttling drugs across the BBB. After synthesis of unmodified PLGA/PLA NPs, surfactants such as polysorbate 80 and polymers such as TMC could be coated on the surface of NPs to enhance brain uptake (i.e., “surface-coated” NPs). Specific ligands which interact with surface receptors on brain endothelial cells could also be covalently linked to NPs to improve brain delivery (i.e., targeting ligand conjugated NPs). PEG-containing NPs were demonstrated to have advantages such as stability, prolonged blood circulation time, reduced liver distribution and enhanced brain uptake as PEG chains can help adsorb less opsonins and immunoglobulin G, but more apolipoprotein (Apo) E/B [3]. PEGylated NPs PEG-PLGA and PEG-PLA were reported in 10 papers [19, 21, 25, 26, 28, 32, 35, 37, 39, 41], and were included in the discussion.
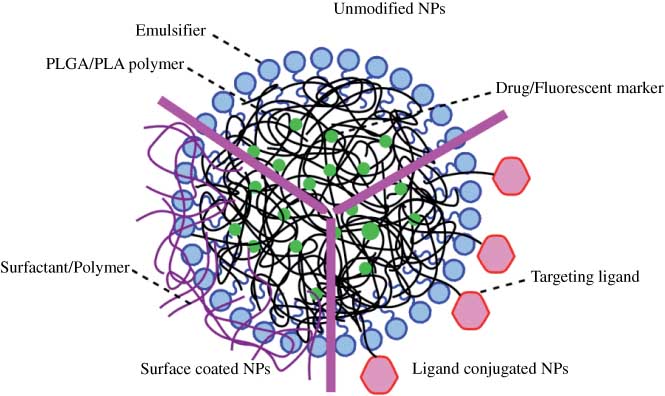
Schematic of three categories of PLGA/PLA NPs. Unmodified NPs, surface-coated NPs and ligand-conjugated NPs.
3.1 Unmodified PLGA/PLA NPs
Brain uptake of PLGA/PLA NPs (100–300 nm) were tested at different doses, ranging between 0.02 and 5.25 mg/animal, by quantifying concentration of entrapped drug [13, 14, 29] or fluorescent marker [17, 18, 26, 30, 34, 39] after administration to rats [13, 14, 17, 18, 30, 34] or mice [26, 29, 39] (Figure 2). In all cases, the percentage of dose administered that was transported to the CNS was below 4%, and the majority of the studies (7 out of 9 reported in this review) showed percentages lower than 1%. The highest brain uptake (4%) was achieved 1 h after carotid artery administration of PLGA-NPs (200 nm) at a dose of 5 mg, coumarin-6 was used as an entrapped fluorescent marker [18]. Carotid artery administration was proven to be more effective than intravenous administration for brain delivery as expected, because NPs could be transported directly to the brain after carotid artery administration [18]. In comparison, oral administered PLGA NPs (134 nm) could only reach the brain at a 0.09% of the initial dose 24 h after administration [14]. Curcumin-loaded PLGA NPs (163 nm) were found to have the lowest brain uptake (approx. 0.02%) according to quantification of drug level in the brain 1 h after intravenous administration [13]. When the level of entrapped drug was used as a measure to determine brain uptake of NPs, values lower than 0.17% were reported [13, 14]. A study that employed PLA NPs loaded with 3H-ritonavir, a radioactive labeled drug (300 nm), indicated that a higher percentage (0.6–1.7% initial dose) reached the brain 24 h post-administration [29]. Considering the potential degradation of drugs in vivo and the sensitivity of the equipment or method used to measure the concentration of drugs, the lower dose of NPs detected in the brain was expected when the method of detection was based on entrapped unmodified drug versus radiolabeled drug.
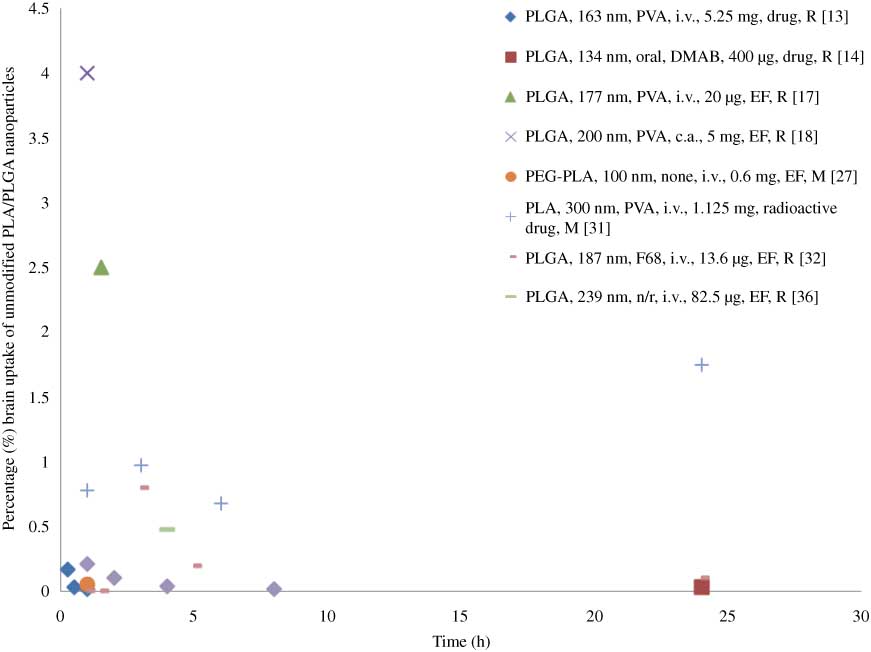
Percentage (%) unmodified NPs brain uptake of dose administered versus time.
Information is presented in the following order: NP type, size, emulsifier (n/r, not reported), administration (i.v., intravenous administration; c.a., carotid artery), dose, quantification method (EF, entrapped fluorescence marker) and animal model (M, mice; R, rats).
Three papers claimed there was no evidence showing that unmodified NPs crossed the BBB by detecting fluorescence in the brain [16, 22, 27]. Two studies reported low intensity of a fluorescent probe loaded in unmodified NPs after administration in the brain [25, 32]. In most cases (11 out of 14 papers), unmodified PLGA/PLA NPs were found to cross the BBB at a relatively low percentage even after intravenous administration of a high NP dose (5 mg/animal). Regardless of the emulsifiers used to prepare NPs, different administration methods and quantification methods (drug or fluorescent marker), the uptake was always <4% and in most cases lower than 1%.
3.2 Surface-coated nanoparticles
3.2.1 Brain uptake
Besides changing the surfactant used in the NP synthesis, the surface of the NP can be modified by the addition of other compounds, such as chitosan or PEG, in an attempt to improve brain NP uptake (Table 2). TPGS has been successfully used to prepare NPs, and it was found that TPGS was a more effective and safer emulsifier than PVA, and could improve adsorption of NPs by the gastrointestinal tract [23]. TPGS-PLGA NPs demonstrated a modest increase of brain uptake (1%) 1 h after intravenous administration compared with PVA-PLGA NPs (0.5%) [34]. Brain delivery of modified PLGA/PLA NPs was improved when positively charged compared with unmodified NPs as brain endothelial cells are negatively charged. The highest brain uptake (16%) was achieved with chitosan (CS)-PLGA NPs (396 nm) 1 h after carotid artery administration [18]. The same group demonstrated that although CS-PLGA NPs (396 nm) reached the highest brain uptake, NPs were only observed on vascular endothelial cells and considering that CS could open intracellular TJs, it was concluded that CS-PLGA NPs might not be a good brain delivery system [18]. However, carotid artery administration was proven to be the most efficient for improved brain uptake, followed by intravenous administration and oral delivery [14, 18].
Percentage (%) brain uptake of surface-coated PLGA/PLA NPsa.
| Percentage brain uptake (%) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NPs | Size (nm) | Admin. | Dose (μg/ml) | Quant. | Animal model | 0.5 h | 1 h | 1.5 h | 3 h | 6 h | 12 h | 24 h |
| 1% T80-PLGA NPs [14] | 138 | Oral | 100/200/400 | Drug | Rats | 0.078/0.052/0.037 | ||||||
| 4% T80-PLGA NPs [14] | 157 | Oral | 100/200/400 | 0.166/0.112/0.088 | ||||||||
| 1% T80-PLGA NPs [14] | 138 | i.v. | 100 | 0.38 | ||||||||
| TPGS-PLGA NPs [17] | 165 | i.v. | 20 | EF | Rats | 4.2 | ||||||
| T80-PLGA NPs [17] | 194 | i.v. | 20 | 4.3 | ||||||||
| F127-PGLA NPs [17] | 188 | i.v. | 20 | 5.5 | ||||||||
| F68-PLGA NPs [17] | 196 | i.v. | 20 | 6.2 | ||||||||
| CS-PLGA NPs [18] | 396 | c.a. | 5000/10,000/25000 | EF | Rats | 16/7.6/4.0 | ||||||
| T80-PLGA NPs [18] | 231 | c.a. | 5000/10,000/25000 | 6.4/6/4.6 | ||||||||
| F68-PLGA NPs [18] | 252 | c.a. | 5000/10,000/25000 | 3.2/3.4/1.5 | ||||||||
| T80-PLA-PEG NPs [22] | 120 | i.v. | 150 | Drug | Mice | 3.52 | 11.9 | 13.5 | 15.7 | 17.3 | 11.9 | |
| TPGS-PLGA NPs [36] | 121 | i.v. | N/A | EF | Rats | 1 | ||||||
aSize (nm), administration, dose (μg/dose), quantification method, animal model and percentage brain uptake are listed.
Abbreviations: Admin., administration; c.a., carotid artery; CS, chitosan; EF, entrapped fluorescence marker; F127, poloxamer 407; F68, poloxamer 188; i.v., intravenous; N/A, not available; Quant., quantification method; T80, Tween 80/polysorbate 80; TPGS, d-α-tocopheryl polyethylene glycol 1000 succinate.
The amount of NPs transported to the brain (μg/g tissue) reported in the literature increased as the administered dose increased, whereas percentage (%) uptake dose did not show a dose-dependent increase. For example, F68-PLGA NPs remaining in the brain 60 min after NP administration via carotid artery were 0.08, 0.17 and 0.19 mg/g tissue at injected doses of 5, 10 and 25 mg/ml, respectively, which corresponded to 3.2%, 3.4% and 1.5% dose.
Among the surfactants used to enhance brain delivery, T80 was the most commonly used. T80 coating could adsorb Apo E/B from the blood onto the NP surface and thus provided a low-density lipoprotein (LDL) receptor-mediated active endocytosis of the NPs. F68 was believed to have a similar effect as T80 [1]. For intravenous administration of surfactant-coated PLGA NPs, F68 was confirmed to be a better targeting agent than T80 [11, 15]. Kulkarni and Feng compared the brain uptake of F68-, poloxamer 407 (F127)- and T80-coated PLGA NPs, and found that F68-PLGA NPs (252 nm) reached the highest brain distribution (6.2%) via intravenous injection [17]. When comparing the anti-tumor effect of drug loaded in T80- and F68-coated PLGA NPs, Gelperina et al. also found that F68-coated NPs (168 nm) had a higher therapeutic efficacy than T80-PLGA NPs after intravenous administration [15]. Kreuter and Kreuter and Gelperina demonstrated that F68 appeared to be a very promising coating agent for both PLGA and polybutylcyanoacrylate (PBCA) NPs, whereas T80 was only effective in the case of PBCA NPs similar to the findings of Kulkarni and Feng [43]. However, carotid artery administration showed a different delivery trend between T80- and F68-coated NPs: T80-PLGA NPs (231 nm) proved to be delivered at a higher percentage (6%) than F68-PLGA NPs (252 nm) (3%) [18]. For the same surfactant and same administration route, the more surfactant coated on the NP surface, the higher the brain NPs uptake [14].
3.2.2 Location of NPs in the brain
Fluorescent microscopy studies focused on identifying the location of the NPs in the brain revealed that T80-PLGA NPs with entrapped coumarin-6 (215 nm) and very small amount of F68-PLGA NPs (252 nm) distributed in the parenchyma [18], and high TMC-PLGA loaded with coumarin-6 (136 nm) NPs were found in the cortex, paracoele, the third ventricle and choroid plexus epithelium [22].
3.2.3 Therapeutic efficacy
Therapeutic efficacy of certain drugs was evaluated in different animal models. T80-PLGA NPs with entrapped estradiol (150 nm) were administered to an ovariectomized (OVX) rat model of AD. Behavior studies and neuropathological examination results indicated enhanced oral delivery of estradiol to rat brain with the NPs [14]. Another AD animal model APP/PS1 double transgenic mice was exposed to NPs introduced to investigate the delivery efficacy of Q10 loaded in TMC-PLGA NPs (146 nm) to the brain; behavior, senile plaque staining and biochemical parameters were studied. Therapeutic efficacy of the drug was greatly enhanced when it was entrapped in TMC-PLGA NPs [22]. Anti-tumor effect of T80- and F68-coated NPs with entrapped doxorubicin was demonstrated in tumor-bearing rats, and the analgesic effect of loperamide loaded T80- and F68-coated NPs was confirmed in mice. The maximal possible effect (% MPE) was determined to evaluate the analgesic effect. F68-PLGA loaded with loperamide provided 80% MPE after 15 min and 70% MPE for at least 60 min after administration, whereas T80-PLGA NPS with entrapped loperamide reached 80% MPE and only 40% MPE after 60 min. Free loperamide showed little ant nociceptive effect [15]. In vivo ant nociception studies of poloxamer 188 (P188)-, polysorbate 80 (P80)-coated and PLGA-PEG-PLGA NPs with entrapped loperamide were conducted after intravenous administration at a dose of 0.5 mg/kg. The results of hot-plate tests and formalin tests showed P188-coated NPs had the best ant nociceptive efficacy [19]. P188 was demonstrated to be a better brain targeting agent for PLGA NPs than P80 after intravenous administration in several reported studies [15, 17, 19, 43].
3.3 Targeting ligand-conjugated nanoparticles
3.3.1 Brain uptake
Brain delivery of specific ligand-conjugated NPs was greatly enhanced compared with that of unconjugated NPs (Figure 3). The percentage of NP dose in the brain reached as high as 29% for g7 (a similopioid peptide)-PLGA NPs (169 nm) at 1.5 h after intravenous administration at a dose of 13.6 μg in rats [30], which is the highest brain uptake reported in the literature. Uptake of g7-PLGA NPs was found to be receptor-mediated endocytosis. The lowest value for % dose NPs in the brain was reported for cationized bovine serum albumin (CBSA)-conjugated NPs: the uptake process was confirmed to be AMT, and the uptake efficiency reached 0.3%/g brain, 2.3-fold more than that of unmodified NPs [26]. Different peptides functionalized NPs were constructed to activate receptor-mediated endocytosis, which could enhance brain-targeted delivery. Four synthetic opioid peptides [27], a 120 amino acid peptide screened from in vivo phage display [28], trans-activating transcriptor (TAT) peptide [29] and g7 [30] were successfully conjugated to PLGA/PLA NPs and the conjugation was shown to improve brain delivery of nanocarriers. Tf is another commonly used ligand for targeted brain delivery and it was proven to interact with the corresponding receptor in the brain to induce RMT [33–35, 40]. Other specific ligands conjugated to successfully increase brain uptake included transferring antibody [37], sialic acid residue [30], lactoferrin [39], poly-(γ-glutamic acid) [40] and DNA aptamer [41].
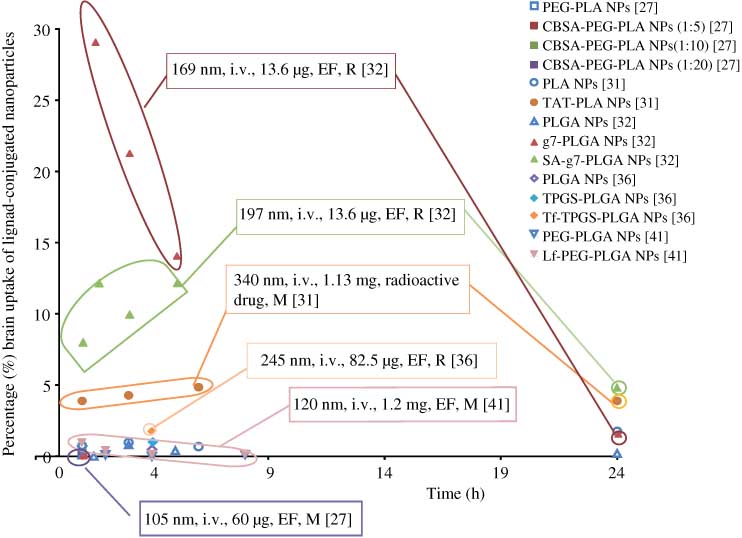
Percentage (%) ligand-conjugated NPs brain uptake of dose administered versus time.
Information is presented in the following order: size, administration (i.v., intravenous administration), dose, quantification method (EF, entrapped fluorescence marker) and animal model (M, mice; R, rats).
In all cases, targeting ligand succeeded to provide a receptor-mediated endocytosis of surface functionalized NPs, and brain uptake of modified NPs greatly increased (the highest reported value was 29%) when compared with unconjugated NPs (<1%) after intravenous administration.
3.3.2 Location of NPs in the brain
Fluorescence studies on the location of NPs in the brain demonstrated that three peptide-derived NPs (162–211 nm) with covalently linked fluorescent probe [27], TAT-PLA NPs with coumarin-6 as fluorescent marker (340 nm) [29], tetramethylrhodamine-linked sialic acid (SA)-g7-PLGA NPs (154 nm) [30] and Tf-PEG-PLGA (121 nm) with entrapped rhodamine 6G [35], all reached cerebral parenchyma, which demonstrated the ability of targeted NPs to cross the BBB.
3.3.3 Therapeutic efficacy
In addition to the direct observation of NPs in the brain, several experiments were performed to prove enhanced brain delivery of the drug in disease animal models. The antinociceptive effects of SA-g7-PLGA NPs with entrapped loperamide was tested and MPE values maintained at 30–50% for 15 h, whereas the effect of loperamide loaded g7-PLGA NPs was 57% for only 5 h after administration [30]. Therapeutic effect of urocortin (UCN) loaded in Lf-NPs on 6-OHDA rat model of Parkinson’s disease was investigated. The behavior, immunohistochemistry and transmitter contents of the brain results confirmed the improved efficiency of UCN loaded Lf-NPs to attenuate the striatum lesion compared with the control group after intravenous administration [39]. In vivo anti-tumor efficacy was carried out on rats bearing glioma xenografts and rats bearing intracranial C6 gliomas. After intravenous administration of paclitaxel (PTX) loaded aptamer-conjugated NPs (Ap-PTX-NP), tumor size, body weight and survival time were recorded to evaluate the anti-glioma efficacy. The average tumor inhibition of Ap-PTX-NP and free PTX (Taxol®) for tumor volume was 81.6% and 68.6%, and for tumor weight was 79.3% and 48.2%. The survival time of Ap-PTX-NP treated rats bearing glioma was 7 days longer than the PTX-treated group and 13 days longer than the saline-treated group [41]. In all cases, therapeutic effect of the drug was greatly improved after the drug was incorporated into surface-modified NPs.
4 Conclusion
In recent years, polymeric NPs were thoroughly investigated and PLGA/PLA NPs (100–300 nm) have been proven as potential carriers for drugs across the BBB, with advantages of enhanced drug efficiency and safety. NPs offer superior alternatives to oral and intravenous administration of free drugs that otherwise cannot cross the BBB, which is definitely a significant breakthrough for brain delivery.
Cytotoxicity, cellular uptake, mechanism of uptake and therapeutic efficiency of drugs delivered with polymeric NPs were investigated in vitro. In vitro toxicity studies showed no toxicity of PLGA NPs for doses of 0.075 to 8000 μg/ml within 72 h. Cellular uptake efficiency was greatly improved with modification of PLGA NPs. The permeation percentages of loperamide entrapped in F68 and T80 surface-modified PLGA NPs (21% and 14.5%) in rat brain endothelial cell lines and C6 co-cultures were significantly increased compared with loperamide loaded unmodified PLGA NPs (4.5%) and free loperamide (0.4%) [19]. A significant decrease in cell viability of glioma cells treated with drug loaded modified NPs was demonstrated, supporting improved uptake and efficacy of drugs loaded in PLGA NPs. Endocytosis mechanisms were unveiled for specific systems. For example, the uptake of Lf-TPGS-PLA NPs was demonstrated to be clathrin-mediated endocytosis and endocytosis of Tf-PLGA NPs was confirmed as caveolae-mediated endocytosis.
In vivo biodistribution studies were conducted in either rats or mice after NP administration (oral, carotid artery and intravenous), to determine brain uptake of various NPs. PLGA/PLA NPs showed very low (<1%) brain uptake without modification, which alleviated the concern that unmodified PLGA/PLA NPs used for applications other than CNS delivery cross the BBB. Surface modifications of PLGA NPs such as surface coating and the addition of targeting ligands improved brain delivery of drugs compared with unmodified PLGA NPs. Ligands, covalently linked to the surface of the particles improved brain uptake of NPs the most. The highest brain uptake was achieved by g7-PLGA NPs (169 nm) (14%/g tissue) which was characterized as instant high brain delivery with a short retention time [30]. The presence of modified PLGA/PLA NPs within rats/mice brain parenchyma after systemic administration was confirmed by microscopy studies. Besides brain uptake, behavior studies of treated animals and therapeutic efficacy studies after NP administration supported the enhancement of brain delivery of drugs loaded in modified PLGA NPs as compared with unmodified NPs or free drug.
It was evident that in vitro and in vivo information on delivery of drugs with PLGA/PLA NPs (100–300 nm) to CNS was available and sufficient to confirm the enhanced brain uptake and therapeutic efficacy of certain drugs entrapped in PLGA NPs. These studies suggest that multiple strategies could be applied to construct an optimum brain delivery system based on PLGA. However, the literature is lacking in several aspects. First, in vivo long-term toxicity studies of modified NPs including their effects on the brain were seldom found in the literature and should be evaluated. It is important to assess the safety and potential risks of brain-targeted PLGA/PLA NPs, especially for systems that are associated with highly improved brain uptake. Second, the amount of data is not sufficient in the literature to understand NP brain uptake as a function of concentration of the NPs administered, or their properties (i.e., morphology hydrophylicity size, charge). It is unclear if brain uptake of NPs is concentration-dependent, and if saturation would be reached as the dose administered was increased. Size appears to affect uptake, with small NP size being more favorable, but it is hard to compare uptake across studies when other parameters (charge, method of administration or detection) are not uniform. Brain uptake is also enhanced for NPs that are more hydrophobic and if they carry a positive charge. However, because of the lack of systematic studies, comparisons between systems reported in the literature so far are not possible. Lastly, information on the location of NPs modified by different strategies in the brain after they cross the BBB is not readily available, as well. Location of the NPs within the brain may affect the efficacy of the entrapped drug.
It is clear that even though much is yet to be done to understand brain uptake of polymeric NPs, and their impact on drug efficacy and safety, the design of safe nanocarriers for improved drug delivery to certain locations in the brain, and an enhanced drug efficacy and low toxicity is certainly possible. Design of such targeted delivery systems to the CNS will aid in treatment of CNS diseases.
About the authors

Jingyan Li received her undergraduate degree in Bioengineering in June 2010 from East China University of Science and Technology. In autumn 2010, she joined the Biological and Agricultural Engineering Department at Louisiana State University, Baton Rouge, LA, USA. Jingyan Li received her Master of Science in Biological and Agricultural Engineering in December 2012. She is an active member of the Phi Kappa Phi Honor Society. She currently resides in China.

Cristina Sabliov, PhD is an Associate Professor in the Biological and Agricultural Engineering Department at Louisiana State University and LSU Agcenter. She came to LSU in 2003 from North Carolina State University where she received a double-major PhD in Food Science and Biological and Agricultural Engineering, and a MS in Chemical Engineering. During her tenure at LSU, Dr. Sabliov quickly developed an expertise in the field of nanotechnology, specifically on polymeric nano particles designed for delivery of bioactive components for improved food quality and human health. Projects pursued in her laboratory range from design and synthesis of multifunctional polymeric nanoparticles for delivery of bioactives of controlled properties (size, surface charge, controlled-release profile and targeting properties) to in vitro and in vivo evaluation of the nanoparticle functiona lity and biotoxicity under the conditions of use. During the past 10 years, Dr. Sabliov’s research was supported by federal (USDA, NSF, ACS, NOAA) and state (LA-BOR, SBB, RRB, ASCL) agencies. She published extensively in reputable journals such as ACS Nano, Nanomedicine, Journal of Biomaterial Science – Polymer Edition, Nanotechnology, and Journal of Physical Chemistry. Dr. Sabliov is a recognized national and international leader in food nanotechno logy. She organized several Institute of Food Technologists (IFT) and American Institute of Chemical Engineers sessions on the topic of Nano technology and Food, and is currently serving on the IFT Food Nanoscience Advisory Panel.
This work was supported by the LSU AgCenter Biotechnology Interdisciplinary Team (BAIT) Program, Pennington Biomedical Research Center/Pennington Biomedical Research Center/Reilly Family Foundation (RFF), and EPA-G2010-STAR-N2 Food Matrices Award # 2010-05269.
References
[1] Wohlfart S, Gelperina S, Kreuter J. Transport of drugs across the blood-brain barrier by nanoparticles. J. Control. Release 2012, 161, 264–273.10.1016/j.jconrel.2011.08.017Search in Google Scholar
[2] Begley DJ. Delivery of therapeutic agents to the central nervous system: the problems and the possibilities. Pharmacol. Ther. 2004, 104, 29–45.10.1016/j.pharmthera.2004.08.001Search in Google Scholar
[3] Chen Y, Liu L. Modern methods for delivery of drugs across the blood-brain barrier. Adv. Drug Deliv. Rev. 2012, 64, 640–665.Search in Google Scholar
[4] Park K. Trojan monocytes for improved drug delivery to the brain. J. Control. Release 2008, 132, 75.10.1016/j.jconrel.2008.10.009Search in Google Scholar
[5] Mistry A, Stolnik S, Illum L. Nanoparticles for direct nose-to-brain delivery of drugs. Int. J. Pharm. 2009, 379, 146–157.Search in Google Scholar
[6] Patel T, Zhou J, Piepmeier JM, Saltzman WM. Polymeric nanoparticles for drug delivery to the central nervous system. Adv. Drug Deliv. Rev. 2012, 64, 701–705.Search in Google Scholar
[7] Rip J, Schenk GJ, de Boer AG. Differential receptor-mediated drug targeting to the diseased brain. Expert Opin. Drug Deliv. 2009, 6, 227–237.Search in Google Scholar
[8] Silva GA. Nanotechnology approaches for drug and small molecule delivery across the blood brain barrier. Surg. Neurol. 2007, 67, 113–116.10.1016/j.surneu.2006.08.033Search in Google Scholar
[9] Roney C, Kulkarni P, Arora V, Antich P, Bonte F, Wu A, Mallikarjuana NN, Manohar S, Liang HF, Kulkarni AR, Sung HW, Sairam M, Aminabhavi TM. Targeted nanoparticles for drug delivery through the blood-brain barrier for Alzheimer’s disease. J. Control. Release 2005, 108, 193–214.10.1016/j.jconrel.2005.07.024Search in Google Scholar
[10] Olivier JC. Drug transport to brain with targeted nanoparticles. NeuroRx. 2005, 2, 108–119.10.1602/neurorx.2.1.108Search in Google Scholar
[11] Kreuter J. Nanoparticulate systems for brain delivery of drugs. Adv. Drug Deliv. Rev. 2001, 47, 65–81.10.1016/S0169-409X(00)00122-8Search in Google Scholar
[12] Costantino L, Boraschi D. Is there a clinical future for polymeric nanoparticles as brain-targeting drug delivery agents? Drug Discov. Today 2012, 17, 367–378.10.1016/j.drudis.2011.10.028Search in Google Scholar PubMed
[13] Tsai YM, Chien CF, Lin LC, Tsai TH. Curcumin and its nano-formulation: the kinetics of tissue distribution and blood-brain barrier penetration. Int. J. Pharm. 2011, 416, 331–338.Search in Google Scholar
[14] Mittal G, Carswell H, Brett R, Currie S, Kumar MN. Development and evaluation of polymer nanoparticles for oral delivery of estradiol to rat brain in a model of Alzheimer’s pathology. J. Control. Release 2011, 150, 220–228.10.1016/j.jconrel.2010.11.013Search in Google Scholar PubMed
[15] Gelperina S, Maksimenko O, Khalansky A, Vanchugova L, Shipulo E, Abbasova K, Berdiev R, Wohlfart S, Chepurnova N, Kreuter J. Drug delivery to the brain using surfactant-coated poly(lactide-co-glycolide) nanoparticles: influence of the formulation parameters. Eur. J. Pharm. Biopharm. 2010, 74, 157–163.Search in Google Scholar
[16] Sun WQ, Xie CS, Wang HF, Hu Y. Specific role of polysorbate 80 coating on the targeting of nanoparticles to the brain. Biomaterials 2004, 25, 3065–3071.10.1016/j.biomaterials.2003.09.087Search in Google Scholar PubMed
[17] Kulkarni SA, Feng SS. Effects of surface modification on delivery efficiency of biodegradable nanoparticles across the blood-brain barrier. Nanomedicine 2011, 6, 377–394.10.2217/nnm.10.131Search in Google Scholar PubMed
[18] Tahara K, Miyazaki Y, Kawashima Y, Kreuter J, Yamamoto H. Brain targeting with surface-modified poly(d,l-lactic-co-glycolic acid) nanoparticles delivered via carotid artery administration. Eur. J. Pharm. Biopharm. 2011, 77, 84–88.Search in Google Scholar
[19] Chen YC, Hsieh WY, Lee WF, Zeng DT. Effects of surface modification of PLGA-PEG-PLGA nanoparticles on loperamide delivery efficiency across the blood-brain barrier. J. Biomater. Appl. 2011, Epub ahead of print.10.1177/0885328211429495Search in Google Scholar PubMed
[20] Xie J, Lei C, Hu Y, Gay GK, Bin Jamali NH, Wang CH. Nanoparticulate formulations for paclitaxel delivery across MDCK cell monolayer. Curr. Pharm. Des. 2010, 16, 2331–2340.Search in Google Scholar
[21] Ren TB, Xu N, Cao CH, Yuan WZ, Yu X, Chen JH, Ren J. Preparation and therapeutic efficacy of polysorbate-80-coated amphotericin B/PLA-b-PEG nanoparticles. J. Biomater. Sci. Polym. Ed. 2009, 20, 1369–1380.Search in Google Scholar
[22] Wang ZH, Wang ZY, Sun CS, Wang CY, Jiang TY, Wang SL. Trimethylated chitosan-conjugated PLGA nanoparticles for the delivery of drugs to the brain. Biomaterials 2010, 31, 908–915.10.1016/j.biomaterials.2009.09.104Search in Google Scholar PubMed
[23] Jalali N, Moztarzadeh F, Mozafari M, Asgari S, Motevalian M, Alhosseini SN. Surface modification of poly(lactide-co-glycolide) nanoparticles by d-alpha-tocopheryl polyethylene glycol 1000 succinate as potential carrier for the delivery of drugs to the brain. Coll. Surf. Physicochem. Eng. Aspects 2011, 392, 335–342.10.1016/j.colsurfa.2011.10.012Search in Google Scholar
[24] Jaruszewski KM, Ramakrishnan S, Poduslo JF, Kandimalla KK. Chitosan enhances the stability and targeting of immuno-nanovehicles to cerebrovascular deposits of Alzheimer’s disease amyloid protein. Nanomed. Nanotechnol. Biol. Med. 2012, 8, 250–260.Search in Google Scholar
[25] Parikh T, Bommana MM, Squillante E, 3rd. Efficacy of surface charge in targeting pegylated nanoparticles of sulpiride to the brain. Eur. J. Pharm. Biopharm. 2010, 74, 442–450.Search in Google Scholar
[26] Lu W, Wan J, She Z, Jiang X. Brain delivery property and accelerated blood clearance of cationic albumin conjugated pegylated nanoparticle. J. Control. Release 2007, 118, 38–53.10.1016/j.jconrel.2006.11.015Search in Google Scholar PubMed
[27] Costantino L, Gandolfi F, Tosi G, Rivasi F, Vandelli MA, Forni F. Peptide-derivatized biodegradable nanoparticles able to cross the blood-brain barrier. J. Control. Release 2005, 108, 84–96.10.1016/j.jconrel.2005.07.013Search in Google Scholar PubMed
[28] Li JW, Feng L, Fan L, Zha Y, Guo LR, Zhang QZ, Chen J, Pang ZQ, Wang YC, Jiang XG, Yang VC, Wen LP. Targeting the brain with PEG-PLGA nanoparticles modified with phage-displayed peptides. Biomaterials 2011, 32, 4943–4950.10.1016/j.biomaterials.2011.03.031Search in Google Scholar PubMed PubMed Central
[29] Rao KS, Reddy MK, Horning JL, Labhasetwar V. TAT-conjugated nanoparticles for the CNS delivery of anti-HIV drugs. Biomaterials 2008, 29, 4429–4438.10.1016/j.biomaterials.2008.08.004Search in Google Scholar PubMed PubMed Central
[30] Tosi G, Vergoni AV, Ruozi B, Bondioli L, Badiali L, Rivasi F, Costantino L, Forni F, Vandelli MA. Sialic acid and glycopeptides conjugated PLGA nanoparticles for central nervous system targeting: in vivo pharmacological evidence and biodistribution. J. Control. Release 2010, 145, 49–57.10.1016/j.jconrel.2010.03.008Search in Google Scholar PubMed
[31] Mathew A, Fukuda T, Nagaoka Y, Hasumura T, Morimoto H, Yoshida Y, Maekawa T, Venugopal K, Kumar DS. Curcumin loaded-PLGA nanoparticles conjugated with Tet-1 peptide for potential use in Alzheimer’s disease. PLoS One 2012, 7, e32616.10.1371/journal.pone.0032616Search in Google Scholar PubMed PubMed Central
[32] Geldenhuys W, Mbimba T, Bui T, Harrison K, Sutariya V. Brain-targeted delivery of paclitaxel using glutathione-coated nanoparticles for brain cancers. J. Drug Target. 2011, 19, 837–845.Search in Google Scholar
[33] Chang J, Jallouli Y, Kroubi M, Yuan XB, Feng W, Kang CS, Pu PY, Betbeder D. Characterization of endocytosis of transferrin-coated PLGA nanoparticles by the blood-brain barrier. Int. J. Pharm. 2009, 379, 285–292.Search in Google Scholar
[34] Gan CW, Feng SS. Transferrin-conjugated nanoparticles of poly(lactide)-d-alpha-tocopheryl polyethylene glycol succinate diblock copolymer for targeted drug delivery across the blood-brain barrier. Biomaterials 2010, 31, 7748–7757.10.1016/j.biomaterials.2010.06.053Search in Google Scholar PubMed
[35] Jain A, Chasoo G, Singh SK, Saxena AK, Jain SK. Transferrin-appended PEGylated nanoparticles for temozolomide delivery to brain: in vitro characterisation. J. Microencapsul. 2011, 28, 21–2810.3109/02652048.2010.522257Search in Google Scholar PubMed
[36] Kuo YC, Lin PI, Wang CC. Targeting nevirapine delivery across human brain microvascular endothelial cells using transferrin-grafted poly(lactide-co-glycolide) nanoparticles. Nanomedicine (Lond.) 2011, 6, 1011–102610.2217/nnm.11.25Search in Google Scholar PubMed
[37] Carroll RT, Bhatia D, Geldenhuys W, Bhatia R, Miladore N, Bishayee A, Sutariya V. Brain-targeted delivery of Tempol-loaded nanoparticles for neurological disorders. J. Drug Target. 2010, 18, 665–67410.3109/10611861003639796Search in Google Scholar PubMed
[38] Grabrucker AM, Garner CC, Boeckers TM, Bondioli L, Ruozi B, Forni F, Vandelli MA, Tosi G. Development of novel Zn2+ loaded nanoparticles designed for cell-type targeted drug release in CNS neurons: in vitro evidences. PLoS One 2011, 6, e1785110.1371/journal.pone.0017851Search in Google Scholar PubMed PubMed Central
[39] Hu K, Shi Y, Jiang W, Han J, Huang S, Jiang X. Lactoferrin conjugated PEG-PLGA nanoparticles for brain delivery: preparation, characterization and efficacy in Parkinson’s disease. Int. J. Pharm. 2011, 415, 273–28310.1016/j.ijpharm.2011.05.062Search in Google Scholar PubMed
[40] Kuo YC, Yu HW. Transport of saquinavir across human brain-microvascular endothelial cells by poly(lactide-co-glycolide) nanoparticles with surface poly-(γ-glutamic acid). Int. J. Pharm. 2011, 416, 365–37510.1016/j.ijpharm.2011.06.037Search in Google Scholar PubMed
[41] Guo J, Gao X, Su L, Xia H, Gu G, Pang Z, Jiang X, Yao L, Chen J, Chen H. Aptamer-functionalized PEG-PLGA nanoparticles for enhanced anti-glioma drug delivery. Biomaterials 2011, 32, 8010–802010.1016/j.biomaterials.2011.07.004Search in Google Scholar PubMed
[42] Rosen GD, Williams AG, Capra JA, Connolly MT, Cruz B, Lu L, Airey DC, Kulkarni K, Williams RW. The Mouse Brain Library @ www.mbl.org. Int. Mouse Genome Conference 2000, 14, 166Search in Google Scholar
[43] Kreuter J, Gelperina S. Use of nanoparticles for cerebral cancer. Tumori 2008, 94, 271–27710.1177/030089160809400220Search in Google Scholar
©2013 by Walter de Gruyter Berlin Boston
Articles in the same Issue
- Masthead
- Masthead
- Graphical abstracts
- In this issue
- Reviews
- PLA/PLGA nanoparticles for delivery of drugs across the blood-brain barrier
- Syntheses and applications of small metallic nanorods from solution and physical vapor deposition
- Functional gold nanoparticles for sensing applications
- Nanostructured phase-changeable heat transfer fluids
- Metal nanoantimicrobials for textile applications
- Synthesis and characterization of elemental iron and iron oxide nano/microcomposite particles by thermal decomposition of ferrocene
- Nanotechnology Institutions
- A review of nanotechnology development in the Arab World
Articles in the same Issue
- Masthead
- Masthead
- Graphical abstracts
- In this issue
- Reviews
- PLA/PLGA nanoparticles for delivery of drugs across the blood-brain barrier
- Syntheses and applications of small metallic nanorods from solution and physical vapor deposition
- Functional gold nanoparticles for sensing applications
- Nanostructured phase-changeable heat transfer fluids
- Metal nanoantimicrobials for textile applications
- Synthesis and characterization of elemental iron and iron oxide nano/microcomposite particles by thermal decomposition of ferrocene
- Nanotechnology Institutions
- A review of nanotechnology development in the Arab World