Genetic advances in neurodevelopmental disorders
-
Shilin Gao
 and
Tianyun Wang
and
Tianyun Wang


Abstract
Neurodevelopmental disorders (NDDs) are a group of highly heterogeneous diseases that affect children’s social, cognitive, and emotional functioning. The etiology is complicated with genetic factors playing an important role. During the past decade, large-scale whole exome sequencing (WES) and whole genome sequencing (WGS) have vastly advanced the genetic findings of NDDs. Various forms of variants have been reported to contribute to NDDs, such as de novo mutations (DNMs), copy number variations (CNVs), rare inherited variants (RIVs), and common variation. By far, over 200 high-risk NDD genes have been identified, which are involved in biological processes including synaptic function, transcriptional and epigenetic regulation. In addition, monogenic, oligogenic, polygenetic, and omnigenic models have been proposed to explain the genetic architecture of NDDs. However, the majority of NDD patients still do not have a definitive genetic diagnosis. In the future, more types of risk factors, as well as noncoding variants, are await to be identified, and including their interplay mechanisms are key to resolving the etiology and heterogeneity of NDDs.
Introduction
Neurodevelopmental disorders (NDDs) are a group of disorders that are highly heterogeneous, primarily including autism spectrum disorder (ASD), developmental delay (DD), attention-deficit/hyperactivity disorder (ADHD), and intellectual disability (ID) [1]. These disorders share some common characteristics, and the phenotypes highly overlap to each other. Sex bias in prevalence is common, with a male-to-female ratio ranging from 1.2:1 to 4:1 [2]. The etiology of NDD is very complex, both genetic and environmental factors contribute to it. In 1977, a twin study conducted by Rutter and Folstein revealed that the concordance rate for ASD was notably elevated in monozygotic twins compared to dizygotic twins, suggesting that genetic factors may play an important role in ASD [3], and the heritability is approximately 60–90 % [4], [5], [6]. However, the actual genetic factors were still largely unknown back then. Mostly in the past decade, the advancement of high throughput sequencing technology enabled us to explore the genetic etiology of NDDs. Among these, WES and WGS have been widely used [7]. Such studies have demonstrated that various forms of genetic variations can contribute to the risk for NDDs, including both rare and common variations [8], 9]. Large-scale genomic studies have identified over 200 high-risk genes and loci, yet only ∼30 % of NDD cases could be attributed to known genetic risk [10], [11], [12], [13], and more genetic factors are yet to be identified. In the future, genetic research still remain crucial to fully decipher the etiology and heterogeneity of NDD.
De novo mutations
In 2007, Sebat and colleagues found that individuals with ASD show an increased frequency of de novo CNVs compared to unaffected individuals [14]. Following in 2012, four landmark studies have shown consistent results with larger sample sizes through WES [13], [15], [16], [17], [18] and found de novo single nucleotide variants (SNVs) and short insertion and deletions (Indels) are also more frequent in individuals with ASD than in their unaffected siblings. Of these, the de novo likely gene-disruptive (dnLGD, including frameshift, stop-gain, splice-donor, and splice-acceptor) variants are particularly significant. It is estimated that the burden of dnLGD variants is nearly two times higher in ASD probands compared to their unaffected siblings, and this enrichment is more pronounced in patients with DD [19], [20], [21]. The majority of dnLGD variants lead to the loss-of-function of genes and have a large impact on the phenotype of carriers [13]. In contrast to dnLGD variants, de novo missense (dnMIS) variants have the potential to cause gain-of-function of genes. Several approaches have been developed to evaluate the effect of missense mutations on genes, such as the CADD [22] and MPC scores [23]. However, the effect of missense mutations on gene function still needs to be further explored. Some studies have reported that missense variants tend to cluster in specific regions of certain genes, and these regions where the clustering of missense variants are defined as “hotspots” [24], [25], [26]. Genes associated with ion channel are examples that are implicated by hotspots, such as GRIN1, GRIN2B, KCNK3, and KCNQ2. The patients with NDDs tend to carry more mutations of hotspot genes and these variants are predicted to be more pathogenic, suggesting that in addition to the enrichment of variants, the clustering of variants is also helpful in identifying candidate genes.
Rare inherited variants
On average, the effect size of inherited variants is much lower than DNM [27]. The overall mutation rate of inherited LGD or CNVs in probands has no significant difference from that of unaffected siblings, however, the private or ultra-rare inherited LGD variants in intolerant genes are significantly enriched in ASD probands [28]. Wilfert et al. found that ASD children in multiplex families are more likely to carry multiple private inherited variants than those in simplex families, supporting a multi-hits model for ASD [29]. By combining DNM with RIVs can also increase the power of identifying NDD genes [30]. TADA, a Bayesian algorithm that integrates de novo and inherited mutations, has been widely used for identifying risk genes [12], [31], [32], [33], [34], [35]. In a recent study with 42,607 ASD cases, a meta-analysis of DNMs and RIVs was performed and identified five novel risk genes (NAV3, ITSN1, MARK2, SCAF1, and HNRNPUL2) [27]. Among these, the association of NAV3 with ASD is mainly driven by RIVs. ITSN1, SCAF1 and HNRNPUL2 have support from both DNM and RIVs. These genes implicated by RIVs have a moderate effect on carriers. This also implies that identifying novel risk genes only through DNM becomes challenging, even in a large sample size.
Copy number variations
CNVs are deletions and duplications of DNA sequence ranging considerably in size from kilobases (Kb) to megabases (Mb), often spanning many different genes. The initial large-scale discovery of CNVs in NDDs, namely CNV MorbidityMap, was facilitated by the array comparative genomic hybridization (arrayCGH) technique around 2010 [36]. An excessive rate of large CNVs (>250 kb) among NDD individuals relative to controls was validated, consistent with previous studies, although a similar trend in smaller CNVs could not be confirmed due to low detection sensitivity [14], 37]. Following this, an integrated analysis of CNV and SNV data pinpointed several genes enriched for putative loss of function, including DYRK1A and SCN1A [38]. The effect size of CNVs is relatively larger than SNVs and indels [9]. Overall, deletions (odds ratio (OR)=5.1) have a larger effect size than duplications (OR=1.8) [38], 39]. The effect of CNVs is primarily driven by the dosage-sensitive genes they contain [40], and the genes affected by CNVs in individuals with NDDs are more likely to be intolerant to variation. Several recurrent CNV loci associated with NDDs have been identified, such as 1q21.1, 3q29, 7q11.23, 15q11.2, 16p11.2, and 22q11.2, with estimates suggesting that 20 % of patients with ASD carry at least one CNV [31]. Of these, 16p11.2 is the most studied risk locus of NDDs. Both deletions and duplications of 16p11.2 can lead to phenotypes associated with NDDs, including social impairment, repetitive behaviors, language delay and intelligence disability [41]. Sometimes, CNVs of a locus have a mirror effect on carriers, with 1q21.2 and 7q11.23 deletions causing microcephaly and 1q21.2 and 7q11.23 duplications causing macrocephaly [42], 43]. The findings of dosage-dependent effects of CNVs have also been demonstrated on social behavior for 7q11.23, where deletions are linked to social disinhibition and duplications to social anxiety [44].
Common variations
Rare variants tend to be more pathogenic under the pressure of natural selection, yet most patients with NDDs do not carry meaningful rare variants. Common variations also contribute to NDDs. It is estimated that nearly 50 % of the heritability of ASD can be explained by single nucleotide polymorphisms (SNPs) in genome-wide association studies (GWAS) [45]. SNP heritability varied greatly across studies due to phenotypic heterogeneity. Generally, autistic individuals without ID had a higher SNP heritability than autistic individuals with ID, and SNP heritability of males with ASD is higher than females [46]. The largest ASD GWAS study including 18,381 individuals of ASD and 27,969 controls identified five genome-wide significant loci (rs910805, rs10099100, rs201910565, rs71190156, and rs111931861) [47]. Another GWAS study with 34,462 cases and 41,201 controls, combining ADHD and ASD into a single phenotype, identified seven genetic loci shared by ADHD and ASD, as well as five loci that differentiate between them [48].
The investigation of common variation can also reveal correlations between NDDs and other diseases or population traits, such as the application of polygenic risk scores (PRS). Multiple studies have demonstrated that individuals with ASD tend to inherit an excess of the polygenic risk of ADHD, schizophrenia, and educational attainment [49]. One of the most intriguing findings is the association between educational attainment and ASD. Consistent with this, epidemiological studies have suggested that there is a significant association between high paternal IQ and offspring risk of ASD without ID/ADHD [50]. A recent study indicates that parental educational attainment might increase the risk of ASD in offspring through its impact on parental reproductive age [51]. Specifically, higher parental educational attainment often correlates with elevated parental reproductive age, resulting in an increased accumulation of variations in reproductive cells, consequently raising the risk of ASD in offspring.
Large cohorts boosted the discovery of NDD genes
Over the past decade, landmark cohorts have been built and largely boosted the discovery of NDD genes, which primarily include SSC, ASC, MSSNG, SPARK, and DDD (Table 1). Early genetic studies on NDDs mainly involved only a few hundred samples (Figure 1). In 2012, four groups identified several high-confidence risk genes (e.g., CHD8, DYRK1A, GRIN2B, PTEN) with recurrent DNMs through WES of nearly 1,000 SSC families in total [16], [17], [18, 52]. These genes were also validated by later large-scale genomic studies, and mutating these genes in animal models can mimic NDD-associated behaviors [53], [54], [55], [56]. In recent years, the sample size has largely increased (Table 2). Kaplanis et al. identified 285 DD genes by integrating DNMs from 31,058 families with DD, and 28 of these genes had not been identified by previous studies [21]. In 2022, ASC, MSSNG, and SPARK all published latest large-scale genomic studies for identifying novel genes associated with NDDs [12], 27], 35].
Major neurodevelopmental disorders cohorts.
| Cohort | Organization | Sample size | NGS | Major publications |
|---|---|---|---|---|
| SSC | Simons Foundation | 2,508 ASD families | WES/WGS | Iossifov et al. 2014 [13] |
| Krumm et al. 2015 [28] | ||||
| Coe et al. 2019 [10] | ||||
| SPARK | Simons Foundation | 10,6744 individuals | WES/WGS | Feliciano et al. 2019 [123] |
| (44,304 ASD cases) | Zhou et al. 2022 [27] | |||
| Wang et al. 2022 [11] | ||||
| ASC | Icahn School of Medicine at Mount Sinai | 6,430 ASD families | WES/WGS | Sanders et al. 2015 [31] |
| Broad Institute | Satterstrom et al. 2020 [34] | |||
| University of California, San Francisco | Fu et al. 2022 [12] | |||
| MSSNG | Autism speaks | 11,312 individuals | WGS | Yuen et al., 2017 [124] |
| Hospital for sick children | (5,100 ASD cases) | Trost et al. 2022 [35] | ||
| KAGD | Seoul National Bundang Hospital | 931 individuals | WGS | Kim et al. 2022 [57] |
| (276 ASD cases) | ||||
| JASD | Yokohama City University | 262 ASD cases | WES | Takata et al. 2018 [32] |
| SHXH&SHMC | Shanghai Xinhua Hospital, | 1,141 ASD families | WES | Wang et al. 2023 [59] |
| Shanghai Mental Health Center | Yuan et al. 2023 [58] | |||
| DDD | Wellcome Sanger Institute | 32,233 individuals | WES | Kaplanis et al. 2020 [21] |
| (9,852 DD cases) | Wright et al. 2023 [125] | |||
| RUMC | Radboud University Medical Center | 7,254 individuals | WES | Kaplanis et al. 2020 [21] |
| (2,417 DD cases) | ||||
| GeneDx | GeneDx | 56,367 individuals | WES | Kaplanis et al. 2020 [21] |
| (18,783 DD cases) |
-
NGS, next generation sequencing; WES, whole exome sequencing; WGS, whole genome sequencing; ASD, autism spectrum disorder; DD, developmental delay.

Progress in NDD research. Number of individuals (A) and genes (B) increasing with cohort studies since 2012. (C) The timeline of major NDD studies since 2007. NDD, neurodevelopmental disorders.
Summary of large-scale genetics studies of neurodevelopmental disorders.
| Studies | Samples | NGS | Transmission type | Variant class | Statistical methods | Statistical threshold | Significant genes |
|---|---|---|---|---|---|---|---|
| Ruzzo et al. 2019 [33] | 13,189 ASD cases | WGS | DNMs & RIVs | SNVs, indels | TADA | FDR<0.1 | 69 |
| Kaplanis et al. 2020 [21] | 31,058 DD trios | WES | DNMs | SNVs, indels | DeNovoWEST | FWER<0.05 | 285 |
| Fu et al. 2022 [12] | 15,036 ASD trios 31,058 DD trios |
WES | DNMs & RIVs | SNVs, indels, CNVs | TADA | FDR<0.05 | 664 |
| Trost et al. 2022 [35] | 16,805 ASD trios, 5,556 cases, 8,809 control |
WGS | DNMs & RIVs | SNVs, indels | TADA | FDR<0.1 | 134 |
| Wang et al. 2022 [11] | 46,612 NDD trios | WES | DNMs | SNVs, indels | CH, DR, DeNovoWEST | FDR<0.05 | 615 |
| Zhou et al. 2022 [27] | 42,607 ASD cases | WES | DNMs & RIVs | SNVs, indels | Meta | FWER<0.05 | 60 |
-
ASD, autism spectrum disorder; DD, developmental delay; NGS, next generation sequencing; WES, whole exome sequencing; WGS, whole genome sequencing; DNMs, de novo mutations; CNVs, copy number variations; RIVs, rare inherited variants; SNVs, single nucleotide variants.
However, these projects primarily consist of samples from European and American regions, lacking representation from Asian and African populations. Recently, Asian regions such as South Korea, Japan, and China have also begun to establish NDD cohorts. KAGD, a South Korean ASD cohort, has completed WGS for 276 ASD families [57]. A Japanese group has also published a WES study containing 262 Japanese families with ASD [32]. In China, Qiu and his team published two WES studies of ASD families [58], 59], the samples for these two studies consisted of more than 1,000 Chinese families with ASD, where they identified ASD genes that have not been identified based on European and American ASD cohorts, suggesting that there may be genetic differences across diverse ethnic populations.
Taken together, over 200 high-confidence risk genes have been identified by large-scale genomics studies in the past decade (Figure 2), it is estimated that more than 1,000 genes are associated with NDDs. To date, SFARI Gene has included 1,176 genes associated with ASD (https://gene.sfari.org/, released on March 28, 2024), of which, 233 genes are listed as “high confidence” [60], and DDG2P (https://www.ebi.ac.uk/gene2phenotype/, retrieved on July 16, 2024) has also included 2,722 DD genes [61]. Collectively, these two major databases have included about 3,000 candidate genes of NDDs, with nearly 500 genes shared in both databases.
![Figure 2:
Genes overlap across major studies on NDDs. The circus layout represents overlapping genes across three main studies on chromosomes. Inner red genes constitute the intersection of the highest-confidence genes identified in all three studies (Fu et al. [12]. “FDR≤0.001”, Wang et al. [11]. “FWER≤0.05”, and Trost et al. [35]. “FDR≤0.1”), while the outer blue genes present in two out of three gene sets. The central Venn diagram illustrates the counts of overlapping genes among the three distinct highest-confidence gene sets. NDD, neurodevelopmental disorders.](/document/doi/10.1515/mr-2024-0040/asset/graphic/j_mr-2024-0040_fig_002.jpg)
Genes overlap across major studies on NDDs. The circus layout represents overlapping genes across three main studies on chromosomes. Inner red genes constitute the intersection of the highest-confidence genes identified in all three studies (Fu et al. [12]. “FDR≤0.001”, Wang et al. [11]. “FWER≤0.05”, and Trost et al. [35]. “FDR≤0.1”), while the outer blue genes present in two out of three gene sets. The central Venn diagram illustrates the counts of overlapping genes among the three distinct highest-confidence gene sets. NDD, neurodevelopmental disorders.
Signaling pathway of NDD genes
NDD genes are involved in signaling pathways primarily including MAPK, mTOR, and Wnt signaling pathways. MAPK signaling pathway plays an important role in extensive development processes, involving neurogenesis, gliogenesis, cortical development, etc [62]. The progenitors undergo either proliferative or neurogenic divisions. Proliferative divisions determine the size of the cortex while neurogenic divisions generating neurons determinescortical thickness [31]. The MAPK pathway modulates neurogenic and proliferative cell division by regulating cell cycle progression via cyclins, such as cyclin D1 and cyclin E [63]. The loss of MAPK1 would cause precocity of G1-phase of neurogenic progenitors, and favor neurogenic divisions in developing telencephalon, resulting in premature depletion of progenitor pools. MAPK knockout mice show reduced pyramidal neuron population and impaired cortical neural circuitry. MAPK3, located in 16p11.2, has been identified as an ASD candidate gene by large-scale sequencing studies [64], 65], as well as other MAPK cascade related genes, such as BRAF, NTRK2, NTRK3.
The PI3K/AKT/mTOR signaling pathway is essential for multiple cellular functions related to brain and neuron development, including cell growth, survival, and autophagy [66]. Germline PTEN mutation can increase phosphorylated Akt in neurons indicating activation of the mTOR signaling pathway [67]. Haploinsufficient Pten+/− mice show brain overgrowth due to neuron hyperplasia, as well as decreased apoptosis and abnormal neuronal migration [68]. Inhibiting mTOC2 demonstrates improvement in both EEG- and ASD-related behavioral deficits in Pten knockout mice [69]. TSC syndrome is mainly caused by mutations in TSC1 and TSC2 genes, leading to increased incidence rates of ASD and DD in patients with TSC [70], 71]. Currently, drug development for TSC primarily focuses on mTOR inhibitors, such as rapamycin and everolimus [72], 73].
Wnt signaling pathway is another well-studied pathway associated with NDDs, which is crucial for the formation and development of neurons. It is involved in regulating the differentiation of neural precursor cells, establishing neuronal architecture, and promoting the growth of neuronal axons and dendrites. The β-catenin is a crucial component of Wnt signaling pathway. The encoding gene of β-catenin, CTNNB1, is a high-confidence gene of NDDs [74]. Various mutations in the CTNNB1 gene have been implicated in patients with NDDs, such as frameshift, stop-gain, and missense [75].
Protein-protein interaction of NDD genes
The functional clusters of NDD genes implicated by protein-protein interaction (PPI) network mainly included synaptic function, transcriptional regulation, and epigenetic regulation [11], 76]. Synapses connect neurons to help transmit information from one neuron to the next. Dysregulation of synaptic function can lead directly to phenotypes associated with NDDs. Mutations in some NDD genes can directly cause abnormal synaptic structure, such as synaptic cell-adhesion molecules (NLGN3, NLGN4, and NRXN1) and postsynaptic scaffolding proteins (SHANK1, SHANK2, and SHANK3). Among these genes, SHANK3 is the most well-studied, which encodes scaffold proteins of the postsynaptic density complex of excitatory synapses. SHANK3 was first implicated in NDDs by studies of PMS (Phelan–McDermid syndrome), a rare genetic condition caused by a deletion or other structural change of the terminal end of chromosome 22 in the 22q13 region or mutation of the SHANK3 [77]. SHANK3 has also been identified as a high-confidence NDD gene by several large-scale genomic studies [12], 31], 34]. Animal models of SHANK3 deficiency generated by various genetic strategies can display multiple NDD-associated behaviors, including social impairment, stereotyped behavior, motor deficits and cognitive impairment [78], 79]. Several NDD genes also encode receptors of neurotransmitters, mostly are ion channels, including GRIN2B, CACNA1C, SCN1A, and GABRB3 [55], [80], [81], [82]. Mutations in these genes lead to abnormal synaptic transmission, resulting in NDD-associated behaviors.
In addition, some NDD genes can not directly affect neuronal function but regulate the expression of other NDD genes through transcriptional regulatory networks. Generally, these genes encode DNA-binding or RNA-binding proteins. For example, CHD8, encoding the protein chromodomain helicase DNA-binding protein 8, is essential to fetal development. Mutations in CHD8 have been linked to macrocephaly of ASD cases [53], 83]. FMR1 encodes the Fragile-X mental retardation protein (FMRP), which is a highly conserved RNA-binding protein and plays a crucial role in regulating synaptic plasticity. FMR1 is the causative gene for fragile X syndrome, which is characterized by intellectual disability and is accompanied by features of ASD including social impairment and abnormal sensory processing [84]. A recent study identified significant overlap in the binding sites of multiple ASD-associated transcriptional regulators (ARID1B, BCL11A, FOXP1, TBR1, and TCF7L2) in the developing human and mouse cortex, particularly within open chromatin regions, suggesting that functional convergence across five ASD-associated transcriptional regulators lead to shared neurodevelopmental outcomes of haploinsufficient disruption [85].
Epigenetic refers to the regulation of gene expression without altering the DNA sequence of genes, including DNA methylation, histone modification, and chromatin remodeling. Epigenetic is involved in the regulation of many biological processes, such as cell differentiation, neural development, tumorigenesis and progression, and is important for linking environmental stress and gene expression. Multiple studies have revealed that epigenetic factors are involved in NDDs. MECP2, an important reader of DNA methylation, is essential for the normal function of nerve cells. Mutations of MECP2 can cause Rett syndrome, a developmental disorder which is characterized by debilitating neurodevelopment, severe mental disability and ASD-like symptoms. Several studies have revealed mutations of MECP2 in animal models can cause NDD phenotypes and reduced MECP2 expression has been found in the frontal cortex of ASD patients [86], [87], [88].
Transcriptomic analyses of NDD genes
The primary mode of action for genetic variation is through altered gene expression, so transcriptomic analysis for disease-related tissues is a complementary approach to genetic studies. NDD genes are highly expressed in early fetal brain development, and DD-predominant genes are expressed earlier compared to ASD-predominant genes [12], 51]. Cell type-specific expression analysis has revealed that NDD genes are enriched in excitatory and inhibitory neurons [11], consistent with the excitatory/inhibitory (E/I) imbalance hypothesis in NDDs [89], 90]. Several studies have performed transcriptomic analyses of post-mortem brain tissues from individuals with ASD, revealing significant differences in gene expression between ASD and neurotypical brains [91], [92], [93], [94]. In a recent RNA-sequencing analysis of 112 post-mortem samples from individuals with ASD and neurotypical controls, they found that broad transcriptomic dysregulation occurs across the cerebral cortex in ASD, especially in the primary visual cortex [95]. Regional patterns of gene expression in ASD brains that normally distinguish between the frontal and temporal lobes are markedly diminished, suggesting abnormal cortical patterns in ASD. One of the most consistent findings in bulk tissue transcriptomic analyses of ASD is the elevated expression of gene modules associated with astrocyte, microglial, and inflammatory function.
However, there is no evidence that ASD-elevated glia/immune-associated modules are enriched for ASD genetic risk factors. In contrast, the downregulated genes are significantly associated with synaptic function and these genes are enriched for both common and rare variants, indicating a genetic etiology for this process [91], 92]. As for sex, higher expression of glial/immune-related gene modules has been observed in males, while in females were neuron-associated gene modules. In general, ASD risk genes are not systematically expressed differentially between sexes, but they may interact with characteristic sexually dimorphic pathways [96], 97]. These results suggest that naturally occurring sexually dimorphic processes modulate the impact of risk variants and contribute to the sex-biased prevalence of ASD. The main challenge of these transcriptomic studies is the scarcity of available samples, especially female donors, for further validation, and they are mainly focused on the cortex. Many subcortical structures associated with NDDs, such as the amygdala, hippocampus, and hypothalamus, remain to be further investigated.
Genetic models of NDD
The phenotypic spectrum of NDDs can be attributable to the combined effects of multiple genetic variations. Risk variants in general population also cause milder phenotypes or lower socioeconomic status, such as decreased intelligence, slower reaction times, lower numeric memory scores and an increase in metrics related to material deprivation [98], 99]. Several genetic models have been proposed to explain the phenotype of NDDs (Figure 3), from monogenic model to omnigenic model [8], 9]. Some variants have a large effect size, such as CNVs, where a single variant can cause the carrier to develop NDDs. This model can be defined as a monogenic model. Under the pressure of natural selection, such variants are often de novo and can’t be transmitted to offspring. In contrast, the polygenic model posits a cumulative effect of thousands of minor variants, where these variants often have an allele frequency greater than 0.01 and can typically be transmitted to offspring. Recently, some studies support an oligogenic model of NDDs [29], 100]. NDD patients from multiplex families tend to carry mutations in multiple genes. Individually, these mutations might not have sufficient impact to cause NDDs. However, when two or more mutations accumulate, they can collectively lead to NDDs. It is estimated that these variants are approximately 2.5 generations old and significantly younger than other variants of similar type and frequency in siblings.
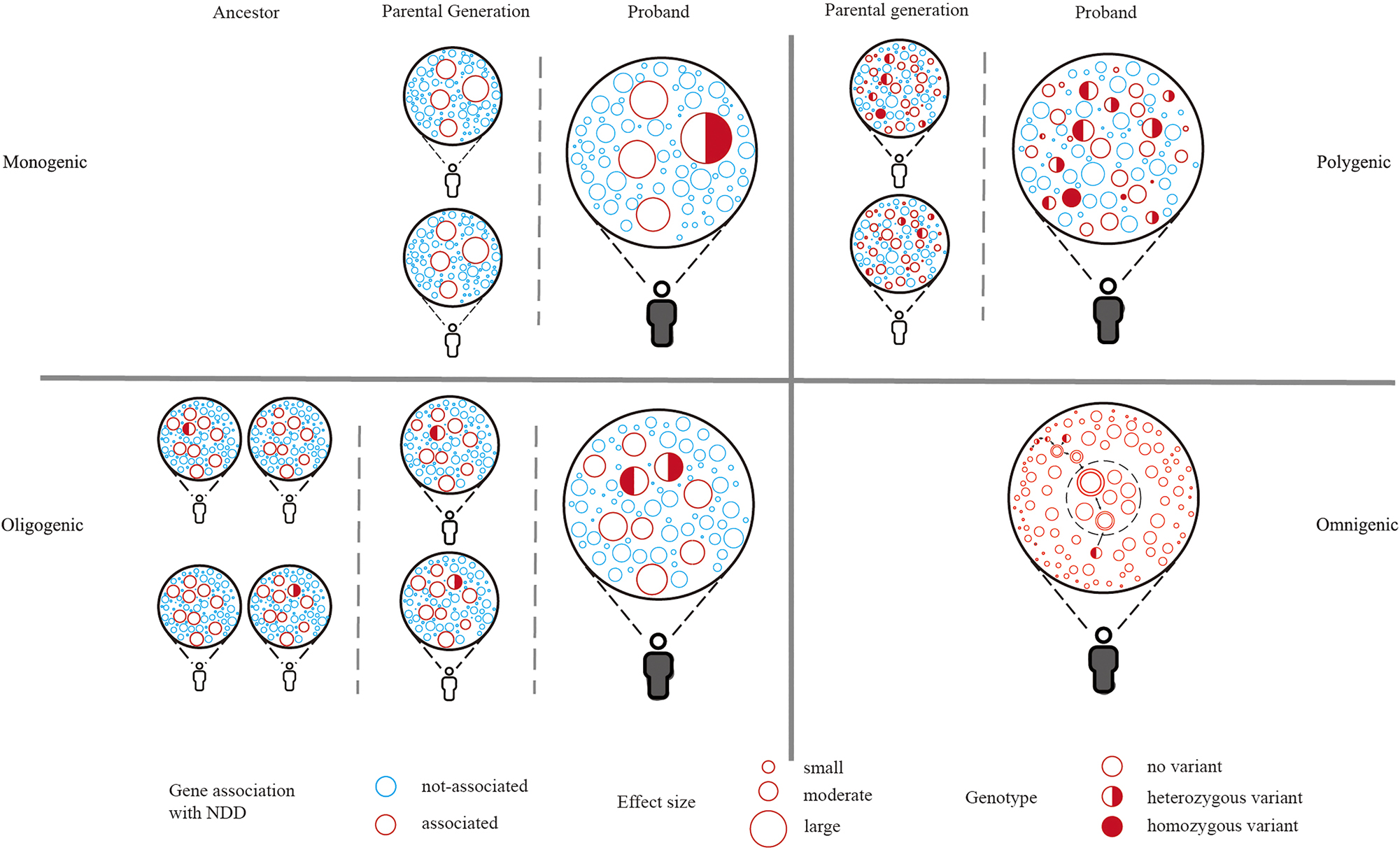
Genetic models of NDD. The diagram illustrates the genetic architecture of NDD, with each circle symbolizing a distinct gene. The monogenic model posits a single variant with a substantial effect size that can precipitate NDD; the oligogenic model proposes that the collection of several moderate variants can lead to NDD; the polygenic model refers to the cumulative effects of a vast array of variants that all contribute to NDD; as in the omnigenic model, all genes are considered to be associated with NDD at some level, the double-bordered circles indicate high-confidence NDD genes within a specific pathway, the genes within the inner dashed circle represent the “core genes” that can directly affect neuronal function and have a direct impact on NDD. NDD, neurodevelopmental disorders.
Furthermore, few studies suggest no NDD-specific risk genes, proposing that almost all genes, directly or indirectly, could contribute to NDDs, termed as an omnigenic model [101], [102], [103], [104], [105]. Specifically, some genes can directly affect neuronal function and have a direct impact on neurodevelopment, such as SHANK3, CTNNB1, and GRIN2B, these genes can be defined as “core genes”. Other genes that indirectly affect neurodevelopment by regulating core genes through gene regulatory networks can be defined as “peripheral genes”. However, due to the high connectivity of gene regulatory networks, peripheral genes can regulate core genes without many procedures. Therefore, thousands of genes can directly or indirectly contribute to NDDs. The omnigenic theory provides a comprehensive frame to understand a large number of NDD genes but remains to be confirmed in the future studies.
NDD and other neuropsychiatric disorders
Patients with NDD often have other neuropsychiatric disorders, such as schizophrenia, indicating that these disorders may share some genetic risk factors. It is well established that schizophrenia has a significant genetic component, with contributions spanning the entire allele frequency spectrum [106], 107]. GWAS have identified nearly 300 common risk loci for schizophrenia, and common variants can explain about 24 % of the variance in disease liability [108], 109]. Several studies found that patients with ASD over inherit polygenic risk for schizophrenia than their unaffected siblings [49]. By conducting a meta-analysis of WES data from 24,248 schizophrenia cases and 97,322 controls [110], Singh et al. have implicated ultra-rare coding variants in 10 genes as significantly increasing the risk for schizophrenia, with odds ratios ranging from 3 to 50 and p-values less than 2.14 × 10−6. They found a significant excess of ultra-rare variants in schizophrenia cases compared to controls within 299 genes associated with DD and 102 genes associated with ASD, suggesting an overlap in rare variant risk among schizophrenia, ASD and DD. Although there is compelling evidence that both common and rare variants contribute to ASD and schizophrenia, a key difference is that much smaller cohorts have been needed to identify large-effect rare variants and risk genes in ASD [111]. In contrast, much larger sample sizes have been necessary to identify the first genome-wide significant common variants in ASD compared to schizophrenia. In addition, NDDs and neurodegenerative diseases also exhibit similar clinical features, such as impairments in language, executive function, and motor skills [112]. Interestingly, some NDD genes are also associated with neurodegenerative disorders, such as Alzheimer’s disease (AD), including APBB1, MARK2, ATP2B2, and NR4A2 [35].
Conclusion and future directions
Recent large-scale genomic studies have led to a better understanding of genetic architecture of NDDs. The number of well-established high-impact genes continues to increase, alongside a broadening collection of comprehensive omics datasets. However, the genetic architecture of NDDs is extremely complex and many genetic risk factors still remain to be explored. For example, studies have suggested mutations in non-coding regions of the genome contribute to NDDs, including non-coding DNMs in promotor region and chromatin interactions [113], 114]. In a recent study, it was estimated that the variants in 18 bp region of small nuclear RNA RNU4-2 can explain 0.4 % of individuals with NDDs, underscoring the importance of non-coding genes in rare disorders [115]. In addition, mitochondria are essential for neurodevelopment, Wang et al., found that mitochondrial genome (mtDNA) heteroplasmies in ASD probands might have elevated pathogenicity [116]. Tandem repeats (TRs) are short lengths of DNA that are repeated multiple times within a gene, anywhere from a handful of times to more than a hundred. A significant genome-wide excess of TR mutations in ASD probands has been identified [117]. Furthermore, most previous studies only focused on one form of variation, ignoring the diversity of genetic backgrounds. Future studies should simultaneously integrate multiple forms of variation in the same sample to understand the combined interplay effects of various variants on NDD phenotypes. Stratifying the samples according to genetic variation may be helpful to further understand the molecular mechanisms underlying various NDD phenotypes and provide deeper insight into clinical therapy.
Researchers have been interested in translating genetic discoveries of NDD into pathogenic mechanisms and drug development, such as the use of iPSC models for understanding risk genes of NDDs [118]. Several studies have compared neurons generated from iPSCs derived from donors with ASD to those from neurotypical individuals and found that iPSC-derived neurons from patients with ASD, in comparison to controls, show increased cellular proliferation, impaired synapse development, and decreased spontaneous [119], 120]. Paulsen et al. utilized human cerebral cortex organoid models to investigate cell-type-specific developmental abnormalities caused by haploinsufficiency in three ASD risk genes: KMT5B, ARID1B, and CHD8 [121]. Their research revealed that each of the three mutations leads to asynchronous development of two primary cortical neuronal lineages – GABAergic neurons and deep-layer excitatory projection neurons through distinct molecular pathways. However, the systematic profiling of iPSCs from individuals with idiopathic autism has faced limitations due to small sample sizes, it remains unclear whether these findings will be confirmed in larger and more comprehensive analyses [122]. Big consortium has also attempted to characterize the contribution of genetic variation to NDDs, such as SSPsyGene consortium, an NIMH-initiated consortium which aims to implement systematic and coordinated assays that generate an accessible catalog of genotypes and phenotypes of neurodevelopmental and psychiatric disorder risk gene deletion models. Overall, translating genetic discoveries into disease biology and actionable drug targets remains important in future NDD studies.
Funding source: National Natural Science Foundation of China
Award Identifier / Grant number: 82201314
Funding source: Fundamental Research Funds for the Central Universities starting fund
Award Identifier / Grant number: BMU2022RCZX038
Acknowledgments
We thank the members of Wang lab for helpful suggestions.
-
Research ethics: The local Institutional Review Board deemed the study exempt from review.
-
Informed consent: Not applicable.
-
Author contributions: Shilin Gao, Chaoyi Shan, Rong Zhang, and Tianyun Wang wrote the paper; Shilin Gao and Chaoyi Shan designed the figures. All authors read and approved the manuscript.
-
Competing interests: The authors declare no conflict of interest.
-
Research funding: This work is supported, in part, by grants from the National Natural Science Foundation of China (82201314) and by the Fundamental Research Funds for the Central Universities starting fund (BMU2022RCZX038) to T.W.
References
1. First, MB. Diagnostic and statistical manual of mental disorders, 5th edition, and clinical utility. J Nerv Ment Dis 2013;201:727–9. https://doi.org/10.1097/nmd.0b013e3182a2168a.Search in Google Scholar PubMed
2. Bölte, S, Neufeld, J, Marschik, PB, Williams, ZJ, Gallagher, L, Lai, MC. Sex and gender in neurodevelopmental conditions. Nat Rev Neurol 2023;19:136–59. https://doi.org/10.1038/s41582-023-00774-6.Search in Google Scholar PubMed PubMed Central
3. Folstein, S, Rutter, M. Infantile autism: a genetic study of 21 twin pairs. J Child Psychol Psychiatry 1977;18:297–321. https://doi.org/10.1111/j.1469-7610.1977.tb00443.x.Search in Google Scholar PubMed
4. Sandin, S, Lichtenstein, P, Kuja-Halkola, R, Hultman, C, Larsson, H, Reichenberg, A. The heritability of autism spectrum disorder. JAMA 2017;318:1182–4. https://doi.org/10.1001/jama.2017.12141.Search in Google Scholar PubMed PubMed Central
5. Faraone, SV, Larsson, H. Genetics of attention deficit hyperactivity disorder. Mol Psychiatry 2019;24:562–75. https://doi.org/10.1038/s41380-018-0070-0.Search in Google Scholar PubMed PubMed Central
6. Gialluisi, A, Andlauer, TFM, Mirza-Schreiber, N, Moll, K, Becker, J, Hoffmann, P, et al.. Genome-wide association study reveals new insights into the heritability and genetic correlates of developmental dyslexia. Mol Psychiatry 2021;26:3004–17. https://doi.org/10.1038/s41380-020-00898-x.Search in Google Scholar PubMed PubMed Central
7. Choi, L, An, JY. Genetic architecture of autism spectrum disorder: lessons from large-scale genomic studies. Neurosci Biobehav Rev 2021;128:244–57. https://doi.org/10.1016/j.neubiorev.2021.06.028.Search in Google Scholar PubMed
8. Iakoucheva, LM, Muotri, AR, Sebat, J. Getting to the cores of autism. Cell 2019;178:1287–98. https://doi.org/10.1016/j.cell.2019.07.037.Search in Google Scholar PubMed PubMed Central
9. Wang, T, Zhao, PA, Eichler, EE. Rare variants and the oligogenic architecture of autism. Trends Genet 2022;38:895–903. https://doi.org/10.1016/j.tig.2022.03.009.Search in Google Scholar PubMed PubMed Central
10. Coe, BP, Stessman, HAF, Sulovari, A, Geisheker, MR, Bakken, TE, Lake, AM, et al.. Neurodevelopmental disease genes implicated by de novo mutation and copy number variation morbidity. Nat Genet 2019;51:106–16. https://doi.org/10.1038/s41588-018-0288-4.Search in Google Scholar PubMed PubMed Central
11. Wang, T, Kim, CN, Bakken, TE, Gillentine, MA, Henning, B, Mao, Y, et al.. Integrated gene analyses of de novo variants from 46,612 trios with autism and developmental disorders. Proc Natl Acad Sci USA 2022;119:e2203491119. https://doi.org/10.1073/pnas.2203491119.Search in Google Scholar PubMed PubMed Central
12. Fu, JM, Satterstrom, FK, Peng, M, Brand, H, Collins, RL, Dong, S, et al.. Rare coding variation provides insight into the genetic architecture and phenotypic context of autism. Nat Genet 2022;54:1320–31. https://doi.org/10.1038/s41588-022-01104-0.Search in Google Scholar PubMed PubMed Central
13. Iossifov, I, O’Roak, BJ, Sanders, SJ, Ronemus, M, Krumm, N, Levy, D, et al.. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014;515:216–21. https://doi.org/10.1038/nature13908.Search in Google Scholar PubMed PubMed Central
14. Sebat, J, Lakshmi, B, Malhotra, D, Troge, J, Lese-Martin, C, Walsh, T, et al.. Strong association of de novo copy number mutations with autism. Science 2007;316:445–9. https://doi.org/10.1126/science.1138659.Search in Google Scholar PubMed PubMed Central
15. O’Roak, BJ, Deriziotis, P, Lee, C, Vives, L, Schwartz, JJ, Girirajan, S, et al.. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet 2011;43:585–9. https://doi.org/10.1038/ng.835.Search in Google Scholar PubMed PubMed Central
16. O’Roak, BJ, Vives, L, Girirajan, S, Karakoc, E, Krumm, N, Coe, BP, et al.. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 2012;485:246–50. https://doi.org/10.1038/nature10989.Search in Google Scholar PubMed PubMed Central
17. Sanders, SJ, Murtha, MT, Gupta, AR, Murdoch, JD, Raubeson, MJ, Willsey, AJ, et al.. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012;485:237–41. https://doi.org/10.1038/nature10945.Search in Google Scholar PubMed PubMed Central
18. Iossifov, I, Ronemus, M, Levy, D, Wang, Z, Hakker, I, Rosenbaum, J, et al.. De novo gene disruptions in children on the autistic spectrum. Neuron 2012;74:285–99. https://doi.org/10.1016/j.neuron.2012.04.009.Search in Google Scholar PubMed PubMed Central
19. Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature 2015;519:223–8. https://doi.org/10.1038/nature14135.Search in Google Scholar PubMed PubMed Central
20. Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017;542:433–8. https://doi.org/10.1038/nature21062.Search in Google Scholar PubMed PubMed Central
21. Kaplanis, J, Samocha, KE, Wiel, L, Zhang, Z, Arvai, KJ, Eberhardt, RY, et al.. Evidence for 28 genetic disorders discovered by combining healthcare and research data. Nature 2020;586:757–62. https://doi.org/10.1038/s41586-020-2832-5.Search in Google Scholar PubMed PubMed Central
22. Kircher, M, Witten, DM, Jain, P, O’Roak, BJ, Cooper, GM, Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–5. https://doi.org/10.1038/ng.2892.Search in Google Scholar PubMed PubMed Central
23. Samocha, KE, Kosmicki, JA, Karczewski, KJ, O’Donnell-Luria, AH, Pierce-Hoffman, E, MacArthur, DG, et al.. Regional missense constraint improves variant deleteriousness prediction. bioRxiv 2017;148353. https://doi.org/10.1101/148353.Search in Google Scholar
24. Geisheker, MR, Heymann, G, Wang, T, Coe, BP, Turner, TN, Stessman, HAF, et al.. Hotspots of missense mutation identify neurodevelopmental disorder genes and functional domains. Nat Neurosci 2017;20:1043–51. https://doi.org/10.1038/nn.4589.Search in Google Scholar PubMed PubMed Central
25. Lelieveld, SH, Wiel, L, Venselaar, H, Pfundt, R, Vriend, G, Veltman, JA, et al.. Spatial Clustering of de Novo Missense Mutations Identifies Candidate Neurodevelopmental Disorder-Associated Genes. Am J Hum Genet 2017;101:478–84. https://doi.org/10.1016/j.ajhg.2017.08.004.Search in Google Scholar PubMed PubMed Central
26. Wiel, L, Hampstead, JE, Venselaar, H, Vissers, L, Brunner, HG, Pfundt, R, et al.. De novo mutation hotspots in homologous protein domains identify function-altering mutations in neurodevelopmental disorders. Am J Hum Genet 2023;110:92–104. https://doi.org/10.1016/j.ajhg.2022.12.001.Search in Google Scholar PubMed PubMed Central
27. Zhou, X, Feliciano, P, Shu, C, Wang, T, Astrovskaya, I, Hall, JB, et al.. Integrating de novo and inherited variants in 42,607 autism cases identifies mutations in new moderate-risk genes. Nat Genet 2022;54:1305–19. https://doi.org/10.1038/s41588-022-01148-2.Search in Google Scholar PubMed PubMed Central
28. Krumm, N, Turner, TN, Baker, C, Vives, L, Mohajeri, K, Witherspoon, K, et al.. Excess of rare, inherited truncating mutations in autism. Nat Genet 2015;47:582–8. https://doi.org/10.1038/ng.3303.Search in Google Scholar PubMed PubMed Central
29. Wilfert, AB, Turner, TN, Murali, SC, Hsieh, P, Sulovari, A, Wang, T, et al.. Recent ultra-rare inherited variants implicate new autism candidate risk genes. Nat Genet 2021;53:1125–34. https://doi.org/10.1038/s41588-021-00899-8.Search in Google Scholar PubMed PubMed Central
30. He, X, Sanders, SJ, Liu, L, De Rubeis, S, Lim, ET, Sutcliffe, JS, et al.. Integrated model of de novo and inherited genetic variants yields greater power to identify risk genes. PLoS Genet 2013;9:e1003671. https://doi.org/10.1371/journal.pgen.1003671.Search in Google Scholar PubMed PubMed Central
31. Sanders, SJ, He, X, Willsey, AJ, Ercan-Sencicek, AG, Samocha, KE, Cicek, AE, et al.. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 2015;87:1215–33. https://doi.org/10.1016/j.neuron.2015.09.016.Search in Google Scholar PubMed PubMed Central
32. Takata, A, Miyake, N, Tsurusaki, Y, Fukai, R, Miyatake, S, Koshimizu, E, et al.. Integrative analyses of de novo mutations provide deeper biological insights into autism spectrum disorder. Cell Rep 2018;22:734–47. https://doi.org/10.1016/j.celrep.2017.12.074.Search in Google Scholar PubMed
33. Ruzzo, EK, Pérez-Cano, L, Jung, JY, Wang, LK, Kashef-Haghighi, D, Hartl, C, et al.. Inherited and de novo genetic risk for autism impacts shared networks. Cell 2019;178:850–66.e26. https://doi.org/10.1016/j.cell.2019.07.015.Search in Google Scholar PubMed PubMed Central
34. Satterstrom, FK, Kosmicki, JA, Wang, J, Breen, MS, De Rubeis, S, An, JY, et al.. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 2020;180:568–84.e23. https://doi.org/10.1016/j.cell.2019.12.036.Search in Google Scholar PubMed PubMed Central
35. Trost, B, Thiruvahindrapuram, B, Chan, AJS, Engchuan, W, Higginbotham, EJ, Howe, JL, et al.. Genomic architecture of autism from comprehensive whole-genome sequence annotation. Cell 2022;185:4409–27.e18. https://doi.org/10.1016/j.cell.2022.10.009.Search in Google Scholar PubMed PubMed Central
36. Cooper, GM, Coe, BP, Girirajan, S, Rosenfeld, JA, Vu, TH, Baker, C, et al.. A copy number variation morbidity map of developmental delay. Nat Genet 2011;43:838–46. https://doi.org/10.1038/ng.909.Search in Google Scholar PubMed PubMed Central
37. Walsh, T, McClellan, JM, McCarthy, SE, Addington, AM, Pierce, SB, Cooper, GM, et al.. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 2008;320:539–43. https://doi.org/10.1126/science.1155174.Search in Google Scholar PubMed
38. Coe, BP, Witherspoon, K, Rosenfeld, JA, van Bon, BW, Vulto-van Silfhout, AT, Bosco, P, et al.. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet 2014;46:1063–71. https://doi.org/10.1038/ng.3092.Search in Google Scholar PubMed PubMed Central
39. Huguet, G, Schramm, C, Douard, E, Tamer, P, Main, A, Monin, P, et al.. Genome-wide analysis of gene dosage in 24,092 individuals estimates that 10,000 genes modulate cognitive ability. Mol Psychiatry 2021;26:2663–76. https://doi.org/10.1038/s41380-020-00985-z.Search in Google Scholar PubMed PubMed Central
40. Collins, RL, Glessner, JT, Porcu, E, Lepamets, M, Brandon, R, Lauricella, C, et al.. A cross-disorder dosage sensitivity map of the human genome. Cell 2022;185:3041–55.e25. https://doi.org/10.1016/j.cell.2022.06.036.Search in Google Scholar PubMed PubMed Central
41. Rein, B, Yan, Z. 16p11.2 copy number variations and neurodevelopmental disorders. Trends Neurosci 2020;43:886–901. https://doi.org/10.1016/j.tins.2020.09.001.Search in Google Scholar PubMed PubMed Central
42. Brunetti-Pierri, N, Berg, JS, Scaglia, F, Belmont, J, Bacino, CA, Sahoo, T, et al.. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet 2008;40:1466–71. https://doi.org/10.1038/ng.279.Search in Google Scholar PubMed PubMed Central
43. Morris, CA, Mervis, CB, Paciorkowski, AP, Abdul-Rahman, O, Dugan, SL, Rope, AF, et al.. 7q11.23 duplication syndrome: physical characteristics and natural history. Am J Med Genet 2015;167a:2916–35. https://doi.org/10.1002/ajmg.a.37340.Search in Google Scholar PubMed PubMed Central
44. Osborne, LR, Mervis, CB. 7q11.23 deletion and duplication. Curr Opin Genet Dev 2021;68:41–8. https://doi.org/10.1016/j.gde.2021.01.013.Search in Google Scholar PubMed
45. Gaugler, T, Klei, L, Sanders, SJ, Bodea, CA, Goldberg, AP, Lee, AB, et al.. Most genetic risk for autism resides with common variation. Nat Genet 2014;46:881–5. https://doi.org/10.1038/ng.3039.Search in Google Scholar PubMed PubMed Central
46. Warrier, V, Zhang, X, Reed, P, Havdahl, A, Moore, TM, Cliquet, F, et al.. Genetic correlates of phenotypic heterogeneity in autism. Nat Genet 2022;54:1293–304. https://doi.org/10.1038/s41588-022-01072-5.Search in Google Scholar PubMed PubMed Central
47. Grove, J, Ripke, S, Als, TD, Mattheisen, M, Walters, RK, Won, H, et al.. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet 2019;51:431–44. https://doi.org/10.1038/s41588-019-0344-8.Search in Google Scholar PubMed PubMed Central
48. Mattheisen, M, Grove, J, Als, TD, Martin, J, Voloudakis, G, Meier, S, et al.. Identification of shared and differentiating genetic architecture for autism spectrum disorder, attention-deficit hyperactivity disorder and case subgroups. Nat Genet 2022;54:1470–8. https://doi.org/10.1038/s41588-022-01171-3.Search in Google Scholar PubMed PubMed Central
49. Weiner, DJ, Wigdor, EM, Ripke, S, Walters, RK, Kosmicki, JA, Grove, J, et al.. Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat Genet 2017;49:978–85. https://doi.org/10.1038/ng.3863.Search in Google Scholar PubMed PubMed Central
50. Gardner, RM, Dalman, C, Rai, D, Lee, BK, Karlsson, H. The association of paternal IQ with autism spectrum disorders and its comorbidities: a population-based cohort study. J Am Acad Child Adolesc Psychiatry 2020;59:410–21. https://doi.org/10.1016/j.jaac.2019.04.004.Search in Google Scholar PubMed
51. Antaki, D, Guevara, J, Maihofer, AX, Klein, M, Gujral, M, Grove, J, et al.. A phenotypic spectrum of autism is attributable to the combined effects of rare variants, polygenic risk and sex. Nat Genet 2022;54:1284–92. https://doi.org/10.1038/s41588-022-01064-5.Search in Google Scholar PubMed PubMed Central
52. Neale, BM, Kou, Y, Liu, L, Ma’ayan, A, Samocha, KE, Sabo, A, et al.. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 2012;485:242–5. https://doi.org/10.1038/nature11011.Search in Google Scholar PubMed PubMed Central
53. Li, B, Zhao, H, Tu, Z, Yang, W, Han, R, Wang, L, et al.. CHD8 mutations increase gliogenesis to enlarge brain size in the nonhuman primate. Cell Discov 2023;9:27. https://doi.org/10.1038/s41421-023-00525-3.Search in Google Scholar PubMed PubMed Central
54. Kim, OH, Cho, HJ, Han, E, Hong, TI, Ariyasiri, K, Choi, JH, et al.. Zebrafish knockout of Down syndrome gene, DYRK1A, shows social impairments relevant to autism. Mol Autism 2017;8:50. https://doi.org/10.1186/s13229-017-0168-2.Search in Google Scholar PubMed PubMed Central
55. Zoodsma, JD, Keegan, EJ, Moody, GR, Bhandiwad, AA, Napoli, AJ, Burgess, HA, et al.. Disruption of grin2B, an ASD-associated gene, produces social deficits in zebrafish. Mol Autism 2022;13:38. https://doi.org/10.1186/s13229-022-00516-3.Search in Google Scholar PubMed PubMed Central
56. Clipperton-Allen, AE, Cohen, OS, Aceti, M, Zucca, A, Levy, J, Ellegood, J, et al.. Pten haploinsufficiency disrupts scaling across brain areas during development in mice. Transl Psychiatry 2019;9:329. https://doi.org/10.1038/s41398-019-0656-6.Search in Google Scholar PubMed PubMed Central
57. Kim, IB, Lee, T, Lee, J, Kim, J, Lee, S, Koh, IG, et al.. Non-coding de novo mutations in chromatin interactions are implicated in autism spectrum disorder. Mol Psychiatry 2022;27:4680–94. https://doi.org/10.1038/s41380-022-01697-2.Search in Google Scholar PubMed
58. Yuan, B, Wang, M, Wu, X, Cheng, P, Zhang, R, Zhang, R, et al.. Identification of de novo mutations in the Chinese autism spectrum disorder cohort via whole-exome sequencing unveils brain regions implicated in Autism. Neurosci Bull 2023;39:1469–80. https://doi.org/10.1007/s12264-023-01037-6.Search in Google Scholar PubMed PubMed Central
59. Wang, J, Yu, J, Wang, M, Zhang, L, Yang, K, Du, X, et al.. Discovery and validation of novel genes in a large Chinese autism spectrum disorder cohort. Biol Psychiatry 2023;94:792–803. https://doi.org/10.1016/j.biopsych.2023.06.025.Search in Google Scholar PubMed
60. Abrahams, BS, Arking, DE, Campbell, DB, Mefford, HC, Morrow, EM, Weiss, LA, et al.. SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol Autism 2013;4:36. https://doi.org/10.1186/2040-2392-4-36.Search in Google Scholar PubMed PubMed Central
61. Wright, CF, Fitzgerald, TW, Jones, WD, Clayton, S, McRae, JF, van Kogelenberg, M, et al.. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet 2015;385:1305–14. https://doi.org/10.1016/s0140-6736(14)61705-0.Search in Google Scholar PubMed PubMed Central
62. Iroegbu, JD, Ijomone, OK, Femi-Akinlosotu, OM, Ijomone, OM. ERK/MAPK signalling in the developing brain: perturbations and consequences. Neurosci Biobehav Rev 2021;131:792–805. https://doi.org/10.1016/j.neubiorev.2021.10.009.Search in Google Scholar PubMed
63. Pucilowska, J, Puzerey, PA, Karlo, JC, Galán, RF, Landreth, GE. Disrupted ERK signaling during cortical development leads to abnormal progenitor proliferation, neuronal and network excitability and behavior, modeling human neuro-cardio-facial-cutaneous and related syndromes. J Neurosci 2012;32:8663–77. https://doi.org/10.1523/jneurosci.1107-12.2012.Search in Google Scholar PubMed PubMed Central
64. De Rubeis, S, He, X, Goldberg, AP, Poultney, CS, Samocha, K, Cicek, AE, et al.. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014;515:209–15. https://doi.org/10.1038/nature13772.Search in Google Scholar PubMed PubMed Central
65. Wang, T, Hoekzema, K, Vecchio, D, Wu, H, Sulovari, A, Coe, BP, et al.. Large-scale targeted sequencing identifies risk genes for neurodevelopmental disorders. Nat Commun 2020;11:4932. https://doi.org/10.1038/s41467-020-18723-y.Search in Google Scholar PubMed PubMed Central
66. Thomas, SD, Jha, NK, Ojha, S, Sadek, B. mTOR signaling disruption and its association with the development of autism spectrum disorder. Molecules 2023;28:1889. https://doi.org/10.3390/molecules28041889.Search in Google Scholar PubMed PubMed Central
67. DeSpenza, TJr., Carlson, M, Panchagnula, S, Robert, S, Duy, PQ, Mermin-Bunnell, N, et al.. PTEN mutations in autism spectrum disorder and congenital hydrocephalus: developmental pleiotropy and therapeutic targets. Trends Neurosci 2021;44:961–76. https://doi.org/10.1016/j.tins.2021.08.007.Search in Google Scholar PubMed PubMed Central
68. Vogt, D, Cho, KKA, Lee, AT, Sohal, VS, Rubenstein, JLR. The parvalbumin/somatostatin ratio is increased in Pten mutant mice and by human PTEN ASD alleles. Cell Rep 2015;11:944–56. https://doi.org/10.1016/j.celrep.2015.04.019.Search in Google Scholar PubMed PubMed Central
69. Chen, CJ, Sgritta, M, Mays, J, Zhou, H, Lucero, R, Park, J, et al.. Therapeutic inhibition of mTORC2 rescues the behavioral and neurophysiological abnormalities associated with Pten-deficiency. Nat Med 2019;25:1684–90. https://doi.org/10.1038/s41591-019-0608-y.Search in Google Scholar PubMed PubMed Central
70. Capal, JK, Williams, ME, Pearson, DA, Kissinger, R, Horn, PS, Murray, D, et al.. Profile of autism spectrum disorder in tuberous sclerosis complex: results from a longitudinal, prospective, multisite study. Ann Neurol 2021;90:874–86. https://doi.org/10.1002/ana.26249.Search in Google Scholar PubMed PubMed Central
71. Curatolo, P, Scheper, M, Emberti Gialloreti, L, Specchio, N, Aronica, E. Is tuberous sclerosis complex-associated autism a preventable and treatable disorder? World J Pediatr 2024;20:40–53. https://doi.org/10.1007/s12519-023-00762-2.Search in Google Scholar PubMed
72. French, JA, Lawson, JA, Yapici, Z, Ikeda, H, Polster, T, Nabbout, R, et al.. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet 2016;388:2153–63. https://doi.org/10.1016/s0140-6736(16)31419-2.Search in Google Scholar PubMed
73. Curatolo, P, Specchio, N, Aronica, E. Advances in the genetics and neuropathology of tuberous sclerosis complex: edging closer to targeted therapy. Lancet Neurol 2022;21:843–56. https://doi.org/10.1016/s1474-4422(22)00213-7.Search in Google Scholar PubMed
74. Zhuang, W, Ye, T, Wang, W, Song, W, Tan, T. CTNNB1 in neurodevelopmental disorders. Front Psychiatry 2023;14:1143328. https://doi.org/10.3389/fpsyt.2023.1143328.Search in Google Scholar PubMed PubMed Central
75. Kayumi, S, Pérez-Jurado, LA, Palomares, M, Rangu, S, Sheppard, SE, Chung, WK, et al.. Genomic and phenotypic characterization of 404 individuals with neurodevelopmental disorders caused by CTNNB1 variants. Genet Med 2022;24:2351–66. https://doi.org/10.1016/j.gim.2022.08.006.Search in Google Scholar PubMed PubMed Central
76. Jiang, CC, Lin, LS, Long, S, Ke, XY, Fukunaga, K, Lu, YM, et al.. Signalling pathways in autism spectrum disorder: mechanisms and therapeutic implications. Signal Transduct Target Ther 2022;7:229. https://doi.org/10.1038/s41392-022-01081-0.Search in Google Scholar PubMed PubMed Central
77. Bonaglia, MC, Giorda, R, Borgatti, R, Felisari, G, Gagliardi, C, Selicorni, A, et al.. Disruption of the ProSAP2 gene in a t(12;22)(q24.1;q13.3) is associated with the 22q13.3 deletion syndrome. Am J Hum Genet 2001;69:261–8. https://doi.org/10.1086/321293.Search in Google Scholar PubMed PubMed Central
78. Monteiro, P, Feng, G. SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat Rev Neurosci 2017;18:147–57. https://doi.org/10.1038/nrn.2016.183.Search in Google Scholar PubMed
79. Delling, JP, Boeckers, TM. Comparison of SHANK3 deficiency in animal models: phenotypes, treatment strategies, and translational implications. J Neurodev Disord 2021;13:55. https://doi.org/10.1186/s11689-021-09397-8.Search in Google Scholar PubMed PubMed Central
80. Navakkode, S, Zhai, J, Wong, YP, Li, G, Soong, TW. Enhanced long-term potentiation and impaired learning in mice lacking alternative exon 33 of Ca(V)1.2 calcium channel. Transl Psychiatry 2022;12:1. https://doi.org/10.1038/s41398-021-01683-2.Search in Google Scholar PubMed PubMed Central
81. Stein, RE, Kaplan, JS, Li, J, Catterall, WA. Hippocampal deletion of Na(V)1.1 channels in mice causes thermal seizures and cognitive deficit characteristic of Dravet Syndrome. Proc Natl Acad Sci USA 2019;116:16571–6. https://doi.org/10.1073/pnas.1906833116.Search in Google Scholar PubMed PubMed Central
82. Babij, R, Ferrer, C, Donatelle, A, Wacks, S, Buch, AM, Niemeyer, JE, et al.. Gabrb3 is required for the functional integration of pyramidal neuron subtypes in the somatosensory cortex. Neuron 2023;111:256–74.e10. https://doi.org/10.1016/j.neuron.2022.10.037.Search in Google Scholar PubMed PubMed Central
83. Bernier, R, Golzio, C, Xiong, B, Stessman, HA, Coe, BP, Penn, O, et al.. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 2014;158:263–76. https://doi.org/10.1016/j.cell.2014.06.017.Search in Google Scholar PubMed PubMed Central
84. Wu, X, Liu, Y, Wang, X, Zheng, L, Pan, L, Wang, H. Developmental impairments of synaptic refinement in the thalamus of a mouse model of fragile X syndrome. Neurosci Bull 2024;40:439–50. https://doi.org/10.1007/s12264-023-01142-6.Search in Google Scholar PubMed PubMed Central
85. Fazel Darbandi, S, An, JY, Lim, K, Page, NF, Liang, L, Young, DM, et al.. Five autism-associated transcriptional regulators target shared loci proximal to brain-expressed genes. Cell Rep 2024;43:114329. https://doi.org/10.1016/j.celrep.2024.114329.Search in Google Scholar PubMed PubMed Central
86. Wu, SH, Li, X, Qin, DD, Zhang, LH, Cheng, TL, Chen, ZF, et al.. Induction of core symptoms of autism spectrum disorder by in vivo CRISPR/Cas9-based gene editing in the brain of adolescent rhesus monkeys. Sci Bull 2021;66:937–46. https://doi.org/10.1016/j.scib.2020.12.017.Search in Google Scholar PubMed
87. Xu, FX, Wang, XT, Cai, XY, Liu, JY, Guo, JW, Yang, F, et al.. Purkinje-cell-specific MeCP2 deficiency leads to motor deficits and autistic-like behavior due to aberrations in PTP1B-TrkB-SK signaling. Cell Rep 2023;42:113559. https://doi.org/10.1016/j.celrep.2023.113559.Search in Google Scholar PubMed
88. Nagarajan, RP, Hogart, AR, Gwye, Y, Martin, MR, LaSalle, JM. Reduced MeCP2 expression is frequent in autism frontal cortex and correlates with aberrant MECP2 promoter methylation. Epigenetics 2006;1:e1–11. https://doi.org/10.4161/epi.1.4.3514.Search in Google Scholar PubMed PubMed Central
89. Foss-Feig, JH, Adkinson, BD, Ji, JL, Yang, G, Srihari, VH, McPartland, JC, et al.. Searching for cross-diagnostic convergence: neural mechanisms governing excitation and inhibition balance in schizophrenia and autism spectrum disorders. Biol Psychiatry 2017;81:848–61. https://doi.org/10.1016/j.biopsych.2017.03.005.Search in Google Scholar PubMed PubMed Central
90. Wang, P, Zhao, D, Lachman, HM, Zheng, D. Enriched expression of genes associated with autism spectrum disorders in human inhibitory neurons. Transl Psychiatry 2018;8:13. https://doi.org/10.1038/s41398-017-0058-6.Search in Google Scholar PubMed PubMed Central
91. Voineagu, I, Wang, X, Johnston, P, Lowe, JK, Tian, Y, Horvath, S, et al.. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011;474:380–4. https://doi.org/10.1038/nature10110.Search in Google Scholar PubMed PubMed Central
92. Gupta, S, Ellis, SE, Ashar, FN, Moes, A, Bader, JS, Zhan, J, et al.. Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat Commun 2014;5:5748. https://doi.org/10.1038/ncomms6748.Search in Google Scholar PubMed PubMed Central
93. Parikshak, NN, Swarup, V, Belgard, TG, Irimia, M, Ramaswami, G, Gandal, MJ, et al.. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 2016;540:423–7. https://doi.org/10.1038/nature20612.Search in Google Scholar PubMed PubMed Central
94. Velmeshev, D, Schirmer, L, Jung, D, Haeussler, M, Perez, Y, Mayer, S, et al.. Single-cell genomics identifies cell type-specific molecular changes in autism. Science 2019;364:685–9. https://doi.org/10.1126/science.aav8130.Search in Google Scholar PubMed PubMed Central
95. Gandal, MJ, Haney, JR, Wamsley, B, Yap, CX, Parhami, S, Emani, PS, et al.. Broad transcriptomic dysregulation occurs across the cerebral cortex in ASD. Nature 2022;611:532–9. https://doi.org/10.1038/s41586-022-05377-7.Search in Google Scholar PubMed PubMed Central
96. Werling, DM, Parikshak, NN, Geschwind, DH. Gene expression in human brain implicates sexually dimorphic pathways in autism spectrum disorders. Nat Commun 2016;7:10717. https://doi.org/10.1038/ncomms10717.Search in Google Scholar PubMed PubMed Central
97. Kissel, LT, Werling, DM. Neural transcriptomic analysis of sex differences in autism spectrum disorder: current insights and future directions. Biol Psychiatry 2022;91:53–60. https://doi.org/10.1016/j.biopsych.2020.11.023.Search in Google Scholar PubMed
98. Kingdom, R, Tuke, M, Wood, A, Beaumont, RN, Frayling, TM, Weedon, MN, et al.. Rare genetic variants in genes and loci linked to dominant monogenic developmental disorders cause milder related phenotypes in the general population. Am J Hum Genet 2022;109:1308–16. https://doi.org/10.1016/j.ajhg.2022.05.011.Search in Google Scholar PubMed PubMed Central
99. Rolland, T, Cliquet, F, Anney, RJL, Moreau, C, Traut, N, Mathieu, A, et al.. Phenotypic effects of genetic variants associated with autism. Nat Med 2023;29:1671–80. https://doi.org/10.1038/s41591-023-02408-2.Search in Google Scholar PubMed PubMed Central
100. Guo, H, Wang, T, Wu, H, Long, M, Coe, BP, Li, H, et al.. Inherited and multiple de novo mutations in autism/developmental delay risk genes suggest a multifactorial model. Mol Autism 2018;9:64. https://doi.org/10.1186/s13229-018-0247-z.Search in Google Scholar PubMed PubMed Central
101. Boyle, EA, Li, YI, Pritchard, JK. An expanded view of complex traits: from polygenic to omnigenic. Cell 2017;169:1177–86. https://doi.org/10.1016/j.cell.2017.05.038.Search in Google Scholar PubMed PubMed Central
102. Wray, NR, Wijmenga, C, Sullivan, PF, Yang, J, Visscher, PM. Common disease is more complex than implied by the core gene omnigenic model. Cell 2018;173:1573–80. https://doi.org/10.1016/j.cell.2018.05.051.Search in Google Scholar PubMed
103. Liu, X, Li, YI, Pritchard, JK. Trans effects on gene expression can drive omnigenic inheritance. Cell 2019;177:1022–34.e6. https://doi.org/10.1016/j.cell.2019.04.014.Search in Google Scholar PubMed PubMed Central
104. Mathieson, I. The omnigenic model and polygenic prediction of complex traits. Am J Hum Genet 2021;108:1558–63. https://doi.org/10.1016/j.ajhg.2021.07.003.Search in Google Scholar PubMed PubMed Central
105. Fóthi, Á, Pintér, C, Pollner, P, Lőrincz, A. Peripheral gene interactions define interpretable clusters of core ASD genes in a network-based investigation of the omnigenic theory. NPJ Syst Biol Appl 2022;8:28. https://doi.org/10.1038/s41540-022-00240-x.Search in Google Scholar PubMed PubMed Central
106. Marshall, CR, Howrigan, DP, Merico, D, Thiruvahindrapuram, B, Wu, W, Greer, DS, et al.. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet 2017;49:27–35. https://doi.org/10.1038/ng.3725.Search in Google Scholar PubMed PubMed Central
107. Singh, T, Walters, JTR, Johnstone, M, Curtis, D, Suvisaari, J, Torniainen, M, et al.. The contribution of rare variants to risk of schizophrenia in individuals with and without intellectual disability. Nat Genet 2017;49:1167–73. https://doi.org/10.1038/ng.3903.Search in Google Scholar PubMed PubMed Central
108. Trubetskoy, V, Pardiñas, AF, Qi, T, Panagiotaropoulou, G, Awasthi, S, Bigdeli, TB, et al.. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 2022;604:502–8. https://doi.org/10.1038/s41586-022-04434-5.Search in Google Scholar PubMed PubMed Central
109. Loh, PR, Bhatia, G, Gusev, A, Finucane, HK, Bulik-Sullivan, BK, Pollack, SJ, et al.. Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis. Nat Genet 2015;47:1385–92. https://doi.org/10.1038/ng.3431.Search in Google Scholar PubMed PubMed Central
110. Singh, T, Poterba, T, Curtis, D, Akil, H, Al Eissa, M, Barchas, JD, et al.. Rare coding variants in ten genes confer substantial risk for schizophrenia. Nature 2022;604:509–16. https://doi.org/10.1038/s41586-022-04556-w.Search in Google Scholar PubMed PubMed Central
111. Willsey, HR, Willsey, AJ, Wang, B, State, MW. Genomics, convergent neuroscience and progress in understanding autism spectrum disorder. Nat Rev Neurosci 2022;23:323–41. https://doi.org/10.1038/s41583-022-00576-7.Search in Google Scholar PubMed PubMed Central
112. Khan, SA, Khan, SA, Narendra, AR, Mushtaq, G, Zahran, SA, Khan, S, et al.. Alzheimer’s disease and autistic spectrum disorder: is there any association? CNS Neurol Disord: Drug Targets 2016;15:390–402. https://doi.org/10.2174/1871527315666160321104303.Search in Google Scholar PubMed
113. An, JY, Lin, K, Zhu, L, Werling, DM, Dong, S, Brand, H, et al.. Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science 2018;362:eaat6576. https://doi.org/10.1126/science.aat6576.Search in Google Scholar PubMed PubMed Central
114. Zhou, J, Park, CY, Theesfeld, CL, Wong, AK, Yuan, Y, Scheckel, C, et al.. Whole-genome deep-learning analysis identifies contribution of noncoding mutations to autism risk. Nat Genet 2019;51:973–80. https://doi.org/10.1038/s41588-019-0420-0.Search in Google Scholar PubMed PubMed Central
115. Chen, Y, Dawes, R, Kim, HC, Ljungdahl, A, Stenton, SL, Walker, S, et al.. De novo variants in the RNU4-2 snRNA cause a frequent neurodevelopmental syndrome. Nature 2024;632:832–40. https://doi.org/10.1038/s41586-024-07773-7.Search in Google Scholar PubMed PubMed Central
116. Wang, Y, Guo, X, Hong, X, Wang, G, Pearson, C, Zuckerman, B, et al.. Association of mitochondrial DNA content, heteroplasmies and inter-generational transmission with autism. Nat Commun 2022;13:3790. https://doi.org/10.1038/s41467-022-30805-7.Search in Google Scholar PubMed PubMed Central
117. Mitra, I, Huang, B, Mousavi, N, Ma, N, Lamkin, M, Yanicky, R, et al.. Patterns of de novo tandem repeat mutations and their role in autism. Nature 2021;589:246–50. https://doi.org/10.1038/s41586-020-03078-7.Search in Google Scholar PubMed PubMed Central
118. Chiaradia, I, Lancaster, MA. Brain organoids for the study of human neurobiology at the interface of in vitro and in vivo. Nat Neurosci 2020;23:1496–508. https://doi.org/10.1038/s41593-020-00730-3.Search in Google Scholar PubMed
119. Mariani, J, Coppola, G, Zhang, P, Abyzov, A, Provini, L, Tomasini, L, et al.. FOXG1-Dependent dysregulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell 2015;162:375–90. https://doi.org/10.1016/j.cell.2015.06.034.Search in Google Scholar PubMed PubMed Central
120. Marchetto, MC, Belinson, H, Tian, Y, Freitas, BC, Fu, C, Vadodaria, K, et al.. Altered proliferation and networks in neural cells derived from idiopathic autistic individuals. Mol Psychiatry 2017;22:820–35. https://doi.org/10.1038/mp.2016.95.Search in Google Scholar PubMed PubMed Central
121. Paulsen, B, Velasco, S, Kedaigle, AJ, Pigoni, M, Quadrato, G, Deo, AJ, et al.. Autism genes converge on asynchronous development of shared neuron classes. Nature 2022;602:268–73. https://doi.org/10.1038/s41586-021-04358-6.Search in Google Scholar PubMed PubMed Central
122. Griesi-Oliveira, K, Fogo, MS, Pinto, BGG, Alves, AY, Suzuki, AM, Morales, AG, et al.. Transcriptome of iPSC-derived neuronal cells reveals a module of co-expressed genes consistently associated with autism spectrum disorder. Mol Psychiatry 2021;26:1589–605. https://doi.org/10.1038/s41380-020-0669-9.Search in Google Scholar PubMed PubMed Central
123. Feliciano, P, Zhou, X, Astrovskaya, I, Turner, TN, Wang, T, Brueggeman, L, et al.. Exome sequencing of 457 autism families recruited online provides evidence for autism risk genes. NPJ Genom Med 2019;4:19. https://doi.org/10.1038/s41525-019-0093-8.Search in Google Scholar PubMed PubMed Central
124. Rk, CY, Merico, D, Bookman, M, J, LH, Thiruvahindrapuram, B, Patel, RV, et al.. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci 2017;20:602–11. https://doi.org/10.1038/nn.4524.Search in Google Scholar PubMed PubMed Central
125. Wright, CF, Campbell, P, Eberhardt, RY, Aitken, S, Perrett, D, Brent, S, et al.. Genomic diagnosis of rare pediatric disease in the United Kingdom and Ireland. N Engl J Med 2023;388:1559–71. https://doi.org/10.1056/nejmoa2209046.Search in Google Scholar PubMed PubMed Central
© 2024 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
Articles in the same Issue
- Frontmatter
- Reviews
- Antimicrobial, remineralization, and infiltration: advanced strategies for interrupting dental caries
- Emerging magic bullet: subcellular organelle-targeted cancer therapy
- Genetic advances in neurodevelopmental disorders
- Biomedical applications of organoids in genetic diseases
- Prevalence of fragile X syndrome in South Asia, and importance of diagnosis
- Perspective
- Embracing a new era of echocardiography-guided percutaneous and non-fluoroscopical procedure for structure heart disease
- Commentary
- When biomarkers for major depressive disorder remain elusive
Articles in the same Issue
- Frontmatter
- Reviews
- Antimicrobial, remineralization, and infiltration: advanced strategies for interrupting dental caries
- Emerging magic bullet: subcellular organelle-targeted cancer therapy
- Genetic advances in neurodevelopmental disorders
- Biomedical applications of organoids in genetic diseases
- Prevalence of fragile X syndrome in South Asia, and importance of diagnosis
- Perspective
- Embracing a new era of echocardiography-guided percutaneous and non-fluoroscopical procedure for structure heart disease
- Commentary
- When biomarkers for major depressive disorder remain elusive