Recent advances in understanding the relationship between lipid metabolism and immune escape in the tumor microenvironment of gastric cancer
-
 ,
,
 and
and

Abstract
Gastric cancer (GC), a leading global malignancy, poses a significant threat to human health. Despite substantial therapeutic advances, drug resistance remains a major challenge for many patients. Aberrant lipid metabolism represents a hallmark in tumors; it critically contributes to GC pathogenesis. This review explores key enzymes and pathways driving lipid metabolic reprogramming within the GC immune microenvironment, which facilitates tumor immune evasion and chemotherapy resistance. This review also addresses the challenges posed by lipid metabolism reprogramming in clinical treatment, explores therapeutic perspectives and novel directions, and discusses metabolism-related mechanisms.
Background
As the leading cause of cancer-related death globally [1], gastric cancer (GC) continues to pose a significant challenge. Despite recent improvements in clinical management, patients still encounter recurrence and chemotherapy resistance, leading to poor prognosis and a major public health burden [2] (Figure 1).

Graphic abstract.
Immune checkpoint therapies, among other tumor immunotherapies, have ushered in a new era of cancer treatment. Immune checkpoint therapies target T-cell regulatory pathways to enhance antitumor immune responses. Recently, immune checkpoint inhibitors (ICIs) have become first-line treatments for various malignant tumors and have been added to surgery, chemotherapy, radiotherapy, and targeted therapies, representing a pillar of antitumor treatment [3]. Although the value of immunotherapy is rapidly recognized in other cancers, the success of ICIs in treating gastric and gastroesophageal adenocarcinomas has been unsatisfactory [4]. The number of patients who benefit from second-line, third-line, and monotherapy is also limited [5].
The tumor immune microenvironment (TIME) encompasses the distinct anatomical region where malignant cells engage in dynamic interplay with resident immune populations. This specialized niche profoundly influences critical oncological processes, including tumorigenesis, invasion, metastatic dissemination, and therapeutic efficacy. Characterized by its complex composition, the TIME integrates tumor cells, diverse immune cell subsets, the extracellular matrix, and signaling molecules such as cytokines and chemokines. Consequently, the bidirectional communication between malignant cells and immune effectors constitutes a primary mechanism underpinning tumor immune tolerance and the capacity for immune evasion [6]. Notably, multiple factors, such as metabolic reprogramming, influence the functions of tumor-infiltrating immune cells [7].
Abnormal lipid accumulation and metabolic reprogramming in the tumor microenvironment (TME) profoundly impact immune cell function and represent a crucial mechanism of tumor immune escape. Aberrant lipid accumulation suppresses T cell function and induces exhaustion through mitochondrial dysfunction [8], endoplasmic reticulum stress [9], epigenetic reprogramming, and inhibition of signaling pathways such as the Sterol regulatory element-binding protein (SREBP)2–cholesterol axis [10]; lipid droplet overload causes lipid peroxidation in tumor-associated macrophages (TAMs), promoting the pro-tumor M2 phenotype and enhancing secretion of immunosuppressive factors [11]. Lipid metabolites act as signaling molecules regulating the differentiation, activation, and expression of immunosuppressive genes in myeloid-derived suppressor cells (MDSCs), with fatty acid oxidation (FAO) serving as a major source of reactive oxygen species (ROS) in MDSCs – where ROS acts as a core weapon to suppress T cell function [12]. TME signals (e.g., Prostaglandin E2[PGE2]) inhibit FAO in dendritic cells (DCs) by downregulating peroxisome proliferator-activated receptor (PPAR)γ/carnitine palmitoyl transferase 1A (CPT1A) while potentially aberrantly activating lipogenesis, thereby blocking DCs from acquiring a fully mature, immunostimulatory phenotype [13].
As a fundamental physiological process, lipid metabolism maintains nutrient equilibrium, hormonal balance, and systemic homeostasis. It drives the synthesis of diverse structural and functional lipids, such as phospholipids, glycolipids, sphingolipids, cholesterol, and prostaglandins. This process is essential for cellular activities and energy regulation. Crucially, dysregulated lipid metabolism constitutes a hallmark feature of tumor cells, with its reprogramming serving as a key driver in GC progression. Studies indicate that lipid metabolism not only provides energy and building blocks for the construction of biomembrane in tumor cells but also plays a crucial role in signal transduction and cell-to-cell communication. Importantly, abnormal activation of lipid metabolism is involved in the immune response within the TIME [14].
This review summarizes key enzymatic mechanisms regulating lipid metabolism in GC cells, alongside biomarkers for tracking metastasis progression, evaluating treatment response, and predicting patient outcomes. Emerging evidence demonstrates a critical interconnection between lipid metabolism and the GC TIME, underscoring its significance for future investigations.
Reprogramming of lipid metabolism in gastric cancer
Dysregulated lipid metabolism and altered signaling pathways represent defining characteristics of aberrant cellular proliferation and tumor advancement. Consequently, deciphering the crosstalk within the lipid-metabolic-signaling network is imperative. As the primary site for nutrient assimilation, the gastrointestinal tract fuels essential physiological processes. Key lipid production routes encompass de novo lipogenesis, cholesterol biosynthesis, ketone body metabolism, fatty acid (FA) β-oxidation, and sterol precursor utilization. Under nutrient scarcity, cancer cells undergo metabolic adaptations that disrupt immune surveillance, fostering an immunosuppressive milieu that crucially facilitates GC progression.
Lipid metabolism is a complex and highly regulated process that includes the synthesis, degradation, and transport of lipids. This process is crucial for maintaining cellular function and energy homeostasis. Key lipid metabolism steps encompass fatty acid synthesis (FAS) from acetyl-CoA via de novo lipogenesis. This process primarily occurs in the cytoplasm and is regulated by enzymes including fatty acid synthase (FASN) and acetyl-CoA carboxylase (ACC) [15]. Beta-oxidation of FAs in mitochondria and peroxisomes produces acetyl-CoA, which enters the citric acid cycle to generate adenosine triphosphate (ATP) as an energy source. Additionally, lipids are transported throughout the body via lipoproteins, and their levels are tightly controlled by various lipases and transfer proteins. Dysregulated lipid metabolism could contribute to cancer development and progression [16]. FA metabolism is a major component of lipid metabolism, with some key enzymes becoming hotspots of research. Within the GC microenvironment, lipid metabolism entails aberrant processes such as abnormal FAS, lipid uptake, and FAO; key enzymes in these pathways play critical roles (Figure 2).

Gastrointestinal energy supply drives cancer cell metabolic reprogramming which interacts with lipid and signaling pathways to induce immune suppression, ultimately promoting gastric cancer progression.
Landscape of lipid metabolism-immune microenvironment interactions in molecular subtypes of gastric cancer
GC, as a highly heterogeneous malignant tumor, has its molecular subtypes profoundly influencing metabolic reprogramming and immune response characteristics within the TME. In recent years, molecular subtyping studies based on multi-omics analyses have revealed complex and intricate interactions between lipid metabolic pathways and immune infiltration features in GC. According to analyses of large-scale databases such as The Cancer Genome Atlas (TCGA), GC can be primarily classified into four molecular subtypes: Epstein-Barr virus (EBV)-positive, microsatellite instability (MSI), genomically stable (GS), and chromosomally unstable (CIN). Each subtype exhibits distinct lipid metabolic signatures and immune microenvironments.
EBV-positive GC refers to tumors where all cancer cells are infected with EBV, characterized by significant activation of the cholesterol biosynthesis pathway alongside intense immune cell infiltration. Overexpression of the cholesterol efflux protein ATP-binding cassette transporter A1 (ABCA1) further reduces MHC-I complex stability, impairing CD8+ T cell recognition [17]. Although intracellular cholesterol accumulation in tumor cells may promote immune synapse formation, EBV simultaneously activates the signal transducer and activator of transcription 3 (STAT3) pathway via latent membrane protein 1 (LMP1), upregulating programmed death ligand 1 (PD-L1) expression and counteracting T cell cytotoxicity [18]. Statins (e.g., simvastatin) restore T cell activity by blocking hydroxymethyl-glutamyl coenzyme A reductase (HMGCR) to reduce intratumoral cholesterol levels [19].
MSI-type GC, a subtype with unique molecular, immune, and clinical features, primarily demonstrates excessive FA accumulation coupled with impaired phospholipid remodeling [20]. This metabolic profile interacts with high tumor neoantigen burden to suppress T cell activity, creating a highly immunogenic microenvironment.
GS-type GC is defined in the TCGA classification by low mutational burden, rare copy number variations, diffuse histology, and frequent CDH1 mutations. Its metabolic reprogramming and immune microenvironment interaction exhibit a unique pattern: Stearoyl-CoA desaturase (SCD) is specifically overexpressed in the GS subtype, driving the conversion of saturated FAs to monounsaturated FAs (e.g., oleic acid) to enhance membrane fluidity and suppress endoplasmic reticulum stress, thereby supporting tumor metastasis. Downregulated ABCA1 expression causes intracellular cholesterol accumulation, activating the liver X receptor (LXR)/RXR inflammatory pathway and further inhibiting T cell function [21].
CIN-type GC exhibits widespread chromosomal segment amplifications or deletions at the whole-genome level [22], accounting for 50 %–70 % of advanced GCs [23]. Its lipid metabolic features include disrupted sphingolipid metabolism [24], altered phospholipid spatial distribution gradients, and hyperactive FAS [25]. These changes directly drive tumor progression by modulating membrane signaling, inflammatory microenvironments, and genomic stability.
Abnormal fatty acid metabolism-related enzymes in gastric cancer
The increase in FA levels is reflected mainly in both increased uptake of exogenous FAs and increased synthesis of endogenous FAs. GC cells enhance lipid uptake from the extracellular microenvironment via the CD36 receptor, promoting FAO to generate ATP, a scavenger receptor that facilitates the high-affinity uptake of long-chain FAs. This process plays a crucial role in cancer cell progression and metastasis [26]. Additionally, FA uptake promotes GC cell metastatic potential through inducing O-GlcNAcylation and activating the NF-κB signaling pathway, which upregulates CD36 expression [27]. FA uptake is also mediated by FABPs, which bind long-chain FAs and increase their solubility. These proteins facilitate lipid metabolism by transferring FAs to cellular or mitochondrial membranes. The expression levels of FABP1, FABP4, and FABP5 are significantly elevated in GC cells, regulating cellular lipid uptake and apoptosis [28]. Experimental evidence demonstrates that the pro-tumorigenic activities of FABP5 and PPARδ are mediated through enhanced EGFR signaling [29]. Suppression of the phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway diminishes the pro-proliferative effects of FABP5 in the SGC-7901 GC cell line [30].
Endogenous FAS is regulated by key enzymes: primarily SREBP1, ATP citrate kinase (ACLY), acetyl-CoA synthases (ACSs), ACC, FASN, and SCD1. In human GC tissues, SREBP-1c activation upregulates FAS-related genes (e.g., SCD1, FASN). Furthermore, SREBP-1c depletion suppresses GC cell migration and invasion [31]. Among these genes, high SCD1 expression is associated with poor prognosis in GC, and some researchers [32], 33] have demonstrated in mouse experiments that SCD1 increases the proliferation and migration capacity of GC cells and has an anti-iron death effect that promotes tumor formation. The progression of FASN and GC is closely related to its associated immunological effects. Zhou et al. [34] demonstrated elevated FASN expression in GC tissues. This overexpression correlates with diminished survival rates and significantly promotes tumor development. Furthermore, FASN levels exhibit a strong association with the infiltration of diverse immune cell populations, including CD8+ T cells, CD4+ T cells, neutrophils, macrophages, and DCs.
Within the cytosol, ACLY catalyzes the conversion of citrate into acetyl coenzyme A and oxaloacetate. Acetyl coenzyme A serves as the key precursor for FA biosynthesis. Zheng [35] performed weighted correlation network analysis (WGCNA) via TCGA and Genotyped Tissue Expression (GTEx) datasets, confirming that ACLY expression in GC tissues is upregulated. Notably, the downregulation of ACLY mRNA levels inhibited the development of GC cells. Acetyl coenzyme A can also be produced from FA catalyzed by ACSs in cells, and Acetyl-CoA synthetase short-chain family member 2 (ACSS2), a member of the ACS short-chain family, uses acetic acid as a substrate to produce acetyl coenzyme A. This process serves as an ACLY auxiliary pathway; thus, the inhibition of ACLY and ACSS2 represents a promising option for inhibiting GC progression and treating GC [36]. Acetyl-CoA synthetase short-chain family member 3 (ACSS3) expression is increased in GC cells; its knockdown suppresses GC cell growth and invasion [37]. However, a study revealed that ectopic expression of long-chain acyl coenzyme A synthetase 4 (ACSL4) inhibited cell growth, colony formation and cell migration, and Ye et al. [38] reported that ACSL4 has tumor suppressor properties and may represent a potential therapeutic target for GC. Therefore, different members of the ACS family do not have the same specific roles in lipid metabolism in GC cells. ACC is an important rate-limiting enzyme in FA metabolism and plays an important role in FA catabolism; it converts acetyl-coenzyme A to malonyl-coenzyme A during de novo production. Moreover, ACC inhibitions improve the antitumor immunity of GC [39].
FAO is also a feature of lipid metabolism in GC cells. Lee et al. [40] reported elevated levels of FAO alongside upregulated expression of aldehyde dehydrogenase Aldehyde dehydrogenase 3 family member A1 (ALDH3A1) in GC. Metabolic investigations indicated that lipid peroxidation, triggered by ROS, resulted in the endogenous generation of 4-hydroxynonenal (4-HNE). Subsequently, ALDH3A1 metabolized 4-HNE, producing NADH that facilitated the conversion of this aldehyde into FAs, thereby promoting further FAO. Collectively, these findings establish ALDH3A1 as a critical enzymatic mediator sustaining FAO dependency within GC cells. Wang et al. [41] reported significant upregulation of CPT1A in both cellular and tissue samples derived from GC. Their findings indicate that CPT1A stimulates FAO within GC cells by elevating the NADP+/NADPH ratio. Critically, pharmacological inhibition of FAO abolished the pro-proliferative and pro-migratory effects mediated by CPT1A in these malignant cells. Regarding the natriuretic peptide receptor A (NPRA), which functions as a guanylate cyclase receptor, investigations in vivo and in vitro established its role in enhancing FAO and facilitating tumor metastasis. Mechanistically, NPRA upregulation sustains PPARα activity. This subsequently activates carnitine palmitoyl transferase 1B (CPT1B), driving FAO and ultimately promoting GC progression [42]. The increase in FAO also promotes GC cell metastasis. Gao et al. [43] performed RNAseq sequencing of the ovarian metastasis of GC. Comparative analysis revealed that CPT1C, a protein-coding gene for FAO rate-limiting enzyme, was significantly upregulated in the ovarian metastasis of primary gastric tumors. A cohort study demonstrated that CPT1C overexpression promoted GC cell migration and invasion by increasing the FAO rate. Wang et al. [44] demonstrated that circ_0024107-driven FAO metabolic reprogramming in gastric cancer tissue-derived mesenchymal stem cells (GC-MSCs) critically contributes to tumor progression. FA metabolic enzymes not only sustain cancer cell metabolic demands but also foster an immunosuppressive microenvironment by altering immune cell function, thereby supporting GC advancement.
Acetyl-CoA acetyltransferase 1 (ACAT1), a mitochondrial enzyme primarily involved in ketone body, isoleucine, and FA biosynthesis/degradation, has been shown to suppress epithelial-mesenchymal transition (EMT) in GC cells through PPAR signaling, propanoate metabolism, and p53 pathways when overexpressed [45]. Fu et al. discovered that NPR1 promotes GC lymph node metastasis by activating lipid droplet lipolysis and enhancing mitochondrial oxidative phosphorylation (OXPHOS), with targeted inhibition of NPR1 effectively suppressing GC metastasis [46] (For details, see Table 1).
Gastric cancer and abnormal fatty acid metabolis.
| Key enzyme | Gastric cancer progression stages | Specific mechanisms |
|---|---|---|
| SREBP-1c | Endogenous fatty acid synthesis and cancer cell progression | SREBP-1c is activated in human gastric cancer tissues, promoting the expression of SCD1 and FASN, affecting the resistance to ferroptosis, and enhancing immune cell infiltration [31]. |
| SCD1 | Poor prognosis | High expression is associated with poor prognosis in gastric cancer, increasing the proliferation and migration ability of GC cells [32], 33]. |
| ACLY | Promotion of growth | Citrate is converted into acetyl-CoA and oxaloacetate, and upregulation of ACLY expression is associated with gastric cancer tissues; inhibiting ACLY mRNA levels can suppress the development of GC cells [35]. |
| ACSL4 | Inhibition of occurrence and growth; potential therapeutic targets | ACSL4 can inhibit cell growth, colony formation, and cell migration through the FAK and P21 signaling pathways, and its ectopic expression possesses tumor-suppressing characteristics [38]. |
| ACSS2 | Fatty acid synthesis | Using acetate as a substrate to produce acetyl-CoA, as an auxiliary pathway for ACLY, inhibiting ACLY and ACSS2 may be a promising choice for the treatment of gastric cancer [36]. |
| ACSS3 | Cancer cell growth and invasion | Expression is increased, and knockdown of ACSS3 inhibits the growth and invasion of GC cells [37]. |
| ALDH3A1 | Fatty acid oxidation and gastric cancer dependence | Activity is increased, promoting fatty acid oxidation, and gastric cancer depends on ALDH3A1 for fatty acid oxidation [40]. |
| ACC | Fatty acid metabolism and anti-tumor immunity | Inhibition of ACC can improve anti-tumor immunity in GC [39]. |
| FABPs | Cancer cell lipid metabolism and apoptosis | Binding to long-chain FAs, increasing the solubility of fatty acids, promoting the transfer of FAs to cell or mitochondrial membranes, affecting cellular lipid uptake and apoptosis [28]. |
| FASN | Carcinogenesis and enhancement of immune infiltration levels | Overexpression is associated with lower survival outcomes, makes a significant contribution to GC carcinogenesis, and is related to the infiltration levels of various immune cells [34]. |
| NPRA | Fatty acid oxidation and tumor cell metastasis | Promotes fatty acid oxidation and tumor cell metastasis, protects PPARα, activates CPT1B, leading to fatty acid oxidation and promoting the development of GC [42]. |
| CPT1A | Fatty acid oxidation and GC cell proliferation, migration | Protein expression is upregulated, activating FAO, affecting the proliferation and migration of GC cells [41]. |
| CPT1C | Gastric cancer cell metastasis and invasion | Overexpression promotes the migration and invasion of GC cells by upregulating the rate of fatty acid oxidation [43]. |
| CD36 | Cancer cell progression and metastasis | Promotes the high-affinity uptake of long-chain fatty acids, generates ATP, and is involved in the progression and metastasis of cancer cells [26]. |
| Upregulates CD36 levels through O-GlcNAcylation glycosylation and activation of the NF-κB signaling pathway, enhancing the metastatic potential of gastric cancer cells [27]. |
-
SREBP, sterol regulatory element-binding protein; SCD, stearoyl-CoA desaturase; FASN, fatty acid synthase; GC, Gastric cancer; ACLY, ATP, citrate kinase; ACSL4, acyl coenzyme A synthetase 4; ACSS2, Acetyl-CoA, synthetase short-chain family member 2; ACSS3, Acetyl-CoA, synthetase short-chain family member 3; ALDH3A1, aldehyde dehydrogenase 3 family member A1; ACC, acetyl-CoA carboxylase; FABP, fatty acid-binding protein; FA, fatty acids; NPRA, natriuretic peptide receptor A; PPAR, peroxisome proliferator-activated receptor; CPT1B, carnitine palmitoyl transferase 1B; FAO, fatty acid oxidation; CPT1A, carnitine palmitoyl transferase 1A; ATP, adenosine triphosphate.
Key mechanisms and therapeutic targets of aberrant cholesterol metabolism driving gastric cancer progression
Beyond abnormal FA levels and oxidation, GC microenvironments exhibit enriched cholesterol/cholesteryl esters. Dysregulated cholesterol synthesis and transport correlate closely with tumorigenesis. The production of cholesterol, which is derived from acetyl-CoA through the mevalonate biosynthetic pathway, is tightly regulated by HMGCR. As the rate-controlling enzyme in this metabolic cascade, HMGCR determines the overall flux of cholesterol synthesis. This enzyme activates Hedgehog/Gli1 signaling, promoting Gli1 target gene expression to drive GC cell growth and migration [47]. Squalene epoxygenase (SQLE), a downstream enzyme that catalyzes the first oxidative step in sterol biosynthesis, is highly expressed in GC tissues [48] and plays an important role in tumorigenesis, progression and prognosis.
Cholesterol transport proteins help maintain the phospholipid balance after cholesterol synthesis. Cholesterol in cells can be exported by the ATP-binding cassette transporters ABCA1 and ATP-binding cassette transporter G1 (ABCG1) or converted by sterol O-acyltransferase 1 (SOAT1). SOAT1 catalyzes the conversion of cholesterol and long-chain FAs into cholesterol esters, crucial for maintaining cellular cholesterol homeostasis. Studies indicate that its high expression elevates lipid availability, enhancing tumor invasiveness [49]. Zhu et al. [50] reported that SOAT1 overexpression in GC tissues modulates genes governing cholesterol metabolism (including SREBP1 and SREBP2) and elevates vascular endothelial growth factor-C (VEGF-C), thereby inducing lymphangiogenesis.
The low-density lipoprotein receptor (LDLR) is a major pathway for extracellular cholesterol uptake. Cancer cells exploit enhanced LDLR-mediated exogenous cholesterol uptake to gain proliferative advantage, establishing LDLR as a tumor prognostic biomarker [51], 52]. A significant inverse relationship exists between low-density lipoprotein receptor-related protein 1B (LRP1B) expression in the cytoplasm and disease aggressiveness in diffuse GC, with higher immunoreactivity levels predicting earlier tumor stage and better prognosis [53]. In clinical populations exhibiting metabolic lipid abnormalities, circulating oxLDL concentrations not only correlate with oncogenesis but also show prognostic value for predicting lymph node metastasis in GC cases [54]. Ma et al. [55] reported that oxLDL activates the NF-κB signaling pathway via LOX-1, which promotes the upregulation of VEGF-C expression, ultimately leading to lymphatic metastasis of GC.
NR1H3 and NR1H2 encode LXRα and LXRβ nuclear receptors, and the LXR is a member of the nuclear receptor superfamily of DNA-binding transcription factors that act as sensors of cholesterol homeostasis [56]. Physiologically, elevated intracellular cholesterol triggers oxysterol synthesis and LXR pathway activation, coordinating cholesterol elimination through efflux stimulation alongside influx and synthesis suppression. During the proliferation of normal and cancerous cells, a net uncoupling occurs between the increase in intracellular cholesterol and LXR activation due to the decrease in the intracellular oxysterol concentration, which results in cholesterol efflux and the transcription of genes related to FA biosynthesis to increase intracellular cholesterol levels [57]. Tang et al. conducted transcriptomic profiling of cholesterol metabolism genes (CMGs) using TCGA and GEO datasets, identifying NR1H3 as a prognostic biomarker for GC outcomes and response to PD-1 inhibitor-based chemotherapy. Immunohistochemistry demonstrated reduced LXRβ expression in GC versus adjacent normal tissues. Functionally, LXRβ agonists suppressed GC proliferation via Wnt pathway inhibition mediated by LXRβ relocalization [58], 59]. Similarly, Liu et al. [60] found that LXRβ expression was low in GC; furthermore, LXRβ activation may inhibit GC cell proliferation, promote apoptosis, and increase chemosensitivity by upregulating Activating transcription factor 4 (ATF4) expression. A striking dichotomy exists in cholesterol acquisition mechanisms between primary tumors and their metastatic derivatives. Contrary to expectations, pharmacological intervention in advanced GC leads to significant downregulation of LDLR expression [61]. This is a challenge in the treatment of GC. Therefore, the abnormal activation of cholesterol metabolism may affect GC progression.
The Helicobacter pylori virulence factor CagA activates the Hippo pathway to drive YAP1 nuclear translocation, upregulating SQLE – the rate-limiting enzyme in cholesterol biosynthesis – which elevates palmitoyl-CoA levels to enhance PD-L1 palmitoylation. This modification stabilizes PD-L1 by reducing its ubiquitination and degradation, enabling tumor immune escape [62]. Other researchers developed oxygen-reduction-responsive supramolecular polyrotaxane nanomedicine (RDPNs@diABZI) co-loaded with STING agonist diABZI and cholesterol-depleting methyl-β-cyclodextrin (MeβCD). Upon TME-specific release, diABZI activates STING in antigen-presenting cells to initiate T-cell immunity while MeβCD depletes membrane cholesterol, stiffening the cancer cell membrane cortex to amplify cytotoxic T lymphocyte (CTL) immune synapse forces. This dual mechanism synergistically overcomes PD-L1-mediated immunosuppression by simultaneously activating antitumor immunity and enhancing CTL mechanical killing efficacy [63] (For details, see Table 2).
Gastric cancer and abnormal cholesterol metabolism.
| Key enzyme | Gastric cancer progression stages | Specific mechanisms |
|---|---|---|
| HMGCR | Tumor initiation, growth, and migration | Activating the Hedgehog/Gli1 signaling pathway, promoting the expression of Gli1 target genes, and enhancing the growth and migration of gastric cancer cells [47]. |
| SQLE | Tumor initiation, progression, and prognosis | Highly expressed in gastric cancer tissues, it catalyzes the first oxidation step in steroid biosynthesis and plays an important role in tumor initiation, progression, and prognosis [48]. |
| SOAT1 | Tumor invasiveness | Catalyzing the conversion of cholesterol and long-chain fatty acids into cholesterol esters, high expression increases lipid availability and promotes tumor invasiveness [49]. |
| ABCA1 and ABCG1 | Maintaining cellular cholesterol homeostasis | Exporting cholesterol from cells to maintain the balance of phospholipids after cholesterol synthesis [49]. |
| LDLR | Cancer cell proliferation | Enhancing LDLR-mediated exogenous cholesterol uptake, providing a proliferative advantage for cancer cells [51], 52]. |
| LRP1B | Associated with low clinical pathological stage and good prognosis | The immunoreactivity of cytoplasmic LDLR-related protein 1B is significantly associated with low clinical pathological stage and good prognosis in patients with diffuse gastric cancer [53]. |
| oxLDL | Predicting lymphatic metastasis | As a risk factor associated with tumor development in patients with abnormal lipid metabolism, predicting lymphatic metastasis of gastric cancer [54]. |
| LOX-1 | Promoting lymphatic metastasis | Activating the NF-κB signaling pathway, promoting the upregulation of VEGF-C expression, leading to lymphatic metastasis of gastric cancer [55]. |
| LXRα and LXRβ | Tumor cell proliferation, apoptosis, and chemosensitivity | Activating the LXR transcription network to drive cholesterol efflux and reduce cholesterol influx and synthesis [56]; low expression of LXRβ is associated with the inhibition of gastric cancer cell proliferation [60]. |
-
HMGCR, hydroxymethyl-glutamyl coenzyme A reductase; SQLE, squalene epoxygenase; SOAT1, sterol O-acyltransferase 1; ABCA1, ATP-binding cassette transporter A1; ABCG1, ATP-binding cassette transporter G1; LDLR, low-density lipoprotein receptor; LRP1B, low-density lipoprotein receptor-related protein 1B; VEGF-C, vascular endothelial growth factor-C; LXR, liver X receptor.
Coordinated sphingolipid-glycerophospholipid metabolic network drives immune tolerance in the gastric cancer microenvironment
Sphingolipids constitute one of the major classes of eukaryotic lipids. Complex sphingolipids include sphingomyelins and glycosphingolipids, the latter encompassing gangliosides. These serve as structural components of plasma membranes and regulators of cell-cell interactions and recognition [64]. Approximately 40 enzymes participate in their metabolic pathways [65]. Sphingolipids have emerged as critical regulators in cancer. Machine learning-based analysis of large GC transcriptomic datasets revealed that a subtype with high expression of sphingolipid metabolism genes – Sphingosine Kinase 1 (SPHK1), Sphingosine 1-phosphate receptor 1 (S1PR1), and N-Acylsphingosine Amidohydrolase 1 (ASAH1) – exhibits a pronounced immune-desert phenotype: reduced CD8+ T cell and M1 macrophage infiltration alongside increased forkhead transcription factor 3 (FOXP3)+ regulatory T cells (Tregs) and M2 macrophages, indicating that S1P-Ceramide imbalance promotes immune escape [66].
Separate studies demonstrate that sphingosine-1-phosphate (S1P) upregulates PD-L1 expression in GC cells via the S1PR1/STAT3 axis and induces M2 polarization of TAMs, thereby suppressing CD8+ T cell function. This effect is reversible by specific S1P synthesis inhibitors or S1PR1 antagonists [67]. Concurrently, SPHK1 upregulation promotes S1P accumulation, activating PI3K/Akt- mammalian target of rapamycin (mTOR) and STAT3 signaling to drive cyclin D1 and matrix metallopeptidase 9 (MMP-9) expression, enhancing proliferation and invasion. Collectively, sphingolipid metabolism critically contributes to GC progression through dual mechanisms: suppressing antitumor immunity via PD-L1 upregulation and M2 polarization, while directly promoting tumor proliferation/invasion via STAT3 and PI3K/Akt-mTOR activation. Targeting this pathway holds therapeutic promise.
Additionally, phospholipid metabolic reprogramming drives immune escape in GC. Glycerophospholipids are synthesized in the endoplasmic reticulum via the Kennedy pathway (CDP-choline/ethanolamine branches) and phosphatidic acid derivatization [68]. High-throughput lipidomics and computational biology identified phosphatidylethanolamine (PE)(36:3), PE(36:2), phosphatidylcholine (PC)(32:0), and sphingomyelin (SM)(d18:0/18:1(9Z)) as enriched in early-stage patients. Abnormal glycerophospholipid accumulation distinguishes chemotherapy-sensitive from resistant cohorts [69]. Lysophosphatidic acid (LPA) – a lipid endogenously synthesized from lysophosphatidylcholine (LPC) by autotaxin (ATX) – impairs T cell activation and cytotoxicity by driving systemic oxidative damage, disrupting perforin and cytokine release at immune synapses, and rewiring CD8+ T cell metabolism to promote tolerance [70]. Thus, dysregulated phospholipid metabolism is a key facilitator of immune evasion.
Lipid metabolism and immune evasion in the gastric cancer environment
The accumulation of and metabolic changes in lipids in the TME significantly affect immune cell function and fate. Tumor cells reprogram lipid metabolism to polarize macrophages toward immunosuppressive phenotypes, thereby shaping an immunosuppressive microenvironment [71]. On the other hand, lipid metabolism abnormalities, such as increased FAO and de novo lipogenesis, provide survival advantages for tumors, affect the antitumor capabilities of immune cells, and even cause immune cells to redifferentiate into protumor phenotypes [72]. Furthermore, the abnormal activation of lipid metabolism and signaling pathways is also considered an important factor in tumor progression and immune evasion. Exploring the effect of abnormal lipid metabolism on immune escape may also provide new directions and ideas for targeted tumor therapy (Figure 3).

During gastric cancer development, aberrant enzyme-driven regulation of lipid metabolism emerges as the core mechanism disrupting normal lipid homeostasis. The resulting metabolic dysregulation profoundly impacts tumor biological behavior, on one hand sustaining malignant proliferation by supplying tumor cells with energy and building blocks, while on the other hand potentiating tumor invasion and distant metastasis. (a) Lipid homeostasis in healthy cells involves balanced processes of de novo synthesis, transport, and degradation, ensuring energy production and membrane integrity. (b) Key enzymes tightly control lipid flux through post-translational modifications, substrate availability, or transcriptional regulation, maintaining metabolic flexibility in response to cellular demands. (c) Tumors exhibit aberrant lipid synthesis, enhanced exogenous uptake, and dependency on fatty acid oxidation to fuel ATP and NADPH production, creating a protumorigenic microenvironment resistant to metabolic stress. (d) Dysregulated lipid metabolism promotes cancer proliferation, invasion, and immune evasion, driving metastatic spread and therapeutic resistance.
Metabolic-immunological crosstalk in gastric cancer: lipid reprogramming drives adaptive immune evasion
Inhibition of T-cell activity and function
Prolonged antigen exposure coupled with an immunosuppressive milieu drives progressive functional exhaustion in tumor-infiltrating CD8+ T lymphocytes [73]. The dysfunctional state of CD8+ T cells is known as exhaustion, and depleted CD8+ T cells highly express suppressor receptors [9]. CD8+ effector T cells (Teffs) use aerobic glycolysis to maintain antitumor effector function [74]. However, within GC’s immune microenvironment, tumor-infiltrating lymphocytes (TILs) face nutrient competition from cancer cells. During conditions of glucose limitation, these cells undergo metabolic reprogramming characterized by enhanced FA absorption and β-oxidation to maintain their functional activity [8]. That results in increased concentrations of free fatty acids (FFAs) in the circulation or within the TME. Moreover, an increase in FFAs correlates with decreased activity of CD8+ CTLs [72]. Moreover, cholesterol accumulation within tumor-infiltrating CD8+ T cells and the tumor milieu correlates with immune checkpoint upregulation and functional exhaustion. Reducing cholesterol in these cellular compartments and/or inhibiting X-box binding protein 1 (XBP1) in CD8+ T lymphocytes may enhance anticancer efficacy of T-cell-based therapies [9]. During initial tumor development, IFN-γ stimulation induces cMyc-mediated metabolic alterations, resulting in enhanced glycolytic flux and suppression of FAO in malignant cells [75]. The elevated expression of cMyc-em confers upon malignant cells the capacity to suppress both cellular senescence and metabolic alterations induced by IFN-γ. This adaptive mechanism not only inhibits the recruitment of CD8+ TILs but also impairs their effector functions within the TME.
In lipid metabolic pathways, ACC occupies a dual regulatory position, serving both as the gatekeeper enzyme for de novo FA production and as a modulator of CPT1-mediated FA catabolism through β-oxidation [76]. In GC, ACC levels show significant inverse correlations with immune signaling. Specifically, its expression negatively associates with both CD8+ T-cell infiltration density and immune cytolytic activity in GC patients. These findings indicate that targeting ACC enhances antitumor immunity in GC [77].
Recent evidence suggests that both checkpoint blockade and inhibition may directly alter metabolism and change the metabolic profile of both T cells and cancer cells [78]. Emerging evidence indicates that PD-1/ligand binding redirects the metabolic phenotype of TILs, suppressing glycolysis while enhancing FAO. This reprogrammed energy metabolism thereby compromises antitumor responses, culminating in diminished cytokine secretion and Teff exhaustion [79].
In the immune microenvironment of GC, Tregs play important roles in immunosuppression, immune escape, and tolerance [80]. Tregs are fundamentally linked to metabolic reprogramming of lipid pathways, which critically maintains their cellular viability and immunosuppressive capacity [81]. FoxP3 is a member of the Forkhead transcriptional regulator family. As a definitive marker for Tregs, FoxP3 modulates immune-targeting gene expression. This functional property establishes its significance in tumor immunology. Elevated FoxP3 expression in Tregs enhances their FA uptake capacity, enabling preferential utilization of mitochondrial oxidative metabolism for lipid and pyruvate breakdown, whereas effector immune cells primarily rely on glycolysis for energy production [82]. This metabolic adaptation sustains Treg immunosuppressive function and drives tumor immune evasion by promoting macrophage M2 polarization and inhibiting CD8+ Teff activity [83]. Through functional inhibition of CTLs, FoxP3+ regulatory T cells compromise the host’s antitumor immune defenses, creating permissive conditions for tumor immune escape [84]. FABPs are a family of lipid chaperone proteins that facilitate FA uptake, transport and metabolic regulation. Among them, FABP5 is expressed in T cells. Studies have shown that in Tregs, inhibition of FABP5 function leads to mitochondrial abnormalities, decreased OXPHOS, and impaired lipid metabolism. Pharmacological blockade of FABP5 in Tregs initiates a cascade of molecular events involving mtDNA release and subsequent activation of the cGAS-STING pathway, culminating in type I interferon production. This process concomitantly enhances IL-10 secretion and amplifies the immunosuppressive capacity of Tregs [85]. This action is closely related to enhanced immunosuppression in the TME.
Fatty acid oxidation and T-cell depletion in the setting of gastric cancer
After the initial immune response, a small proportion of T cells persist as memory T cells (Trm), which are antigen-specific T cells involved in the antitumor immune response. Instead of using glucose, Trm cells rely on FAO for survival, and a lack of FAs leads to Trm cells death. In gastric adenocarcinoma, approximately 30 % of TILs are CD69+CD103+ Trm cells. PD-L1 targeting suppresses fatty acid-binding proteins (FABPs) 4 and 5 in tumor cells, impairing Trm cell survival in vitro and in vivo. Moreover, lipid uptake blockade induces apoptosis in Trm cells, effectively depleting this population within the TME [86].
Lin et al. [87] investigated whether lipid metabolism in cancer cells impacts lipid uptake and apoptosis in Trm cells. Studies revealed significantly elevated expression of FABP4 and FABP5 in gastric adenocarcinoma tissues compared to normal gastric mucosa [86]. Mechanistically, tumor cells induce apoptosis in tissue-resident Trm by depriving them of lipid uptake; additional evidence confirms this process activates the p38 kinase pathway via CD36-dependent lipid peroxidation, ultimately leading to CD8+ T cell death [88]. PPARs critically regulate lipid metabolism transcriptionally. Their three isoforms – PPARα, PPARγ, and PPARδ – perform distinct functions upon ligand activation. PPAR-mediated upregulation of peripheral apolipoprotein A-I/II (ApoA-I/II) indirectly affects T-cell cholesterol levels through high-density lipoprotein (HDL) levels and increases cholesterol efflux. This indirectly affects T-cell cholesterol levels, which influence cell survival and proliferation. In helper T (Th) cells, PPARγ activation suppresses proliferation, downregulates IL-2, and triggers apoptosis [89].
Activated T cells display augmented metabolic needs during proliferation, demanding intensified de novo FAS via glucose transformation into FAs [90]. Research indicates that while activated and memory T cells employ FAO as their primary metabolic pathway, SREBPs induce de novo FAS in activated T cells through mechanistic target of rapamycin complex 1 (mTORC1) [91]. FAS requires multiple enzyme-mediated steps to execute lipid synthesis. Certain key enzymes in this process modulate T-cell reattachment dynamics. For example, CD8+ T cells fail to expand during viral infection without SREBP signaling. SREBP in Teffs induces the expression of FASN, acetyl coenzyme A carboxylase and HMGCR [90]. Lim et al. [92] performed studies on TME-bearing mice and reported that inhibiting SREBP-dependent lipid synthesis and metabolic reprogramming in Tregs elicited potent antitumor responses while avoiding autoimmune dysregulation. Similarly, Wen et al. [93] demonstrated that inhibiting tumor growth required knockdown of SREBP1/2 target genes essential for lipid synthesis. In addition, depleting SREBPs in both cancer cells and Tregs conferred anticancer benefits; however, Treg-specific SREBP depletion promoted immune evasion. In summary, by modulating lipid metabolism and targeting specific metabolic pathways to enhance T-cell-mediated antitumor immune responses, new strategies for the treatment of GC may be developed.
Lipid metabolism in gastric cancer-associated B cells
Currently, research on tumor-associated B cells is limited compared with T cells, and B cell metabolism at all stages is understudied. TMEs, such as those with hypoxia and local deficiencies in glutamine, glucose, and tryptophan nutrients, may interfere with B-cell maturation, affecting B-cell differentiation and normal function [94]. A seminal study by Weisel et al. demonstrated that germinal center B (GCB) cells preferentially utilize FAO over glycolysis for energy. Isotope tracing revealed that while B cells in vivo exhibited increased FA uptake, GCB cells in vitro employed FAs, rather than glucose, to generate elevated acetyl-coenzyme A levels. Concurrently, RNA sequencing revealed significant downregulation in GCB cells of multiple glycolytic pathway genes, along with nuclear protein Hypoxia Inducible Factor 1 Subunit Alpha (HIF-1α), a key mediator of hypoxia-induced glycolysis [95]. HIFs in the TME regulate multiple cellular pathways, including cellular metabolism, to adapt to hypoxic stress [87]. HIF1α promotes glycolysis; enhances FA uptake, lipogenesis, and storage; while diminishing FAO within the cell. FAS is catalyzed by acetyl coenzyme A produced within the cytoplasm by mitochondrial processes, and occurs via ACLY. Conversely, inhibiting ACLY activity in B cells leads to impaired proliferation, defective endosomal expansion, and diminished CD138 and Blimp-1 expression. Thus, FAs present in the TME undergo uptake by cells expressing FA importers, such as B cells and metastatic cancer cells.
Other studies have shown that FAO provides essential energy requirements for B cells [95]. Zhou et al. [96] demonstrated that SCD1, the rate-limiting enzyme in FAS, catalyzes the conversion of SFAs to MUFAs including oleic acid and palmitoleic acid. In in vitro experiments, SCD1 activity was significantly increased during B-cell activation, and its products, oleic acid and palmitoleic acid, were enriched in activated cells. These factors support oxidative phosphorylation and mTOR activity and prevent excessive autophagy and endoplasmic reticulum stress to maintain the metabolic fitness of B cells. In addition, Rockett et al. [97] reported that SFA inhibits B-cell activation in vitro via lipid-mediated apoptosis, but MUFA (oleic acid) inhibits lipid-mediated apoptosis in specific cell types and that exposure of B cells to omega-3 PUFAs increases the production of key immunomodulatory cytokines (IL-10, TNF-α and IFN-γ). Thus, GC B cells contribute not only to tumor cell biosynthesis and energy supply via lipid metabolism but also participate in GC treatment and prognosis by influencing the TME and immune response (Figure 4).

The interplay between lipid metabolism and CD8+ T cell exhaustion/treg-mediated immunosuppression in the gastric cancer immune microenvironment. Treg, regulatory T cells.
Myeloid and innate immune cell dysfunction in gastric cancer: lipid reprogramming polarizes immunosuppressive landscapes
Lipid metabolism and myeloid-derived suppressor cells
MDSCs comprise a heterogeneous cell population encompassing both granulocytic (G-MDSCs/PMN-MDSCs) and monocytic myeloid-derived suppressor cells (M-MDSCs) [98]. These cells suppress adaptive antitumor immunity via multiple mechanisms, such as impairing the activation and function of CD4+ and CD8+ T cells, while also driving and recruiting regulatory T cells. They can also suppress innate immunity by polarizing macrophages toward a phenotype that promotes tumor growth and inhibits the cytotoxicity of natural killer (NK) cells [99]. Additionally, they can alter FAO, resulting in an increase in lipids and other metabolic products in the TME [100]. Altered lipid metabolism in the TME drives MDSC generation. Tumor-infiltrating MDSCs enhance FA uptake and oxidation, while intracellular lipid accumulation augments oxidative metabolism to potentiate their immunosuppressive function [101]. Al-Khami et al. [102] demonstrated that tumor-derived cytokines and granulocyte colony-stimulating factors utilize STAT3 and signal transducer and activator of transcription 5 (STAT5) signaling to enhance lipid uptake while augmenting the immunosuppressive properties of MDSCs. Therefore, by blocking STAT3 or STAT5 signaling, the oxidative metabolism of lipids is inhibited, preventing the immunosuppressive function of MDSCs, inducing T-cell-mediated antitumor activity, enhancing CD8+ T-cell function and delaying tumor cell growth.
Lipid metabolism and tumor-associated macrophages
TAMs, among the most common immune infiltrating cells in the TME, are classified into the antitumor M1 and protumor M2 subtypes, and these cells play key roles in tumor development. M1 TAMs preferentially use glucose for glycolysis to provide an energy supply, whereas M2 TAMs tend to rely on mitochondrial FAO to maintain energy homeostasis [103], 104]. Tumor development is dominated by lipid metabolism due to nutritional constraints of the tumor, hypoxia, etc. Lipid metabolism in TAMs is strongly associated with immune escape and chemoresistance. This impairs phagocytosis while upregulating PD-L1 expression in TAMs, which attenuates antitumor T cell immune responses and consequently enhances immune escape [105]. This finding also suggests that resistance to chemotherapy is a serious challenge in current tumor therapy [103]. In addition, Su et al. [104] also explored the possible mechanisms of lipid metabolism by TAMs in tumor immunity. Researchers have reported that TAMs from human and mouse tumor tissues are enriched with high lipid levels. Inhibiting lipid uptake by blocking CD36, knocking down CD36, or inhibiting FAO in MΦs blocked TAM production, thereby preventing their immune function in vitro and in vivo. These results suggest that lipid metabolism may play an important role in TAM differentiation and function.
Lipid metabolism in dendritic cells during gastric cancer progression
DCs, as antigen-presenting cells, capture pathogens/tumor antigens and present them to T cells to initiate immune responses. Their metabolic programming governs development, polarization, maturation, and functional bioenergetic demands. However, DC immunoreactivity is typically suppressed within the TME. Aberrant tumor metabolism induces TME alterations – including elevated glycolysis, lactate/lipid accumulation, acidification, and tryptophan deficiency – undermining DC function and facilitating tumor immune escape. PGE2, a potent lipid mediator abundant in inflammatory milieus, critically regulates lipid metabolism [106].
Other studies have shown that high FA concentrations in the TME, the upregulation of FASN [105], inositol-requiring kinase 1-X-box binding protein 1 (IRE1-XBP1) signaling pathway stimulation [107], TLR stimulation [108], etc., can promote FAS and lipid accumulation in DCs. First, lipid accumulation in DCs can reduce antigen processing ability, downregulate the costimulatory molecule CD86, induce the overexpression of the tolerogenic cytokine IL-10, and weaken the ability to stimulate T-cell responses, leading to DC dysfunction [108]. The accumulation of oxidized lipids, especially triacylglycerols, in lymphoma, ovarian cancer, thymic lymphoma and mesothelioma lead to DC dysfunction and failure to properly induce antitumor responses in T cells. Here, these cells can covalently bind to heat shock protein 70 (Hsp70), preventing the transport of peptide‒MHC complexes to the cell surface. This leads to the accumulation of peptide‒MHC complexes in late-stage endosomes/lysosomes of the nucleus [106]. Finally, FA metabolism in the TME affects DC development, maturation, and function. Increased Low-Density Lipoprotein (LDL) uptake during the early stages of DC maturation inhibits maturation itself [109]. In addition, downregulation of SREBP-1 and PPAR-γ activator expression induced by methotrexate secreted by tumor cells inhibits FAS and mitochondrial metabolism in DCs, which also prevents DC maturation [110].
In terms of cholesterol metabolism, cholesterol, hydroxysteroids and cholesterol transporter proteins affect the differentiation and maturation of DCs. For example, 27-Hydroxycholesterol (27-HC) induces the differentiation of monocytes into mature DCs that express several surface signature molecules, such as MHC-II, C-C chemokine receptor type 7 (CCR7), CD40 and CD80, thereby promoting the immune response [111]. Cyclosporin A, a broad immunosuppressant, effectively affects 27-HC-induced monocyte differentiation and downregulates DC-specific markers, in addition to inhibiting T-cell activation by interacting with calmodulin neural phosphatase [112]. Apolipoprotein E (ApoE) deficiency leads to cholesterol accumulation on DC membranes. This improves antigen presentation by enhancing the aggregation of MHC-II molecules in the membranes of DCs, thereby promoting the CD4+ T-cell-mediated immune response and CD4+ T-cell-mediated immune responses [113]. In addition, oxidized lipids play a role in the cross-presentation of DCs in cancer: tumor-derived factors promote the intracellular accumulation of triacylglycerols, cholesterol esters and FAs in DCs, which reduces MHC-I expression on the cell surface of DCs and blocks exogenous antigen presentation [114].
Lipid metabolism of NK cells in gastric cancer progression
In tumor immunity, NK cells directly secrete perforin and apoptosis-inducing granzymes to kill tumor cells and recruit other immune cells when activated [115]. Studies have shown that exogenous lipids disrupt lipid metabolism in NK cells, thereby inhibiting their effector functions and responsiveness to immune stimuli [116]. Michelet et al. [117] reported that enhanced PPAR family-mediated lipid metabolism in NK cells drove the accumulation of intracellular lipids, leading to impaired metabolism and, in addition, lipid accumulation induces the upregulation of PPARα/δ, which is associated with a reduction in the number of NK cells. Here, NK cells take up FAs for their own defense and store them in lipid droplets to reduce lipotoxicity, but the concentration of FFAs in NK cells is still relatively high, and the expression of FAO-associated transporter proteins and enzymes is still relatively high. This may further limit mTOR-mediated glycolysis, thus further limiting mTOR-mediated glycolysis. mediated glycolysis, thereby limiting granzyme and IFN-γ production, and further research confirms that lipid droplet accumulation leads to reduced perforin levels.
In the GC microenvironment, significant alterations in lipid metabolism directly interfere with CD1d-mediated lipid antigen presentation. Research reveals that abnormal accumulation of sphingolipids (e.g., ceramide) in GC cells competitively occupies the CD1d antigen-binding groove, shifting CD1d-presented antigens from immunogenic lipids to immunosuppressive lipids [118]. This lipid replacement markedly reduces TCR recognition efficiency by NKT cells and weakens their activation. Additionally, dysregulation of the acidic environment in lysosomes impacts lipid antigen loading. Accumulation of saturated FAs (e.g., palmitate) in GC cells suppresses lysosomal acidification, impairing the binding efficiency between lipid antigens and CD1d [119]. This antigen-loading defect results in CD1d molecules exhibiting an “empty-loaded” state, rendering them incapable of effectively activating NKT cells.
In innate immune cells, lipid metabolism plays a crucial role, affecting the activation, function, and response of immune cells to pathogens. These findings reveal the importance of lipid metabolism in innate immune cells and provide potential targets for future immunotherapy (For details, see Supplementary Table 1) (Figures 5 and 6).

MDSCs and M2-TAMs coordinate with DC dysfunction and NK cell exhaustion, forming a lipid-metabolic immunosuppressive axis that fuels tumor immune evasion via T-cell suppression, checkpoint activation, and cytotoxic impairment. MDSC, myeloid-derived suppressor cell; TAM, tumor-associated macrophage; DC, dendritic cell; NK cell, natural killer cell.

Abnormal lipid metabolism and immune response in gastric cancer.
Convergence of lipid metabolism and antitumor immunity may provide a platform for biomarker-driven studies of new combination therapies
Despite advances in both basic and clinical immunotherapy, GC patient prognosis remains poor. Therefore, further exploration of combination therapies with immunotherapy is crucial. Targeting lipid metabolism in combination with immunotherapy and its effect on clinical prognosis has gained considerable attention.
Lipid metabolism biomarkers for predicting immunotherapy response in gastric cancer
Reprogrammed lipid metabolism drives GC progression by altering the TIME, with related biomarkers offering critical value for predicting immunotherapy efficacy. Transcriptomic analysis of TCGA and GEO datasets reveals that lipid metabolism-related genes (LMRGs) such as GPX3 and Nicotinamide N-methyltransferase (NNMT) effectively stratify patient risk: high-risk groups show increased infiltration of activated NK cells, resting monocytes, macrophages, mast cells, and CD4+ T cells, indicating an immunosuppressive TIME and poor prognosis [120]. A lipid metabolism score (LM score) constructed via single-cell sequencing and random forest algorithms predicts survival outcomes across multiple cohorts and reflects sensitivity to ICIs and chemotherapy [121]. Moreover, fatty acid amide hydrolase (FAAH) is overexpressed in GC tissues, driving lipid metabolic reprogramming via anandamide (AEA)/LPA signaling, while miR-1275 – which targets FAAH – serves as a potential diagnostic biomarker or therapeutic target [122]. Mechanistically, bioinformatics analyses demonstrate that lipid metabolic reprogramming modulates ICI response by regulating immune cell infiltration (e.g. CD8+ T cell exhaustion, Treg expansion), with BCHE identified as a key bridging molecule: its expression positively correlates with immunosuppressive cell infiltration and predicts poor PD-1 inhibitor response [123]. A combined lipid-immune prognostic model further confirms that high-risk patients exhibit activated arachidonic acid metabolism and prostaglandin synthesis alongside enriched metastasis markers, leading to ICI resistance (risk score positively correlates with TIDE score). Conversely, low-risk patients may benefit from anti-PD-1/CTLA-4 bispecific antibodies (e.g., cadonilimab), achieving a median overall survival of up to 20.24 months [124], [125], [126].
The application of lipid metabolism inhibitors
Changes in lipid metabolism are associated with resistance to anticancer drugs
Accumulating evidence indicates that metabolic reprogramming, particularly lipid metabolism alterations affecting cancer cell drug sensitivity, drives tumor resistance to chemotherapy and targeted agents. Lipid metabolic changes represent central mediators of antitumor resistance. In drug-resistant cancer cells, lipid reprogramming encompasses modified de novo lipogenesis and lipolysis pathways. Specifically in GC, MaCC1 (an upstream FASN regulator) reduces oxaliplatin chemosensitivity through FASN upregulation [127]. FASN is an important enzyme involved in the de novo biosynthesis of FAs, and FASN inhibition has been demonstrated [128] to impair, to a certain extent, the expression of human epidermal growth factor receptor 2 (HER2). These findings support the hypothesis that FASN inhibition improves the benefits of anti-HER2 biopharmaceuticals and thus suggests that FASN plays an important role in the therapeutic resistance of GC cells to anti-HER2 therapies. Glutathione peroxidase 4 (GPX4) is considered to be the most important member of the GPX family, which reduces lipid peroxidation. In addition, SREBP-1a has been shown to regulate GPX4 expression, upregulate GPX4 levels in GC cells exposed to the drug apatinib and promote drug resistance in GC cells [129]. SCD1 is one of the key rate-limiting metabolic enzymes that regulates unsaturated FA and lipid synthesis. Changes in FA composition and lipid metabolism were investigated in drug-resistant organisms. Adenosine deaminase (ADAR1), which acts on RNA1, mediates the upregulation of SCD1 and drives lipid droplets to inhibit chemotherapy-induced ER stress, thereby promoting tumor survival and enhancing chemotolerance during chemotherapy [130]. Furthermore, mesenchymal stem cells (MSCs) also develop resistance to chemotherapeutic agents via FAO. In this study, FTO was found to promote chemoresistance in GC cells via ex vivo MSCs. The secretion of transforming growth factor β1 (TGF-β1) by MSCs induces the expression of the long-chain noncoding RNA (lncRNA) MACC1-AS1 in GC cells, which promotes chemoresistance [131]. Given that NPRA promotes FAO and tumor cell metastasis, Chen et al. [132] reported that NPRA was upregulated in GC cells cocultured with MSCs and that NPRA knockdown reversed MSC-induced chemotolerance. Additionally, NPRA ameliorates FAO-promoted chemoresistance by protecting mitochondrial fusion protein 2 (Mfn2) from protein degradation and promoting its mitochondrial localization.
Cholesterol critically drives drug resistance. Blocking cellular cholesterol synthesis and uptake represents a novel therapeutic approach against cancer. Platinum (IV) complexes FP and DFP – utilizing fenofibric acid as an axial ligand – overcome drug resistance by dual mechanisms: DNA damage and cholesterol metabolism regulation. Beyond generating DNA-damaging reactive oxygen species and platinum (II) compounds, FP/DFP suppress cholesterol accumulation, enhance efflux, upregulate PPARα, and induce caspase-1 activation with gasdermin D (GSDMD) cleavage, triggering synergistic apoptosis and pyroptosis. Critically, cholesterol reduction substantially reverses cancer cell drug resistance [133]. Cholesterol is a major component of lipid rafts, and many transduction signals related to cell survival and proliferation are associated with lipid rafts, such as receptor tyrosine kinases, platelet-derived growth factor receptors, and EGFR. Notably, the number of lipid rafts in cancer cells is greater than that in normal cells, and increased cholesterol levels are noted in the lipid rafts of drug-resistant cancer cells are compared with drug-sensitive cancer cells [134]. Changes in cholesterol levels in the membranes of cancer cells significantly affect their permeability to therapeutic drugs; therefore, cholesterol-modulated uptake of chemotherapeutic drugs is considered one of the mechanisms by which drug resistance is initiated. It has been suggested that in the presence of cholesterol, the transport activity of p-glycoprotein is increased, affecting the ability of p-glycoprotein and drug transport [135] (Figure 7).
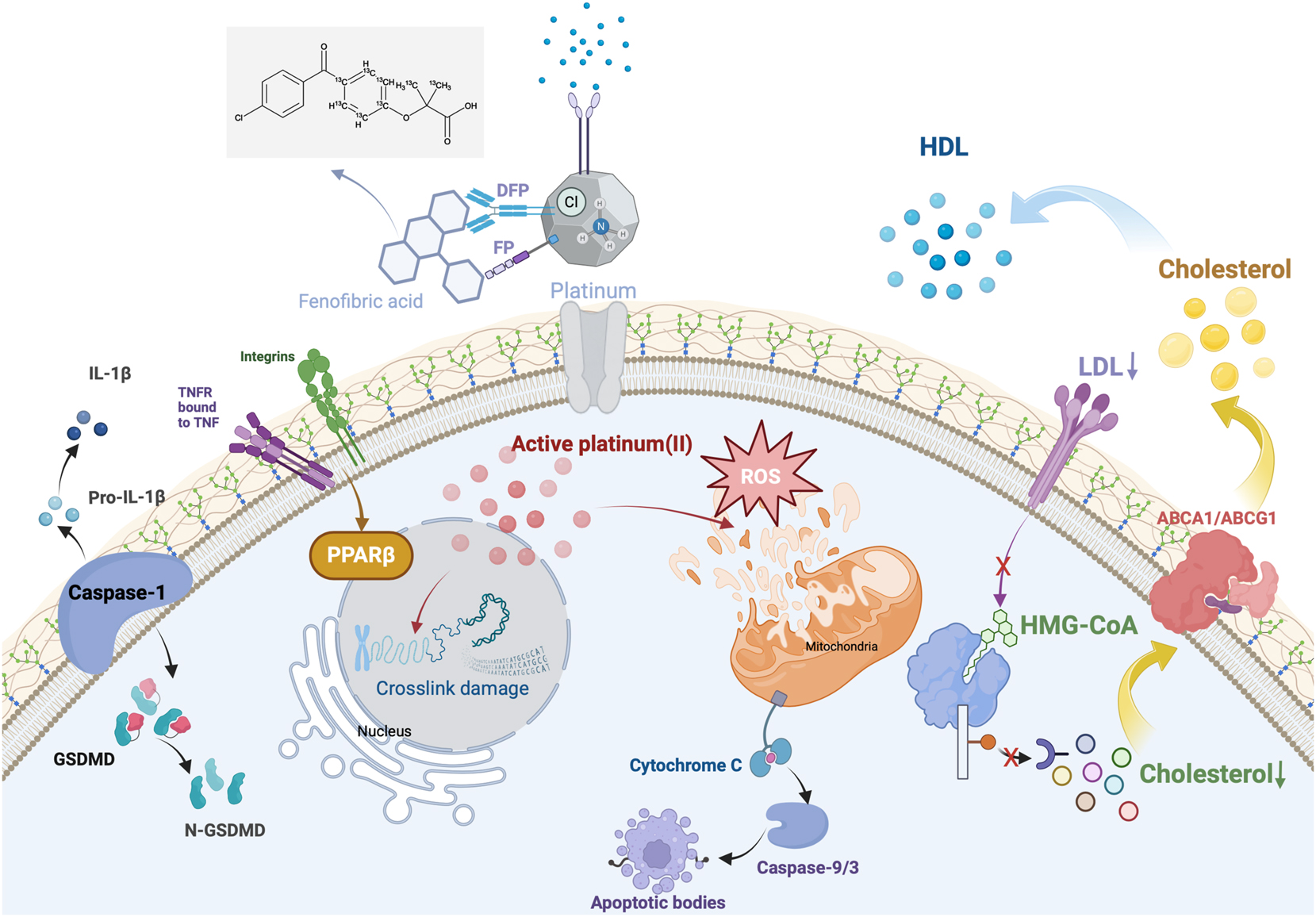
Platinum(IV)-fenofibrate conjugates exert dual anticancer effects by releasing cytotoxic Pt(II) for DNA and ROS damage and reprogramming cholesterol metabolism via HMG-CoA and LDLR inhibition, ABCA1 and G1-mediated efflux, and PPARα activation, while triggering pyroptosis-apoptosis crosstalk to overcome chemoresistance in gastric cancer. ROS, reactive oxygen species; LDLR, low-density lipoprotein receptor; ABCA1, ATP-binding cassette transporter A1; PPAR, peroxisome proliferator-activated receptor; LDLR, low-density lipoprotein receptor.
The therapeutic effect of lipid metabolism inhibitors on gastric cancer
In recent years, significant progress has been made in the treatment of GC. Lipid metabolism plays an important role in the immunological microenvironment of GC; thus, therapies targeting lipid metabolism represent a potential new direction for cancer treatment.
The key regulatory enzyme of endogenous FAS, SREBP1, has a significant effect on lipid metabolism in GC. It coordinates cellular lipid synthesis programs and inhibitory receptor signals in Tregs. Research has shown that SREBP1-dependent intracellular lipid metabolic homeostasis plays a key role in promoting the survival of M2-like TAMs and their oncogenic phenotype and that SREBP1 inhibition enhances the efficacy of immune checkpoint blockade, suppresses M2 TAMs, and promotes antitumor immune responses [136].
A previous study [137] explored the potential of histone deacetylase (HDAC) inhibitors in the treatment of GC. One study revealed that HDAC inhibitors activate lipid peroxidation and ferroptosis. In different GC cell lines, after treatment with HDAC inhibitors, the level of lipid peroxidation increased, and significant changes in the expression of proteins related to the induction of ferroptosis were observed. These findings provide evidence for a new mechanism of HDAC inhibitors in GC treatment.
Another study reported the effectiveness of lipid metabolism inhibitors such as etomoxir (an irreversible CPT1 inhibitor) in inhibiting the growth, migration, and EMT of GC cells overexpressing CPT1A. In addition, this study highlights the potential of other lipid metabolism pathway inhibitors, such as statins, in GC treatment and the role of inhibitors that target TAG production, DGAT enzymes, and PF06424439 stored in lipid droplets (LDs) in GC treatment [77].
The mevalonate pathway is an important metabolic pathway for cholesterol. Several inhibitors of the mevalonate pathway, such as zoledronic acid, farnesyl, and geranylgeranyl transferase inhibitors [138], 139], have been tested in GC, and statins that lower plasma cholesterol by inhibiting HMGCR and exerting anti-proliferative and pro-apoptotic effects in vitro [140] have also been assessed. The activity of SOAT1, a key enzyme in the cholesterol ester synthesis step, affects cholesterol concentrations in GC tissues. The SOAT1 inhibitor avasimibe was first identified in 1996. Importantly, the proliferation of GC cells, cholesterol ester synthesis, and lymphangiogenesis are inhibited upon inhibition of SOAT1 with avasimibe [50]. Studies reveal distinct drug sensitivity in primary versus metastatic GC cells toward agents targeting isoprenoid synthesis, cholesterol metabolism, and uptake, prompting exploration of avasimibe-based combination therapies. For primary GC, statins and avasimibe, which target isoprenoid and cholesterol metabolism in GC tissues, are promising potential new antitumor drug candidates [61].
Lipid metabolism reprogramming mediates GC treatment resistance by regulating FAO, cholesterol metabolism, and lipid droplet accumulation. Studies have shown that chemotherapy drugs such as oxaliplatin activate the NFATc3-CPT2 axis in GC cells, promoting FAO and clearing ROS, leading to drug resistance. Meanwhile, inhibiting CPT2 (such as with piperacillin) can block FAO and increase ROS accumulation, achieving an antitumor rate exceeding 60 % in preclinical studies [141]. Additionally, studies have shown that in apatinib resistance, upregulation of the cholesterol synthase HMGCR drives lipid raft formation and inhibits ferroptosis. Statins (such as simvastatin) reverse resistance by inhibiting HMGCR, with an objective response rate (ORR) improvement of 2.1-fold [142].
These studies suggest that lipid metabolism inhibitors may affect GC cell survival and metastasis through multiple mechanisms, providing new strategies and potential drug targets for GC treatment.
The combination of targeted lipid metabolism and immunotherapy offers more options for gastric cancer patients
Mounting evidence indicates lipid metabolism contributes to GC’s intricate metabolic network, offering key targets for novel therapeutic development. Therefore, in the field of GC treatment, an in-depth analysis of the combination of lipid metabolism and immunotherapy can reveal the potential for developing new anti-inflammatory therapies by targeting metabolic pathways [143]. Determining optimal combination strategies for lipid metabolism and GC immunotherapy demands multifaceted consideration: targeting specific lipid pathways in immune cells, combining lipid-targeted therapies with immunotherapy, and assessing how lipid metabolic reprogramming impacts immune functionality.
Research demonstrates that Tregs suppress antitumor immunity while FAS promotes their functional maturation. Absence of CD36 reduces Treg lipid uptake, impeding tumor growth. Researchers have reported that CD36 antibodies and PD-1 antibodies exhibit synergistic antitumor effects; therefore, blocking CD36 could represent an immunotherapy strategy targeting the lipid metabolism of Treg cells [144]. A recent study identified reduced phosphatidylcholine and phosphatidylethanolamine levels in tumor-infiltrating CD8+ T cells, concomitant with significant downregulation of PLPP1 – the metabolic enzyme mediating their synthesis [145]. PLPP1 overexpression inhibits the phospholipid metabolism of chimeric antigen receptor T cell (CAR-T) cells and improves antitumor functions.
In addition, a clinical study demonstrated that, in GC immunotherapy research, the median overall survival in the entire population of the combination therapy group reached 17.41 months, the median progression-free survival reached 9.2 months, the 12-month overall survival rate was 61.4 %, the ORR reached 68.2 %, and the disease control rate (DCR) reached 92 %. In the population with a PD-L1 CPS≥5, the median overall survival time is as high as 20.24 months [126]. These findings provide a new strategy for future GC treatment. In summary, the combination of GC immunotherapy and targeted lipid metabolism offers a new therapeutic strategy that can potentially enhance the antitumor immune response, improve treatment efficacy, reduce side effects, and provide more treatment options for patients with GC.
Challenges in lipid metabolism therapy
Targeting lipid metabolism in combination with immunotherapy is a promising therapy; however, little is known about the relevant pathways and key enzymes involved in the regulation of lipid metabolism in GC. These still need to be explored and investigated. The vast majority of information comes from studies in tumor cells or experimental animals, with few clinical applications in humans. Although in vitro experiments demonstrate superior efficacy of lipid metabolism inhibitor combination therapy for GC, multidrug treatment requires further clinical validation to establish synergy, safety, efficacy, and side-effect profiles. Lipid distribution across GC subtypes exhibits significant heterogeneity influenced by study design, population, methodology, and analytical approaches. Current clinical evidence incompletely defines subtype-specific lipid characteristics and metabolic patterns. Furthermore, the mechanistic roles of lipid-associated enzymes/proteins and standardized lipid metabolism assessment metrics remain unestablished.
GC biomarkers can be important tools for the early detection, diagnosis, and prognosis of this disease. Important enzymes involved in lipid metabolism can be used as biomarkers to predict GC development and progression and play important roles in clinical diagnosis. Current evidence linking lipid biomarkers to clinical outcomes is experimentally derived and predominantly retrospective, thus restricting their application. Consequently, lipid biomarker development remains nascent, with safety, efficacy, and adverse effects uncharacterized. These constraints impede extending biomarker-related clinical trial results to the entire GC patient population.
The regulation of lipid metabolism offers new strategies and targets for cancer treatment. Therapeutic strategies may target key enzymes or transport proteins in lipid metabolic pathways, aiming to reverse or prevent the development of drug resistance through combination therapies and enhance or restore sensitivity to therapeutic drugs. These studies provide an important theoretical foundation and potential targets for the development of new anticancer drugs and treatment strategies.
Conclusions
This review comprehensively elucidates the pivotal role of dysregulated lipid metabolism in the TIME of GC, highlighting how lipid metabolic reprogramming drives immune evasion and chemoresistance by modulating immune cell functions (e.g., T-cell exhaustion, Treg-mediated immunosuppression, macrophage polarization). Although fundamental studies have identified key metabolic enzymes (e.g., SREBP-1c, FASN, CPT1A) and pathways involved in GC progression, translating these findings into clinical applications remains a significant challenge. Future research should prioritize the development of precision therapies targeting lipid metabolism, such as small-molecule inhibitors against SREBP-1c or FAO rate-limiting enzymes like CPT1A, which have shown promising antitumor effects and enhanced chemosensitivity in preclinical models. Combining these agents with ICIs (e.g., PD-1/PD-L1 blockers) may yield synergistic efficacy, as evidenced by recent clinical trials where PD-1/CTLA-4 bispecific antibodies extended median overall survival beyond 20 months in select patients. Furthermore, lipid metabolism biomarkers (e.g., plasma oxLDL, cholesteryl ester levels) identified through metabolomics and multi-omics technologies hold potential for early diagnosis and personalized treatment. Interdisciplinary collaboration to integrate basic research with clinical data will accelerate the translation of metabolic-targeted therapies and enhance clinicians’ understanding of lipid metabolism modulation in oncology, ultimately improving patient outcomes.
Funding source: Characteristic Discipline – Gastroenterology
Award Identifier / Grant number: ML12202323
Funding source: Clinical study on digestive endoscopic ultrasound microprobes and imaging systems
Award Identifier / Grant number: ML12203223
-
Research ethics: This article is a review and does not involve human participants, animal experiments, or primary data collection. Therefore, no ethical approval was required.
-
Informed consent: Not applicable, as this is a review article synthesizing existing published studies.
-
Author contributions: All authors meet the ICMIE criteria for authorship. Contributions are as follows: Qin Zhan: study design, literature collection, and manuscript writing. Haoxiang Ni: content modification and manuscript writing. Ming Zhou: literature collection and manuscript writing. Xiaozhe Mao: data organization and manuscript writing. Yifan Ouyang: literature collection, manuscript writing, and review. Tonggao Shi: manuscript writing and critical review. Rui Li: manuscript writing, data collection, and supervision. All authors reviewed and approved the final version of the manuscript.
-
Use of Large Language Models, AI and Machine Learning Tools: None declared.
-
Conflict of interest: All authors declare no financial or non-financial conflicts of interest related to this work.
-
Research funding: This work was supported by Characteristic Discipline Gastroenterology (ML12202323) and Clinical study on digestive endoscopic ultrasound microprobes and imaging systems (ML12203223).
-
Data availability: The data supporting this review are derived from publicly available studies cited in the references.
References
1. Chen, J, Liu, K, Luo, Y, Kang, M, Wang, J, Chen, G, et al.. Single-cell profiling of tumor immune microenvironment reveals immune irresponsiveness in gastric signet-ring cell carcinoma. Gastroenterology 2023;165:88–103. https://doi.org/10.1053/j.gastro.2023.03.008.Search in Google Scholar PubMed
2. Fang, X, Xu, J, Jin, K, Qian, J. Combining of immunotherapeutic approaches with chemotherapy for treatment of gastric cancer: achievements and limitations. Int Immunopharmacol 2023;118:110062. https://doi.org/10.1016/j.intimp.2023.110062.Search in Google Scholar PubMed
3. Mou, P, Ge, QH, Sheng, R, Zhu, TF, Liu, Y, Ding, K. Research progress on the immune microenvironment and immunotherapy in gastric cancer. Front Immunol 2023;14:1291117. https://doi.org/10.3389/fimmu.2023.1291117.Search in Google Scholar PubMed PubMed Central
4. Janjigian, YY, Shitara, K, Moehler, M, Garrido, M, Salman, P, Shen, L, et al.. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet 2021;398:27–40. https://doi.org/10.1016/s0140-6736(21)00797-2.Search in Google Scholar PubMed PubMed Central
5. Alcindor, T. Immunotherapy in gastric cancer-choosing methods or results. JAMA Oncol 2024;10:704–5. https://doi.org/10.1001/jamaoncol.2023.7262.Search in Google Scholar PubMed
6. Pereira, MA, Ramos, M, Cardili, L, de Moraes, RDR, Dias, AR, Szor, DJ, et al.. Prognostic implications of tumor-infiltrating lymphocytes within the tumor microenvironment in gastric cancer. J Gastrointest Surg 2024;28:151–7. https://doi.org/10.1016/j.gassur.2023.12.002.Search in Google Scholar PubMed
7. Bui, TM, Yalom, LK, Sumagin, R. Tumor-associated neutrophils: orchestrating cancer pathobiology and therapeutic resistance. Expert Opin Ther Targets 2021;25:573–83. https://doi.org/10.1080/14728222.2021.1954162.Search in Google Scholar PubMed
8. Zhang, Y, Kurupati, R, Liu, L, Zhou, XY, Zhang, G, Hudaihed, A, et al.. Enhancing CD8+ T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell 2017;32:377–91.e9. https://doi.org/10.1016/j.ccell.2017.08.004.Search in Google Scholar PubMed PubMed Central
9. Ma, X, Bi, E, Lu, Y, Su, P, Huang, C, Liu, L, et al.. Cholesterol induces CD8+ T cell exhaustion in the tumor microenvironment. Cell Metab 2019;30:143–56.e5. https://doi.org/10.1016/j.cmet.2019.04.002.Search in Google Scholar PubMed PubMed Central
10. Zhu, L, Shi, Y, Feng, Z, Yuan, D, Guo, S, Wang, Y, et al.. Fatostatin promotes anti-tumor immunity by reducing SREBP2 mediated cholesterol metabolism in tumor-infiltrating T lymphocytes. Eur J Pharmacol 2024;971:176519. https://doi.org/10.1016/j.ejphar.2024.176519.Search in Google Scholar PubMed
11. Najt, CP, Khan, SA, Heden, TD, Witthuhn, BA, Perez, M, Heier, JL, et al.. Lipid droplet-derived monounsaturated fatty acids traffic via PLIN5 to allosterically activate SIRT1. Mol Cell 2020;77:810–24.e8. https://doi.org/10.1016/j.molcel.2019.12.003.Search in Google Scholar PubMed PubMed Central
12. Wang, Y, Jia, A, Bi, Y, Wang, Y, Liu, G. Metabolic regulation of myeloid-derived suppressor cell function in cancer. Cells 2020;9. https://doi.org/10.3390/cells9041011.Search in Google Scholar PubMed PubMed Central
13. You, Z, Chi, H. Lipid metabolism in dendritic cell biology. Immunol Rev 2023;317:137–51. https://doi.org/10.1111/imr.13215.Search in Google Scholar PubMed PubMed Central
14. Yu, W, Lei, Q, Yang, L, Qin, G, Liu, S, Wang, D, et al.. Contradictory roles of lipid metabolism in immune response within the tumor microenvironment. J Hematol Oncol 2021;14:187. https://doi.org/10.1186/s13045-021-01200-4.Search in Google Scholar PubMed PubMed Central
15. Zeng, Q, Gong, Y, Zhu, N, Shi, Y, Zhang, C, Qin, L. Lipids and lipid metabolism in cellular senescence: emerging targets for age-related diseases. Ageing Res Rev 2024;97:102294. https://doi.org/10.1016/j.arr.2024.102294.Search in Google Scholar PubMed
16. Lukasiewicz, M, Zwara, A, Kowalski, J, Mika, A, Hellmann, A. The role of lipid metabolism disorders in the development of thyroid cancer. Int J Mol Sci 2024;25:7129. https://doi.org/10.3390/ijms25137129.Search in Google Scholar PubMed PubMed Central
17. Guo, R, Liang, JH, Zhang, Y, Lutchenkov, M, Li, Z, Wang, Y, et al.. Methionine metabolism controls the B cell EBV epigenome and viral latency. Cell Metab 2022;34:1280–97.e9. https://doi.org/10.1016/j.cmet.2022.08.008.Search in Google Scholar PubMed PubMed Central
18. Lee, SH, Khoo, AS, Griffiths, JR, Mat Lazim, N. Metabolic regulation of the tumour and its microenvironment: the role of epstein-barr virus. Int J Cancer 2025;156:488–98. https://doi.org/10.1002/ijc.35192.Search in Google Scholar PubMed
19. Müller-Durovic, B, Jäger, J, Engelmann, C, Schuhmachers, P, Altermatt, S, Schlup, Y, et al.. A metabolic dependency of EBV can be targeted to hinder B cell transformation. Science 2024;385:eadk4898. https://doi.org/10.1126/science.adk4898.Search in Google Scholar PubMed
20. Balakrishnan, K, Ganesan, K. Identification of oncogenic signaling pathways associated with the dimorphic metabolic dysregulations in gastric cancer subtypes. Med Oncol 2022;39:132. https://doi.org/10.1007/s12032-022-01717-9.Search in Google Scholar PubMed
21. Huang, R, Lu, TL, Zhou, R. Identification and immune landscape analysis of fatty acid metabolism genes related subtypes of gastric cancer. Sci Rep 2023;13:20443. https://doi.org/10.1038/s41598-023-47631-6.Search in Google Scholar PubMed PubMed Central
22. Nanki, K, Toshimitsu, K, Takano, A, Fujii, M, Shimokawa, M, Ohta, Y, et al.. Divergent routes toward Wnt and R-spondin niche independency during human gastric carcinogenesis. Cell 2018;174:856–69.e17. https://doi.org/10.1016/j.cell.2018.07.027.Search in Google Scholar PubMed
23. Janjigian, YY, Cecchini, M, Shitara, K, Enzinger, PC, Wainberg, ZA, Chau, I, et al.. Genomic landscape of late-stage gastric cancer: analysis from KEYNOTE-059, KEYNOTE-061, and KEYNOTE-062 studies. JCO Precis Onco 2025;9:e2400456. https://doi.org/10.1200/po-24-00456.Search in Google Scholar PubMed PubMed Central
24. Tsai, CK, Yeh, TS, Wu, RC, Lai, YC, Chiang, MH, Lu, KY, et al.. Metabolomic alterations and chromosomal instability status in gastric cancer. World J Gastroenterol 2018;24:3760–9. https://doi.org/10.3748/wjg.v24.i33.3760.Search in Google Scholar PubMed PubMed Central
25. Hung, CY, Yeh, TS, Tsai, CK, Wu, RC, Lai, YC, Chiang, MH, et al.. Glycerophospholipids pathways and chromosomal instability in gastric cancer: global lipidomics analysis. World J Gastrointest Oncol 2019;11:181–94. https://doi.org/10.4251/wjgo.v11.i3.181.Search in Google Scholar PubMed PubMed Central
26. Ladanyi, A, Mukherjee, A, Kenny, HA, Johnson, A, Mitra, AK, Sundaresan, S, et al.. Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 2018;37:2285–301. https://doi.org/10.1038/s41388-017-0093-z.Search in Google Scholar PubMed PubMed Central
27. Jiang, M, Wu, N, Xu, B, Chu, Y, Li, X, Su, S, et al.. Fatty acid-induced CD36 expression via O-GlcNAcylation drives gastric cancer metastasis. Theranostics 2019;9:5359–73. https://doi.org/10.7150/thno.34024.Search in Google Scholar PubMed PubMed Central
28. Li, C, Zhang, L, Qiu, Z, Deng, W, Wang, W. Key molecules of fatty acid metabolism in gastric cancer. Biomolecules 2022;12. https://doi.org/10.3390/biom12050706.Search in Google Scholar PubMed PubMed Central
29. Levi, L, Lobo, G, Doud, MK, von Lintig, J, Seachrist, D, Tochtrop, GP, et al.. Genetic ablation of the fatty acid-binding protein FABP5 suppresses HER2-induced mammary tumorigenesis. Cancer Res 2013;73:4770–80. https://doi.org/10.1158/0008-5472.can-13-0384.Search in Google Scholar PubMed PubMed Central
30. Zhao, G, Wu, M, Wang, X, Du, Z, Zhang, G. Effect of FABP5 gene silencing on the proliferation, apoptosis and invasion of human gastric SGC-7901 cancer cells. Oncol Lett 2017;14:4772–8. https://doi.org/10.3892/ol.2017.6748.Search in Google Scholar PubMed PubMed Central
31. Sun, Q, Yu, X, Peng, C, Liu, N, Chen, W, Xu, H, et al.. Activation of SREBP-1c alters lipogenesis and promotes tumor growth and metastasis in gastric cancer. Biomed Pharmacother 2020;128:110274. https://doi.org/10.1016/j.biopha.2020.110274.Search in Google Scholar PubMed
32. Gao, Y, Li, J, Xi, H, Cui, J, Zhang, K, Zhang, J, et al.. Stearoyl-CoA-desaturase-1 regulates gastric cancer stem-like properties and promotes tumour metastasis via Hippo/YAP pathway. Br J Cancer 2020;122:1837–47. https://doi.org/10.1038/s41416-020-0827-5.Search in Google Scholar PubMed PubMed Central
33. Wang, C, Shi, M, Ji, J, Cai, Q, Zhao, Q, Jiang, J, et al.. Stearoyl-CoA desaturase 1 (SCD1) facilitates the growth and anti-ferroptosis of gastric cancer cells and predicts poor prognosis of gastric cancer. Aging (Albany NY) 2020;12:15374–91. https://doi.org/10.18632/aging.103598.Search in Google Scholar PubMed PubMed Central
34. Zhou, Y, Su, W, Liu, H, Chen, T, Höti, N, Pei, H, et al.. Fatty acid synthase is a prognostic marker and associated with immune infiltrating in gastric cancers precision medicine. Biomark Med 2020;14:185–99. https://doi.org/10.2217/bmm-2019-0476.Search in Google Scholar PubMed
35. Zheng, X, Wang, X, Zheng, L, Zhao, H, Li, W, Wang, B, et al.. Construction and analysis of the tumor-specific mRNA-miRNA-lncRNA network in gastric cancer. Front Pharmacol 2020;11:1112. https://doi.org/10.3389/fphar.2020.01112.Search in Google Scholar PubMed PubMed Central
36. Zaidi, N, Royaux, I, Swinnen, JV, Smans, K. ATP citrate lyase knockdown induces growth arrest and apoptosis through different cell- and environment-dependent mechanisms. Mol Cancer Ther 2012;11:1925–35. https://doi.org/10.1158/1535-7163.mct-12-0095.Search in Google Scholar PubMed
37. Chang, WC, Cheng, WC, Cheng, BH, Chen, L, Ju, LJ, Ou, YJ, et al.. Mitochondrial Acetyl-CoA Synthetase 3 is biosignature of gastric cancer progression. Cancer Med 2018;7:1240–52. https://doi.org/10.1002/cam4.1295.Search in Google Scholar PubMed PubMed Central
38. Ye, X, Zhang, Y, Wang, X, Li, Y, Gao, Y. Tumor-suppressive functions of long-chain acyl-CoA synthetase 4 in gastric cancer. IUBMB Life 2016;68:320–7. https://doi.org/10.1002/iub.1486.Search in Google Scholar PubMed
39. Fang, W, Cui, H, Yu, D, Chen, Y, Wang, J, Yu, G. Increased expression of phospho-acetyl-CoA carboxylase protein is an independent prognostic factor for human gastric cancer without lymph node metastasis. Med Oncol 2014;31:15. https://doi.org/10.1007/s12032-014-0015-7.Search in Google Scholar PubMed
40. Lee, JS, Kim, SH, Lee, S, Kang, JH, Lee, SH, Cheong, JH, et al.. Gastric cancer depends on aldehyde dehydrogenase 3A1 for fatty acid oxidation. Sci Rep 2019;9:16313. https://doi.org/10.1038/s41598-019-52814-1.Search in Google Scholar PubMed PubMed Central
41. Wang, L, Li, C, Song, Y, Yan, Z. Inhibition of carnitine palmitoyl transferase 1A-induced fatty acid oxidation suppresses cell progression in gastric cancer. Arch Biochem Biophys 2020;696:108664. https://doi.org/10.1016/j.abb.2020.108664.Search in Google Scholar PubMed
42. Cao, T, Wang, S, Qian, L, Wu, C, Huang, T, Wang, Y, et al.. NPRA promotes fatty acid metabolism and proliferation of gastric cancer cells by binding to PPARα. Transl Oncol 2023;35:101734. https://doi.org/10.1016/j.tranon.2023.101734.Search in Google Scholar PubMed PubMed Central
43. Gao, J, Song, J, Zhang, Y, Zhu, Z. CPT1C promotes the potential of gastric cancer ovarian metastasis through up-regulating fatty acid oxidation. Acta Biochim Biophys Sin (Shanghai) 2022;54:752–5. https://doi.org/10.3724/abbs.2022027.Search in Google Scholar PubMed PubMed Central
44. Wang, L, Wu, C, Xu, J, Gong, Z, Cao, X, Huang, J, et al.. GC-MSC-derived circ_0024107 promotes gastric cancer cell lymphatic metastasis via fatty acid oxidation metabolic reprogramming mediated by the miR-5572/6855-5p/CPT1A axis. Oncol Rep 2023;50. https://doi.org/10.3892/or.2023.8575.Search in Google Scholar PubMed PubMed Central
45. He, W, Li, Y, Liu, SB, Chang, Y, Han, S, Han, X, et al.. From mitochondria to tumor suppression: ACAT1’s crucial role in gastric cancer. Front Immunol 2024;15:1449525. https://doi.org/10.3389/fimmu.2024.1449525.Search in Google Scholar PubMed PubMed Central
46. Fu, H, Zhang, J, Chen, H, Hou, H, Chen, H, Xie, R, et al.. NPR1 promotes lipid droplet lipolysis to enhance mitochondrial oxidative phosphorylation and fuel gastric cancer metastasis. Adv Sci (Weinh) 2025:e03233. https://doi.org/10.1002/advs.202503233.Search in Google Scholar PubMed PubMed Central
47. Chushi, L, Wei, W, Kangkang, X, Yongzeng, F, Ning, X, Xiaolei, C. HMGCR is up-regulated in gastric cancer and promotes the growth and migration of the cancer cells. Gene 2016;587:42–7. https://doi.org/10.1016/j.gene.2016.04.029.Search in Google Scholar PubMed
48. Ma, YC, Jin, SJ, Gu, GJ, Zhao, LF, Xu, ST. The expression of squalene epoxidase in human gastric cancer and its clinical significance. Indian J Pathol Microbiol 2023;66:799–803. https://doi.org/10.4103/ijpm.ijpm-1183_21.Search in Google Scholar
49. Tu, T, Zhang, H, Xu, H. Targeting sterol-O-acyltransferase 1 to disrupt cholesterol metabolism for cancer therapy. Front Oncol 2023;13:1197502. https://doi.org/10.3389/fonc.2023.1197502.Search in Google Scholar PubMed PubMed Central
50. Zhu, T, Wang, Z, Zou, T, Xu, L, Zhang, S, Chen, Y, et al.. SOAT1 promotes gastric cancer lymph node metastasis through lipid synthesis. Front Pharmacol 2021;12:769647. https://doi.org/10.3389/fphar.2021.769647.Search in Google Scholar PubMed PubMed Central
51. Gu, J, Zhu, N, Li, HF, Zhao, TJ, Zhang, CJ, Liao, DF, et al.. Cholesterol homeostasis and cancer: a new perspective on the low-density lipoprotein receptor. Cell Oncol (Dordr) 2022;45:709–28. https://doi.org/10.1007/s13402-022-00694-5.Search in Google Scholar PubMed
52. Nam, SY, Jeong, J, Jeon, SW. Constant association between low high-density lipoprotein cholesterol and gastric cancer regardless of site. J Obes Metab Syndr 2023;32:141–50. https://doi.org/10.7570/jomes22045.Search in Google Scholar PubMed PubMed Central
53. Yasufuku, I, Saigo, C, Kito, Y, Yoshida, K, Takeuchi, T. Prognostic significance of LDL receptor-related protein 1B in patients with gastric cancer. J Mol Histol 2021;52:165–72. https://doi.org/10.1007/s10735-020-09932-2.Search in Google Scholar PubMed
54. Lorenzano, S. Comment: oxidized low-density lipoprotein: a plasma biomarker predicting stroke recurrence. Neurology 2018;91:443. https://doi.org/10.1212/wnl.0000000000006127.Search in Google Scholar PubMed
55. Ma, C, Xie, J, Luo, C, Yin, H, Li, R, Wang, X, et al.. OxLDL promotes lymphangiogenesis and lymphatic metastasis in gastric cancer by upregulating VEGF-C expression and secretion. Int J Oncol 2019;54:572–84. https://doi.org/10.3892/ijo.2018.4648.Search in Google Scholar PubMed PubMed Central
56. Russo-Savage, L, Schulman, IG. Liver X receptors and liver physiology. Biochim Biophys Acta Mol Basis Dis 2021;1867:166121. https://doi.org/10.1016/j.bbadis.2021.166121.Search in Google Scholar PubMed PubMed Central
57. Bovenga, F, Sabbà, C, Moschetta, A. Uncoupling nuclear receptor LXR and cholesterol metabolism in cancer. Cell Metab 2015;21:517–26. https://doi.org/10.1016/j.cmet.2015.03.002.Search in Google Scholar PubMed
58. Tang, W, Li, G, Lin, Q, Zhu, Z, Wang, Z, Wang, Z. Multiplex immunohistochemistry defines two cholesterol metabolism patterns predicting immunotherapeutic outcomes in gastric cancer. J Transl Med 2023;21:887. https://doi.org/10.1186/s12967-023-04758-4.Search in Google Scholar PubMed PubMed Central
59. Wang, Q, Feng, F, Wang, J, Ren, M, Shi, Z, Mao, X, et al.. Liver X receptor activation reduces gastric cancer cell proliferation by suppressing Wnt signalling via LXRβ relocalization. J Cell Mol Med 2019;23:789–97. https://doi.org/10.1111/jcmm.13974.Search in Google Scholar PubMed PubMed Central
60. Liu, J, Qi, YB. Activation of LXRβ inhibits proliferation, promotes apoptosis, and increases chemosensitivity of gastric cancer cells by upregulating the expression of ATF4. J Cell Biochem 2019;120:14336–47. https://doi.org/10.1002/jcb.28558.Search in Google Scholar PubMed
61. Ortiz, N, Delgado-Carazo, JC, Díaz, C. Importance of mevalonate pathway lipids on the growth and survival of primary and metastatic gastric carcinoma cells. Clin Exp Gastroenterol 2021;14:217–28. https://doi.org/10.2147/CEG.S310235.Search in Google Scholar PubMed PubMed Central
62. Liu, S, Zhang, N, Ji, X, Yang, S, Zhao, Z, Li, P. Helicobacter pylori CagA promotes gastric cancer immune escape by upregulating SQLE. Cell Death Dis 2025;16:17. https://doi.org/10.1038/s41419-024-07318-w.Search in Google Scholar PubMed PubMed Central
63. Luo, H, Lv, J, Wen, P, Zhang, S, Ma, W, Yang, Z. Supramolecular polyrotaxane-based nano-theranostics enable cancer-cell stiffening for enhanced T-cell-mediated anticancer immunotherapy. Nat Commun 2025;16:2331. https://doi.org/10.1038/s41467-025-57718-5.Search in Google Scholar PubMed PubMed Central
64. Hannun, YA, Obeid, LM. Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol 2018;19:175–91. https://doi.org/10.1038/nrm.2017.107.Search in Google Scholar PubMed PubMed Central
65. Hannun, YA, Obeid, LM. Many ceramides. J Biol Chem 2011;286:27855–62. https://doi.org/10.1074/jbc.r111.254359.Search in Google Scholar
66. Yan, J, Yu, X, Li, Q, Miao, M, Shao, Y. Machine learning to establish three sphingolipid metabolism genes signature to characterize the immune landscape and prognosis of patients with gastric cancer. BMC Genom 2024;25:319. https://doi.org/10.1186/s12864-024-10243-z.Search in Google Scholar PubMed PubMed Central
67. Lee, M, Lee, SY, Bae, YS. Functional roles of sphingolipids in immunity and their implication in disease. Exp Mol Med 2023;55:1110–30. https://doi.org/10.1038/s12276-023-01018-9.Search in Google Scholar PubMed PubMed Central
68. Zhu, Y, Tong, X, Xue, J, Qiu, H, Zhang, D, Zheng, DQ, et al.. Phospholipid biosynthesis modulates nucleotide metabolism and reductive capacity. Nat Chem Biol 2025;21:35–46. https://doi.org/10.1038/s41589-024-01689-z.Search in Google Scholar PubMed
69. Chen, M, Zhang, C, Li, H, Zheng, S, Li, Y, Yuan, M, et al.. PLA2G4A and ACHE modulate lipid profiles via glycerophospholipid metabolism in platinum-resistant gastric cancer. J Transl Med 2024;22:249. https://doi.org/10.1186/s12967-024-05055-4.Search in Google Scholar PubMed PubMed Central
70. Turner, JA, Fredrickson, MA, D’Antonio, M, Katsnelson, E, MacBeth, M, Van Gulick, R, et al.. Lysophosphatidic acid modulates CD8 T cell immunosurveillance and metabolism to impair anti-tumor immunity. Nat Commun 2023;14:3214. https://doi.org/10.1038/s41467-023-38933-4.Search in Google Scholar PubMed PubMed Central
71. Lim, CX, Redl, A, Kleissl, L, Pandey, RV, Mayerhofer, C, El Jammal, T, et al.. Aberrant lipid metabolism in macrophages is associated with granuloma formation in sarcoidosis. Am J Respir Crit Care Med 2024;209:1152–64. https://doi.org/10.1164/rccm.202307-1273oc.Search in Google Scholar PubMed PubMed Central
72. Corn, KC, Windham, MA, Rafat, M. Lipids in the tumor microenvironment: from cancer progression to treatment. Prog Lipid Res 2020;80:101055. https://doi.org/10.1016/j.plipres.2020.101055.Search in Google Scholar PubMed PubMed Central
73. Noyes, D, Bag, A, Oseni, S, Semidey-Hurtado, J, Cen, L, Sarnaik, AA, et al.. Tumor-associated tregs obstruct antitumor immunity by promoting T cell dysfunction and restricting clonal diversity in tumor-infiltrating CD8+ T cells. J Immunother Cancer 2022;10. https://doi.org/10.1136/jitc-2022-004605.Search in Google Scholar PubMed PubMed Central
74. Cao, J, Liao, S, Zeng, F, Liao, Q, Luo, G, Zhou, Y. Effects of altered glycolysis levels on CD8+ T cell activation and function. Cell Death Dis 2023;14:407. https://doi.org/10.1038/s41419-023-05937-3.Search in Google Scholar PubMed PubMed Central
75. Tsai, CH, Chuang, YM, Li, X, Yu, YR, Tzeng, SF, Teoh, ST, et al.. Immunoediting instructs tumor metabolic reprogramming to support immune evasion. Cell Metab 2023;35:118–33.e7. https://doi.org/10.1016/j.cmet.2022.12.003.Search in Google Scholar PubMed PubMed Central
76. Wang, Y, Yu, W, Li, S, Guo, D, He, J, Wang, Y. Acetyl-CoA carboxylases and diseases. Front Oncol 2022;12:836058. https://doi.org/10.3389/fonc.2022.836058.Search in Google Scholar PubMed PubMed Central
77. Cui, MY, Yi, X, Zhu, DX, Wu, J. The role of lipid metabolism in gastric cancer. Front Oncol 2022;12:916661. https://doi.org/10.3389/fonc.2022.916661.Search in Google Scholar PubMed PubMed Central
78. Park, J, Hsueh, PC, Li, Z, Ho, PC. Microenvironment-driven metabolic adaptations guiding CD8+ T cell anti-tumor immunity. Immunity 2023;56:32–42. https://doi.org/10.1016/j.immuni.2022.12.008.Search in Google Scholar PubMed
79. Kouidhi, S, Ben Ayed, F, Benammar Elgaaied, A. Targeting tumor metabolism: a new challenge to improve immunotherapy. Front Immunol 2018;9:353. https://doi.org/10.3389/fimmu.2018.00353.Search in Google Scholar PubMed PubMed Central
80. Negura, I, Pavel-Tanasa, M, Danciu, M. Regulatory T cells in gastric cancer: key controllers from pathogenesis to therapy. Cancer Treat Rev 2023;120:102629. https://doi.org/10.1016/j.ctrv.2023.102629.Search in Google Scholar PubMed
81. Shan, Y, Xie, T, Sun, Y, Lu, Z, Topatana, W, Juengpanich, S, et al.. Lipid metabolism in tumor-infiltrating regulatory T cells: perspective to precision immunotherapy. Biomark Res 2024;12:41. https://doi.org/10.1186/s40364-024-00588-8.Search in Google Scholar PubMed PubMed Central
82. Beier, UH, Angelin, A, Akimova, T, Wang, L, Liu, Y, Xiao, H, et al.. Essential role of mitochondrial energy metabolism in Foxp3+ T-regulatory cell function and allograft survival. FASEB J 2015;29:2315–26. https://doi.org/10.1096/fj.14-268409.Search in Google Scholar PubMed PubMed Central
83. Miska, J, Lee-Chang, C, Rashidi, A, Muroski, ME, Chang, AL, Lopez-Rosas, A, et al.. HIF-1α is a metabolic switch between glycolytic-driven migration and oxidative phosphorylation-driven immunosuppression of tregs in glioblastoma. Cell Rep 2019;27:226–37.e4. https://doi.org/10.1016/j.celrep.2019.03.029.Search in Google Scholar PubMed PubMed Central
84. Hu, L, Zhu, M, Shen, Y, Zhong, Z, Wu, B. The prognostic value of intratumoral and peritumoral tumor-infiltrating FoxP3+Treg cells in of pancreatic adenocarcinoma: a meta-analysis. World J Surg Oncol 2021;19:300. https://doi.org/10.1186/s12957-021-02420-1.Search in Google Scholar PubMed PubMed Central
85. Field, CS, Baixauli, F, Kyle, RL, Puleston, DJ, Cameron, AM, Sanin, DE, et al.. Mitochondrial integrity regulated by lipid metabolism is a cell-intrinsic checkpoint for treg suppressive function. Cell Metab 2020;31:422–37.e5. https://doi.org/10.1016/j.cmet.2019.11.021.Search in Google Scholar PubMed PubMed Central
86. Lin, R, Zhang, H, Yuan, Y, He, Q, Zhou, J, Li, S, et al.. Fatty acid oxidation controls CD8+ tissue-resident memory T-cell survival in gastric adenocarcinoma. Cancer Immunol Res 2020;8:479–92. https://doi.org/10.1158/2326-6066.cir-19-0702.Search in Google Scholar
87. Yuan, X, Ruan, W, Bobrow, B, Carmeliet, P, Eltzschig, HK. Targeting hypoxia-inducible factors: therapeutic opportunities and challenges. Nat Rev Drug Discov 2024;23:175–200. https://doi.org/10.1038/s41573-023-00848-6.Search in Google Scholar PubMed PubMed Central
88. Xu, S, Chaudhary, O, Rodríguez-Morales, P, Sun, X, Chen, D, Zappasodi, R, et al.. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8+ T cells in tumors. Immunity 2021;54:1561–77.e7. https://doi.org/10.1016/j.immuni.2021.05.003.Search in Google Scholar PubMed PubMed Central
89. Robinson, GA, Waddington, KE, Pineda-Torra, I, Jury, EC. Transcriptional regulation of T-Cell lipid metabolism: implications for plasma membrane lipid rafts and T-Cell function. Front Immunol 2017;8:1636. https://doi.org/10.3389/fimmu.2017.01636.Search in Google Scholar PubMed PubMed Central
90. Kidani, Y, Elsaesser, H, Hock, MB, Vergnes, L, Williams, KJ, Argus, JP, et al.. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol 2013;14:489–99. https://doi.org/10.1038/ni.2570.Search in Google Scholar PubMed PubMed Central
91. Hukelmann, JL, Anderson, KE, Sinclair, LV, Grzes, KM, Murillo, AB, Hawkins, PT, et al.. The cytotoxic T cell proteome and its shaping by the kinase mTOR. Nat Immunol 2016;17:104–12. https://doi.org/10.1038/ni.3314.Search in Google Scholar PubMed PubMed Central
92. Lim, SA, Wei, J, Nguyen, TM, Shi, H, Su, W, Palacios, G, et al.. Lipid signalling enforces functional specialization of T(reg) cells in tumours. Nature 2021;591:306–11. https://doi.org/10.1038/s41586-021-03235-6.Search in Google Scholar PubMed PubMed Central
93. Wen, YA, Xiong, X, Zaytseva, YY, Napier, DL, Vallee, E, Li, AT, et al.. Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell Death Dis 2018;9:265. https://doi.org/10.1038/s41419-018-0330-6.Search in Google Scholar PubMed PubMed Central
94. Wang, Y. Applications of lipidomics in tumor diagnosis and therapy. Adv Exp Med Biol 2021;1316:25–39. https://doi.org/10.1007/978-981-33-6785_2_2.Search in Google Scholar
95. Weisel, FJ, Mullett, SJ, Elsner, RA, Menk, AV, Trivedi, N, Luo, W, et al.. Germinal center B cells selectively oxidize fatty acids for energy while conducting minimal glycolysis. Nat Immunol 2020;21:331–42. https://doi.org/10.1038/s41590-020-0598-4.Search in Google Scholar PubMed PubMed Central
96. Zhou, X, Zhu, X, Li, C, Li, Y, Ye, Z, Shapiro, VS, et al.. Stearoyl-CoA desaturase-mediated monounsaturated fatty acid availability supports humoral immunity. Cell Rep 2021;34:108601. https://doi.org/10.1016/j.celrep.2020.108601.Search in Google Scholar PubMed PubMed Central
97. Rockett, BD, Salameh, M, Carraway, K, Morrison, K, Shaikh, SR. n-3 PUFA improves fatty acid composition, prevents palmitate-induced apoptosis, and differentially modifies B cell cytokine secretion in vitro and ex vivo. J Lipid Res 2010;51:1284–97. https://doi.org/10.1194/jlr.m000851.Search in Google Scholar
98. Chen, HJ, Zhang, S, Luo, QD, Li, J. Research advance on the immune escape of acute myeloid leukemia--review (in Chinese). J Exp Hematol 2020;28:1429–32. https://doi.org/10.19746/j.cnki.issn.1009-2137.2020.04.060.Search in Google Scholar PubMed
99. Ostrand-Rosenberg, S, Fenselau, C. Myeloid-derived suppressor cells: immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J Immunol 2018;200:422–31. https://doi.org/10.4049/jimmunol.1701019.Search in Google Scholar PubMed PubMed Central
100. Veglia, F, Sanseviero, E, Gabrilovich, DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol 2021;21:485–98. https://doi.org/10.1038/s41577-020-00490-y.Search in Google Scholar PubMed PubMed Central
101. Le Bourgeois, T, Strauss, L, Aksoylar, HI, Daneshmandi, S, Seth, P, Patsoukis, N, et al.. Targeting T cell metabolism for improvement of cancer immunotherapy. Front Oncol 2018;8:237. https://doi.org/10.3389/fonc.2018.00237.Search in Google Scholar PubMed PubMed Central
102. Al-Khami, AA, Zheng, L, Del Valle, L, Hossain, F, Wyczechowska, D, Zabaleta, J, et al.. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology 2017;6:e1344804. https://doi.org/10.1080/2162402x.2017.1344804.Search in Google Scholar
103. Luo, Q, Zheng, N, Jiang, L, Wang, T, Zhang, P, Liu, Y, et al.. Lipid accumulation in macrophages confers protumorigenic polarization and immunity in gastric cancer. Cancer Sci 2020;111:4000–11. https://doi.org/10.1111/cas.14616.Search in Google Scholar PubMed PubMed Central
104. Su, P, Wang, Q, Bi, E, Ma, X, Liu, L, Yang, M, et al.. Enhanced lipid accumulation and metabolism are required for the differentiation and activation of tumor-associated macrophages. Cancer Res 2020;80:1438–50. https://doi.org/10.1158/0008-5472.can-19-2994.Search in Google Scholar PubMed PubMed Central
105. Jiang, L, Fang, X, Wang, H, Li, D, Wang, X. Ovarian cancer-intrinsic fatty acid synthase prevents anti-tumor immunity by disrupting tumor-infiltrating dendritic cells. Front Immunol 2018;9:2927. https://doi.org/10.3389/fimmu.2018.02927.Search in Google Scholar PubMed PubMed Central
106. Peng, X, He, Y, Huang, J, Tao, Y, Liu, S. Metabolism of dendritic cells in tumor microenvironment: for immunotherapy. Front Immunol 2021;12:613492. https://doi.org/10.3389/fimmu.2021.613492.Search in Google Scholar PubMed PubMed Central
107. Cubillos-Ruiz, JR, Silberman, PC, Rutkowski, MR, Chopra, S, Perales-Puchalt, A, Song, M, et al.. ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell 2015;161:1527–38. https://doi.org/10.1016/j.cell.2015.05.025.Search in Google Scholar PubMed PubMed Central
108. Pearce, EJ, Everts, B. Dendritic cell metabolism. Nat Rev Immunol 2015;15:18–29. https://doi.org/10.1038/nri3771.Search in Google Scholar PubMed PubMed Central
109. Menzner, AK, Rottmar, T, Voelkl, S, Bosch, JJ, Mougiakakos, D, Mackensen, A, et al.. Hydrogen-peroxide synthesis and LDL-uptake controls immunosuppressive properties in monocyte-derived dendritic cells. Cancers (Basel) 2021;13. https://doi.org/10.3390/cancers13030461.Search in Google Scholar PubMed PubMed Central
110. Santos, PM, Menk, AV, Shi, J, Tsung, A, Delgoffe, GM, Butterfield, LH. Tumor-derived α-fetoprotein suppresses fatty acid metabolism and oxidative phosphorylation in dendritic cells. Cancer Immunol Res 2019;7:1001–12. https://doi.org/10.1158/2326-6066.cir-18-0513.Search in Google Scholar PubMed
111. Son, Y, Kim, SM, Lee, SA, Eo, SK, Kim, K. Oxysterols induce transition of monocytic cells to phenotypically mature dendritic cell-like cells. Biochem Biophys Res Commun 2013;438:161–8. https://doi.org/10.1016/j.bbrc.2013.07.046.Search in Google Scholar PubMed
112. Son, Y, Choi, J, Kim, B, Park, YC, Eo, SK, Cho, HR, et al.. Cyclosporin A inhibits differentiation and activation of monocytic cells induced by 27-hydroxycholesterol. Int Immunopharmacol 2019;69:358–67. https://doi.org/10.1016/j.intimp.2019.01.045.Search in Google Scholar PubMed
113. Bonacina, F, Coe, D, Wang, G, Longhi, MP, Baragetti, A, Moregola, A, et al.. Myeloid apolipoprotein E controls dendritic cell antigen presentation and T cell activation. Nat Commun 2018;9:3083. https://doi.org/10.1038/s41467-018-05322-1.Search in Google Scholar PubMed PubMed Central
114. Ramakrishnan, R, Tyurin, VA, Veglia, F, Condamine, T, Amoscato, A, Mohammadyani, D, et al.. Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J Immunol 2014;192:2920–31. https://doi.org/10.4049/jimmunol.1302801.Search in Google Scholar PubMed PubMed Central
115. Chen, Y, Sui, M. Lipid metabolism in tumor-associated natural killer cells. Adv Exp Med Biol 2021;1316:71–85. https://doi.org/10.1007/978-981-33-6785_2_5.Search in Google Scholar
116. Viel, S, Besson, L, Charrier, E, Marçais, A, Disse, E, Bienvenu, J, et al.. Alteration of natural killer cell phenotype and function in obese individuals. Clin Immunol 2017;177:12–7. https://doi.org/10.1016/j.clim.2016.01.007.Search in Google Scholar PubMed
117. Michelet, X, Dyck, L, Hogan, A, Loftus, RM, Duquette, D, Wei, K, et al.. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat Immunol 2018;19:1330–40. https://doi.org/10.1038/s41590-018-0251-7.Search in Google Scholar PubMed
118. Melum, E, Jiang, X, Baker, KD, Macedo, MF, Fritsch, J, Dowds, CM, et al.. Control of CD1d-restricted antigen presentation and inflammation by sphingomyelin. Nat Immunol 2019;20:1644–55. https://doi.org/10.1038/s41590-019-0504-0.Search in Google Scholar PubMed PubMed Central
119. Nardi, F, Fitchev, P, Brooks, KM, Franco, OE, Cheng, K, Hayward, SW, et al.. Lipid droplet velocity is a microenvironmental sensor of aggressive tumors regulated by V-ATPase and PEDF. Lab Invest 2019;99:1822–34. https://doi.org/10.1038/s41374-019-0296-8.Search in Google Scholar PubMed PubMed Central
120. Zhang, J, Wang, H, Tian, Y, Li, T, Zhang, W, Ma, L, et al.. Discovery of a novel lipid metabolism-related gene signature to predict outcomes and the tumor immune microenvironment in gastric cancer by integrated analysis of single-cell and bulk RNA sequencing. Lipids Health Dis 2023;22:212. https://doi.org/10.1186/s12944-023-01977-y.Search in Google Scholar PubMed PubMed Central
121. Zeng, J, Tan, H, Huang, B, Zhou, Q, Ke, Q, Dai, Y, et al.. Lipid metabolism characterization in gastric cancer identifies signatures to predict prognostic and therapeutic responses. Front GenetA 2022;13:959170. https://doi.org/10.3389/fgene.2022.959170.Search in Google Scholar PubMed PubMed Central
122. Yang, Q, Kong, S, Yu, J, Xu, Y, Tao, M, Ma, S, et al.. MicroRNA miR-1275 coordinately regulates AEA/LPA signals via targeting FAAH in lipid metabolism reprogramming of gastric cancer. Cell Death Dis 2023;14:62. https://doi.org/10.1038/s41419-023-05584-8.Search in Google Scholar PubMed PubMed Central
123. Wang, S, Huang, X, Zhao, S, Lv, J, Li, Y, Wang, S, et al.. Progressions of the correlation between lipid metabolism and immune infiltration characteristics in gastric cancer and identification of BCHE as a potential biomarker. Front Immunol 2024;15:1327565. https://doi.org/10.3389/fimmu.2024.1327565.Search in Google Scholar PubMed PubMed Central
124. Dai, J, Li, Q, Quan, J, Webb, G, Liu, J, Gao, K. Construction of a lipid metabolism-related and immune-associated prognostic score for gastric cancer. BMC Med Genom 2023;16:93. https://doi.org/10.1186/s12920-023-01515-w.Search in Google Scholar PubMed PubMed Central
125. Zhou, X, Meng, F, Xiao, L, Shen, H. CYP19A1 promotes gastric cancer as part of a lipid metabolism-related gene signature related to the response of immunotherapy and prognosis. BMC Med Genom 2023;16:228. https://doi.org/10.1186/s12920-023-01664-y.Search in Google Scholar PubMed PubMed Central
126. Gao, X, Xu, N, Li, Z, Shen, L, Ji, K, Zheng, Z, et al.. Safety and antitumour activity of cadonilimab, an anti-PD-1/CTLA-4 bispecific antibody, for patients with advanced solid tumours (COMPASSION-03): a multicentre, open-label, phase 1b/2 trial. Lancet Oncol 2023;24:1134–46. https://doi.org/10.1016/s1470-2045-23-00411-4.Search in Google Scholar
127. Duan, J, Chen, L, Zhou, M, Zhang, J, Sun, L, Huang, N, et al.. MACC1 decreases the chemosensitivity of gastric cancer cells to oxaliplatin by regulating FASN expression. Oncol Rep 2017;37:2583–92. https://doi.org/10.3892/or.2017.5519.Search in Google Scholar PubMed PubMed Central
128. Castagnoli, L, Corso, S, Franceschini, A, Raimondi, A, Bellomo, SE, Dugo, M, et al.. Fatty acid synthase as a new therapeutic target for HER2-positive gastric cancer. Cell Oncol (Dordr) 2023;46:661–76. https://doi.org/10.1007/s13402-023-00769-x.Search in Google Scholar PubMed PubMed Central
129. Zhao, L, Peng, Y, He, S, Li, R, Wang, Z, Huang, J, et al.. Apatinib induced ferroptosis by lipid peroxidation in gastric cancer. Gastric Cancer 2021;24:642–54. https://doi.org/10.1007/s10120-021-01159-8.Search in Google Scholar PubMed
130. Wong, TL, Loh, JJ, Lu, S, Yan, HHN, Siu, HC, Xi, R, et al.. ADAR1-mediated RNA editing of SCD1 drives drug resistance and self-renewal in gastric cancer. Nat Commun 2023;14:2861. https://doi.org/10.1038/s41467-023-38581-8.Search in Google Scholar PubMed PubMed Central
131. He, W, Liang, B, Wang, C, Li, S, Zhao, Y, Huang, Q, et al.. MSC-regulated lncRNA MACC1-AS1 promotes stemness and chemoresistance through fatty acid oxidation in gastric cancer. Oncogene 2019;38:4637–54. https://doi.org/10.1038/s41388-019-0747-0.Search in Google Scholar PubMed PubMed Central
132. Chen, Z, Xu, P, Wang, X, Li, Y, Yang, J, Xia, Y, et al.. MSC-NPRA loop drives fatty acid oxidation to promote stemness and chemoresistance of gastric cancer. Cancer Lett 2023;565:216235. https://doi.org/10.1016/j.canlet.2023.216235.Search in Google Scholar PubMed
133. Wang, Y, Cai, L, Li, H, Chen, H, Yang, T, Tan, Y, et al.. Overcoming cancer resistance to platinum drugs by inhibiting cholesterol metabolism. Angew Chem Int Ed Engl 2023;62:e202309043. https://doi.org/10.1002/ange.202309043.Search in Google Scholar
134. Yan, A, Jia, Z, Qiao, C, Wang, M, Ding, X. Cholesterol metabolism in drug-resistant cancer (Review). Int J Oncol 2020;57:1103–15. https://doi.org/10.3892/ijo.2020.5124.Search in Google Scholar PubMed
135. Subramanian, N, Schumann-Gillett, A, Mark, AE, O’Mara, ML. Understanding the accumulation of P-glycoprotein substrates within cells: the effect of cholesterol on membrane partitioning. Biochim Biophys Acta 2016;1858:776–82. https://doi.org/10.1016/j.bbamem.2015.12.025.Search in Google Scholar PubMed
136. Liu, C, Chikina, M, Deshpande, R, Menk, AV, Wang, T, Tabib, T, et al.. Treg cells promote the SREBP1-dependent metabolic fitness of tumor-promoting macrophages via repression of CD8+ T cell-derived interferon-γ. Immunity 2019;51:381–97.e6. https://doi.org/10.1016/j.immuni.2019.06.017.Search in Google Scholar PubMed PubMed Central
137. Jenke, R, Oliinyk, D, Zenz, T, Körfer, J, Schäker-Hübner, L, Hansen, FK, et al.. HDAC inhibitors activate lipid peroxidation and ferroptosis in gastric cancer. Biochem Pharmacol 2024;225:116257. https://doi.org/10.1016/j.bcp.2024.116257.Search in Google Scholar PubMed
138. Iannelli, F, Lombardi, R, Milone, MR, Pucci, B, De Rienzo, S, Budillon, A, et al.. Targeting mevalonate pathway in cancer treatment: repurposing of statins. Recent Pat Anticancer Drug Discov 2018;13:184–200. https://doi.org/10.2174/1574892812666171129141211.Search in Google Scholar PubMed
139. Ahmadi, Y, Ghorbanihaghjo, A, Argani, H. The balance between induction and inhibition of mevalonate pathway regulates cancer suppression by statins: a review of molecular mechanisms. Chem Biol Interact 2017;273:273–85. https://doi.org/10.1016/j.cbi.2017.06.026.Search in Google Scholar PubMed
140. Sharpe, LJ, Brown, AJ. Controlling cholesterol synthesis beyond 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR). J Biol Chem 2013;288:18707–15. https://doi.org/10.1074/jbc.r113.479808.Search in Google Scholar PubMed PubMed Central
141. Wang, Y, Lu, JH, Wang, F, Wang, YN, He, MM, Wu, QN, et al.. Inhibition of fatty acid catabolism augments the efficacy of oxaliplatin-based chemotherapy in gastrointestinal cancers. Cancer Lett 2020;473:74–89. https://doi.org/10.1016/j.canlet.2019.12.036.Search in Google Scholar PubMed
142. Li, Z, Liu, C, Wang, M, Wei, R, Li, R, Huang, K, et al.. Cholesterol confers resistance to Apatinib-mediated ferroptosis in gastric cancer. Cell Biosci 2025;15:95. https://doi.org/10.1186/s13578-025-01435-5.Search in Google Scholar PubMed PubMed Central
143. Fan, S, Han, H, Yan, Z, Lu, Y, He, B, Zhang, Q. Lipid-based nanoparticles for cancer immunotherapy. Med Rev (2021) 2023;3:230–69. https://doi.org/10.1515/mr-2023-0020.Search in Google Scholar PubMed PubMed Central
144. Zhang, M, Wei, T, Zhang, X, Guo, D. Targeting lipid metabolism reprogramming of immunocytes in response to the tumor microenvironment stressor: a potential approach for tumor therapy. Front Immunol 2022;13:937406. https://doi.org/10.3389/fimmu.2022.937406.Search in Google Scholar PubMed PubMed Central
145. Ping, Y, Shan, J, Qin, H, Li, F, Qu, J, Guo, R, et al.. PD-1 signaling limits expression of phospholipid phosphatase 1 and promotes intratumoral CD8+ T cell ferroptosis. Immunity 2024;57:2122–39.e9. https://doi.org/10.1016/j.immuni.2024.08.003.Search in Google Scholar PubMed
Supplementary Material
This article contains supplementary material (https://doi.org/10.1515/mr-2025-0035).
© 2025 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
Articles in the same Issue
- Frontmatter
- Reviews
- Artificial intelligence-driven transformative applications in disease diagnosis technology
- Recent advances in understanding the relationship between lipid metabolism and immune escape in the tumor microenvironment of gastric cancer
- Transcript diversity in aging: cryptic transcription and splicing
- Original Article
- Outcomes of revised portoenterostomy for postoperative bile lakes in patients with biliary atresia
- Perspectives
- Orchestration of tertiary lymphoid structures: decoding developmental mechanisms for next-generation cancer immunotherapies
- Trophoblast-derived exosomes containing PD-L1 may have protective effects on preeclampsia by regulating Tregs
- Lac-Phe: a central metabolic regulator and biomarker
- Unfolding ecDNA as a pan-cancer therapeutic target
Articles in the same Issue
- Frontmatter
- Reviews
- Artificial intelligence-driven transformative applications in disease diagnosis technology
- Recent advances in understanding the relationship between lipid metabolism and immune escape in the tumor microenvironment of gastric cancer
- Transcript diversity in aging: cryptic transcription and splicing
- Original Article
- Outcomes of revised portoenterostomy for postoperative bile lakes in patients with biliary atresia
- Perspectives
- Orchestration of tertiary lymphoid structures: decoding developmental mechanisms for next-generation cancer immunotherapies
- Trophoblast-derived exosomes containing PD-L1 may have protective effects on preeclampsia by regulating Tregs
- Lac-Phe: a central metabolic regulator and biomarker
- Unfolding ecDNA as a pan-cancer therapeutic target