X-chromosome linked genes associated with myeloid cell CNS trafficking contributes to female–male differences in the disease outcome for neuroinflammatory diseases
-
Sopiko Darchiashvili



Abstract
Certain diseases such as Multiple Sclerosis (MS), a chronic demyelinating disease, affect more women than men, despite males appearing to be predisposed to infections and malignancies. X-linked genes contribute to increased MS susceptibility. Currently, an immense body of research exists that explores the complexity surrounding underlying risk factors for MS development including X-chromosome-linked inflammatory processes. Female–male disparities in disease susceptibility have been found at both the gene and chromosomal level. Genes such as CXORF21 and DDX3X can escape X-chromosome inactivation (XCI) and contribute to various disease pathogenesis. Additionally, blocking immune cell entry to the central nervous system (CNS) can have a major impact on MS. Prior research on MS has shown that immune cells such as T cells and dendritic cells (DCs) infiltrate the CNS. Due to persistent tissue stress, these cells may induce local inflammation and autoimmunity, subsequent neurodegeneration, and both the onset and progression of MS. Chemokines are signaling proteins which regulate leukocyte trafficking to the site of injury, contributing to cell recruitment, CNS inflammation, and disease severity. Some chemokine receptors (CXCR3) are X-linked and may escape XCI. This review provides an account of the contribution of x-linked genes in MS in relation to the chemotaxis of myeloid cells into CNS and subsequent neuroinflammation. The impact of the X-chromosome on autoimmunity, including XCI and the expression of X-linked genes is evaluated. Collectively, the analyses from this review seek to advance both our understanding of MS and advocate for more patient-specific therapies.
Introduction
Autoimmune diseases, including those related to the central nervous system (CNS) such as multiple sclerosis (MS), are a leading cause of morbidity and mortality in women, who can have up to a four-fold increased risk than men [1]. MS is characterized by many typical and atypical attributes that are found in autoimmune disorders, such as organ-specific or systemic immune reactions [2]. MS particularly afflicts women between the ages of 20–40 years. Remission during pregnancy can be observed, with transient reduction or augmented presence in the puerperium, which shows a diverse response to immunosuppressive therapies. The production of autoantibodies is well-documented in MS; however, this examination lacks significant impact due to the absence of linkage between autoantibodies and MS clinical manifestation [2].
This female–male bias is not unique to MS but applies to other autoimmune conditions too. Of the approximately 8 percent of the human population affected by autoimmune illnesses, 78 percent are women. Connective tissue diseases like Hashimoto’s thyroiditis, Sjogren’s syndrome (SS), systemic lupus erythematosus (SLE), dermatomyositis (DM), and systemic sclerosis (SSc) also disproportionately affect women compared to men (reviewed in [3]). This is particularly apparent in SLE where females are at greatest risk factor for the disease, and like other illnesses, can disparagingly affect the well-being of a patient across significant areas of life, including mental health and social functioning. Consequences of a disease can range from depression to illness-linked disability and can be associated with worse health outcomes [4]. Depending on the condition, these can include adverse maternal and fetal consequences for pregnant women [5], hypertensive disorders during pregnancy leading to longer hospitalizations [6], pulmonary arterial hypertension [7], negative self-perceptions due to body mass index (BMI) [8], lower quality of life (QoL) [9], and both resilience, and health literacy scores [10]. Comorbidities like major depressive disorder (MDD) and anxiety further complicate treatment and affect progression and cognition, resulting in earlier mortality [11]. Table 1 enlists common female–male biased diseases in addition to their incidence and ratios, emphasizing the importance of studying female–male differences.
Common female-male biased autoimmune illnesses with incidence rate, common abnormalities, and female-male ratio
| Disease | Incidence among 100,000 people/year | Common abnormalities | Male to female ratio | References |
|---|---|---|---|---|
| Addison disease | 1.0 | Inadequate production of cortisol, aldosterone, and androgens, can lead to Addisonian crisis, presented by hypotension | 1:2–4 | [12, 13] |
| Celiac disease | 13 | An immune response is triggered by the consumption of gluten, leading to inflammation of the small intestine, anemia, and headaches | 1:3 | [14–16] |
| Dermatomyositis | 1.0 | Muscle weakness and skin lesions | 1:2 | [17, 18] |
| Graves’ disease | 20 | Overactivity of the thyroid | 2–10 | [19–21] |
| Hashimoto thyroiditis | 30–150 | Error of the immune system attacking the thyroid gland | 1:10 | [22–24] |
| Multiple sclerosis | 363 | Immune system attacks the protective portion of nerves, yielding nerve damage | 1:4 | [25, 26] |
| Myasthenia gravis | 0.4–3 | Antibodies terminate existing communication between nerves and muscles, contributing to muscle weakness | 1:2 | [27, 28] |
| Pernicious anemia | 25 | Insufficient vitamin B12 absorption, resulting to its deficiency | 1:2 | [29, 30] |
| Rheumatoid arthritis | 40 | Immune system attack on tissues and at times, internal organs | 1:3 | [31, 32] |
| Sjogren syndrome | 4 | Dry eyes and mouth | 1:9 | [33, 34] |
| Systemic lupus erythematosus | 5 | Immune system attacks on healthy tissues, leading to their inflammation and damage | 1:9 | [35, 36] |
To balance gene products, a process known as X-chromosome inactivation (XCI) occurs. Among its regulatory functions, XCI ensures that X-chromosomal genes are not overly expressed in males [37]. While XCI occurs primarily in females, this process is also been observed in males with female–male dimorphic developmental timing. In females, inactivation occurs during embryonic development [38]; however, in males, it is transient and is confined to the late phases of initial meiotic prophase during spermatogenesis [39]. This phenomenon is called meiotic sex chromosome inactivation (MSCI) [39]. Interestingly, mRNA genes are not known to escape MSCI in males, but about 80 percent of X-linked miRNA genes are shown to escape this process. Thus, it has been suggested that RNA interference may participate in controlling XCI in female cells.
Interestingly, female–male bias is linked at the gene as well as chromosome level since males and females possess different copies of the X-chromosome (1:2). However, to maintain the balance, one X-chromosome copy is inactivated (explained in detail later). Nevertheless, some disease-related genes especially those escaping X-inactivation are present in different ratios between females and males. Table 2 summarizes those genes along with their disease prevalence. For example, LAMP-2 and IRAK-1 that escape X-chromosome inactivation are involved in Danon disease and inflammatory autoimmune disorders like SLE, respectively [40, 41]. Another gene of interest is TLR7, which participates in lupus [42]. This condition has a particularly high female-to-male prevalence ratio (9:1). Additionally, USP27X has been suggested to participate in various neurodegeneration diseases like Parkinson’s as well as infection and cancer, along with other disorders [43]. Interestingly, this gene has also been attributed to X-linked intellectual disability. DDX3X participates in various antiviral signaling pathways and its role has been implicated in DDX3X syndrome [44]. Individuals impacted by this disease experience both developmental and intellectual delays that can range from moderate to severe. Notably, a variation on the DDX3X gene may contribute to X-linked mental disability [45]. Furthermore, CXORF21 is another gene that escapes the inactivation process. Like TLR7, this gene’s abnormalities can promote SLE with a high female-to-male ratio (9:1) as discussed later in further detail.
Genes that escape X-chromosome inactivation.
| Gene | Location on chromosome | Normal function | Disease association | Prevalence women vs. men | References |
|---|---|---|---|---|---|
| LAMP-2 | Xq24 | Autophagy, lysosomal integrity | Danon disease | 5:9 | [37, 46] |
| IRAK-1 | Xq28 | Encode IL-1 receptor-associated kinase | Inflammatory autoimmune disorders | 4:1 | [38, 47–50] |
| TLR7 | Xp22.2 | Pathogen recognition, innate immunity | Lupus | 9:1 | [39, 51] |
| USP27X | Xp11 | Regulation of apoptosis and protein stability | Parkinson’s disease | 1:2 | [52, 53] |
| DDX3X | p11.3 | Antiviral signaling pathways | DDX3X syndrome | 3:1 | [41, 42] |
| CXORF21 | Xp21.2 | Positive regulation of innate immune system | Systemic lupus erythematosus | 9:1 | [36, 54] |
Particularly in MS, there is an underlying dysregulation of neurological inflammation significantly contributing to disease onset. However, the initiating pathology and why it is more prevalent in women is a growing area of research [55]. One of the most common neurological illnesses among young adults, particularly women, is MS and a primary element of such neurodegenerative diseases includes persistent neuroinflammation [56]. As previously mentioned, disease symptoms are attenuated during pregnancy [2]. This is evidenced by the significantly diminished rate and potency of relapses over time, especially in the final trimester [57]. It is a chronic, prevalent rising inflammatory condition with accumulations of neurological deficits, progressing over time being a key characteristic of its pathogenicity [58]. Particularly, MS results in the demyelination of neuron fibers and reduced muscular mobility associated with underlying inflammation mounted by infiltrating leukocytes following a cascade of events for CNS entry [59]. In both MS and a commonly used animal model (experimental autoimmune encephalomyelitis; EAE), infiltrating macrophages and CNS-resident microglia both demonstrate increased reactivity that is known to contribute to both the onset and maintenance of a proinflammatory environment [60–63]. Sustained inflammation can then both initiate and exacerbate demyelination, axonal damage, and oligodendrocyte cell death [64–66]. Figure 1 demonstrates how immune cells behave in the leukocyte trafficking cascade. This review discusses markers and genes related to each stage of leukocyte trafficking in particular interest to those associated with X-chromosome and MS pathogenesis.

The process of leukocyte trafficking is illustrated, demonstrating the behavior of immune cells during chemoattraction, initial contact, firm adhesion, rolling, and transmigration. Specifically, markers interleukin 2 receptor subunit alpha, forkhead box protein 3 (FoxP3) gene, and C-X-C motif chemokine receptor 3 are X-chromosome linked and are shown with an “X” in the figure, along with other genes and markers who are not known to be X-linked.
Among several drugs with differing mechanisms of action, it has been shown that blocking immune cell entry into the CNS can have a major impact on MS [47, 67]. FDA-approved drugs, such as Tysabri and Gilenya, that target immune cell invasion at the BBB have shown efficacy in treating autoimmune disorders like MS. In addition, antibodies and disease-modifying therapies such as glatiramer acetate, block the leukocyte integrin VLA-4 [68]. However, current marketed drugs have several limitations that include cost inefficiency, they only have a moderate effect in alleviating disease symptoms, and may even be linked with opportunistic infections like progressive multifocal leukoencephalopathy (PML) [67]. Since disease culminates in severe disability, patients experience significant financial and symptom burdens. Consequently, advancing therapies that would ameliorate these issues may be highly beneficial. Thus, addressing the female–male bias present in MS in the context of therapies is needed.
Recent evidence suggests dendritic cells (DCs) are critically involved in the initiation and progression of autoimmune disorders, including MS. In both EAE and MS, they are essential for the effector phase and immune response cascade. DCs, one of the most potent antigen-presenting cells (APCs), play a crucial role in inducing inflammatory or tolerogenic responses contingent upon local environmental cues and lineage [69]. DC accumulation within the CNS has been observed in ongoing MS and EAE [70, 71], Notably, DCs are detected within inflammatory demyelinating lesions of MS patients and EAE animals, indicative of a role in both disease initiation and progression [70]. We too have demonstrated the most efficient recruitment of DCs into EAE lesions compared to other leukocytes [71]. This is why contemporary therapies for MS, such as disease-modifying drugs, target the function of these APCs [72]. Chemokines and their receptors play a crucial role in regulating the migration of immune cells to inflammatory sites and thus, their contribution to autoimmune diseases like MS, is well-studied [73].
This paper focuses on the female–male bias seen in X-linked genes in the context of myeloid cell trafficking in MS. We also elaborate on the steps involved in leukocyte trafficking following neuroinflammatory induced immune cell entry along with chemokines and their receptors. Other topics of inquiry that we touch upon include X-chromosome inactivation, dosage compensation, and how X-linked differences in both DCs and T cells affect disease progression. Overall, the paper offers unique and novel perspectives on the mechanism of myeloid cell trafficking across the BBB, examining uncontrolled activation and antigen presentation.
Female–male biased autoimmune and neuroinflammatory diseases
Heightened immune response in women may result in a predisposition for loss of self-tolerance, leading to the development of skin disorders as well as autoimmunity [1]. Female skin is “autoimmunity prone,” with close examinations depicting the upregulation of proinflammatory genes. This section will discuss autoimmune disorders, including MS, highlighting the role of skin, autoantibodies, gut microbiota, and sex hormones.
One possibility that can elucidate female–male bias seen in autoimmune disorders, such as lupus, is skin processes that include mediating inflammation, immune responses, wound healing, and angiogenesis [74]. Vestigial-like 3 (VGLL3) is a component of the VGLL family of proteins and despite the absence of a precise and transparent physiological role [75, 76], its augmented expression in females is evident and also encourages autoimmunity [76]. A previous study has identified VGLL3 as a putative transcription cofactor enriched in female skin. Markedly, this study found that the skin-directed overexpression of murine VGLL3 causes a severe lupus-like rash due to autoantibody production and immune complex deposition. It is hypothesized that this sex-linked increase in VGLL3 transcription contributes to increased lupus rates in females. The autoantibodies are associated with autoimmune diseases and female–male differences are also seen with antibody production. It has been shown that generally, women possess higher CD3+ and CD4+ T-cell counts, increased basal and stimulated antibody reactions to viruses and vaccines, and enhanced cytokine production in response to infection [77]. This difference may be due to an evolutionary development to protect offspring from infection [78]. Meanwhile, another notion regarding increased autoimmunity occurrence in women includes the pregnancy compensation hypothesis (PCH), a concept that assumes female–male specific immune differences arise in response to challenges of maintaining protection against pathogens, simultaneously tolerating an invasive placenta, and the introduction of foreign fetal antigens [77].
While the origins of increased female–male differences in antibody production still need further study, it has been shown that B cells play a major role in autoimmunity, especially for women. Several studies have concluded the integral role of B cells and the CD40 gene in autoimmune disorders. One study found that the administration of an anti-B-cell agent called rituximab, delayed disease onset in pre-Rheumatoid arthritis (RA) patients [78]. For individuals with SLE, there is an overexpression of CD40L on T-cell surfaces as well as in serum. Also, CD40 serves as a pathogenic factor in many other autoimmune illnesses, including MS [78]. Similarly, it has been noted that expression of CD40L on activated CD4+ T cells was greater in patients with Sjogren’s syndrome compared to healthy controls [79]. CD40 has been found to play a pathogenic role in other autoimmune diseases including type 1 diabetes [80], inflammatory bowel disease [81], MS [82], and lupus [83] as epigenetic deregulation of genes occurring on the X-chromosome. The CD40L gene is X-linked, its duplication can yield an overexpression of CD40L and has been associated with various autoimmune illnesses, including lupus [84].
Another X-linked dysregulation that is associated with autoimmune diseases is gut microbiota dysfunction. SLE disease progression rates in humans have been associated with certain compositions of gut microbiota [85]. It has been demonstrated that people with MS have altered microbiome and gut dysbiosis, possessing both enrichment and/or depletion of certain types of bacteria [86]. Common trends show that those with MS have decreased amounts of Prevotella, Parabacteroides, and Lactobacillus, among others. Meanwhile, there was heightened concentrations of Akkermansia, Eggerthella, and Blautia. There is some evidence that females are more effected by gut dysbiosis. Utilizing lupus-prone mice, one study found that a pro-inflammatory immune response in the gut mucosa of female lupus-prone mice can be detected at a juvenile age [85]. Correspondingly, a microbiota-dependent pro-inflammatory immune response is seen in female mice gut mucosa at a juvenile age. Alternatively, there seems to be androgen-dependent protection of males, as these SNF1 mice are safeguarded from this disease through this hormone, indirectly and/or gut microbiota dependently [85]. These observations can contribute to female–male differences in the intestinal immune phenotype and systemic autoimmune progression. Along with gut microbiota, another significant factor contributing to MS development is sex hormones. Generally, sex hormones partake in B-cell development, function in physiology, and have a significant role in the abnormalities that occur in autoimmune diseases [87]. The most prominent female sex hormone, estrogen, modulates humoral responses, B-cell differentiation, and immunoglobulin production. Several complex roles are attributed to estrogen and its protective contribution in MS and RA is evident in other published work. However, its pathogenic effects can be noted in other conditions such as SLE. While progesterone and androgen have more immune-suppressive effects, estrogen is more dose and cell-type dependent increasing cell activation, intracellular signaling, and antibody production in the context of autoimmunity [88]. Additionally, estrogen has been demonstrated to produce an anti-inflammatory response and decrease inflammation in the CNS via ERalpha, and its activation of ERbeta results in myelin and axonal restitution [89]. To better understand how female–male differences influence autoimmunity in general as well as in MS, the elucidation of complex and intricate interactions transpiring between sex hormones, X-chromosomes, and immune response genes is indispensable.
MS pathogenesis
MS pathogenesis can be characterized by a multitude of factors that include activated microglia, astrocyte proliferation, and neuronal damage [90]. Infiltrating neutrophils have been observed in the brains of MS patients, while animal models have demonstrated their accumulation in the brain before the appearance of clinical manifestations. Neutrophils continue to accumulate in the CNS throughout MS progression, implying a greater contribution to disease progression and chronicity [90]. Neurons and oligodendrocytes, myelinating cells of the CNS, are particularly affected in MS as is evident in their reduced population in disease [91] and leads to clinical symptoms in MS. The microvasculature of the brain facilitates crosstalk with neighboring astrocytes, perivascular microglia, and neurons to maintain the BBB [92]. Immune cells such as DCs, investigate brain microvasculature for imbalance in homeostatic signaling and mobilize resident microglial cells within the CNS and infiltrating peripheral cells. Particularly, CD11c+ DCs of myeloid lineage are present in the CNS of EAE-induced mice, indicating infiltration of DCs into the CNS from systemic circulation under inflammatory conditions [92]. Additionally, T lymphocytes have been implicated in MS pathogenesis [93]. Both CD4+ and CD8+ T cells have been found in MS lesions. More specifically, CD4+ T cells predominantly accumulated in acute lesions and CD8+ T cells are more common in chronic lesions [93].
The conditions that lead to MS are established, but tracking its progression is complex and unreliable due to various genetic susceptibilities [94]. The most common form of MS is relapsing remitting (RRMS) [95], which represents a pattern of disease activity followed by a decrease or absence of symptoms for some time. The following secondary progressive (SPMS) phase is accompanied by neuronal death and synaptic dysfunction, culminating with motor disability [96]. Continuous microglia and macrophage activation are seen in the CNS of MS patients, ranging from short to long-term duration, especially evident in SPMS [96]. The prevalence of MS is about 20 percent greater in individuals who have a family member with the disease compared to those in the general population, suggestive of genetic predisposition [97].
Both genetic sensitivity and environmental immune triggers have been implicated in MS pathogenesis [98]. Although its etiology is unclear, MS is believed to be a T-cell dependent condition driven by activated CD8+ T cells located in disease-associated lesions [99] and observed by inflammatory neutrophils and demyelinated CNS lesions, involving immune cell infiltrates and glial cells. Environmental risk factors include Epstein-Barr virus (EBV), UV radiation exposure, and vitamin D deficiency [100]. Data suggests a link between EBV infection and developing MS, with the risk being 15 times greater in childhood and approximately 30 times higher during adolescence or thereafter [100]. Greater sun exposure is linked to a lower MS risk, especially during childhood and the years before the onset. Studies indicate that epigenetic alterations during MS can be mediated by vitamin D deficiency [100]. However, diagnosis is not limited to genetic and environmental influences, as other contributors to increases in diagnosis in women include improvements in access to healthcare and being more likely to undergo consultation than men for mild symptoms [100].
T cells possess proinflammatory functions in EAE and are kept in check via regulatory T (Treg) cells, a subset of CD4+ T cells, capable of reducing the severity of EAE and providing recuperation from CNS inflammation [101]. Generally, Th17 cells can induce CNS inflammation through a critically reliant mechanism of Ca2+ influx. This mechanism is facilitated through Ca2+ release activated Ca2+ (CRAC) channel, which is located in the plasma membrane and drives this Ca2+ signaling. Consequently, inhibiting the CRAC channel function or knocking out calcium release-activated calcium channel protein 1, also known as ORAI, and stromal interaction molecule 1 (STIM) family proteins have attenuated EAE onset as well as progression. This occurs through the reduction of encephalitogenic T-cell advancement in CNS. Studies have assessed the role of Ca2+ signals in T-cell migration in the CNS and have demonstrated T-cells to be motile in the parenchyma with influx contingent on TCR-major histocompatibility complex (MHC) exchange between T cells and APCs. Another study noted high frequencies in addition to amplitudes of Ca2+ signals at the early stage of EAE; however, a significant reduction is observed at the disease apex. Tregs in the spinal cord attenuate EAE pathological progression EAE through suppression of Ca2+ influx present in Th17 cells. They are also responsible for displacing Th17 cells from APCs during the chronic stage of EAE. Altogether, findings suggest that Tregs suppress the influx signals in encephalitogenic Th17 cells once EAE has resolved. Dual immunoglobulin domain-containing cell adhesion molecule (DICAM) is a transmembrane protein critical in angiogenesis, bone formation, and degradation; identified as a molecule involved in TH17 trafficking into the CNS [102]. An up-regulation of circulating DICAM+ CD4+ T cells was noted in MS patients, on BBB endothelium as well as disease lesions. DICAM’s connection to neuroinflammation was recognized considering the molecule fostering the migration of Th17 lymphocytes across the BBB endothelium. MS also affects microglia, the most common resident immune cell in the brain/CNS. Under physiological conditions, microglia mediate synaptic pruning, neuron survival, and plasticity, and apoptotic cell clearance; however, under pathological conditions microglia can exacerbate a pro-inflammatory environment [103]. These cells promote initiation of inflammation via pattern-recognition receptors (PRRs), recognizing noxious stimuli located in the CNS and prompt transcription of proinflammatory genes through NF-KB or interferon-regulatory elements. Certain PRRs, such as toll-like receptors (TLR), which includes TLR-4 and TLR-2, Nod-like receptors (NLRs), and C-type lectins encompassing CLEC-7A introduce neuroinflammation, with further mechanisms in place such as “inflammasomes”. Importantly, inflammasome coordination by intracellular sensor NLRP3 has arisen to be a significant factor in neuroinflammation [103]. Microglia are able to adapt is inflammatory response under various conditions so as to not induce pathological consequences; however, this ability is impaired and can lead to neurodegenerative states as a consequence of aging or disease. Collectively, MS can result from DCs and T-cell infiltration into the CNS. Women due to a variety of factors including X-linked genes and autoimmunity, are more prone to its development. Since it is more common in women, we wanted to understand the connection between female–male linked immune cell migration and MS. Therefore, the next section highlights the X-linked genes regarding leukocyte trafficking, noting their function and location on a chromosome.
Impact of immune cell CNS entry on neuroinflammation
Although there is no solid explanation for the female–male bias in autoimmune diseases, theories with respect to a more comprehensive immune response, the microbiome, sex hormones, and the X-chromosome, have been proposed. Due to increased phagocytic activity, more effective antigen presentation, and higher expression of pathways linked to PRRs such as TLRs, women have been shown to produce a more robust innate response. Changes in the gut microbiota composition called gut dysbiosis can disrupt homeostasis and put individuals at risk for various inflammatory and neurological conditions like MS [104]. Increasing evidence suggests that along with genetic factors, numerous hormonal elements influence autoimmune susceptibility [105]. Sex hormone outcomes can differ contingent upon their concentration and target cell and receptor subtype expressed on any cell form. Due to the existence of hormone receptors on immune cells, sex hormones like progesterone and estrogens can impact various components of the immune system function, with the likelihood of influencing risk, activity, as well as the progression of the condition. Of these factors, X-linked genes, and markers play an important role in determining female–male bias vs. diseases. Specifically, X-linked gene expression such as TLR7 and CD40L in healthy females and Klinefelter syndrome (KS) individuals implies that X-chromosomes may function as major factors in enhancing both innate and adaptive immune responses, possibly explaining female–male based differences in immune biology as well as female–male bias in autoimmune disease predisposition [106]. XCI (also known as lyonization) is an epigenetic process that occurs in females and results in the silencing of one X-chromosome through dosage compensation to balance X-associated gene expression levels among females and males [107].
Recent evidence suggests that innate, as well as adaptive, immune responses are vital contributors to neuroinflammation [108]. The role of T cells was examined in the previous section, but here we will concentrate on innate immune cells, particularly DCs. There is evidence that neuroinflammation attracts both mature and immature DCs. In previous studies, we reported CNS trafficking of DCs across the BBB during EAE via intravital video microscopy (IVM) which has enabled direct visualization of the inflamed white matter (WM) of the spinal cord [71]. DCs can present neuroantigens (i.e. myelin basic protein, MBP) to prime naïve autoreactive CD4+ T cells in the peripheral immune system, and within the CNS. Depending upon the pathological conditions, these cells can mature into T helper type 1 (Th1), Th2, and Th17 effector cells or regulatory T cells (Tregs) [109]. In MS/EAE, circulating neuroantigen-specific T cells gain access to the CNS and promote neurodegeneration contributing to the disease progression. The source of the inflammation, however, appears to be the DCs, which not only prime T cells, but also maintain a chronic inflammation within MS lesions. Furthermore, the influx of leukocytes contributes to inflammation and their accumulation can lead to abnormalities observed in MS, such as the dysfunction of the BBB [110].
During MS and EAE, accumulation of DCs is observed within inflammatory infiltrates in the CNS parenchyma and as well as the CSF [69]. Its presence in the CSF of affected patients suggests that DCs might play a key role in determining the outcome of CNS inflammation. This notion of EAE pathogenesis is underlined by observations that CD11c−/− mice are resistant to EAE and that CD11c+ DCs are sufficient to present Ag to primed myelin-reactive T cells in vivo to mediate CNS inflammation and clinical EAE [71]. In addition, DCs have been implicated in epitope spreading during chronic EAE and are particularly well-suited to polarize myelin-specific T cells toward Th1 and Th17 effector T cells in vitro. Our observations in prior studies confirm the conviction of the migratory ability of DCs being intrinsically linked to their maturation state and differences in traffic signals displayed on the surface of immature vs. mature DCs might influence their efficiency to enter certain inflammatory sites [71].
In addition to the role of DCs and neutrophils in leukocyte trafficking, T cell involvement is also apparent, especially in MS. CD4+ T cells are essential in the pathogenesis of MS, and they depart from the lymphoid organs and move through the BBB following activation and differentiation [59]. This step is vital for CD4+ T-cell entry into the CNS. Contemporary data depict that the paths that regulate the departure and migration of these cells are perhaps contingent on their type. Additionally, their hindrance could have ramifications outside of migration. In MS, abnormally activated autoimmune T cells recognize components of the CNS myelin and initiate the disease [111]. EAE models have shown that the representation of the disease may be passively transferred by activated myelin-specific T cells. Thus, it may be suggested that T-cell autoimmune reactions to myelin can be involved in the inflammatory response inception observed in the CNS. Likewise, the pathogenic mechanisms of MS development include Ag-specific T-cell activation and Th1 differentiation, followed by T-cell and macrophage migration into the CNS.
Chemoattraction, initial contact, firm adhesion, rolling, and diapedesis are the complex steps involved in the leukocyte trafficking cascade (Figure 1 and [112]). The final step of the leukocyte extravasation cascade, migration, is attributed to multiple cell trafficking molecules, located on surfaces of moving leukocytes as well as BBB endothelial cells. Participation of molecules such as very late antigen 4 (VLA4) and vascular cell adhesion molecule 1 (VCAM1) is evident, as they engage in advancing the movement of various immune cell subsets. Other contributors such as IL-17 cytotoxic CD8+ T (TC17) cells and activated leukocyte cell adhesion molecule (ALCAM) observed in both monocytes and lymphocytes, partake in expression and cell trafficking. Leukocyte trafficking also involves other genes that are not associated with the X-chromosome. Details on each are provided below in accordance with the step of the cascade.
Chemoattraction
In particular, CXCL8, also termed IL-8, is a member of the CXC chemokine family. It is located on chromosome 4 and serves as a critical mediator of inflammatory responses [113]. By guiding neutrophils to the site of inflammation, it operates as a chemotactic factor and is secreted by various types of cells including, macrophages, eosinophils, T-lymphocytes, and fibroblasts, among others. For instance, platelet factor 4 (PF4) is a common chemokine, released from platelet alpha-granules following activation [114]. Located on chromosome 4, the protein is chemotactic for various cell types and plays a role as an inhibitor of hematopoiesis, angiogenesis, and T-cell function [115]. CXCL1, also known as GRO1, is a chemokine ligand, observed on chromosome 4. This protein partakes in inflammation and functions as a chemoattractant for neutrophils, encoding a member of the CXC subfamily of chemokines [116]. CXCL12, also known as SDF-1, is characterized as an antimicrobial gene present on chromosome 10 [117]. Encoding protein is crucial in diverse cellular processes including embryogenesis, immune surveillance, inflammation retorts, tissue homeostasis, and tumor growth as well as metastasis.
Initial contact
P selectin glycoprotein ligand 1 (PSGL-1) encodes a glycoprotein that serves as a high-affinity counter-receptor for cell adhesion molecules P, E, as well as L-selectins [118]. Located on chromosome 12, this protein plays a crucial role in leukocyte trafficking during inflammation, especially by tethering. The atypical expression of this gene is linked to inadequate innate and adaptive immune responses. Selectin L can be observed on chromosome 1 and encodes a cell surface adhesion molecule that is part of adhesion/homing receptors [119]. Its role in leukocyte trafficking is apparent. The gene product is mandatory for binding and ensuing rolling of leukocytes on endothelial cells and assisting their migration to other lymphoid organs in addition to inflammatory sites.
Firm adhesion
Integrin subunit alpha 4 (ITGA4) seen on chromosome 2 encodes a unit of the integrin alpha chain family of proteins [120]. Integrins are known to partake in cell surface adhesion and signaling. This gene in particular is also aimed as a therapeutic target for treating MS. Integrin subunit alpha 1 (ITGA1) also known as VLA1 and CD49a encodes the alpha 1 subunit of integrin receptors [121]. Present on chromosome 5, this protein undergoes heterodimerization with beta1 subunit to generate a cell-surface receptor for collagen as well as laminin and the formation of this receptor participates in cell-to-cell adhesion and its involvement in inflammation and fibrosis is plausible. Intercellular adhesion molecule 1 (ICAM-1) can be found on chromosome 19 and the gene encodes a cell surface glycoprotein that is normally observed on endothelial and immune system cells [122]. It is capable of binding to integrins like CD11a or CD11b and its expression is noted in various organs of the body including the lung, bone marrow, and 20 other tissues. Vascular adhesion molecule (VCAM-1), is located on chromosome 1 and the gene is a member of the Ig superfamily and mediates leukocyte-endothelial cell adhesion as well as signal transduction [123]. Interestingly, its role in the progression of atherosclerosis and RA has been theorized, supporting the notion of its involvement in inflammatory diseases.
Rolling
P-selectin is classified as a protein created by activated platelets and endothelial cells that serves as a cell adhesion molecule [124]. It is known to play a critical role in the initial recruitment of leukocytes as well as their recruitment and aggregation of platelets to the affected inflamed area. Selectin E, also known by the names ELAM, ESEL, CD62E, ELAM1, and LECAM2, encodes a protein that can be found in cytokine-stimulated endothelial cells. This selectin is theorized to be responsible for the accumulation of blood leukocytes at inflammation sites [125]. Found on chromosome 1, it is part of the selectin family of adhesion molecules and engages in the interaction between leukocytes and the endothelium. Its participation in the pathogenesis of atherosclerosis is apparent. The integrin VLA-4 is known as a receptor for the vascular cell adhesion molecule 1 (VCAM-1). It is expressed on monocytes and lymphocytes and partakes in lymphocyte homing [126]. Integrin is well-known to play a vital role in the transfer of hematopoietic stem cells to the bone marrow. Opposing antibodies have been utilized to hinder leukocyte infiltration to the inflamed CNS in EAE.
Transmigration
C-X-C motif chemokine receptor 2 also known as CXCR2 is located on chromosome 2. The protein encoded by this gene is part of the G-protein-coupled receptor family [127]. The receptor is responsible for neutrophil diapedesis to the sites of inflammation and binds to chemokine (C-X-C motif) ligand 1 and IL-8. Found on chromosome 1, the F11R receptor is also known as junctional adhesion molecule (JAM). The protein encoded by this gene is a crucial mediator of tight junction assembly seen in epithelia [128]. The encoded protein also participates in leukocyte transmigration and as a platelet receptor. Its expression is observed in the lung, small intestine, and 24 other tissues. Platelet and endothelial cell adhesion molecule (PECAM-1) is noted on chromosome 17. Additionally, the protein encoded by this gene is located on the surface of a diverse set of cell types, including monocytes, platelets, neutrophils, and certain T-cells [129]. Furthermore, it makes up a large amount of endothelial cell intercellular junctions. Interestingly, it is regarded as a participant in leukocyte diapedesis, angiogenesis, as well as integrin activation.
It has been demonstrated that the recruitment of leukocytes following tissue injury does not cease randomly [130]. Instead, through a process termed resolution, the action of activated various cell types is resolved through different pathways [130]. Evidence suggests that the resolution of inflammation is an intricate, active process that is governed by the spatial-temporal generation of pro-resolving mediators. These mediators act on certain receptors with the aim of cell regulation and tissue reactivity. Pro-resolving mediators can include canonical mediators like cortisol and adenosine and recent lipid, protein, and peptide agonists like lipoxins (LXs), resolvins (Rvs), annexin (AnxA1) as well as melanocortins (MCs) in addition to others. Such mediators can serve as stimulators and activate endogenous pathways, regulating cellular trafficking, and modulating myeloid cell life expectancy and phenotype. The actions taken by these mediators can contribute to improving tissue restoration to quicken resolution. When key processes involved in inflammation resolution are impaired, there is a delayed restoration of tissue homeostasis. Consequently, these impairments can lead to fibrosis and/or chronic inflammation.
Cannabinoids have demonstrated their utility in the treatment of MS as is supported by their effectiveness in not only improving EAE symptoms but also neuroprotective properties [131]. They can advance oligodendrocyte survival, decrease demyelinating lesions, and lessen the loss of neurons. Cannabinoids contribute to these protective factors via decreases in immune cell activation and infiltration. Specifically, CB2 receptors are known to be immensely expressed in bone marrow immune cells and myeloid progenitor cells can be assembled into the neuroinflammatory areas of the CNS. Additionally, infiltrating T-cells and monocytes can express CB2 receptors in the control of neuroinflammation. One study explored the role of the peripheral CB2 cannabinoid receptor in directing myeloid progenitor cell trafficking to the site of injury in EAE [131]. CB2 receptor knock-out mice display worse EAE clinical scores compared to wild-type counterparts. Pronounced EAE clinical scores were associated with extensive axonal loss, T-cell (CD4+) infiltration, as well as microglial (CD11b+) activation. Immature bone marrow-originated CD34+ myeloid progenitor cells exhibited expression of CB2 receptors and were immensely present in the spinal cords of CB2 knock-out EAE mice. Likewise, selective pharmacological CB2 activation tremendously reduced EAE symptoms in addition to axonal loss and microglial activation. Consequently, findings suggest the protective role of CB2 receptors in EAE pathology, illustrating support for a new location of CB2 receptor action [131].
Chemokine and chemokine receptors
Chemokines are proteins expressed by various cells. They are a critical contributor to leukocyte extravasation, guiding cells to their eventual migration across the endothelium and into the inflamed tissue. They, along with their receptors, will be discussed in detail in the following section. Clinical manifestations of autoimmune illnesses arise from a strong immune response to a self-antigen, leading to leukocyte activation and accumulation. Through the recruitment of chemokine-dependent cells, structural cells potentially exhibit a role in chronic inflammation [132]. Chemokines play a crucial role in the regulation of leukocyte trafficking. By binding to their G protein-coupled receptors (GPCR), they guide defined leukocyte subsets to inflammatory sites or coordinate homing and recirculation of lymphocytes [133]. These proteins are critical in sustaining the typical function of immune responses, including instances of cellular destruction and inflammatory reactions. As a result, they impact leukocyte cell survival, and effector functions, and act on tumor cells to contribute to angiogenesis, tumor growth, and metastasis [132, 133]. Upon tissue injury or infection, there is an inflammatory response involving the recruitment of inflammatory leukocytes by chemokines, guiding appropriate cell migrations to the site of injury. An upregulation of chemokine receptor expression and function is essential for a significant leukocyte response. However, influxes of leukocytes require monitoring to prevent excessive accumulations, as unrestrained buildup may lead to a continuous inflammatory response that can originate both tissue damage as well as chronic inflammation. Figure 2 illustrates the mechanism and signaling pathways associated with CXCR3 activation.
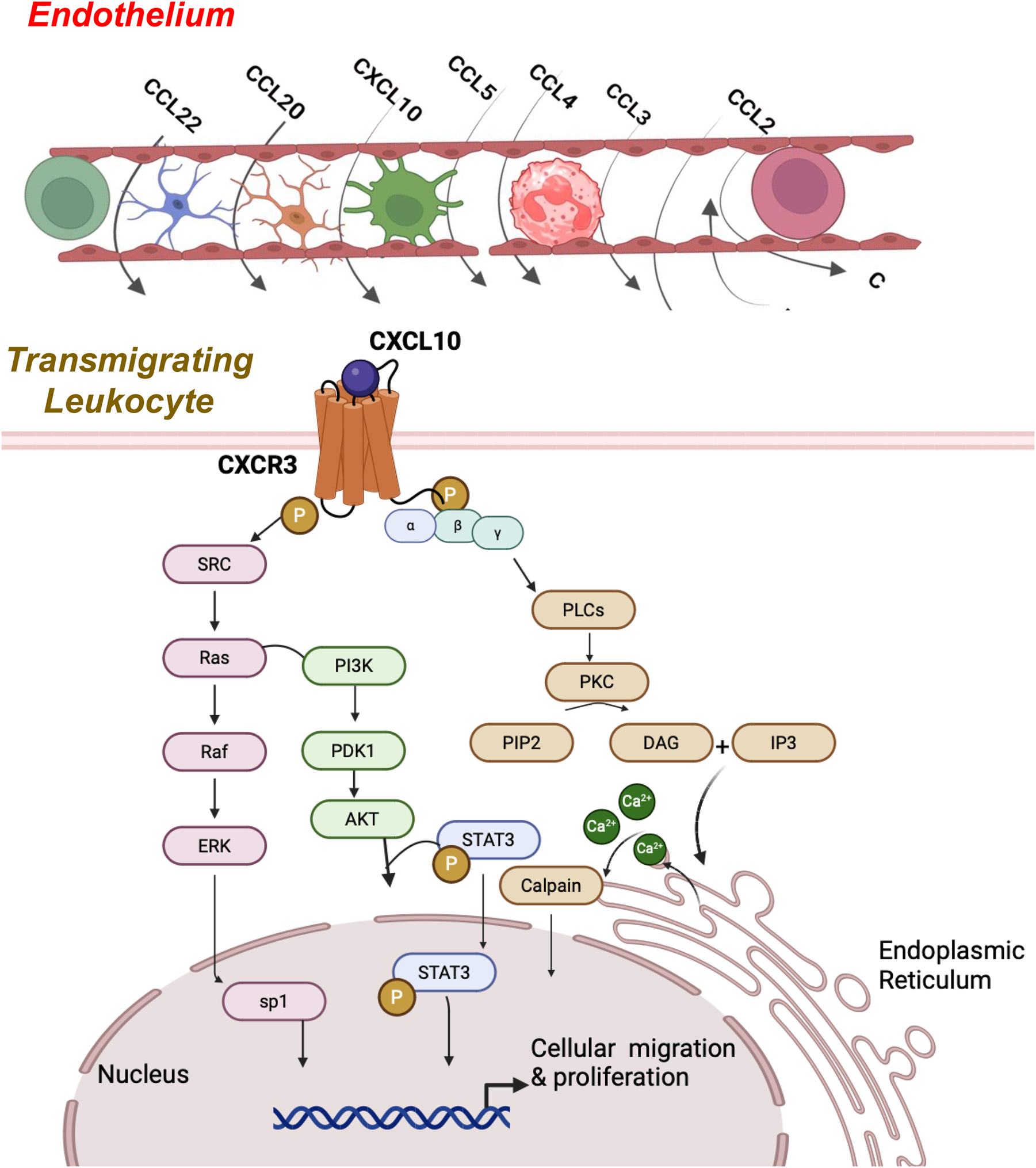
Cellular migration and proliferation via CXCL10 and CCR3 signaling. CXCL10 binding to CXCR3 activates the G-couple protein receptor via the Gαi subunit and induces the activation of SRC/Ras pathways and downstream PI3K/ERK/AkT signaling. These pathways are responsible for promoting cell proliferation. At the same time, via the Gαq subunit, activation triggers downstream phospholipase C-beta (PlCβ) signaling. This results in the cleavage of phosphatidylinositol 4,5-bisphosphate (PIP2) into diacylglycerol (DAG) and inositol-triphosphate (IP3). This promotes the release of calcium from the endoplasmic reticulum, which activates the calpain protease, which is responsible for cell mobility and increased migration.
Chemokines can increase immune cell infiltration across the BBB and cause uncontrolled activation and antigen presentation [134]. They can also modulate neuroinflammation and neurodegenerative processes and are crucial to the activation and migration of immune cells to lesion sites. C–C chemokine ligand 2 (CCL2), a potent chemoattractant released by astrocytes and microvascular endothelial cells, permits chemoattraction of patrolling immune cells of the BBB vasculature. CCR2 is a receptor for CCL2 which is commonly known as monocyte chemoattraction protein 1 (MCP-1). MCP-1 participates in monocyte infiltration seen in inflammatory conditions like RA [135]. Increased CCL2 expression has been noted in several EAE models and a reduced accumulation of CD11c+ DCs was seen within the spinal cord homogenates of CCL2−/− chimeric mice induced with EAE. Interestingly, these mice also display less demyelination than wild-type EAE controls [134].
In a prior study, we imaged and evaluated the ability of endogenous DCs to transmigrate across the BBB during EAE and assessed both mechanism and extent of chemotaxis and diapedesis in the presence of CCL2 [92]. Our transmigration and immunofluorescence studies determined that DCs were more potent responders to CCL2 than T cells. Furthermore, we noted that the DC transmigration pattern was primarily paracellular and dependent on signaling molecule ERK1/2 phosphorylation compared to that of T cells. This observation was transcellular and dependent on phosphorylation or signaling molecule p38-mitogen-activated protein kinase (MAPK). DCs also transmigrated with greater efficiency than T cells, potentially due to specific adhesion molecules used to tether to endothelia. We previously determined that DC transmigration correlates with the severity of inflammation during EAE, and confirmed via ex vivo histology that CCL2 was present in EAE lesions. Additionally, CCL2facilitates DC transmigration in an ERK1/2-dependent manner. In one study, we used an EAE model known to exhibit peak leukocyte activation between days 12 and 30 post-inoculation. The sclerotic lesions of this model are known to induce CCL2 generation and release [92].
A 2013 genome-wide association study conducted by the International Multiple Sclerosis Genetics Consortium (IMSGC), highlighted the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) pathway as a key player in MS pathology [136]. Accordingly, its activation results in the rise of inflammation [136]. Comparably, CCL2 expression is dependent on NF-kB signaling, and its mRNA has been noted to increase within the demyelinated MS hippocampus. However, there are discrepancies regarding the spatial and quantitative distribution concerning gray matter (GM) and WM lesions. This chemokine-induced monocyte migration also results in the breakdown of the BBB via the downregulation of endothelial tight junction proteins. CCL2 is a pro-inflammatory chemokine that attracts DCs, monocytes, T cells, and natural killer (NK) cells to inflammatory sites via its CCR2 receptor. CCL2 can increase the permeability of the BBB allowing for infiltration of circulating systemic immune cells and potentially recruits monocytes and microglia to CNS inflammatory sites [137]. This chemokine induces migration of monocytes, memory T cells, and DCs [138]. Picomolar levels of CCL2 have been shown to limit inflammation in inflammatory bowel disease models. CCR2 is expressed on immune cells and mediates monocyte recruitment. Inadequate quantities of CCR2 within T cells promote the buildup of FoxP3+ regulatory T cells (Tregs) and simultaneously reduce levels of Th17 cells in vivo. Low doses of CCL2 are influential in repressing MOG-affected EAE and can downregulate severe EAE. EAE is modulated by CCL2 and was linked to the downregulation of Th1/Th17 cells as well as upregulation of TGF-β and induction of regulatory CD4+ FoxP3 T cells. Accordingly, high levels of CCL2 and receptor CCR2 in the CNS have been associated with EAE relapse.
Chemokine receptor CXCR3 is a GPCR expressed by T cells that participates in mediating chemotactic migration, cell proliferation, and survival; studies have identified its expression in CNS diseases [139]. Intriguingly, increased presence of CXCR3 is observed in neurological disorders like MS, Alzheimer’s disease, and bipolar disorder. Meanwhile, its antagonistic agents have demonstrated therapeutic effects. CXCL10, a ligand of CXCR3, was expressed by astrocytes in MS brain lesions, however, an equivalent observation was not made in control patients’ WM. Another study displayed IFN-β’s therapeutic effects where following treatment, CXCR3 expression on CD4+ and CD8+ T cells is significantly decreased [140].
CCR7 is critical in eliciting an immune response and initiation of immunological tolerance and its chemokine ligands include CCL19 and CCL21 [141]. Under physiological conditions in the human brain, CCL19 can be transcribed and measured as a protein in both tissue lysates and CSF. Notably, in both active and inactive relapsing-remitting and secondary progressive MS patients, this chemokine ligand is elevated in the CSF and it has been implicated to participate in both normal and abnormal immunosurveillance [141].
CCR1 and CCR2 are chemokine receptors regulating monocyte trafficking to the location of inflammation [142]. Particularly, CCR1 is essential for the control of the acute phase of inflammation that transpires in both EAE and MS. It responds to inflammation through CCL3 and CCL5 and targeting the actions of the former in vivo via antibody or genomic deletion significantly reduces MS. Current therapies target leukocyte trafficking and inflammation in the CNS. However, despite showing adequate safety profiles in the treatment of MS, CCR2-targeted drugs have failed in efficacy and continued research into their development has ceased [142].
Astrocytes as the major source of chemokines within the CNS
Astrocytes represent one of the major intrinsic sources of chemokines within the CNS [143]. Interestingly, the involvement of astrocytes in MS has been well-documented through astrogliosis, altered gene expression, cellular function, cell structure [144]. They participate in neural tissue reaction to inflammation and injury [145]. They promote innate inflammation and neurodegeneration through cytokine, chemokine, and neurotoxic metabolite production [146]. These cells enhance MS through different pathways, such as the production of neurotoxic molecules like nitric oxide and the ushering of neurotoxic inflammatory monocytes toward the CNS [147]. The loss of astrocyte function appears to possess contrasting results, depending on disease type. While astrocyte deletion can exacerbate the disease in acute EAE, astrocyte deletion in progressive EAE can augment the disorder. Pharmacological treatments targeting astrocytes in ameliorating disease burden have been proposed. The aryl hydrocarbon receptor (AHR) is crucial in being the driving force in both peripheral immune cells and CNS-resident cells [148]. Laquinimod, a drug introduced for the treatment of MS that targets astrocytic AHR, mitigated EAE symptoms. Afatinib, a tyrosine kinase inhibitor, promotes anti-inflammatory polarization of astrocytes as characterized by the downregulation of proinflammatory genes such as Csf2 and TNF [149]. An upregulation of epidermal growth factor receptor (EGFR) on astrocytes during the final phases of CNS inflammation can be emblematic of their sensitivity to Afatinib treatment in progressive disease. X-chromosome-linked genes are pertinent and crucial in the development of autoimmune illnesses, including MS. Disturbances in their alleles can enable their overexpression, putting individuals at a greater risk of autoimmune disease pathogenesis. These genes will be presented in detail in the forthcoming section, highlighting their role in the context of leukocyte trafficking.
X-linked genes in the context of leukocyte trafficking
Several genes on the X-chromosome have been implicated in increasing susceptibility to MS and other autoimmune disorders. Some of these genes include members of the interleukin family of cytokines and receptors such as Interleukin 2 receptor A (IL-2RA) and Interleukin 3 receptor A (IL-3A), which are discussed in detail as follows.
Interleukin 2 receptor A
The gene that encodes the alpha chain of the IL-2 receptor, known as IL-2RA, possesses alleles that increases risk of developing MS as well as other immune conditions [150]. This gene is X-linked and is therefore, known to cause immunodeficiency with autoimmunity [151]. Evidence of abundant autoimmune disease anomalies is present in patients, including extensive autoimmune diseases involving lung inflammation as well as infiltration. Impressively, these repercussions of IL-2RA in combination with IPEX-like illnesses are also associated with T-cell specific immunodeficiency. An inadequate supply of the gene can yield a decrease in RICD and a reduction in Treg function. These events occur due to IL-2 being pertinent in Treg survival and effector function. Another perspective gene of interest is the IL2R y chain, a component of interleukin receptors, whose increased presence is shown in MS affected brain tissues [150].
Interleukin 3 receptor A
The communication pathways between the peripheral immune system and the CNS in MS are not well understood [152]. While Interleukin-3 (IL-3), a growth factor and cytokine implicated in inflammatory diseases, has been found to play a protective role in Alzheimer’s disease, its function in neuroinflammatory contexts like MS remains unclear. One prior study revealed that IL-3 produced by astrocytes and infiltrating T cells stimulates microglia and myeloid cells in the CNS [152]. As a result, immune cell recruitment, CNS inflammation, and disease severity in MS are promoted. In humans, CSF IL-3 levels correlated with MS diagnosis and acuteness. Strikingly, deletion of IL-3 or its receptor in mice improved the animal model of MS and restricted immune cell recruitment. Additionally, IL3RA-expressing myeloid cells were found to accumulate in MS plaques and exhibit migratory and chemotactic properties [152]. In addition, they contributed to immune cell migration, highlighting the role of IL-3 in central-peripheral immune crosstalk and CNS inflammation. In MS, both resident astrocytes and infiltrating CD44(hi)CD4(+) effector T cells are sources of IL-3 in the inflamed CNS. IL-3 signaling induces a chemotactic program in IL-3Rα-expressing myeloid cells such as (microglia, MHCII+ macrophages, MoDCs, and monocytes), leading to immune cell recruitment to the CNS and worsening clinical severity. Single-nuclei RNA sequencing (snRNA-seq) of brain tissue from control individuals and MS patients reveals increased expression of IL3RA by myeloid cells in WM and GM, as well as in MS plaque tissue. It is important to note that this study focused on a murine model of progressive MS and further investigations are required to explore the role of IL-3 in RRMS. Additionally, more research is needed to understand the stimuli and factors that induce astrocytic IL-3 production, as well as the contribution of IL3RA-expressing non-myeloid cells (neurons, oligodendrocytes, and endothelial cells) to MS pathology [152].
CXCR3
The chemokine receptor, CXCR3, is involved in mediating the migration, proliferation, and survival of T cells [111] and its expression is elevated in the peripheral blood and CSF of patients with neurological diseases, including MS. CXCR3 has a clear linkage with the X-chromosome, with greater overexpression observed in females than males [153] and is subjected to XCI, an epigenetic attribute aimed to balance X-linked genes to prevent their overexpression [48]. However, it remains unclear how this gene bypasses this mechanism in immune cells. Nevertheless, it has been demonstrated that XCI escapes by CXCR3, possibly advancing female–male bias noted during infection. Intriguingly, strong experimental and clinical evidence suggests that CXCR3 pathway involvement is evident in the development of autoimmune diseases such as RA and systemic lupus erythematosus (SLE), particularly by establishing local amplification loops in specific organs and thus, inducing worsening clinical symptoms [49].
Table 3 provides a list of genes related to leukocyte trafficking and X-chromosome linkage. One such gene is TLR7, identified on the X-chromosome [154]. Nevertheless, its skewing was not recorded [154]. Other potential genes include FoxP3, CD40 ligand, and CD154. The former is critical in regulating T cells with its reduction observed in MS patients, whereas the latter’s overexpression is exhibited on CD4+ T cells in individuals possessing the disease. CXCR3, a noteworthy protein-encoding gene, pathologically participates in neuroinflammation in MS. Several immune-linked genes such as TLR7 and TLR8 did not detect single-nucleotide polymorphisms (SNPs), although they had gene lengths similar to the observable genes [155]. However, genes that were noticed with SNPs included BCOR, FTX, and ACSL4, and did not demonstrate skewing in the sample. Thus, a better mechanistic understanding of these genes and associated ligands can enhance available treatment options for various CNS disorders [156].
List of X-linked leukocyte trafficking genes that escape from X-inactivation.
| Cascade | Associated Factor | Gene | X-linkage | Escape (Y/N) | Associated diseases | Reference |
|---|---|---|---|---|---|---|
| Initial contact and adhesion | Cellular adhesion molecules (CAMs) | IL-2RA | X-linked | Yes | Multiple sclerosis, Diabetes I, immuno-deficiency 41, Rheumatoid arthritis | [157–159] |
| Firm adhesion | Integrins | ITGA-4 | Not X-linked, on Chr 2 | No | Multiple sclerosis, Crohn’s disease, Inflammatory bowel disease, Alzheimer’s disease | [109, 160] |
| VLA-1 | Not X-linked, on Chr 5 | No | Colon carcinoma, intestine carcinoma | [110, 161] | ||
| Activation and firm adhesion | CD11a/CD18 integrin (αLβ2 or LFA-1) | ICAM-1 | Not X-linked, on Chr 19 | No | Inflammatory bowel disease, multiple sclerosis, rheumatoid arthritis, atherosclerosis, Ulcerative colitis | [111, 162] |
| Integrins α4β1 or α4β7 | VCAM- 1 | Not X-linked, on Chr 1 | No | Rheumatoid arthritis, asthma, stroke, cancer, atherosclerosis | [112, 163] | |
| Adhesion trans-migration | CD99 molecule | MIC2 | X-linked | Yes | Multiple sclerosis, cancer | [164, 165] |
| Rolling adhesion | Selectins | P & E selectin | Not X-linked, on Chr 1 | No | Coronary heart disease, atherosclerosis, Hypercholesterolemia, hypertension | [114, 166, 167] |
| Leukocyte migration | Chemokine receptor | CXCR3 | X-linked | Yes | Multiple sclerosis, rheumatoid arthritis, Transplantation rejection, Atherosclerosis, inflammatory skin diseases | [141, 168, 169] |
| CXCR2 | Not X-linked, on Chr 2 | No | Chronic obstructive pulmonary disease, Asthma, Acute respiratory distress syndrome, Acute lung injury cancer | [116, 170, 171] | ||
| Leukocyte migration | CCL5-CCR5 pathway | FOXP3 | X-linked | No | Multiple sclerosis, IPEX syndrome, cancer | [172–174] |
| Trans-migration | JAMs | F11R | Not X-linked, on Chr 1 | No | Congenital myopathy 23, Bardet-Biedel syndrome 20 | [117, 175, 176] |
The process of XCI ensures cellular mosaicism where half of the cells in tissues express maternal X-chromosome genes and half is from the paternal X-chromosome [177]. The X-chromosome also possesses pseudo-autosomal regions (PAR), while PAR1 and PR2 do not participate in XCI. Genes like CSF2RA, SLC25A6, IL3RA on PAR1, or IL9R on PAR2 belong in the minority stemming from these PAR regions. Most genes outside the pseudo-autosomal regions are distinctive of the X-chromosome where two copies are observed in women and only one is in men. Many of these genes encode proteins and microRNA that can mediate immune responses. Some genes like BTK, WAS, IL2RG, and FoxP3 can have loss-of-function mutations that can initiate primary X-associated abnormalities. The X-chromosome has a diverse array of immune-related genes. The endosomal Toll-like receptor (TLR) genes, TLR-7 and TLR-8, express from the non-pseudo-autosomal area of Xp. However, XCI is not an infallible process and as a result, genes tend to escape it, contributing to their overexpression from active (Xa) as well as inactive (Xi) X-chromosomes in some tissues [177].
Mineralocorticoid receptors in myeloid cells (My-MR) have been shown to advance inflammatory cell infiltration at the site of injury in an animal model of atherosclerosis, but its mechanism is not well elucidated [50]. One study evaluated the role of My-MR in leukocyte trafficking and female–male influence [50] and results confirmed that My-MR deletion (My-MR-KO) in apolipoprotein E-knockout (ApoE-KO) mice reduced plaque magnitude. Intravital microscopy images displayed significantly attenuated monocyte recruitment like slow-rolling and adhesion to mesenteric vessels. It also produced fewer peritoneal infiltration of myeloid cells regarding inflammatory stimuli, but only in males. Few p-selectin glycoprotein ligand 1 (PSGL1) were noted in peritoneal macrophages and surface PSGL1 protein in the monocytes of male subjects. Similarly, MR antagonist spironolactone inhibited PSGL1 expression in the monocytes of humans. Data suggests that PSGL1 can be utilized as a My-MR target gene that could drive leukocyte trafficking to augment atherosclerotic plaque formation. Therefore, these findings provide a greater understanding of ischemia risk seen in MR activation, as well as cardiovascular protection in women. Furthermore, the role of MR in atherosclerosis in addition to tissue inflammation is evident [50]. A connection between chemokines and EAE exists. In this respect, CCL22 gene is a component of a chemokine cluster, also encompassing thymus and activation-regulated chemokine (TARC/CCL17) [51]. The occurrence of AT haplotype was significantly decreased in MS patients. No significant differences were noted in females; however, the AT haplotype in the chromosome 16 chemokine cluster presents a lower risk of developing MS in males [51].
Inflammasomes are large multiprotein complexes known to play a role in autoimmune disease progression [178]. They can contain NOD-like receptor family members NLRP1 and NLRP3 in addition to AIM2-like receptor family member AIM2. These multiprotein complexes are continually evaluated in the context of inflammatory skin illnesses. Abnormal activation of the inflammasome can influence both the largest organ in the body and can contribute to disorders like SLE and SSc. The inflammasome aberrations can also affect SS, and RA, along with a myriad of other conditions including metabolic and cardiovascular diseases like diabetes, atherosclerosis, as well as gout. Importantly, IFN signaling is evident in most of the mentioned autoimmune diseases. Particularly, in SLE and SS, IFN signaling can result in heightened expression and enhanced activation of both the inflammasome and a lytic form of cell death called pyroptosis, further exacerbating the diseases [178].
Gut microbiota also possesses an association with autoimmune illnesses. By employing multiple genome-wide linkages, relationships between gut microbiota and complex illnesses were evaluated [179]. The assessed autoimmune disorders included celiac disease, inflammatory bowel diseases, MS, primary biliary cirrhosis, type 1 diabetes, primary sclerosing cholangitis, and osteoporosis. Results demonstrated that the family Oxalobacteraceae and genus Candidatus Soleaferrea were all observed to be correlated with these six diseases [179]. Except for primary biliary cirrhosis, shared features were shown including the family Peptostreptococcaceae, order Gastranaerophilales, and genus Romboutsia. An association between the six illnesses and the genus Candidatus Soleaferrea was seen. Conclusions about the influence of gut microbiota’s metabolites and its plausible role in the communication of the immune-gut axis were drawn [179].
Various autoimmune disorders have demonstrated their linkage to the X-chromosome. One of these illnesses, SLE is characterized by autoreactive T-cell governed inflammation [52]. Markedly, individuals possessing multiple X-chromosomes are more susceptible to SLE [52]. Mechanisms behind this genetic basis remain ambiguous. Nonetheless, it has been demonstrated that there is modified X-chromosome inactivation maintenance in T cells of SLE patients as well as female mice. Thus, further exhibiting that X-linked genes are atypically regulated in SLE patient T cells [52]. Additionally, XIST RNA interactome genes have an altered expression in SLE T cells, contributing to Xi epigenetic attribute disturbances. Therefore, it has been suggested that XCI maintenance is a part of SLE disease and theorizing that Xist RNA localization at the XI may be a defining feature in sustaining X-linked gene T cell dosage compensation. SS is another autoimmune disorder known to chronically affect exocrine glands, resulting in mouth and eye dryness [180]. About 58 upregulated X-chromosome genes, including 22 genes previously shown to avoid XCI, were identified. This was based on an evaluation of the SS patient salivary gene of interest, GEO2R gene expression data sets [180]. It was discovered that XIST and its cis regulators RLIM, FTX, as well as CHIC1, and polycomb repressor genes of the PRC1 and PRC2 complexes, were upregulated. X-chromosome genes that participate in the pathogenesis of SS may be regulated by transcription factors whose overexpression is evident and are also differentially methylated in affected SS individuals [180]. Autoimmune thyroid diseases (AITD) like Grave’s Disease and HD affect females predominately, with studies concentrated on female–male dimorphism in immune responses [181]. It has been proposed that IL-1 receptor-associated kinase 1 (IRAK1) gene on the X-chromosome is a strong autoimmune disease-prone locus. IRAK1 has been suggested to have a role in the pathogenesis of HD and AITD [181]. Interestingly, an association between IRAK1 and SSc has also been demonstrated. In discovery and replication sets, the rs1059702 TT genotype was observed to be linked to certain SSc subsets, exhibiting its prospective role in disease prognosis [54]. IRAK1 and SSc’s connection has been established, highlighting that a X-chromosome gene undeviatingly affects SSc susceptibility as well as its phenotypic heterogeneity [54]. The X-chromosome is a noteworthy participant in the processes of XCI, dosage compensation, and escape. Anomalies in these processes contribute to the initiation of various autoimmune illnesses, encompassing MS.
X-chromosome inactivation/dosage compensation and escape
Remarkably, the human X-chromosome has a peculiar inheritance pattern and unique biology [182]. Genes on the X-chromosomes and the X-inactivation process are the root cause of a considerable portion of the health disparities between females and males. Autoimmunity has been linked to X-chromosome structural and number abnormalities, as deletions and translocations of the X autosome are associated with early ovarian failure. Disturbances in the XCI process are one possibility for these deviations. Interestingly, the Kast-Stewart hypothesis is based on two observations: first, women are disproportionately affected by autoimmune disorders, and second, X-inactivation is a crucial biological regulator that primarily affects women [182]. Notably, XCI occurs early, particularly during embryonic development [38]. Following fertilization, the X-chromosome inherited from either parent is silenced. This process is perpetual, and every daughter cell can recognize which X-chromosome is to be inactivated. Additionally, the level of expression from Xi is not as high compared to the Xa and a large portion of Xi genes are silenced [183, 184]. However, studies suggest that silencing is not invariably successful; genes below 20 percent can avoid silencing and variability exists in activation between females [38]. Interestingly, as mentioned before, in MSCI, the XCI process in males, no mRNA genes are found to be escapees [39]. Thus, this process is incomplete in humans as up to one-third of X-chromosomal genes are expressed from the active as well as the inactive chromosome, separately [185]. As a result of vast number of genes present on the X-chromosome, it has been suggested that female–male differences in MS susceptibility are plausibly due to X [12]. Figure 3 depicts the process of X-chromosome inactivation.

Process of X inactivation or lyonization during embryonic development (A). Noncoding RNAs, known as XIST and TSIX begin and regulate this complex process. Of the two X-chromosomes, one is randomly selected for inactivation. (B) Inactive chromosome condenses and forms a Barr body structure. (C) X-chromosome remains inactive throughout subsequent cell divisions.
The X-chromosome, unlike the gene-poor Y-chromosome, contains over 1,000 genes essential to development and cell viability. Additionally, it can be subjected to X-chromosome dosage compensation (XDC). XDC occurs when an X-chromosome associated gene attains equivalent expression between females and males [13]. It has been proposed that XDC is realized by two-fold upregulations of X-associated genes in both males and females. This transpires by the silencing of one X-chromosome, hence XCI in females [13]. Interference in the X inactivation process can produce a loss of dosage compensation, imbalance of gene products, as well as changed endogenous material originating from typical epigenetic context [13]. Consequently, these anomalies can lead to the initiation of autoimmune disorders [48]. Importantly, the gravity of such diseases is reliant upon the status of the XCI and whether it is X-linked, influenced by XCI escape, or due to X-chromosome aneuploidy. The human X-chromosome is unique in comparison to other species. Thus, the alterations that accompany such differences can both instances and intensities of autoimmune illnesses in humans in addition to encountered autoantigens [48]. Particularly, even non-autoimmune disorders characterized by X-chromosome aberrations can contribute to autoimmune disorders. This notion has been supported by the presence of lupus-like symptoms. However, the underlying mechanism behind this process is still not well understood. Reports exist in which dosage alterations could solely influence X-linked dosage-sensitive genes like the protein complex coding genes (PCGs) [13]. In one study, it has been exhibited that human PCGs have a greater probability of escaping XCI and avoiding PCGs (EsP) depicting a two-fold greater expression vs. the inactivated PCGs (InP) or other X-linked genes at the level of both RNA and protein. Therefore, this suggests that EsP could potentially achieve upregulations as well as XDC [13]. The same study also indicated that EsP genes are enhanced in the TLR pathway, NF-kB pathway, and apoptotic pathway and many phenotypes including abnormal, mental, and developmental. Findings from this study provide the conviction that anomalies of EsP can be a decisive contributing factor in autoimmune, reproductive, as well as neurological disorders [13]. XCI is a mechanism of dosage compensation in mammalian females, flies, and worms [14]. Through XCI, female mammals transcriptionally silence one of the two Xs, and the inactivated X-chromosome condenses into a compact structure called a Barr body, stabilized in a silent state. Two noncoding RNAs, XIST and TSIX, initiate and control the inactivation process [14].
One study looked at whether female–male differences were caused by differences in X-chromosomes using EAE [105]. An in-depth look at FoxP3 suggested that maternal compared to paternal imprinting of X-chromosome genes may be responsible for female–male differences seen in autoimmunity. EAE mice with XY chromosomes in the CNS vs. EAE mice with XX exhibited worse clinical symptoms. This was apparent in various brain regions like the cerebellum as demonstrated by Purkinje cell and myelin loss and cerebral cortex via synaptic deprivation. The mapping of the transcriptome, as well as methylome in T cells and neurons, has the capability of better comprehending underlying female–male differences seen in both autoimmunity and neurodegeneration.
Overall, there is a supported notion that demonstrates that women suffer higher immunoreactivity and a more vigorous humoral and cell-mediated immune response to microbial infection and vaccination than men [106]. Genes encoded by the Xi may “escape” inactivation and lead to the expression of X-related genes from the active and inactive chromosome female cells. One study observed the expression of the X-linked gene CD40L, involved in adaptive immunity, and TLR7, a participant in innate immunity. Levels of expression were determined in varying karyotypes including males, females, and those with X-chromosome aneuploidy. An increase in these X-linked genes was observed in healthy females and patients with KS. There seems to be a role for X-chromosome gene dosage expression in innate and adaptive immune responses and a cause for female–male bias in autoimmune disease predisposition. TLR7 plays a major role in SLE development, and it can escape XCI in about 30 percent of immune cells in healthy women. Various genes can escape XCI and ultimately contribute to the development of X-linked diseases. These genes include LAMP-2, IRAK-1, TLR7, USP27X, DDX3X, and CXORF21 and are presented in detail in the next subsections.
LAMP-2
LAMP2 belongs to the LAMP protein family and in humans, it originates from a single gene that possesses nine coding exons and three other last exons, 9a, 9b, and 9c [40]. Consequently, isoforms like LAMP2A, LAMP2B, and LAMP2C are generated. Abnormalities in the lysosome-linked membrane protein-2 (LAMP-2) gene which encodes for the LAMP protein initiate Danon disease pathogenesis. The disease is characterized as an X-linked chief lysosomal storage condition that presents cardiomyopathy, skeletal myopathy as well as intellectual stability as its principal symptoms. Females are affected later in life and less severely than males. Danon disease results in an accumulation of lysosomal material that is secondary to the LAMP2 gene variants impeding LAMP2 normal function. The gene’s pathogenicity can be attributed to a variety of factors including overabundance of autophagy materials and augmented apoptosis [40].
IRAK-1
This gene enables the encoding of interleukin-1 receptor-associated kinase 1 (IRAK1), which is one of the presumed serine/threonine kinases that become linked with the interleukin-1 receptor (IL1R) following stimulation [15]. IL-1 induced upregulation of the transcription factor NF-kappa B is enabled through this gene. Furthermore, spliced transcript variants encoding various isoforms have been discovered for the IRAK-1 gene [15]. Several studies have demonstrated this gene’s association with SLE in both murine and human models [41]. For example, IRAK1 may be involved in controlling nuclear factor κB (NFκB) in TLR activation and TCR signaling in animal models. Additionally, it is a crucial participant in the induction of IFN-α and IFN-γ. Moreover, in humans, five SNPs spanning this gene indicated disease linkage in adult-onset as well as childhood-onset SLE [41].
TLR-7
TLR-7 is a vital participant in the innate immune system, playing a crucial role in pathogen recognition and activation of the immune response [16, 17]. The human TLR-7 gene is approximately 23 kbp in size and is present on the X-chromosome, possessing three exons [16]. Sequence variability as well as the expression of this gene has been linked to SLE pathogenesis. Notably, males with another X-chromosome are more susceptible to developing this condition which is about 14 times greater than XY males [16]. Contrarily, females with only one chromosome and SLE are uncommon. The TLR-7 gene is largely expressed in the lung, placenta, and spleen and is related to and is in close position to TLR-8 on chromosome X [17]. It has been suggested that the copy number variation (CNV) surge of the TLR7 gene may intensify the autoimmune response to nuclear material [42]. Furthermore, the CNV of target genes has the potential to contribute to SLE development.
USP27X
This gene encodes is responsible for encoding a constituent of the peptidase protein family [18]. The encoded protein serves as a deubiquitinase, participating in the upregulation of the pro-apoptotic protein termed Bim [18]. Numerous diseases including cancer, inflammation, neurodegeneration, and infection, have been related to deubiquitinating enzymes (DUBs) [40]. Pathogenic variations in the ubiquitin-specific protease gene, USP27X can initiate X-linked intellectual disability [43]. This condition results due to USP27X deficiency. It has also been reported that USP27X controls histone mono-ubiquitination and promotes epithelial-to-mesenchymal transition (EMT) [19]. However, USP27 is not completely understood as different protein sizes have been suggested and currently, there is no antibody against human USP27X.
DDX3X
The gene encodes a protein that is a component of the large DEAD-box protein family [20]. This protein has multiple functions partaking roles in the nucleus as well as the cytoplasm. Some of the nuclear functions include transcriptional regulation and mRNP assembly [20]. In the cytoplasm, this protein may be involved in translation, cellular signaling, and viral replication. Abnormalities in this gene have been shown in tumorigenesis. The gene possesses a paralog found in the nonrecombining portion of the Y-chromosome. Additionally, pseudogenes that share a similarity to this gene and the DDX3Y paralog are located on both chromosome 4 and X [20]. Furthermore, this gene is involved in DDX3X disorder. Interestingly, DDX3X-related neurodevelopmental disorder (DDX3X-NDD) transpires much more in females compared to males [44]. Affected individuals experience developmental and intellectual disabilities that are mild to severe. A case report found that X-linked mental disability can be due to a variant in the DDX3X gene [45]. This may suggest a basis for clinical as well as prenatal diagnosis for these pedigrees [45].
CXORF21
This gene is located on the endolysosome membrane and is also known as TLR adapter interacting with endolysosomal SLC15A4 [21]. CXORF21 is identified as a protein-encoding gene without a verified function. Nevertheless, it appears to be involved in positive regulation of the innate immune response, participating in various cellular activities such as toll-like receptor signaling pathways and regulating lysosomal lumen pH [21]. Moreover, the gene has been demonstrated to escape XCI in lymphoblastoid cell lines (LCLs) and is expressed differentially in males with KS in LCLs [22]. In addition, CXORF21 is also associated with SLE as evidenced by a study that demonstrated its gene’s upregulation in female SLE patients [22]. Also, CXORF21 is an IFN-responder and following cellular activation, it is expressed in a cell type specific and female–male dimorphic manner [22]. While there have been positive conclusions about autoimmune disease and X inactivation, some have found contrasting results. Earlier studies have attempted to explain this process but have failed to find the significance of skewed inactivation or the degree of skewing in patients compared to controls [23, 38]. Furthermore, X-inactivation did not explain the overwhelming representation of MS in women. A recent study on a mother and daughter showed an X-linked disorder possessing skewed X inactivation [24]. While it is evident that the X-chromosome is involved in autoimmune diseases there needs to be more research to aid in illuminating X-inactivation involvement in the disease.
Conclusion and perspective
Women are disproportionately affected by autoimmune diseases that cause significant morbidity and mortality such as SLE, SSc, SS, and MS. This is evident through occurrences of greater incidence and prevalence among females. The explanations behind this female–male bias can vary, ranging from the gut microbiota to the expression of certain genes, i.e., FoxP3. It is through a variety of factors that developments of autoimmune conditions transpire and there is no sole source that can be attributed to them. Consequently, a combination of genetic and environmental factors can lead to their pathogenic initiation and eventual progression. Leukocyte trafficking via immune cell entry and migration is critical in disease pathogenesis. It is through this cascade that immune cells, with chemokines serving as their mediators, travel to the site of inflammation and contribute to their accumulation. There is a integral role for immune cells, particularly T cells and DCs, in the development and progression of MS and EAE. Proinflammatory Th1, as well as Th17, have been associated with MS pathology [133]. CD4 Tregs are also involved by restricting pathogenic T cell responses via mechanisms such as inhibitory cytokine production, metabolic disruption, and cell-contact-dependent cytolysis. Dysregulation of autoimmune T cell response linked with MS pathology is thought to be due to a variety of factors such as reduced Treg suppressive activity and a decline in Treg numbers. CD8+ T cells have one of the most paramount roles in MS pathology. Large quantities of CD8+ T cells are observed in the injured WM and GM of MS patients. CD8+ T-cell participation in CNS damage is evident through the production of granzymes and perforins as well as secretion of proinflammatory cytokines. The innate immune response encompasses the activation of DCs [25]. These cells take up and process antigens while traveling through lymphatic vessels to tissue-draining lymph nodes, advancing T-cell activation. Therefore, they prepare the immune response and immune memory against future attacks of the same antigen. Numerous amounts of DCs are detected in brain lesions of MS patients and modified phenotype or function is shown vs. healthy controls [26]. Studies indicate the robust presence of DCs in the CSF, where plasmacytoid DCs (pDCs) are reliant on disease terms. Once relapses occur, the amount of pDCs in the CSF increases vs. patients in remission. Thus, based on these T cell and DCs functions, it can be suggested that these cells have a critical role to play in the disease progression of MS. The X-chromosome is another element to consider, as anomalies in chromosome structure and number are linked to autoimmune symptoms. Whether they’re random or imprinted, XCI disturbances may contribute to autoimmune disorders. Genes on the X-chromosome have essential roles in development and cell viability, and XCI helps balance their expression. X-linked genes may escape XCI and be expressed on both the active and inactive chromosomes in female cells. This phenomenon could affect the immune response and contribute to the female–male bias observed in autoimmune diseases. For instance, TLR7, an X-linked gene, plays a significant role in the development of SLE, and its escape from XCI has been observed in the immune cells of healthy women. Thus, a greater understanding of the mechanism of XCI is warranted.
MS is a demyelinating CNS disease with various explanations as to its origins/causation. In this review, we highlighted these topics and describe the risk factors, gene involvement, how X-linked and non-X-linked genes contribute to autoimmune diseases, and how they can escape from dosage compensation. We have also discussed in detail various immune cells such as DCs and T cells, the X-chromosome, chemokines and their receptors, and the role of leukocyte trafficking, among other areas of interest. We have identified the importance of these topics and their cumulative contribution of knowledge to autoimmunity and female–male biased disorders. Taken together, they provide a more comprehensive outlook on autoimmune disorders and their overall pathogenicity, yielding more robust explanations for autoimmunity.
Autoimmune disorders inordinately impacting women can be very concerning; it has been observed that several areas of life are negatively affected, including overall quality. Additionally, it can be very important to investigate the mechanisms underlying female–male bias in autoimmune illnesses since they can highlight and showcase features of diseases that would otherwise go neglected. As more research is conducted into the emergence as well as the mechanism of autoimmune disorders, we expect that over time, the female–male bias observed in these conditions could be further illuminated and ultimately addressed so that fewer women will be affected compared to the present incidence and prevalence. Greater comprehension of autoimmune diseases could pave the way for more advanced remedial options for patients, potentially improving daily life and standard of care.
Even if there is no one specific cause for autoimmune disease, there needs to be a greater understanding of pathogenesis and the reasons for the higher frequency of these illnesses in some regions of the world than others need to be better understood. For instance, certain environmental factors like exposure to EBV, vitamin D deficiency, and lack of sunlight have been demonstrated to increase risk of developing MS. As previously mentioned, females display a more robust immune response and combined with genetic factors are more likely to develop female–male biased diseases emphasizing the need to identify causative factors. X-chromosome inactivation provides another avenue of research worth further exploration in the context of disease etiology and pathogenesis. Additionally, targeting the recruitment and accumulation of immune cells could be beneficial as mechanisms of leukocyte trafficking remains poorly understood. Based on the growing acknowledgment amongst researchers and current literature, there is a recognized need for further research on understanding the observed female–male bias among autoimmune disorders. Future studies should address the aforementioned research gaps discussed in this review so as to better develop targeted pharmacological treatments that ease disease burden and improve quality of life among those affected.
Acknowledgments
Images were produced with Biorender.com. Creative assistance provided by Ms. Julie Joseph in the mechanistic figure is also acknowledged.
-
Research ethics: Not applicable since a review.
-
Informed consent: All authors agree to publish the content of this study as formulated.
-
Author contributions: SD and RK prepared manuscript, tables and figures under the direct supervision and guidance of PJ. PJ conceptualized the idea for the review and worked with SD to finalize the paper for final submission. PD and RT edited the paper and provided constructive comments.
-
Competing interests: The authors declare that there is no potential conflict of interest.
-
Research funding: None declared.
-
Data availability: Not applicatble.
References
1. Billi, AC, Gharaee-Kermani, M, Fullmer, J, Tsoi, LC, Hill, BD, Gruszka, D, et al.. The female-biased factor VGLL3 drives cutaneous and systemic autoimmunity. JCI Insight 2019;4:e127291. https://doi.org/10.1172/jci.insight.127291. 30996136.Search in Google Scholar PubMed PubMed Central
2. Broadley, SA, Deans, J, Sawcer, SJ, Clayton, D, Compston, DAS. Autoimmune disease in first-degree relatives of patients with multiple sclerosis. A UK survey. Brain 2000;123:1102–11. https://doi.org/10.1093/brain/123.6.1102.Search in Google Scholar PubMed
3. Fairweather, D, Rose, NR. Women and autoimmune diseases. Emerg Infect Dis 2004;10:2005–11. https://doi.org/10.3201/eid1011.040367.Search in Google Scholar PubMed PubMed Central
4. Sawyer, AT, Harris, SL, Koenig, HG. Illness perception and high readmission health outcomes. Health Psychol Open 2019;6:Article no. 2055102919844504. https://doi.org/10.1177/2055102919844504.Search in Google Scholar PubMed PubMed Central
5. Elliott, B, Spence, AR, Czuzoj-Shulman, N, Abenhaim, HA. Effect of Sjogren’s syndrome on maternal and neonatal outcomes of pregnancy. J Perinat Med 2019;47:637–42. https://doi.org/10.1515/jpm-2019-0034.Search in Google Scholar PubMed
6. Kolstad, KD, Fiorentino, D, Li, S, Chakravarty, EF, Chung, L. Pregnancy outcomes in adult patients with dermatomyositis and polymyositis. Semin Arthritis Rheum 2018;47:865–9. https://doi.org/10.1016/j.semarthrit.2017.11.005.Search in Google Scholar PubMed PubMed Central
7. Ninagawa, K, Kato, M, Ohira, H, Tsuneta, S, Iwano, H, Kono, M, et al.. The assessment of left heart disease in patients with systemic sclerosis and pulmonary hypertension. Clin Exp Rheumatol 2021;39 (131 Suppl):103–10. https://doi.org/10.55563/clinexprheumatol/c1j9gb.Search in Google Scholar PubMed
8. Borg, A, Gomez, A, Cederlund, A, Cobar, F, Qiu, V, Lindblom, J, et al.. Contribution of abnormal BMI to adverse health-related quality of life outcomes after a 52-week therapy in patients with SLE. Rheumatology 2021;60:4205–17. https://doi.org/10.1093/rheumatology/keaa909.Search in Google Scholar PubMed PubMed Central
9. Mitropoulos, A, Boström, C, Mattsson, M, Kouidi, E, Dimitroulas, T, Liem, SIE, et al.. Exploring the effects of a combined exercise programme on pain and fatigue outcomes in people with systemic sclerosis: study protocol for a large European multi-centre randomised controlled trial. Trials 2022;23:962. https://doi.org/10.1186/s13063-022-06853-1.Search in Google Scholar PubMed PubMed Central
10. Claflin, SB, Klekociuk, S, Campbell, JA, Bessing, B, Palmer, AJ, van der Mei, I, et al.. Association between MS-related knowledge, health literacy, self-efficacy, resilience, and quality of life in a large cohort of MS community members: a cross-sectional study. Mult Scler Relat Disord 2021;54:103158. https://doi.org/10.1016/j.msard.2021.103158.Search in Google Scholar PubMed
11. Goischke, HK. Comorbidities in multiple sclerosis-a plea for interdisciplinary collaboration to improve the quality of life of MS patients. Degener Neurol Neuromuscul Dis 2019;9:39–53. https://doi.org/10.2147/dnnd.s204555.Search in Google Scholar PubMed PubMed Central
12. Voskuhl, RR. The effect of sex on multiple sclerosis risk and disease progression. Mult Scler 2020;26:554–60. https://doi.org/10.1177/1352458519892491.Search in Google Scholar PubMed PubMed Central
13. Xing, Z, Zhang, Y, Tian, Z, Wang, M, Xiao, W, Zhu, C, et al.. Escaping but not the inactive X-linked protein complex coding genes may achieve X-chromosome dosage compensation and underlie X chromosome inactivation-related diseases. Heliyon 2023;9:e17721. https://doi.org/10.1016/j.heliyon.2023.e17721.Search in Google Scholar PubMed PubMed Central
14. Galupa, R, Heard, E. X-Chromosome inactivation: a crossroads between chromosome architecture and gene regulation. Annu Rev Genet 2018;52:535–66. 30256677.10.1146/annurev-genet-120116-024611Search in Google Scholar PubMed
15. Zheng, Y, He, JQ. Interleukin receptor associated kinase 1 signaling and its association with cardiovascular diseases. Rev Cardiovasc Med. 2022;23:97. PMID: 35345264; PMCID: PMC9637324.10.31083/j.rcm2303097Search in Google Scholar PubMed PubMed Central
16. Skonieczna, K, Woźniacka, A, Czajkowski, R, Styczyński, J, Krenska, A, Robak, E, et al.. X-linked TLR7 gene polymorphisms are associated with diverse immunological conditions but not with discoid lupus erythematosus in Polish patients. Postepy Dermatol Alergol 2018;35:26–32. https://doi.org/10.5114/pdia.2017.69984.Search in Google Scholar PubMed PubMed Central
17. Noreen, M, Shah, M, Mall, S, Choudhary, S, Hussain, T, Ahmed, I, et al.. TLR4 polymorphisms and disease susceptibility. Inflamm Res 2012;61:177–88. 22277994.10.1007/s00011-011-0427-1Search in Google Scholar PubMed
18. Koch, I, Slovik, M, Zhang, Y, Liu, B, Rennie, M, Konz, E, et al.. USP27X variants underlying X-linked intellectual disability disrupt protein function via distinct mechanisms. Life Sci ALliance 2024;7:e202302258. 38182161.10.26508/lsa.202302258Search in Google Scholar PubMed PubMed Central
19. Dold, MN, Ng, X, Alber, C, Gentle, IE, Häcker, G, Weber, A. The deubiquitinase Usp27x as a novel regulator of cFLIP(L) protein expression and sensitizer to death-receptor-induced apoptosis. Apoptosis 2022;27:112–32. https://doi.org/10.1007/s10495-021-01706-9.Search in Google Scholar PubMed PubMed Central
20. Lacroix, M, Beauchemin, H, Fraszczak, J, Ross, J, Shooshtarizadeh, P, Chen, R, et al.. The X-linked helicase DDX3X is required for lymphoid differentiation and MYC-driven lymphomagenesis. Cancer Res 2022;82:3172–86. 35815807.10.1158/0008-5472.CAN-21-2454Search in Google Scholar PubMed
21. Heinz, L, Lee, J, Kapoor, U, Kartnig, F, Sedlyarov, V, Papkostas, K, et al.. TASL is the SLC15A4-associated adaptor for IRF5 activation by TLR7-9. Nature 2020;581:316–22. 32433612.10.1038/s41586-020-2282-0Search in Google Scholar PubMed PubMed Central
22. Odhams, CA, Roberts, AL, Vester, SK, Duarte, CST, Beales, CT, Clarke, AJ, et al.. Interferon inducible X-linked gene CXorf21 may contribute to sexual dimorphism in systemic lupus erythematosus. Nat Commun 2019;10:2164. https://doi.org/10.1038/s41467-019-10106-2.Search in Google Scholar PubMed PubMed Central
23. Knudsen, GP, Harbo, HF, Smestad, C, Celius, EG, Åkesson, E, Oturai, A, et al.. X chromosome inactivation in females with multiple sclerosis. Eur J Neurol 2007;14:1392–6. https://doi.org/10.1111/j.1468-1331.2007.01987.x.Search in Google Scholar PubMed
24. Rubegni, A, Battisti, C, Tessa, A, Cerase, A, Doccini, S, Malandrini, A, et al.. SPG2 mimicking multiple sclerosis in a family identified using next generation sequencing. J Neurol Sci 2017;375:198–202. https://doi.org/10.1016/j.jns.2017.01.069.Search in Google Scholar PubMed
25. Mapunda, JA, Tibar, H, Regragui, W, Engelhardt, B. How does the immune system enter the brain? Front Immunol 2022;13:805657. https://doi.org/10.3389/fimmu.2022.805657.Search in Google Scholar PubMed PubMed Central
26. Nuyts, AH, Lee, W, Bashir-Dar, R, Berneman, Z, Cools, N. Dendritic cells in multiple sclerosis: key players in the immunopathogenesis, key players for new cellular immunotherapies? Mult Scler 2013;19:995–1002. https://doi.org/10.1177/1352458512473189.Search in Google Scholar PubMed
27. Bubuioc, A, Kudebayeva, A, Turuspekova, S, Lisnic, V, Leone, MA. The epidemiology of myasthenia gravis. J Med Life 2021;14:7–16. 33767779.Search in Google Scholar
28. Rodrigues, E, Umeh, E, Aishwarya, Navaratnarajah, N, Cole, A, Moy, K. Incidence and prevalence of myasthenia gravis in the United States: a claims-based analysis. Muscle Nerve 2024;69:166–71. 38040629.10.1002/mus.28006Search in Google Scholar PubMed
29. Vaqar, S, Shackelford, KB. Pernicious anemia. In: StatPearls [Internet]. Treasure Island (FL): StatPearls. Publishing 2024; PMID: 31082033.Search in Google Scholar
30. Carmel, R. Prevalence of undiagnosed pernicious anemia in the elderly. Arch Intern Med 1996;156:1097–100. 8638997.10.1001/archinte.156.10.1097Search in Google Scholar
31. Cai, Y, Zhang, J, Liang, J, Xiao, M, Zhang, G, Jing, Z, et al.. The burden of rheumatoid arthritis: findings from the 2019 global burden of diseases study and forecasts for 2030 by Bayesian age-period-cohort analysis. J Clin Med 2023;12:1291. 36835827.10.3390/jcm12041291Search in Google Scholar PubMed PubMed Central
32. van Vollenhoven, RF. Sex differences in rheumatoid arthritis: more than meets the eye. BMC Med 2009;7:12. 19331649.10.1186/1741-7015-7-12Search in Google Scholar PubMed PubMed Central
33. Patel, R, Shahane, A. The epidemiology of Sjögren’s syndrome. Clin Epidemiol 2014;6:247–55. 25114590.10.2147/CLEP.S47399Search in Google Scholar PubMed PubMed Central
34. Zhang, Y, Chen, J-Q, Yang, J-Y, Liao, J-H, Wu, T-H, Yu, X-B, et al.. Sex difference in primary sjögren syndrome: a medical records review study. J Clin Rheumatol 2023;29:e78–85. 37068269.10.1097/RHU.0000000000001962Search in Google Scholar PubMed PubMed Central
35. Tian, J, Zhang, D, Yao, X, Huang, Y, Lu, Q. Global epidemiology of systemic lupus erythematosus: a comprehensive systematic analysis and modelling study. Ann Rheum Dis 2023;82:351–6. 36241363.10.1136/ard-2022-223035Search in Google Scholar PubMed PubMed Central
36. Rider, V, Abdou, NI, Kimler, BF, Lu, N, Brown, S, Fridley, BL. Gender bias in human systemic Lupus Erythematosus: a problem of steroid receptor action? Front Immunol 2018;9:611. 29643853.10.3389/fimmu.2018.00611Search in Google Scholar PubMed PubMed Central
37. Shvetsova, E, Sofronova, A, Monajemi, R, Gagalova, K, Draisma, HHM, White, SJ, et al.. Skewed X-inactivation is common in the general female population. Eur J Hum Genet 2019;27:455–65. https://doi.org/10.1038/s41431-018-0291-3.Search in Google Scholar PubMed PubMed Central
38. Knudsen, GP. Gender bias in autoimmune diseases: X chromosome inactivation in women with multiple sclerosis. J Neurol Sci 2009;286:43–6. https://doi.org/10.1016/j.jns.2009.04.022.Search in Google Scholar PubMed
39. Yan, W, McCarrey, JR. Sex chromosome inactivation in the male. Epigenetics 2009;4:452–6. https://doi.org/10.4161/epi.4.7.9923.Search in Google Scholar PubMed PubMed Central
40. Shalata, A, Bar-Shai, M, Hadid, Y, Mahroum, M, Mintz, H, Shalata, ZE, et al.. Danon disease: entire LAMP2 gene deletion with unusual clinical presentation-case report and review of the literature. Genes 2023;14:1539. https://doi.org/10.3390/genes14081539.Search in Google Scholar PubMed PubMed Central
41. Aranoq, C, Diamond, B, Mackay, M. Clinical immunology: principles and practice. In: Chapter 77 – systemic lupus erythematosus, 5th ed. Amsterdam: Elsevier; 2019:685–704 pp.10.1016/B978-0-7020-6896-6.00051-XSearch in Google Scholar
42. Pacheco, GV, Cruz, DC, Herrera, LJG, Mendoza, GJP, Amaro, GIA, Ueji, YEN, et al.. Copy number variation of TLR-7 gene and its association with the development of systemic lupus erythematosus in female patients from Yucatan Mexico. Genet Epigenet 2014;6:31–6. https://doi.org/10.4137/geg.s16707.Search in Google Scholar
43. Santiago-Sim, T, Burrage, LC, Ebstein, F, Tokita, MJ, Miller, M, Bi, W, et al.. Biallelic variants in OTUD6B cause an intellectual disability syndrome associated with seizures and dysmorphic features. Am J Hum Genet 2017;100:676–88. https://doi.org/10.1016/j.ajhg.2017.03.001.Search in Google Scholar PubMed PubMed Central
44. Johnson-Kerner, B, Snijders Blok, L, Suit, L, Thomas, J, Kleefstra, T, Sherr, EH. DDX3X-related neurodevelopmental disorder. 2020 Aug 27. In: Adam, MP, Feldman, J, Mirzaa, GM, Pagon, RA, Wallace, SE, Bean, LJH, et al.., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2024. PMID: 32852922.Search in Google Scholar
45. Wang, Q, Yang, Y, Liu, L, Tie, X, Lei, H, Zhang, L, et al.. Analysis of a child with X-linked mental retardation due to a de novo variant of DDX3X gene. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2022;39:1111–15.Search in Google Scholar
46. Brambatti, M, Caspi, O, Maolo, A, Koshi, E, Greenberg, B, Taylor, MRG, et al.. Danon disease: Gender differences in presentation and outcomes. Int J Cardiol 2019;286:92–8. 30857840.10.1016/j.ijcard.2019.01.020Search in Google Scholar PubMed
47. Petratos, S, Ozturk, E, Azari, MF, Kenny, R, Young Lee, J, Magee, KA, et al.. Limiting multiple sclerosis related axonopathy by blocking Nogo receptor and CRMP-2 phosphorylation. Brain 2012;135:1794–818. https://doi.org/10.1093/brain/aws100.Search in Google Scholar PubMed PubMed Central
48. Brooks, WH, Renaudineau, Y. Epigenetics and autoimmune diseases: the X chromosome-nucleolus nexus. Front Genet 2015;6:22. https://doi.org/10.3389/fgene.2015.00022.Search in Google Scholar PubMed PubMed Central
49. Lacotte, S, Brun, S, Muller, S, Dumortier, H. CXCR3, inflammation, and autoimmune diseases. Ann N Y Acad Sci 2009;1173:310–7. https://doi.org/10.1111/j.1749-6632.2009.04813.x.Search in Google Scholar PubMed
50. Man, JJ, Lu, Q, Moss, ME, Carvajal, B, Baur, W, Garza, AE, et al.. Myeloid mineralocorticoid receptor transcriptionally regulates P-selectin glycoprotein ligand-1 and promotes monocyte trafficking and atherosclerosis. Arterioscler Thromb Vasc Biol 2021;41:2740–55. https://doi.org/10.1161/atvbaha.121.316929.Search in Google Scholar
51. Galimberti, D, Scalabrini, D, Fenoglio, C, De Riz, M, Comi, C, Venturelli, E, et al.. Gender-specific influence of the chromosome 16 chemokine gene cluster on the susceptibility to multiple sclerosis. J Neurol Sci 2008;267:86–90. https://doi.org/10.1016/j.jns.2007.10.001.Search in Google Scholar PubMed
52. Syrett, CM, Paneru, B, Sandoval-Heglund, D, Wang, J, Banerjee, S, Sindhava, V, et al.. Altered X-chromosome inactivation in T cells may promote sex-biased autoimmune diseases. JCI Insight 2019;4:e126751. https://doi.org/10.1172/jci.insight.126751.Search in Google Scholar PubMed PubMed Central
53. Cerri, S, Mus, L, Blandini, F. Parkinson’s disease in women and men: what’s the difference? J Parkinsons Dis 2019;9:501–15. 31282427.10.3233/JPD-191683Search in Google Scholar PubMed PubMed Central
54. Dieude, P, Bouaziz, M, Guedj, M, Riemekasten, G, Airò, P, Müller, M, et al.. Evidence of the contribution of the X chromosome to systemic sclerosis susceptibility: association with the functional IRAK1 196Phe/532Ser haplotype. Arthritis Rheum 2011;63:3979–87. https://doi.org/10.1002/art.30640.Search in Google Scholar PubMed
55. Amor, S, Puentes, F, Baker, D, Van Der Valk, P. Inflammation in neurodegenerative diseases. Immunology 2010;129:154–69. https://doi.org/10.1111/j.1365-2567.2009.03225.x.Search in Google Scholar PubMed PubMed Central
56. Papiri, G, D’Andreamatteo, G, Cacchiò, G, Alia, S, Silvestrini, M, Paci, C, et al.. Multiple sclerosis: inflammatory and neuroglial aspects. Curr Issues Mol Biol 2023;45:1443–70. https://doi.org/10.3390/cimb45020094.Search in Google Scholar PubMed PubMed Central
57. Passaponti, S, Ermini, L, Acconci, G, Severi, FM, Romagnoli, R, Cutrupi, S, et al.. Rank-Rankl-Opg axis in multiple sclerosis: the contribution of placenta. Cells 2022;11:1357. https://doi.org/10.3390/cells11081357.Search in Google Scholar PubMed PubMed Central
58. Sahakyan, AY, Mavelyan, HM. Specifics of relationship within a complex of neurological deficit, quality of life and depression in the phenotypic spectrum of multiple sclerosis. Neurol India 2022;70:1083–90. https://doi.org/10.4103/0028-3886.349619.Search in Google Scholar PubMed
59. Oukka, M, Bettelli, E. Regulation of lymphocyte trafficking in central nervous system autoimmunity. Curr Opin Immunol 2018;55:38–43. https://doi.org/10.1016/j.coi.2018.09.008.Search in Google Scholar PubMed PubMed Central
60. Vogel, DY, Vereyken, EJ, Glim, JE, Heijnen, PD, Moeton, M, van der Valk, P, et al.. Macrophages in inflammatory multiple sclerosis lesions have an intermediate activation status. J Neuroinflammation 2013;10:35. https://doi.org/10.1186/1742-2094-10-35.Search in Google Scholar PubMed PubMed Central
61. Montilla, A, Zabala, A, Er-Lukowiak, M, Rissiek, B, Magnus, T, Rodriguez-Iglesias, N, et al.. Microglia and meningeal macrophages depletion delays the onset of experimental autoimmune encephalomyelitis. Cell Death Dis 2023;14:16. https://doi.org/10.1038/s41419-023-05551-3.Search in Google Scholar PubMed PubMed Central
62. Chu, F, Shi, M, Zheng, C, Shen, D, Zhu, J, Zheng, X, et al.. The roles of macrophages and microglia in multiple sclerosis and experimental autoimmune encephalomyelitis. J Neuroimmunol 2018;318:1–7. https://doi.org/10.1016/j.jneuroim.2018.02.015.Search in Google Scholar PubMed
63. Murphy, KL, Fischer, R, Swanson, KA, Bhatt, IJ, Oakley, L, Smeyne, R, et al.. Synaptic alterations and immune response are sexually dimorphic in a non-pertussis toxin model of experimental autoimmune encephalomyelitis. Exp Neurol 2020;323:113061. https://doi.org/10.1016/j.expneurol.2019.113061.Search in Google Scholar PubMed PubMed Central
64. Komiyama, Y, Nishikado, H, Masuda, M, Egawa, H, Kobayashi, N, Kobatake, S, et al.. A rapid enzyme-linked immunosorbent assay for human factor XI. Thromb Res 1988;50:329–34. https://doi.org/10.1016/0049-3848(88)90234-4.Search in Google Scholar PubMed
65. Pukoli, D, Vecsei, L. Smouldering lesion in MS: microglia, lymphocytes and pathobiochemical mechanisms. Int J Mol Sci 2023;24:12631. https://doi.org/10.3390/ijms241612631.Search in Google Scholar PubMed PubMed Central
66. van den Bosch, AMR, van der Poel, M, Fransen, NL, Vincenten, MCJ, Bobeldijk, AM, Jongejan, A, et al.. Profiling of microglia nodules in multiple sclerosis reveals propensity for lesion formation. Nat Commun 2024;15:1667. https://doi.org/10.1038/s41467-024-46068-3.Search in Google Scholar PubMed PubMed Central
67. Zeitelhofer, M, Adzemovic, MZ, Moessinger, C, Stefanitsch, C, Strell, C, Muhl, L, et al.. Blocking PDGF-CC signaling ameliorates multiple sclerosis-like neuroinflammation by inhibiting disruption of the blood-brain barrier. Sci Rep 2020;10:22383. https://doi.org/10.1038/s41598-020-79598-z.Search in Google Scholar PubMed PubMed Central
68. Lopes Pinheiro, MA, Kooij, G, Mizee, MR, Kamermans, A, Enzmann, G, Lyck, R, et al.. Immune cell trafficking across the barriers of the central nervous system in multiple sclerosis and stroke. Biochim Biophys Acta 2016;1862:461–71. https://doi.org/10.1016/j.bbadis.2015.10.018.Search in Google Scholar PubMed
69. Liu, C, Zhu, J, Mi, Y, Jin, T. Impact of disease-modifying therapy on dendritic cells and exploring their immunotherapeutic potential in multiple sclerosis. J Neuroinflammation 2022;19:298. https://doi.org/10.1186/s12974-022-02663-z.Search in Google Scholar PubMed PubMed Central
70. Sie, C, Perez, LG, Kreutzfeldt, M, Potthast, M, Ohnmacht, C, Merkler, D, et al.. Dendritic cell accumulation in the gut and central nervous system is differentially dependent on alpha4 integrins. J Immunol 2019;203:1417–27. https://doi.org/10.4049/jimmunol.1900468.Search in Google Scholar PubMed PubMed Central
71. Jain, P, Coisne, C, Enzmann, G, Rottapel, R, Engelhardt, B. Alpha4beta1 integrin mediates the recruitment of immature dendritic cells across the blood-brain barrier during experimental autoimmune encephalomyelitis. J Immunol 2010;184:7196–206. https://doi.org/10.4049/jimmunol.0901404.Search in Google Scholar PubMed PubMed Central
72. Luessi, F, Kraus, S, Trinschek, B, Lerch, S, Ploen, R, Paterka, M, et al.. FTY720 (fingolimod) treatment tips the balance towards less immunogenic antigen-presenting cells in patients with multiple sclerosis. Mult Scler 2015;21:1811–22. https://doi.org/10.1177/1352458515574895.Search in Google Scholar PubMed
73. Ghafouri-Fard, S, Honarmand, K, Taheri, M. A comprehensive review on the role of chemokines in the pathogenesis of multiple sclerosis. Metab Brain Dis 2021;36:375–406. https://doi.org/10.1007/s11011-020-00648-6.Search in Google Scholar PubMed
74. Diotallevi, F, Offidani, A. Skin, autoimmunity and inflammation: a comprehensive exploration through scientific research. Int J Mol Sci 2023;24:15857. https://doi.org/10.3390/ijms242115857.Search in Google Scholar PubMed PubMed Central
75. Hori, N, Okada, K, Takakura, Y, Takano, H, Yamaguchi, N, Yamaguchi, N. Vestigial-like family member 3 (VGLL3), a cofactor for TEAD transcription factors, promotes cancer cell proliferation by activating the Hippo pathway. J Biol Chem 2020;295:8798–807. https://doi.org/10.1074/jbc.ra120.012781.Search in Google Scholar PubMed PubMed Central
76. Pagenkopf, A, Liang, Y. Immunometabolic function of the transcription cofactor VGLL3 provides an evolutionary rationale for sexual dimorphism in autoimmunity. FEBS Lett 2020;594:3371–83. https://doi.org/10.1002/1873-3468.13911.Search in Google Scholar PubMed PubMed Central
77. Xing, E, Billi, AC, Gudjonsson, JE. Sex bias and autoimmune diseases. J Invest Dermatol 2022;142:857–66. https://doi.org/10.1016/j.jid.2021.06.008.Search in Google Scholar PubMed
78. Kronzer, VL, Bridges, SLJr., Davis, JM3rd. Why women have more autoimmune diseases than men: an evolutionary perspective. Evol Appl 2021;14:629–33. https://doi.org/10.1111/eva.13167.Search in Google Scholar PubMed PubMed Central
79. Belkhir, R, Gestermann, N, Koutero, M, Seror, R, Tost, J, Mariette, X, et al.. Upregulation of membrane-bound CD40L on CD4+ T cells in women with primary Sjogren’s syndrome. Scand J Immunol 2014;79:37–42. https://doi.org/10.1111/sji.12121.Search in Google Scholar PubMed
80. Vaitaitis, GM, Waid, DM, Yussman, MG, Wagner, DHJr. CD40-mediated signalling influences trafficking, T-cell receptor expression, and T-cell pathogenesis, in the NOD model of type 1 diabetes. Immunology 2017;152:243–54. https://doi.org/10.1111/imm.12761.Search in Google Scholar PubMed PubMed Central
81. Senhaji, N, Kojok, K, Darif, Y, Fadainia, C, Zaid, Y. The contribution of CD40/CD40L axis in inflammatory bowel disease: an update. Front Immunol 2015;6:529. https://doi.org/10.3389/fimmu.2015.00529.Search in Google Scholar PubMed PubMed Central
82. Balashov, KE, Smith, D, Khoury, S, Hafler, D, Weiner, H. Increased interleukin 12 production in progressive multiple sclerosis: induction by activated CD4+ T cells via CD40 ligand. Proc Natl Acad Sci U S A 1997;94:599–603. https://doi.org/10.1073/pnas.94.2.599.Search in Google Scholar PubMed PubMed Central
83. Zhu, L, Song, G, Chen, X, Zhang, Y, Cui, Y, Qiao, J, et al.. Higher CD4(+)CD40(+) T cells (Th40 cells) associate with systemic lupus erythematosus activity. Sci Rep 2023;13:10702. https://doi.org/10.1038/s41598-023-37749-y.Search in Google Scholar PubMed PubMed Central
84. Syrett, CM, Sierra, I, Beethem, ZT, Dubin, AH, Anguera, MC. Loss of epigenetic modifications on the inactive X chromosome and sex-biased gene expression profiles in B cells from NZB/W F1 mice with lupus-like disease. J Autoimmun 2020;107:102357. https://doi.org/10.1016/j.jaut.2019.102357.Search in Google Scholar PubMed PubMed Central
85. Johnson, BM, Gaudreau, MC, Gudi, R, Brown, R, Gilkeson, G, Vasu, C. Gut microbiota differently contributes to intestinal immune phenotype and systemic autoimmune progression in female and male lupus-prone mice. J Autoimmun 2020;108:102420. https://doi.org/10.1016/j.jaut.2020.102420.Search in Google Scholar PubMed PubMed Central
86. Mangalam, AK, Giri, S. Role of microbiome and metabolome in the pathobiology of MS. Clin Immunol 2022;235:108934. https://doi.org/10.1016/j.clim.2022.108934.Search in Google Scholar PubMed
87. Moulton, VR. Sex hormones in acquired immunity and autoimmune disease. Front Immunol 2018;9:2279. https://doi.org/10.3389/fimmu.2018.02279.Search in Google Scholar PubMed PubMed Central
88. Ysrraelit, MC, Correale, J. Impact of sex hormones on immune function and multiple sclerosis development. Immunology 2019;156:9–22. https://doi.org/10.1111/imm.13004.Search in Google Scholar PubMed PubMed Central
89. Collongues, N, Patte-Mensah, C, De Seze, J, Mensah-Nyagan, AG, Derfuss, T. Testosterone and estrogen in multiple sclerosis: from pathophysiology to therapeutics. Expert Rev Neurother 2018;18:515–22. https://doi.org/10.1080/14737175.2018.1481390.Search in Google Scholar PubMed
90. Rossi, B, Santos-Lima, B, Terrabuio, E, Zenaro, E, Constantin, G. Common peripheral immunity mechanisms in multiple sclerosis and Alzheimer’s disease. Front Immunol 2021;12:639369. https://doi.org/10.3389/fimmu.2021.639369.Search in Google Scholar PubMed PubMed Central
91. Hayes, CE, Donald Acheson, E. A unifying multiple sclerosis etiology linking virus infection, sunlight, and vitamin D, through viral interleukin-10. Med Hypotheses 2008;71:85–90. https://doi.org/10.1016/j.mehy.2008.01.031.Search in Google Scholar PubMed
92. Sagar, D, Lamontagne, A, Foss, CA, Khan, ZK, Pomper, MG, Jain, P. Dendritic cell CNS recruitment correlates with disease severity in EAE via CCL2 chemotaxis at the blood-brain barrier through paracellular transmigration and ERK activation. J Neuroinflammation 2012;9:245. https://doi.org/10.1186/1742-2094-9-245.Search in Google Scholar PubMed PubMed Central
93. Chitnis, T. The role of CD4 T cells in the pathogenesis of multiple sclerosis. Int Rev Neurobiol 2007;79:43–72. https://doi.org/10.1016/s0074-7742(07)79003-7.Search in Google Scholar
94. Ford, H. Clinical presentation and diagnosis of multiple sclerosis. Clin Med 2020;20:380–3. https://doi.org/10.7861/clinmed.2020-0292.Search in Google Scholar PubMed PubMed Central
95. Etta, I, Elballushi, R, Kolesnyk, V, Sia, KP, Rehman, S, Arif, S, et al.. Comparison of pharmacological therapies in relapse rates in patients with relapsing-remitting multiple sclerosis. Cureus 2023;15:e45454. https://doi.org/10.7759/cureus.45454.Search in Google Scholar PubMed PubMed Central
96. Connick, P, De Angelis, F, Parker, RA, Plantone, D, Doshi, A, John, N, et al.. Multiple Sclerosis-Secondary Progressive Multi-Arm Randomisation Trial (MS-SMART): a multiarm phase IIb randomised, double-blind, placebo-controlled clinical trial comparing the efficacy of three neuroprotective drugs in secondary progressive multiple sclerosis. BMJ Open 2018;8:e021944. https://doi.org/10.1136/bmjopen-2018-021944.Search in Google Scholar PubMed PubMed Central
97. Mescheriakova, JY, Broer, L, Wahedi, S, Uitterlinden, AG, van Duijn, CM, Hintzen, RQ. Burden of genetic risk variants in multiple sclerosis families in the Netherlands. Mult Scler J Exp Transl Clin 2016;2:Article no. 2055217316648721. https://doi.org/10.1177/2055217316648721.Search in Google Scholar PubMed PubMed Central
98. Milo, R, Kahana, E. Multiple sclerosis: geoepidemiology, genetics and the environment. Autoimmun Rev 2010;9:A387–94. https://doi.org/10.1016/j.autrev.2009.11.010.Search in Google Scholar PubMed
99. Leffler, J, Trend, S, Gorman, S, Hart, PH. Sex-specific environmental impacts on initiation and progression of multiple sclerosis. Front Neurol 2022;13:835162. https://doi.org/10.3389/fneur.2022.835162.Search in Google Scholar PubMed PubMed Central
100. Leray, E, Moreau, T, Fromont, A, Edan, G. Epidemiology of multiple sclerosis. Rev Neurol 2016;172:3–13. https://doi.org/10.1016/j.neurol.2015.10.006.Search in Google Scholar PubMed
101. Wang, L, Feske, S. Seeing is believing: visualizing immune cells and calcium signals at different stages of neuroinflammation. Proc Natl Acad Sci U S A 2020;117:20360–2. https://doi.org/10.1073/pnas.2013377117.Search in Google Scholar PubMed PubMed Central
102. Charabati, M, Grasmuck, C, Ghannam, S, Bourbonnière, L, Fournier, AP, Lécuyer, MA, et al.. DICAM promotes T(H)17 lymphocyte trafficking across the blood–brain barrier during autoimmune neuroinflammation. Sci Transl Med 2022;14:eabj0473. https://doi.org/10.1126/scitranslmed.abj0473.Search in Google Scholar PubMed
103. Fernandez, D, Geisse, A, Bernales, JI, Lira, A, Osorio, F. The unfolded protein response in immune cells as an emerging regulator of neuroinflammation. Front Aging Neurosci 2021;13:682633. https://doi.org/10.3389/fnagi.2021.682633.Search in Google Scholar PubMed PubMed Central
104. Probstel, AK, Baranzini, SE. The role of the gut microbiome in multiple sclerosis risk and progression: towards characterization of the “MS microbiome”. Neurotherapeutics 2018;15:126–34. https://doi.org/10.1007/s13311-017-0587-y.Search in Google Scholar PubMed PubMed Central
105. Voskuhl, RR, Sawalha, AH, Itoh, Y. Sex chromosome contributions to sex differences in multiple sclerosis susceptibility and progression. Mult Scler 2018;24:22–31. https://doi.org/10.1177/1352458517737394.Search in Google Scholar PubMed PubMed Central
106. Sarmiento, L, Svensson, J, Barchetta, I, Giwercman, A, Cilio, CM. Copy number of the X-linked genes TLR7 and CD40L influences innate and adaptive immune responses. Scand J Immunol 2019;90:e12776. https://doi.org/10.1111/sji.12776.Search in Google Scholar PubMed
107. Sun, Z, Fan, J, Wang, Y. X-chromosome inactivation and related diseases. Genet Res 2022;2022:1391807. https://doi.org/10.1155/2022/1391807.Search in Google Scholar PubMed PubMed Central
108. Arias, C, Sepúlveda, P, Castillo, RL, Salazar, LA. Relationship between hypoxic and immune pathways activation in the progression of neuroinflammation: role of HIF-1alpha and Th17 cells. Int J Mol Sci 2023;24:3073. https://doi.org/10.3390/ijms24043073.Search in Google Scholar PubMed PubMed Central
109. Karulin, AY, Quast, S, Hesse, MD, Lehmann, PV. Neuroantigen-specific CD4 cells expressing interferon-gamma (IFN-gamma), interleukin (IL)-2 and IL-3 in a mutually exclusive manner prevail in experimental allergic encephalomyelitis (EAE). Cells 2012;1:576–96. https://doi.org/10.3390/cells1030576.Search in Google Scholar PubMed PubMed Central
110. Prajjwal, P, Shree, A, Das, S, Inban, P, Ghosh, S, Senthil, A, et al.. Vascular multiple sclerosis: addressing the pathogenesis, genetics, pro-angiogenic factors, and vascular abnormalities, along with the role of vascular intervention. Ann Med Surg 2023;85:4928–38. https://doi.org/10.1097/ms9.0000000000001177.Search in Google Scholar PubMed PubMed Central
111. Satoh, J, Nanri, Y, Tabunoki, H, Yamamura, T. Microarray analysis identifies a set of CXCR3 and CCR2 ligand chemokines as early IFNbeta-responsive genes in peripheral blood lymphocytes in vitro: an implication for IFNbeta-related adverse effects in multiple sclerosis. BMC Neurol 2006;6:18. https://doi.org/10.1186/1471-2377-6-18.Search in Google Scholar PubMed PubMed Central
112. Nathan, P, Gibbs, JE, Rainger, GE, Chimen, M. Changes in circadian rhythms dysregulate inflammation in ageing: focus on leukocyte trafficking. Front Immunol 2021;12:673405. https://doi.org/10.3389/fimmu.2021.673405.Search in Google Scholar PubMed PubMed Central
113. Cambier, S, Gouwy, M, Proost, P. The chemokines CXCL8 and CXCL12: molecular and functional properties, role in disease and efforts towards pharmacological intervention. Cell Mol Immunol 2023;20:217–51. 36725964.10.1038/s41423-023-00974-6Search in Google Scholar PubMed PubMed Central
114. Buka, RJ, Montague, SJ, Moran, LA, Martin, EM, Slater, A, Watson, SP, et al.. PF4 activates the c-Mpl-Jak2 pathway in platelets. Blood 2024;143:64–9. https://doi.org/10.1182/blood.2023020872. 37883794.Search in Google Scholar PubMed PubMed Central
115. Greinacher, A, Warkentin, T. Platelet factor 4 triggers thrombo-inflammation by bridging innate and adaptive immunity. Int J Lab Hematol 2023;45:11–22. 37150909.10.1111/ijlh.14075Search in Google Scholar PubMed
116. Korbecki, J, Bosiacki, M, Barczak, K, Lagocka, R, Brodowska, A, Chlubek, D, et al.. Involvement in tumorigenesis and clinical significance of CXCL1 in reproductive cancers: breast cancer, cervical cancer, endometrial cancer, ovarian cancer and prostate cancer. Int J Mol Sci 2023;24:7262. 37108425.10.3390/ijms24087262Search in Google Scholar PubMed PubMed Central
117. Yin, X, Xia, K, Peng, S, Tan, B, Huang, Y, Wang, M, et al.. ABCF1/CXCL12/CXCR4 enhances glioblastoma cell proliferation, migration, and invasion by activating the PI3K/AKT signal pathway. Dev Neurosci 2024;46:210–20. 37757768.10.1159/000533130Search in Google Scholar PubMed
118. Zhang, X, Zhu, M, Jiang, X-L, Liu, X, Liu, X, Liu, P, et al.. P-selectin glycoprotein ligand 1 deficiency prevents development of acute pancreatitis by attenuating leukocyte infiltration. World J Gastroenterol 2020;26:6361–77. 33244198.10.3748/wjg.v26.i41.6361Search in Google Scholar PubMed PubMed Central
119. Boziki, M, Karapanayotides, T, Papdopoulos, G, Lagoudaki, R, Melo, P, Bakirtzis, C, et al.. Reduced expression of L-selectin in T-cells correlates with relative lymphocyte increase in patients with RRMS treated with natalizumab - functional implication towards PML risk. Neurol Res 2020;42:209–21. 32048570.10.1080/01616412.2020.1722913Search in Google Scholar PubMed
120. Núñez, L, Marrón-Liñares, G, Crespo-Leiro, M, Barge-Caballero, E, Álvarez-López, E, Suarez-Fuentetaja, N, et al.. AGT haplotype in ITGA4 gene is related to antibody-mediated rejection in heart transplant patients. PLoS One 2019;14:e0219345. 31335901.10.1371/journal.pone.0219345Search in Google Scholar PubMed PubMed Central
121. Gharibi, A, Kim, S, Molnar, J, Brambilla, D, Adamian, Y, Hoover, M, et al.. ITGA1 is a pre-malignant biomarker that promotes therapy resistance and metastatic potential in pancreatic cancer. Sci Rep 2017;7:10060. 28855593.10.1038/s41598-017-09946-zSearch in Google Scholar PubMed PubMed Central
122. Chen, M, Wu, C, Fu, Z, Liu, S. ICAM1 promotes bone metastasis via integrin-mediated TGF-β/EMT signaling in triple-negative breast cancer. Cancer Sci 2022;113:3751–65. 35969372.10.1111/cas.15532Search in Google Scholar PubMed PubMed Central
123. VanHeyst, K, Choi, SH, Kingsley, D, Huang, A. Ectopic tumor VCAM-1 expression in cancer metastasis and therapy resistance. Cells 2022;11:3922. 36497180.10.3390/cells11233922Search in Google Scholar PubMed PubMed Central
124. Ruiz-Ojeda, F, Olza, J, Gil, Á, Aguilera, C. Chapter 1 - oxidative stress and inflammation in obesity and metabolic syndrome. In: Obesity: oxidative stress and dietary antioxidants. Amsterdam: Elsevier; 2018:1–15 pp.10.1016/B978-0-12-812504-5.00001-5Search in Google Scholar
125. Liarmakopoulos, E, Gazouli, M, Aravantinos, G, THeodoropoulos, G, Rizos, S, Vaiopoulou, A, et al.. E-Selectin S128R gene polymorphism in gastric cancer. Int J Biol Markers 2013;28:38–42. 23015400.10.5301/JBM.2012.9582Search in Google Scholar PubMed
126. Timmerman, I, Daniel, AE, Kroon, J, van Buul, JD. Chapter five – leukocytes crossing the endothelium: a matter of communication. In: International review of cell and molecular biology. Amsterdam: Elsevier; 2016, 322:281–329 pp.10.1016/bs.ircmb.2015.10.005Search in Google Scholar PubMed
127. Omari, K, John, G, Lango, R, Raine, C. Role for CXCR2 and CXCL1 on glia in multiple sclerosis. Glia 2006;53:24–31. 16086366.10.1002/glia.20246Search in Google Scholar PubMed
128. Czubak-Prowizor, K, Babinska, A, Swiatkowska, M. The F11 receptor (F11R)/junctional adhesion molecule-A (JAM-A) (F11R/JAM-A) in cancer progression. Mol Cell Biochem 2022;477:79–98. 34533648.10.1007/s11010-021-04259-2Search in Google Scholar PubMed PubMed Central
129. Ren, Q, Ren, L, Ren, C, Liu, X, Dong, C, Zhang, X. Platelet endothelial cell adhesion molecule-1 (PECAM1) plays a critical role in the maintenance of human vascular endothelial barrier function. Cell Biochem Funct 2015;33:560–5. 26607202.10.1002/cbf.3155Search in Google Scholar PubMed
130. Norling, LV, Perretti, M. Control of myeloid cell trafficking in resolution. J Innate Immun 2013;5:367–76. https://doi.org/10.1159/000350612.Search in Google Scholar PubMed PubMed Central
131. Palazuelos, J, Davoust, N, Julien, B, Hatterer, E, Aguado, T, Mechoulam, R, et al.. The CB(2) cannabinoid receptor controls myeloid progenitor trafficking: involvement in the pathogenesis of an animal model of multiple sclerosis. J Biol Chem 2008;283:13320–9. https://doi.org/10.1074/jbc.m707960200.Search in Google Scholar PubMed
132. Baecher-Allan, C, Kaskow, BJ, Weiner, HL. Multiple sclerosis: mechanisms and immunotherapy. Neuron 2018;97:742–68. https://doi.org/10.1016/j.neuron.2018.01.021.Search in Google Scholar PubMed
133. Rodriguez Murua, S, Farez, MF, Quintana, FJ. The immune response in multiple sclerosis. Annu Rev Pathol 2022;17:121–39. https://doi.org/10.1146/annurev-pathol-052920-040318.Search in Google Scholar PubMed
134. Opdenakker, G, Van Damme, J. Probing cytokines, chemokines and matrix metalloproteinases towards better immunotherapies of multiple sclerosis. Cytokine Growth Factor Rev 2011;22:359–65. https://doi.org/10.1016/j.cytogfr.2011.11.005.Search in Google Scholar PubMed
135. Matoba, T, Egashira, K. Anti-inflammatory gene therapy for cardiovascular disease. Curr Gene Ther 2011;11:442–6. 22023473.10.2174/156652311798192888Search in Google Scholar PubMed
136. Fazia, T, Nova, A, Gentilini, D, Beecham, A, Piras, M, Saddi, V, et al.. Investigating the causal effect of brain expression of CCL2, NFKB1, MAPK14, TNFRSF1A, CXCL10 genes on multiple sclerosis: a two-sample Mendelian randomization approach. Front Bioeng Biotechnol 2020;8:397. https://doi.org/10.3389/fbioe.2020.00397.Search in Google Scholar PubMed PubMed Central
137. Semple, BD, Kossmann, T, Morganti-Kossmann, MC. Role of chemokines in CNS health and pathology: a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J Cerebr Blood Flow Metabol 2010;30:459–73. https://doi.org/10.1038/jcbfm.2009.240.Search in Google Scholar PubMed PubMed Central
138. Chu, HX, Arumugam, TV, Gelderblom, M, Magnus, T, Drummond, GR, Sobey, CG. Role of CCR2 in inflammatory conditions of the central nervous system. J Cerebr Blood Flow Metabol 2014;34:1425–9. https://doi.org/10.1038/jcbfm.2014.120.Search in Google Scholar PubMed PubMed Central
139. Zhou, YQ, Liu, DQ, Chen, SP, Sun, J, Zhou, XR, Xing, C, et al.. The role of CXCR3 in neurological diseases. Curr Neuropharmacol 2019;17:142–50. https://doi.org/10.2174/1570159x15666171109161140.Search in Google Scholar PubMed PubMed Central
140. Sorensen, TL, Sellebjerg, F. Selective suppression of chemokine receptor CXCR3 expression by interferon-beta1a in multiple sclerosis. Mult Scler 2002;8:104–7. https://doi.org/10.1191/1352458502ms781oa.Search in Google Scholar PubMed
141. Krumbholz, M, Theil, D, Steinmeyer, F, Cepok, S, Hemmer, B, Hofbauer, M, et al.. CCL19 is constitutively expressed in the CNS, up-regulated in neuroinflammation, active and also inactive multiple sclerosis lesions. J Neuroimmunol 2007;190:72–9. https://doi.org/10.1016/j.jneuroim.2007.07.024.Search in Google Scholar PubMed
142. Cui, LY, Chu, SF, Chen, NH. The role of chemokines and chemokine receptors in multiple sclerosis. Int Immunopharm 2020;83:106314. https://doi.org/10.1016/j.intimp.2020.106314.Search in Google Scholar PubMed PubMed Central
143. Park, MH, Lee, YK, Lee, YH, Kim, YB, Yun, YW, Nam, SY, et al.. Chemokines released from astrocytes promote chemokine receptor 5-mediated neuronal cell differentiation. Exp Cell Res 2009;315:2715–26. https://doi.org/10.1016/j.yexcr.2009.06.017.Search in Google Scholar PubMed
144. Pappalardo, LW, Samad, OA, Liu, S, Zwinger, PJ, Black, JA, Waxman, SG. Nav1.5 in astrocytes plays a sex-specific role in clinical outcomes in a mouse model of multiple sclerosis. Glia 2018;66:2174–87. https://doi.org/10.1002/glia.23470.Search in Google Scholar PubMed
145. Chistyakov, DV, Azbukina, NV, Astakhova, AA, Goriainov, SV, Chistyakov, VV, Sergeeva, MG. Sex-mediated differences in LPS induced alterations of TNFalpha, IL-10 expression, and prostaglandin synthesis in primary astrocytes. Int J Mol Sci 2018;19:2793. https://doi.org/10.3390/ijms19092793.Search in Google Scholar PubMed PubMed Central
146. Qiu, D, Li, XN. Pioglitazone inhibits the secretion of proinflammatory cytokines and chemokines in astrocytes stimulated with lipopolysaccharide. Int J Clin Pharmacol Therapeut 2015;53:746–52. https://doi.org/10.5414/cp202339.Search in Google Scholar PubMed
147. Ponath, G, Park, C, Pitt, D. The role of astrocytes in multiple sclerosis. Front Immunol 2018;9:217. https://doi.org/10.3389/fimmu.2018.00217.Search in Google Scholar PubMed PubMed Central
148. Rothhammer, V, Kenison, JE, Li, Z, Tjon, E, Takenaka, MC, Chao, CC, et al.. Aryl hydrocarbon receptor activation in astrocytes by laquinimod ameliorates autoimmune inflammation in the CNS. Neurol Neuroimmunol Neuroinflamm 2021;8:e946. https://doi.org/10.1212/nxi.0000000000000946.Search in Google Scholar
149. Linnerbauer, M, Lößlein, L, Vandrey, O, Tsaktanis, T, Beer, A, Naumann, UJ, et al.. Intranasal delivery of a small-molecule ErbB inhibitor promotes recovery from acute and late-stage CNS inflammation. JCI Insight 2022;7:e154824. https://doi.org/10.1172/jci.insight.154824.Search in Google Scholar PubMed PubMed Central
150. Maier, LM, Anderson, DE, Severson, CA, Baecher-Allan, C, Healy, B, Liu, DV, et al.. Soluble IL-2RA levels in multiple sclerosis subjects and the effect of soluble IL-2RA on immune responses. J Immunol 2009;182:1541–7. https://doi.org/10.4049/jimmunol.182.3.1541.Search in Google Scholar PubMed PubMed Central
151. Comrie, WA, Lenardo, MJ. Molecular classification of primary immunodeficiencies of T lymphocytes. Adv Immunol 2018;138:99–193. https://doi.org/10.1016/bs.ai.2018.02.003.Search in Google Scholar PubMed PubMed Central
152. Kiss, MG, Mindur, JE, Yates, AG, Lee, D, Fullard, JF, Anzai, A, et al.. Interleukin-3 coordinates glial-peripheral immune crosstalk to incite multiple sclerosis. Immunity 2023;56:1502–14.e8. https://doi.org/10.1016/j.immuni.2023.04.013.Search in Google Scholar PubMed PubMed Central
153. Oghumu, S, Varikuti, S, Stock, JC, Volpedo, G, Saljoughian, N, Terrazas, CA, et al.. Cutting edge: CXCR3 escapes X chromosome inactivation in T cells during infection: potential implications for sex differences in immune responses. J Immunol 2019;203:789–94. https://doi.org/10.4049/jimmunol.1800931.Search in Google Scholar PubMed PubMed Central
154. Fox, R. X-linked genes exhibit skewed expression in Sjogren’s disease (SjD): a further step toward understanding the female predominance of autoimmune disease. J Mol Med 2022;100:1267–9. https://doi.org/10.1007/s00109-022-02223-1.Search in Google Scholar PubMed PubMed Central
155. Shaw, TM, Zhang, W, McCoy, SS, Pagenkopf, A, Carp, DM, Garg, S, et al.. X-linked genes exhibit miR6891-5p-regulated skewing in Sjogren’s syndrome. J Mol Med 2022;100:1253–65. https://doi.org/10.1007/s00109-022-02205-3.Search in Google Scholar PubMed PubMed Central
156. Koper, OM, Kamińska, J, Sawicki, K, Kemona, H. CXCL9, CXCL10, CXCL11, and their receptor (CXCR3) in neuroinflammation and neurodegeneration. Adv Clin Exp Med 2018;27:849–56. https://doi.org/10.17219/acem/68846.Search in Google Scholar PubMed
157. Buhelt, S, Søndergaard, HB, Oturai, A, Ullum, H, von Essen, MR, Sellebjerg, F. Relationship between multiple sclerosis-associated IL2RA risk allele variants and circulating T cell phenotypes in healthy genotype-selected controls. Cells 2019;8. 31242590.10.3390/cells8060634Search in Google Scholar PubMed PubMed Central
158. Qu, H-Q, Bradfield, JP, Bélisle, A, Grant, SFA, Hakonarson, H, Polychronakos, C, Type I Diabetes Genetics Consortium. The type I diabetes association of the IL2RA locus. Genes Immun 2009;10(Suppl 1):S42–8. 19956099.10.1038/gene.2009.90Search in Google Scholar PubMed PubMed Central
159. van Steenbergen, HW, van Nies, JAB, Ruyssen-Witrand, A, Huizinga, TWJ, Cantagrel, Al, Berenbaum, F, et al.. IL2RA is associated with persistence of rheumatoid arthritis. Arthritis Res Ther 2015;17:244. 26350950.10.1186/s13075-015-0739-6Search in Google Scholar PubMed PubMed Central
160. Durmanova, V, Parnicka, Z, Javor, J, Minarik, G, Vrazda, L, Vaseckova, B, et al.. A novel association of polymorphism in the ITGA4 gene encoding the VLA-4 α4 subunit with increased risk of Alzheimer’s disease. Mediators Inflamm 2018;2018:7623823. 29769839.10.1155/2018/7623823Search in Google Scholar PubMed PubMed Central
161. Koretz, K, Schlag, P, Boumsell, L, Möller, P. Expression of VLA-alpha 2, VLA-alpha 6, and VLA-beta 1 chains in normal mucosa and adenomas of the colon, and in colon carcinomas and their liver metastases. Am J Pathol 1991;138:741–50. 2000944.Search in Google Scholar
162. Kirman, I, Nielsen, OH. LFA-1 subunit expression in ulcerative colitis patients. Dig Dis Sci 1996;41:670–6. 8674386.10.1007/BF02213121Search in Google Scholar PubMed
163. Supanc, V, Biloglav, Z, Kes, VB, Demarin, V. Role of cell adhesion molecules in acute ischemic stroke. Ann Saudi Med 2011;31:365–70. 21808112.10.4103/0256-4947.83217Search in Google Scholar PubMed PubMed Central
164. Becker, KG, Mattson, DH, Powers, JM, Gado, AM, Biddison, WE. Analysis of a sequenced cDNA library from multiple sclerosis lesions. J Neuroimmunol 1997;77:27–38. 9209265.10.1016/S0165-5728(97)00045-3Search in Google Scholar PubMed
165. Zhang, PJ, Barcos, M, Stewart, CC, Block, AW, Sait, S, Brooks, JJ. Immunoreactivity of MIC2 (CD99) in acute myelogenous leukemia and related diseases. Mod Pathol 2000;13:452–8. 10786814.10.1038/modpathol.3880077Search in Google Scholar PubMed
166. Tailor, A, Granger, D. Hypercholesterolemia promotes P-selectin-dependent platelet-endothelial cell adhesion in postcapillary venules. Arterioscler Thromb Vasc Biol 2003;23:675–80. 12615684.10.1161/01.ATV.0000056742.97580.79Search in Google Scholar PubMed
167. Sanada, H, Midorikawa, S, Yatabe, J, Yatabe, MS, Katoh, T, Baba, T, et al.. Elevation of serum soluble E- and P-selectin in patients with hypertension is reversed by benidipine, a long-acting calcium channel blocker. Hypertens Res 2005;28:871–8. 16555575.10.1291/hypres.28.871Search in Google Scholar PubMed
168. Hancock, WW, Lu, B, Gao, W, Csizmadia, V, Faia, K, King, JA, et al.. Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J Exp Med 2000;192:1515–20. 11085753.10.1084/jem.192.10.1515Search in Google Scholar PubMed PubMed Central
169. Lacotte, S, Brun, S, Muller, S, Dumortier, H. CXCR3, inflammation, and autoimmune diseases. Ann N Y Acad Sci 2009;1173:310–7. 19758167.10.1111/j.1749-6632.2009.04813.xSearch in Google Scholar PubMed
170. O’Byrne, PM, Metev, H, Puu, M, Richter, K, Keen, C, Uddin, M, et al.. Efficacy and safety of a CXCR2 antagonist, AZD5069, in patients with uncontrolled persistent asthma: a randomised, double-blind, placebo-controlled trial. Lancet Respir Med 2016;4:797–806. 27574788.10.1016/S2213-2600(16)30227-2Search in Google Scholar PubMed
171. Cheng, Y, Mo, F, Li, Q, Han, X, Shi, H, Chen, S, et al.. Targeting CXCR2 inhibits the progression of lung cancer and promotes therapeutic effect of cisplatin. Mol Cancer 2021;20:62. 33814009.10.1186/s12943-021-01355-1Search in Google Scholar PubMed PubMed Central
172. Ma, B, Miao, W, Xiao, J, Chen, X, Xu, J, Li, Y. The role of FOXP3 on tumor metastasis and Its interaction with traditional Chinese medicine. Molecules 2022;27:6706. 36235242.10.3390/molecules27196706Search in Google Scholar PubMed PubMed Central
173. Verma, ND, Lam, AD, Chiu, C, Tran, GT, Hall, BM, Hodgkinson, SJ. Multiple sclerosis patients have reduced resting and increased activated CD4+CD25+FOXP3+T regulatory cells. Sci Rep 2021;11:10476. 34006899.10.1038/s41598-021-88448-5Search in Google Scholar PubMed PubMed Central
174. van der Vliet, HJJ, Nieuwenhuis, EE. IPEX as a result of mutations in FOXP3. Clin Dev Immunol 2007;2007:89017. 18317533.10.1155/2007/89017Search in Google Scholar PubMed PubMed Central
175. Compton, AG, Albrecht, DE, Seto, JT, Cooper, ST, Ilkovski, B, Jones, KJ, et al.. Mutations in contactin-1, a neural adhesion and neuromuscular junction protein, cause a familial form of lethal congenital myopathy. Am J Hum Genet 2008;83:714–24. 19026398.10.1016/j.ajhg.2008.10.022Search in Google Scholar PubMed PubMed Central
176. Neřoldová, M, Ciara, E, Slatinská, J, Fraňková, S, Lišková, P, Kotalová, R, et al.. Exome sequencing reveals IFT172 variants in patients with non-syndromic cholestatic liver disease. PLoS One 2023;18:e0288907. 37471416.10.1371/journal.pone.0288907Search in Google Scholar PubMed PubMed Central
177. Youness, A, Miquel, CH, Guery, JC. Escape from X chromosome inactivation and the female predominance in autoimmune diseases. Int J Mol Sci 2021;22:1114. https://doi.org/10.3390/ijms22031114.Search in Google Scholar PubMed PubMed Central
178. Fetter, T, de Graaf, DM, Claus, I, Wenzel, J. Aberrant inflammasome activation as a driving force of human autoimmune skin disease. Front Immunol 2023;14:1190388. https://doi.org/10.3389/fimmu.2023.1190388.Search in Google Scholar PubMed PubMed Central
179. Cao, RR, He, P, Lei, SF. Novel microbiota-related gene set enrichment analysis identified osteoporosis associated gut microbiota from autoimmune diseases. J Bone Miner Metabol 2021;39:984–96. https://doi.org/10.1007/s00774-021-01247-w.Search in Google Scholar PubMed
180. Mougeot, JL, Noll, BD, Bahrani Mougeot, FK. Sjogren’s syndrome X-chromosome dose effect: an epigenetic perspective. Oral Dis 2019;25:372–84. https://doi.org/10.1111/odi.12825.Search in Google Scholar PubMed
181. Shin, HR, Cho, WK, Baek, IC, Lee, NY, Lee, YJ, Kim, SK, et al.. Polymorphisms of IRAK1 gene on X chromosome is associated with Hashimoto thyroiditis in Korean children. Endocrinology 2020;161:bqaa088. https://doi.org/10.1210/endocr/bqaa088.Search in Google Scholar PubMed
182. Ozcelik, T. X chromosome inactivation and female predisposition to autoimmunity. Clin Rev Allergy Immunol 2008;34:348–51. https://doi.org/10.1007/s12016-007-8051-0.Search in Google Scholar PubMed
183. Navarro-Cobos, MJ, Balaton, BP, Brown, CJ. Genes that escape from X-chromosome inactivation: potential contributors to Klinefelter syndrome. Am J Med Genet C Semin Med Genet 2020;184:226–38. https://doi.org/10.1002/ajmg.c.31800.Search in Google Scholar PubMed PubMed Central
184. Berletch, JB, Yang, F, Xu, J, Carrel, L, Disteche, CM. Genes that escape from X inactivation. Hum Genet 2011;130:237–45. https://doi.org/10.1007/s00439-011-1011-z.Search in Google Scholar PubMed PubMed Central
185. Tukiainen, T, Villani, AC, Yen, A, Rivas, MA, Marshall, JL, Satija, R, et al.. Landscape of X chromosome inactivation across human tissues. Nature 2017;550:244–8. https://doi.org/10.1038/nature24265.Search in Google Scholar PubMed PubMed Central
© 2024 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Frontmatter
- Review Article
- X-chromosome linked genes associated with myeloid cell CNS trafficking contributes to female–male differences in the disease outcome for neuroinflammatory diseases
- Short Review Article
- Something to talk about; crosstalk disruption at the neurovascular unit during HIV infection of the CNS
- Research Articles
- Sera from people with HIV and depression induce commensurate metabolic alterations in astrocytes: toward precision diagnoses and therapies
- Neuroprotective mushrooms
- HIV-1 and methamphetamine co-treatment in primary human astrocytes: TAARgeting ER/UPR dysfunction
- Viral-mediated inflammation by Poly I:C induces the chemokine CCL5 in NK cells and its receptors CCR1 and CCR5 in microglia in the neonatal rat cerebellum
Articles in the same Issue
- Frontmatter
- Review Article
- X-chromosome linked genes associated with myeloid cell CNS trafficking contributes to female–male differences in the disease outcome for neuroinflammatory diseases
- Short Review Article
- Something to talk about; crosstalk disruption at the neurovascular unit during HIV infection of the CNS
- Research Articles
- Sera from people with HIV and depression induce commensurate metabolic alterations in astrocytes: toward precision diagnoses and therapies
- Neuroprotective mushrooms
- HIV-1 and methamphetamine co-treatment in primary human astrocytes: TAARgeting ER/UPR dysfunction
- Viral-mediated inflammation by Poly I:C induces the chemokine CCL5 in NK cells and its receptors CCR1 and CCR5 in microglia in the neonatal rat cerebellum