Genetically-engineered hamster models: applications and perspective in dyslipidemia and atherosclerosis-related cardiovascular disease
-
George Liu



Abstract
Cardiovascular disease is the leading cause of morbidity and mortality in both developed and developing countries, in which atherosclerosis triggered by dyslipidemia is the major pathological basis. Over the past 40 years, small rodent animals, such as mice, have been widely used for understanding of human atherosclerosis-related cardiovascular disease (ASCVD) with the advantages of low cost and ease of maintenance and manipulation. However, based on the concept of precision medicine and high demand of translational research, the applications of mouse models for human ASCVD study would be limited due to the natural differences in metabolic features between mice and humans even though they are still the most powerful tools in this research field, indicating that other species with biological similarity to humans need to be considered for studying ASCVD in future. With the development and breakthrough of novel gene editing technology, Syrian golden hamster, a small rodent animal replicating the metabolic characteristics of humans, has been genetically modified, suggesting that gene-targeted hamster models will provide new insights into the precision medicine and translational research of ASCVD. The purpose of this review was to summarize the genetically-modified hamster models with dyslipidemia to date, and their potential applications and perspective for ASCVD.
Introduction
Atherosclerosis, a chronic inflammatory disease leading to the occlusion of arteries by atherosclerotic plaques, is a major pathological basis of cardiovascular disease (CVD), including coronary heart disease, stroke and peripheral vascular disease, which is the first leading cause of morbidity and mortality worldwide. In China, the epidemiological evidence shows that the prevalence of CVD has been increasing continuously, contributing to –45% of death based on the China Cardiovascular Disease Report 2018 [1]. Given the burden of CVD and its devastating consequence of national public health, considerable efforts have been made to study the molecular mechanisms underlying the pathogenesis of CVD. However, inasmuch as CVD, especially atherosclerosis-related CVD (ASCVD), is multifactorial disease, the incomplete understanding of human atherosclerosis limits the development of therapeutic approaches for clinical trials.
Although there are many risk factors influencing the progress of atherosclerosis, to date, the relationship between dyslipidemia and atherosclerosis has been well documented, suggesting that dyslipidemia is an important culprit of atherosclerosis [2]. Dyslipidemia is classically caused by environmental factors and genetic factors. The former can be easily corrected through dietary adjustment and increased physical activity; however, the latter is difficult to normalize through environmental intervention, in which pharmaceutical treatment or even gene therapy will be required. To our knowledge, familial hypercholesterolemia (FH) with elevated low density lipoprotein-cholesterol (LDL-C), familial hypertriglyceridemia with increased triglyceride and familial low high density lipoprotein-cholesterol (HDL-C) are 3 common rare diseases with dysfunctional mutations in genes involved in lipid regulation, which have been reported to be independent strong risk factors of atherosclerosis. Therefore, targeting dyslipidemia has been proposed to be a promising treatment for atherosclerosis. Over the past 40 years, mouse models have been widely used for human atherosclerosis study due to the advantages of low cost and ease of maintenance and manipulation [3]. Genetically-engineered mouse models with lipid disorders provide valuable insights into the mechanisms of lipid metabolism and atherosclerosis based on the similarities of mouse genes to those risk genes identified by human genome-wide association study (GWAS), extrapolating the findings using mouse models to human study. Yet, the failure of therapeutic treatment for the patients with lipid disorders and atherosclerosis in different clinical trials demonstrates that the favorable results and theories from mouse study need to be revisited when they are translated into human disease because mice cannot completely mimic the metabolic profiles of humans. Numerous studies with more species that better replicate humans should be considered.
Like mouse, Syrian golden hamster is also a small laboratory rodent animal model showing high similarities of metabolic features to human, which has been largely introduced into the research field of virology and cancer [4, 5]. Recently, emerging data have reported that Syrian golden hamsters exhibit clinical manifestations observed in corona virus disease 2019 (COVID-19)-infected patients [6], [7], [8], [9], suggesting that Syrian golden hamster will be an ideal tool for human disease. With more advantages of Syrian golden hamster used for basic research and the development of novel gene editing technology, genetically-modified hamster models have been successfully created and applied to different research areas. Due to the importance of dyslipidemia and atherosclerosis for CVD, this review focuses on the selected discoveries using the genetically-modified hamster models with dyslipidemia that we consider to be critical for human atherosclerosis study, which will help us better understand the substantial contributions of genetically-modified hamster models to human ASCVD.
History of the application of Syrian golden hamster for dyslipidemia and atherosclerosis studies
Compared with large experimental animals, such as rabbits, minipigs and non-human primates, small rodent animals, including mice, rats and hamsters, have obvert advantages in terms of low cost, ease of maintenance and manipulation, and short reproductive cycle. Interestingly, accumulating lines of evidence showed that Syrian golden hamsters possess human-like characteristics of lipid metabolism, circadian rhythm and immune response compared to mice, the most widely used animal models for basic medical research (Table 1), indicating that Syrian golden hamsters would be a potentially ideal animal model used for the study of dyslipidemia, atherosclerosis and ASCVD without any needs of gene modification.
Comprehensive characteristics of lipid metabolism and immune response among human, hamster and mouse.
| Characteristics |
 Human Human |
 Hamster Hamster |
 Mouse Mouse |
Refs |
|---|---|---|---|---|
| Dominant plasma lipoproteins | LDL | LDL | HDL | [37] |
| CETP gene expression | Abundant | Abundant | None | [35] |
| ApoB editing | Intestine | Intestine | Intestine/liver | [120] |
| ApoB-48 | Chylomicron | Chylomicron | Chylomicrons/VLDL | [120] |
| Hepatic LDL receptor abundance | Low | Low | High | [38] |
| Extrahepatic cholesterol synthesis | 90% | 85% | 50% | [36] |
| High cholesterol diet | Susceptible | Susceptible | Resistant | [39] |
| Immune response | Comparable infectious disease progression to human, even the only lethal model of some virus infection. | Not suitable candidate models for some infectious diseases. | [121], [122], [123], [124] | |
-
LDL: low density lipoprotein; VLDL: very low density lipoprotein; HDL: high density lipoproteincholesterol.
Based on the high similarity of metabolic features between hamsters and humans, in the 1950s, Altschul [10] at University of Saskatchewan in Canada initiated an important study to observe the relationship between cholesterol and atherosclerosis in hamsters, in which showed that hamster fed with a diet containing milk and egg yolk displayed vascular responses and consequential thrombosis. Although consistent results confirming the impact of dietary cholesterol on atherosclerosis using hamsters have been reported in the next decades [11], [12], [13], until 1987, Nistor and the colleagues [14] from Romania systemically described the characteristics and progression of atherosclerosis in male Syrian golden hamsters on high fat diet feeding. They challenged hamster with a high-fat diet containing 3% cholesterol and 15% butter for 10 months. As expected, they found that plasma total cholesterol and LDL-C levels in high fat diet fed hamsters were markedly increased at week 4, which is as high as 17 and 13 folds after 10 months. Pathological analysis demonstrated that large amounts of inflammatory cells were accumulated on the surface of endothelium and lipids were largely deposited in the aortic arch and thoracic aorta with smooth muscle cell infiltration in the intima as early as 4 weeks after diet intervention, which is so called “early stage of atherosclerosis”. With the development of atherosclerosis in those hamsters on high fat diet feeding, lipid deposition was also observed in smooth muscle cells mainly localized in the intima-media, which is called “advanced atherosclerosis”, suggesting that the atherosclerotic development characterized in high fat diet fed hamsters is similarly to those observed in human atherosclerotic lesions. In 1990s, Chen et al. [15] and Wollett et al. [16] worked out the effects of dietary cholesterol on lipoprotein profiles in hamsters through enhancing hepatic very low density lipoprotein (VLDL) secretion and LDL-C production without affecting high density lipoprotein (HDL) transport. Meanwhile, a team led by Schaefer from the Tufts University in Boston discovered that Lovastatin, a lipid lowering agent, attenuated diet-induced atherosclerosis with Syrian golden hamsters with the F1B strain through reducing non-high-density lipoprotein cholesterol [17], indicating the good responsiveness of Syrian golden hamsters to statins, which could be used for a drug test.
Since 2000s, using Syrian golden hamster models, Adeli’s group at University of Toronto in Canada has extensively investigated lipid metabolism and the related disease. When treating hamsters with a fructose diet, they identified new molecular mechanisms regulating lipid metabolism in the setting of insulin resistance [18], [19], [20], showing that high fructose intake elicited hyperinsulinemia, then leading to hypertriglyceridemia by overproduction of hepatic VLDL, which could be ameliorated by statins [21]. Furthermore, the recent work from the same team has made substantial contributions to ApoB metabolism and its role in obesity, type II diabetes and metabolic syndrome when using the hamster models [22], [23], [24], [25], and verified glucagon-like peptides (GLP-1 and GLP-2) as novel regulators of lipids in intestine and liver [26], [27], [28], [29], [30], [31].
In 1990s, two independent groups led by Breslow at University of Rockefeller and Maeda at University of North Carolina at Chapel Hill reported the first genetically targeted mouse mode in the world using homologous recombination technology, apolipoprotein E (ApoE) knockout mouse, which showed hyperlipidemia and spontaneous atherosclerosis under regular chow diet condition [32, 33]. Whereas at the most same time, Brown and Goldstein from University of Texas Southwestern Medical Center at Dallas, two 1985 Nobel Laureates for the discoveries of cholesterol metabolism through LDL receptor (LDLR) pathway, reported the second dyslipidemic mouse model, LDLR knockout mice showing hypercholesterolemia to mimic human familial hypercholesterolemia (FH) [34]. Since these two mouse models are prone to diet-induced hyperlipidemia and atherosclerosis, making them strong tools that have been widely used for the research of lipid metabolism and ASCVD to date, accumulating evidence has revealed more advantages, suggesting that wild type Syrian golden hamster model is still an ideal supplement for ApoE and LDLR knockout mice. For example, similar to humans, hamsters possess endogenous cholesteryl ester transfer protein (CETP), a key protein responsible for exchange of triglycerides carried on VLDLs and LDLs and cholesterols on HDLs [35], and reverse cholesterol transport (RCT) [35], whereas mice lack CETP, implying that mice cannot replicate the physiological process of lipid metabolism in vivo. Furthermore, many human-like metabolic features in Syrian golden hamsters have been demonstrated by previous critical work when compared to mouse models: (1) Higher endogenous CETP activity [35]; (2) Apolipoprotein B (ApoB) editing only in small intestine, but not in liver, leading to a truncated ApoB, so called ApoB48, present on small intestine-derived chylomicrons (CMs), and full length ApoB100 carried on hepatic VLDLs, but mice have ApoB48 in both CMs and VLDLs due to the ApoB editing observed in both small intestine and liver [35]; (3) Extrahepatic cholesterol synthesis with more than 85% is a major sources for total cholesterol in hamsters, which is much closer to 90% observed in humans when compared to mice that only have 50% [36]; (4) ∼48% of total plasma cholesterol is carried on LDL particles and ∼49% is distributed on HDL, yet most of plasma cholesterol is present on HDLs in mice [37]; (5) Relative low hepatic LDLR activity in hamsters [38]; (6) More susceptible to hypercholesterolemia, hypertriglyceridemia, and atherosclerosis, and then a development of coronary artery disease under dietary interventions [39]; (7) Insulin resistance and type 2 diabetes can also be induced by high-sugar diet [40]. Collectively, these key findings and overt advantages suggest that hamster model will play a crucial role in the study of lipid metabolism disorders and ASCVD.
However, it is should be noted that although wild-type hamsters possess many advantages described above, poor reproducibility and a large degree of variation of consistency in aortic lesions induced by high fat diets limit the further use for the study of diet-induced atherosclerosis [41], indicating that genetically modified hamsters would represent better animal models to mimic human diseases and investigate the impact of monogenic risk on occurrence and development of human genetic rare diseases, such as familial hypercholesterolemia, familial hypertriglyceridemia and low HDL. With the development of gene editing technology and the breakthrough of in vitro fertilized egg culture technology, several lines of genetically-modified hamster models have been successfully generated in the past years. In this review, we will summarize the crucial work on genetically-engineered hamster models with elevated plasma cholesterol, triglyceride or low HDL using CRISPR/Cas9, a powerful gene editing system awarded for Nobel Prize in chemistry 2020, and their applications and the perspective in the field of lipid metabolism disorders and ASCVD.
LDL, LDLR and LDLR knockout hamsters
In 1950s, two major cholesterol-rich compositions, low density lipoprotein (LDL) and high density lipoprotein (HDL), were identified by John Gofman at the University of California, Berkeley [42]. Unlike low HDL levels that remains controversial in the study of CVD in the following decades, the positive relationship between elevated plasma LDL concentration and atherosclerosis has been consistently reported, suggesting that LDL is a strong and reliable risk factor for ASCVD [43], [44], [45], [46]. However, as LDL particle was pathogenic and gained increased attention, the molecular mechanisms of LDL regulation and by which LDL elicited atherosclerosis were unknown yet until the discoveries by Michael Brown and Joseph Goldstein in 1970s, who identified a lipoprotein receptor so called LDLR in fibroblast cells from the patients with FH [47] (Figure 1). FH is an autosomal dominant genetic disease characterized by increased plasma total cholesterol and LDL-C levels, then leading to premature coronary heart disease (CHD) [48], [49], [50], which is primarily caused by the defect of LDLR attributable to variable mutations in LDLR (c.85A>T, c.97C>T, c.191_313del, c.195dupT…etc.), ApoB (c.523 + 2T>G, c.1069C>T, c.1120G>A, c.1399C>G) and proprotein convertase subtilisin/kexin 9 (PCSK9) (c.7057C>T, c.10579C>T, c.10580G>A, c.12739C>T) [51]. According to the number of mutations in allele, there are two forms reported for FH. Heterozygous FH (HeFH) caused by one mutation in an allele with a prevalence of 1:250–500 is relatively common, whereas homozygous FH (HoFH) with two mutations in two alleles show an incidence of 1:160,000–1,000,000. Compared to normal individuals, the former shows moderate hypercholesterolemia and increased risk for CHD by 20 folds without any treatment or prevention; however, the latter exhibits severe hypercholesterolemia with markedly elevated LDL-C, leading to early onset CHD observed in children. Although total plasma cholesterol levels in Chinese populations are much lower than the individuals in Western countries due to ethnicity and dietary habit, recent epidemiological data show that there are 687,728 cases of global FH in 2017, in which China accounts for 8% of FH with predicted 7 million HeFH cases and 5,000 HoFH cases, respectively, suggesting that FH is a devastating disease and systemically basic and clinical investigations are still warranted in China.
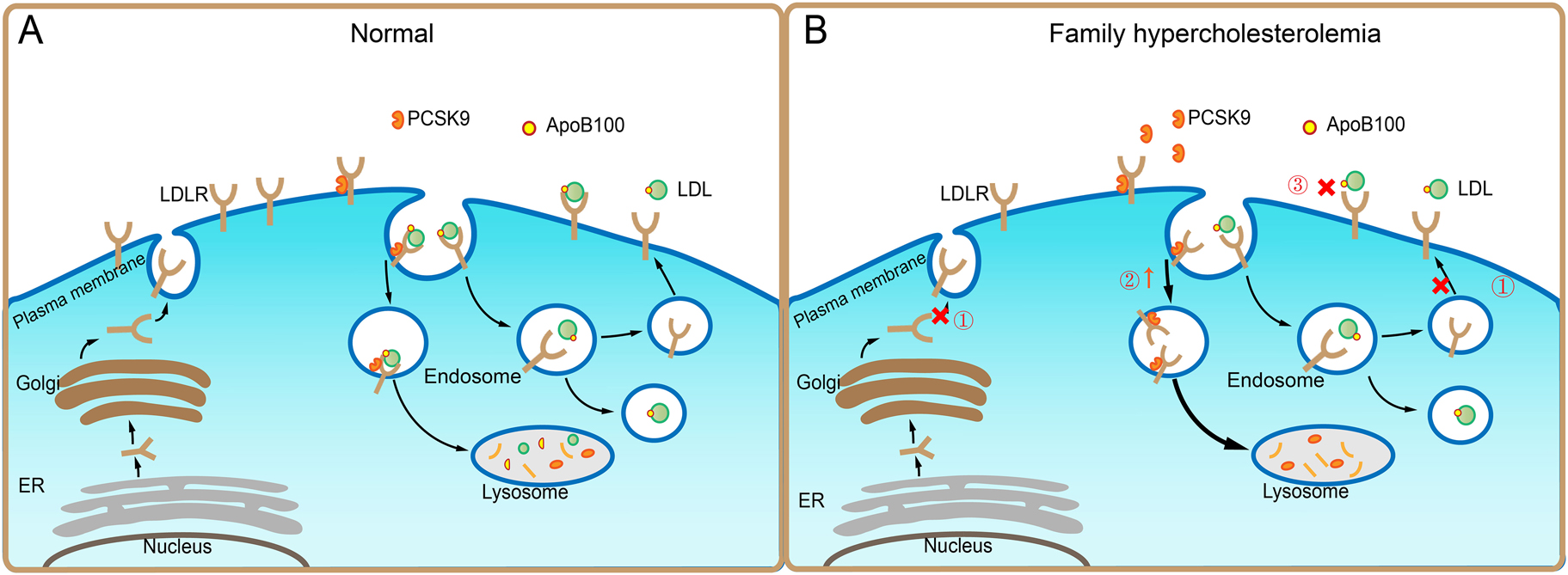
Genetic causes of familial hypercholesterolemia. A: After synthesized and secreted from endoplasmic reticulum (ER), low density lipoprotein (LDL) receptor (LDLR) molecules are processed in the Golgi, and finally transported to the plasma membrane of cells, where LDLR binds apolipoprotein B (ApoB)100-containing LDL particles. The complex undergoes endocytosis. In the endosomes, LDL particle and LDLR are disassociated, leading to a direct delivery of LDL to lysosomes for degradation, whereas LDLR is recycled to the cell surface for future use. In the meantime, intracellular cholesterol upregulates the expression of the proprotein convertase subtilisin/kexin 9 (PCSK9), which binds to and internalizes LDLR for lysosomal degradation to maintain lipid homeostasis under normal condition. B: In familial hypercholesterolemia (FH), ① The LDLR mutations, namely LDLR (c.85A>T, c.97C>T, c.191_313del, c.195dupT…etc.) inhibit the product and recycle of LDLR; ② The gain-of-function of mutations in PCSK9, namely PCSK9(c.7057C>T, c.10579C>T, c.10580G>A, c.12739C>T) cause increased degradation of LDLR; ③ The ApoB100 mutations, namely ApoB (c.523 + 2T>G, c.1069C>T, c.1120G>A, c.1399C>G) block the interaction between LDL and LDLR, all of which impair LDLR-mediated cholesterol transport, thus leading to hypercholesterolemia.
To better understand the mechanism underlying the pathogenesis of FH in vivo, several animal models with different species have been created to mimic FH, including LDLR knockout mice [52], LDLR knockout rats [53], Watanabe hereditary hyperlipidemia (WHHL) rabbits [54], LDLR knockout rabbits [55] and LDLR knockout minipigs [56, 57]. Because LDLR knockout mice are the most commonly used models for FH by far, which have been fully characterized in different reviews and other species possess different disadvantages of FH study, limiting the further applications for human FH and atherosclerosis. Herein, we only listed a head to head comparison among these FH animal models in Table 2 and mainly introduced CRISPR/Cas9 targeted-hamster with LDLR deficiency, a novel small rodent model of FH.
Current mammal animal models with FH.
| Animal model |
 Advantages Advantages |
 Disadvantages Disadvantages |
Refs |
|---|---|---|---|
| LDLR KO mouse | 1. Amenable to genetic alterations. 2. Atherosclerotic plaques formation within short time. |
1. Modest hypercholesterolemia in homozygotes. 2. Hard to develop atherosclerotic lesions under normal condition. 3. Atherosclerotic lesions distinct from those of human. 4. CETP deficiency. |
[34, 52] |
| LDLR KO rat | 1. Useful for studying not only atherogenesis, but obesity and insulin resistance as well. 2. More capable of surgical operations than mouse. |
Without gallbladder, more resistant to diet induced atherosclerosis compared to mouse. | [53] |
| WHHL rabbit | 1. Human-like lipoprotein metabolism and atherosclerotic lesion features. 2. Smaller individual heterogeneity. 3. Hypercholesterolemia at birth. |
Breeding is difficult, limiting its further application. | [54] |
| LDLR KO rabbit | 1. Human-like lipoprotein metabolism profile. 2. Spontaneous atherosclerosis when fed a normal chow diet. 3. Abundant plasma CETP activity. |
Rabbits are more expensive than mice and they also have no hepatic lipase activity and ApoA2. | [55] |
| LDLR KO minipig | 1. Similar to humans in physiology, anatomy, genetics, and size. 2. Spontaneous atherosclerosis on a normal chow diet. |
1. Require specialized housing, trained staff, unique study facilities, and specially formulated diets. 2. Absence of CETP. 3. No phenotypes in heterozygous minipig fed with HCHF diet. 4. High variability caused by genetic backgrounds. |
[56, 57] |
-
KO: knock out.
Consistent with the phenotypes observed in human FH, LDLR knockout hamsters on regular chow diet displayed hypercholesterolemia to different extent in the gene dose dependent manner [58, 59]. The levels of plasma total cholesterol were moderately increased to ∼500 mg/dL in heterozygotes and significantly elevated to ∼1,000 mg/dL in homozygotes. However, only homozygous LDLR knockout mice exhibited a slight increase in plasma total cholesterol levels, which were normal in heterozygous LDLR knockout mice fed with chow diet. Cholesterol distribution analyzed by fast protein liquid chromatography (FPLC) further confirmed that most of plasma cholesterol was present on LDL to form a dominant peak contributing to hypercholesterolemia in both two forms of FH, whereas HDLs were major components of plasma lipoproteins in heterozygous LDLR knockout mice and comparable to LDLs in homozygous LDLR knockout mice, demonstrating that LDLR-deficient hamsters, but not mice, replicated the characteristics of cholesterol metabolism described in FH without any interventions. Previous studies reported the difficulties to induce hypercholesterolemia in heterozygous LDLR knockout mice, but when LDLR knockout hamsters with heterozygous form were fed with a high fat diet containing 0.5% cholesterol and 15% lard for only 2 weeks, they showed combined hyperlipidemia with increased total cholesterol and triglyceride, which have not been seen in mice with the same genotype. As expected, high fat diet resulted in a more severe hyperlipidemia in homozygous LDLR knockout hamsters, thus leading to high mortality rate caused. Taken together, loss of LDLR predisposes only hamsters, but not mice, to diet-induced hyperlipidemia.
The development of atherosclerotic plaques in coronary arteries is a major consequence of hypercholesterolemia in FH, who always dies from heart attacks, suggesting that atherosclerotic progress should be closely monitored. The lesions were observed in aged homozygous LDLR knockout hamsters (–18 months) under a normal chow diet, whereas animals with the same genotype at the age of 12 months did not develop obvious atherosclerotic lesions in whole aorta and aortic root. Importantly, homozygous LDLR knockout hamsters with hypercholesterolemia and hypertriglyceridemia rapidly developed atherosclerotic lesions not only throughout whole aorta, but also in coronary artery, which resembles human ASCVD. As we have known that HeFH is relatively common in human populations, the use of heterozygous LDLR knockout hamsters for atherosclerosis study will provide new insights into the pathogenesis of HeFH and the development of lipid lowering agents because no species lacking one copy of LDLR gene have been suggested due to a normal lipid profile when maintained on normal chow diet and the difficulties to induce hyperlipidemia by dietary intervention. Guo and the colleagues [58] found that in response to a high cholesterol/high fat feeding for 12 weeks, heterozygous LDLR knockout hamsters displayed accelerated atherosclerotic lesions in whole aorta, aortic roots and coronary artery. In a following study, Wang et al. [60] reported that in heterozygous LDLR knockout hamsters fed with diets containing variable contents of cholesterol for different indicated time points, severity of atherosclerosis was positively associated with plasma cholesterol levels, which were easily modulated by dietary cholesterol. Given that previous work using both ApoE and LDLR-deficient mice showed that atherosclerotic lesion sizes were not dependent on plasma cholesterol levels and time [61], these crucial findings are encouraging that heterozygous LDLR knockout hamsters will be an ideal animal model used for atherosclerosis study.
Many novel genes and/or proteins modulating plasma LDL through LDLR pathway have been identified over the past decades, such as PCSK9 and inducible degrader of LDLR (IDOL), but their functions are still not completely understood. Based on that LDLR is a master regulator of LDL and the key discoveries of lipid metabolism and atherosclerosis observed in heterozygous LDLR knockout hamsters, it is important to test the agonist or inhibitor targeting PCSK9 or IDOL. Recently, a collaborative study led by Liu at Peking University and Chen at Novo Nordisk Research Centre China, Beijing, confirmed a good response of heterozygous LDLR knockout hamsters to Evolocumab, a PCSK9 antibody, with lower plasma cholesterol and reduced atherosclerotic lesions after high cholesterol/high fat diet feeding [62]. Moreover, Ezetimibe, an inhibitor of intestinal cholesterol absorption, also significantly reduced plasma cholesterol in heterozygous LDLR knockout hamsters. Of note that both Evolocumab and Ezetimibe have been applied to the patients with hyperlipidemia for clinical trials, including FH, the positive results from heterozygous LDLR knockout hamsters treated with Evolocumab or Ezetimibe suggest that this model also be a helpful tool for drug development. Furthermore, Sun et al. [63] found that lack of one copy of LDLR gene in hamsters promoted pathological cardiac remodeling in the setting of diabetes through Galectin 3-mediated pathway, providing new ideas on the prevention of diabetic cardiac remodeling. Interestingly, recent work by Gu et al. [63] demonstrated that heterozygous LDLR knockout hamsters showed cognitive impairment similar to that reported in elderly HeFH patients attributed to impaired blood brain barrier (BBB) caused by elevated LDL-C, which could be attenuated by Yangxue qingnao wan and silibinin capsules, the two Chinese medicines. In summary, LDLR knockout hamsters targeted by CRISPR/Cas9, including both heterozygous and homozygous form, represent a valuable animal model with the characteristics of FH, which will be a promising platform for the study of lipid metabolism disorders and the related diseases, such ASCVD, diabetes and neurodegeneration.
Triglyceride metabolism, hypertriglyceridemia and ASCVD
Triglycerides (TGs) are major molecules carried on liver-derived VLDLs and intestine-derived chylomicrons (CMs) required for the usage and storage of energy and transport of fatty acids. The process of TG metabolism is complex, in which lipoprotein lipase (LPL) plays a central role. Under normal conditions, TG-rich lipoproteins (TRLs) are hydrolyzed by LPL complex at the vascular surface of adipose tissue, heart, muscle and other tissues, leading to the formation of large lipoprotein remnants and lipid transfer between TRLs and HDL (Figure 2). When this LPL-mediated hydrolysis process is dysfunctional, plasma TG levels are significantly increased to different extents, eventually resulting in hypertriglyceridemia (HTG). To our knowledge, HTG is a multifactorial disease classically divided into hereditary and non-hereditary disorders caused by gene mutations and environmental intervention, respectively (Table 3). Unlike hypercholesterolemia that has been recognized as a risk factor of ASCVD, whether HTG is an independent contributor to ASCVD still remains conflicting and elusive for a long time. It is proposed that the size of TRLs is a major determinant of their proatherogenic property because TRL particles with large sizes, such as CMs, cannot enter the subendothelial space to form foam cells, a key component of atherosclerotic plaques [64]. However, recent emerging data with lipidomics and proteomics show that TRL associated apolipoprotein C2 (ApoC2) and apolipoprotein C3 (ApoC3), two crucial modulators required for LPL activity, are closely linked to the risk of CVD [65, 66], suggesting that they are potential predictors for ASCVD. Considering the importance of ApoC2 and ApoC3 in TG metabolism and ASCVD, in this review we mainly discussed the Syrian golden models lacking these two gene.
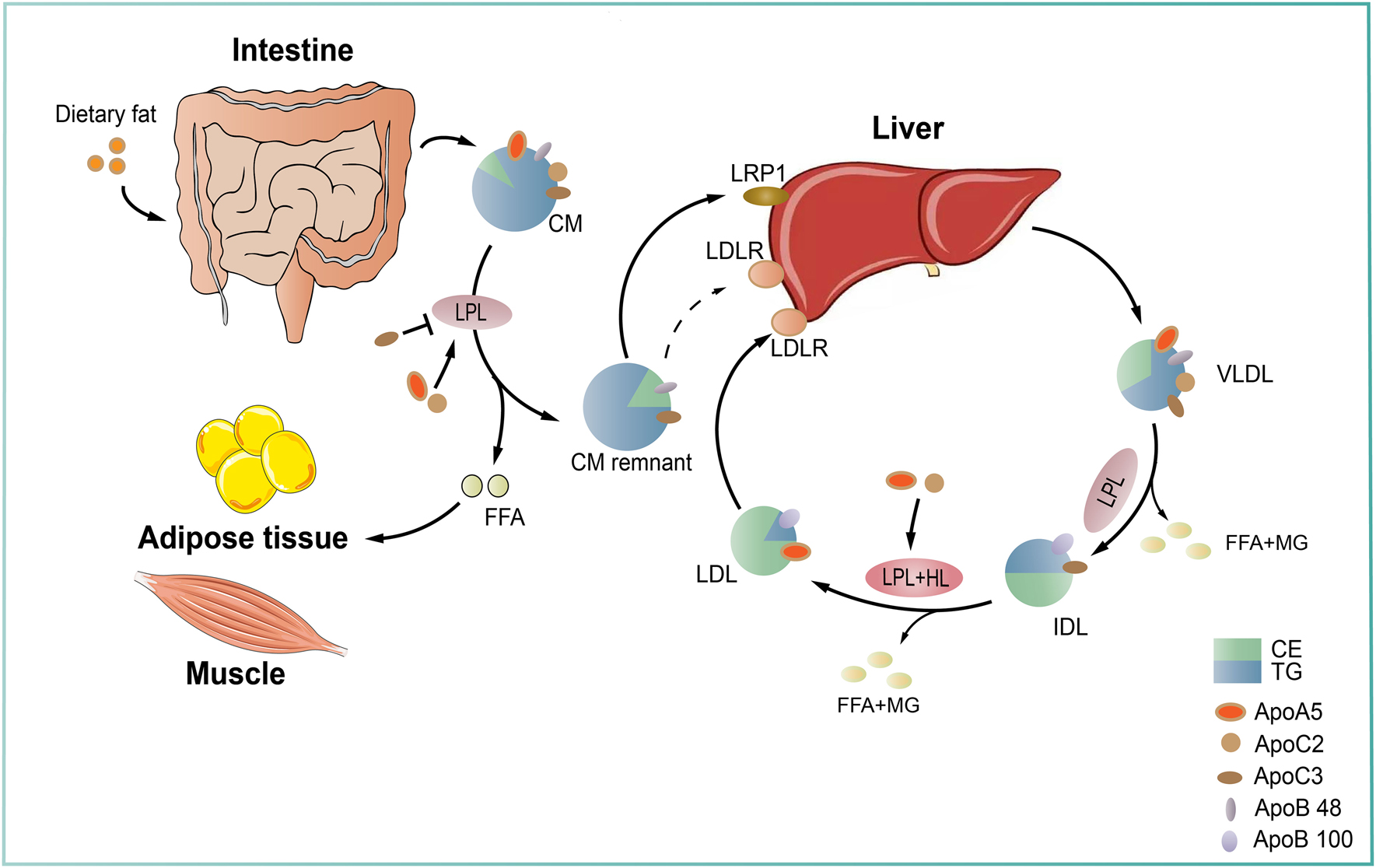
Triglyceride-rich lipoprotein metabolism. Triglyceride-rich lipoproteins (TGRLs) are classified into endogenous and exogenous, which are derived from the liver and the small intestine, respectively. In the endogenous pathway, triglycerides (TGs) are synthesized in hepatocytes using free fatty acids and then incorporated into the core of ApoB-containing very low density lipoprotein (VLDL) particles. After secreted into plasma, VLDLs are hydrolyzed by lipoprotein lipase (LPL) generate smaller VLDLs and then intermediate density lipoproteins (IDLs). IDL particles can be further catabolized by LPL and hepatic lipase (HL) to produce LDL particles. In the exogenous pathway, dietary fats are absorbed by enterocytes and incorporated into ApoB48-containing chylomicrons (CMs), which then enter the circulation. Circulating CMs are quickly hydrolyzed by LPL to form CM remnants that are cleared from circulation through receptor mediated pathway by the liver. The released free fatty acids (FFAs) can be taken up by muscle for energy usage and by adipose tissue for lipid storage, respectively. LPL-mediated TG hydrolysis is the rate-limiting step during TG metabolism, which is highly regulated by other factors, including apolipoprotein C2 (ApoC2) and apolipoprotein A5 (ApoA5), two critical activators required for LPL hydrolytic activity, and apolipoprotein C3 (ApoC3), an inhibitor. CE: cholesteryl ester; LRP1: LDLR related receptor 1; MG: monoglyceride; TG: triglyceride.
A summary of primary and secondary causes of HTG.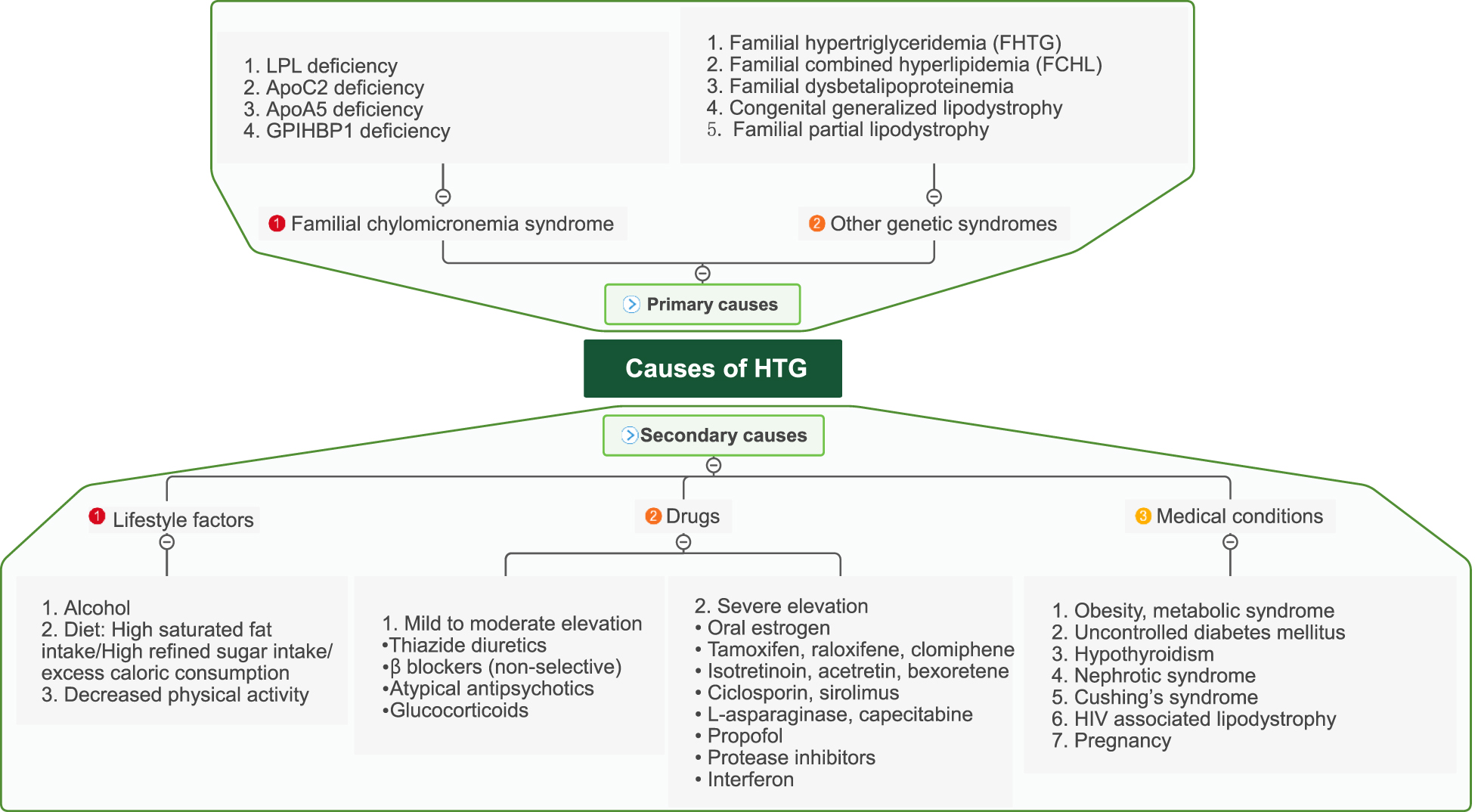
Familial hypertriglyceridemia and Syrian golden hamsters with ApoC2 and ApoA5 deficiency
Familial chylomicronemia (FCS), one type of severe HTG, largely caused by loss-of-function mutations in Lpl gene, is a rare genetic disorder with a prevalence of one per million [67, 68]. It has been reported that ApoC2 functions as a key activator required for LPL catalytic activity, thus playing a central role in the TG hydrolysis process mediated by LPL [69]. Until now, there are more than 30 ApoC2 mutations reported in different clinical cases worldwide [68]. Clinical data show that hypertriglyceridemic patients with TG>1,000 mg/dL always present a high risk of acute pancreatitis. Therefore, individuals with severe HTG (TG>10,000 mg/dL) caused by ApoC2 mutations are predisposed to recurrent acute pancreatitis, a life-threatening disease seriously influencing life quality, who need repeated hemodialysis all life.
Given the crucial role of ApoC2 in TG metabolism, it is rational to establish an experimental animal model lacking ApoC2 to investigate the impact of ApoC2 on human disease and build up a powerful tool for the screening of TG lowering compounds. However, Shachter et al. [70] found that overexpression of human ApoC2 also present unexpected HTG in mice similar to the patients with ApoC2 mutations, in which the amounts of plasma ApoC2 were reduced, indicating that the role of ApoC2 in TG metabolism is complex and more animal models will be needed. Unfortunately, no homozygous ApoC2-deficient mice with HTG were obtained due to embryonic lethality using homologous recombination. Recently, a team from Remaley’s laboratory at the National Institutes of Health (NIH) applied zinc finger nuclease technology to target mouse ApoC2 [71]. They observed that this mouse model has a full length and immature ApoC2 protein that cannot be efficiently cleaved to form an active structural conformation and largely accumulates on cholesteryl ester-rich HDLs, but not TRLs, leading to the inability to hydrolyze plasma TG and thus HTG [71]. Further investigation discovered that ApoC2 mutant mice showed an increase in circulating ApoC2 concentration similar to that observed in mice overexpressing human ApoC2, but ApoC2 deficient patients, meaning that this is not an ApoC2 knockout mouse model [71]. At the same time, Liu et al. [72] at the University of California, San Diego, successfully deleted the Apoc2 gene from zebrafish and an experimental model with ApoC2 deficiency, showing reduced LPL activity and then FCS, a phenotype that is similar to ApoC2-deficient patients in clinics. Although zebrafish as a laboratory animal model has many advantages in biological study, including lipid research, it is not a mammal with an obvious difference in vascular biology and its application for atherosclerosis study is limited.
To better understand the precious function of ApoC2 in TG metabolism, we used CRISPR/Cas9 technology for the first time to generate a small rodent animal model with ApoC2 deficiency using Syrian golden hamster [73]. Unlike ApoC2 deficient mice with embryonic lethality, ApoC2 knockout hamsters could survive up to 9 days after birth, showing small size, pink blood vessels and milky plasma with marked elevation of TG (∼20,000 mg/dL) and total cholesterol (∼2,000 mg/dL) during lactation. Further biochemical and pathological analysis revealed that ApoC2 pups exhibited morphological changes in multiple organs, such as liver, kidney and pancreas with a significant reduction in blood glucose level. However, repeated injection of glucose could not rescue the early death, demonstrating that multiple organ injury caused by severe HTG, but not low glucose, probably contributed to this death. To prevent neonates with ApoC2 deficiency from early death, we continuously administered WT hamster serum containing endogenous ApoC2 to ApoC2 pups though intravenous injection every 3 days till weaning and found that 80% of ApoC2 knockout pups could survive to adulthood without any overt morphological changes in different tissues, but still showing severe HTG because administration of WT hamster serum only temporarily corrects severe HTG due to a fast catabolic rate of endogenous ApoC2 by kidney in vivo. Thus, adult ApoC2 knockout hamsters rescued using WT hamster serum present no endogenous ApoC2. In the following experiments, we found that adult ApoC2 knockout hamsters with severe HTG showed a resistance to different lipid lowering agents. A special diet containing medium chain fatty acids could partially reduce TG levels, whereas recombinant human ApoC2 expressed by adeno-associated virus (AAV-hApoC2) completely corrected this refractory hyperlipidemia in both neonatal and adult ApoC2 knockout animals in a long-term manner without any adverse effects [74], suggesting that AAV-mediated gene therapy will be a potential therapeutic approach for severe HTG caused by ApoC2 mutations. Furthermore, a causal link between HCS with very large TRLs and atherogenesis was discovered in our model because ApoC2 knockout hamsters developed spontaneous atherosclerotic lesions through whole aorta and aortic toot at the age of 8 months on a regular chow diet; however, before that the relationship between ApoC2 and atherosclerosis was not clear due to lacking of a proper animal model and a very small size of case number. It is worth to note that WT and LDLR knockout hamsters receiving AAV-hApoC2 showed moderate HTG and severe HTG, while as in another study, Pulawa et al. [75] at the University of Colorado at Denver found that HTG in ApoC2 transgenic mice was improved by overexpressing LPL in muscle, suggesting that the ratio of ApoC2 and LPL should be considered as a more important determinant factor of plasma TG levels when ApoC2 is targeted for the treatment of severe HTG.
Additionally, apolipoprotein A5 (ApoA5), another protein secreted by liver, is also important for LPL-mediated TG hydrolysis in plasma. Clinical data reported that ApoA5 variants always affect LPL activity to elicit HTG and then increase the incidence of atherosclerosis in different populations [76], [77], [78]. Transgenic mice overexpressing human ApoA5 had a lower plasma TG levels and reduced atherosclerotic lesions in the atherosclerosis prone mice compared to control mice [79]; however, hepatic TG contents were significantly increased [80]. In ApoA5 knockout mice, Berg et al. [81] from Leiden University Medical Center, Netherlands, reported that ApoA5 deficiency exacerbated high fat diet-induced obesity, but Shulman and his team [82] from Yale University, New Haven, found that knocking down hepatic ApoA5 using an antisense oligonucleotide (ASO) improved whole-body insulin sensitivity in high fat-diet fed mice.These controversial observations from mice indicate a puzzlement over the role of ApoA5 in TG metabolism, leading to no conclusion on whether ApoA5 deficiency accelerated atherosclerotic development to date. Based on this unsolved problem, we constructed ApoA5 knockout hamsters using CRISPR/Cas9 and found that ApoA5 deficiency in hamsters resulted in a phenotype of HTG resembling the patients with decreased ApoA5 caused by loss-of-function mutations (data unpublished). Future comprehensive studies will be conducted using this hypertriglyceridemic model to investigate TG metabolism and atherosclerosis, and we believe that ApoA5 knockout hamster will provide new insights into the relationship between HTG cause by ApoA5 deficiency and atherosclerosis.
ApoC3, hypolipoproteinemia and ApoC3 knockout hamsters
Unlike ApoC2 and ApoA5 described above, which activate LPL function, the key function of ApoC3 is to inhibit the hydrolysis of TG on TRLs mediated by LPL [83], impair the clearance of TRL remnants by lipoprotein receptors in liver [84]. Therefore, ApoC3 is an important regulator of plasma TG, and the relationship between ApoC3 and HTG has been well documented. Large scale population-based cohort studies have found that the elevated levels of plasma ApoC3 are positively correlated with higher concentrations of circulating triglyceride and the incidence of CHD [85]. In agreement with the findings observed in human studies, transgenic mice overexpressing human Apoc3 gene show overt HTG, which predisposes animals to diet-induced atherosclerosis in the ApoE or LDLR deficient background [86, 87]. Moreover, some epidemiological cases report that patients carrying loss-of-function of Apoc3 mutants show both reduced levels of plasma ApoC3 protein and triglyceride associated with a decreased risk of ASCVD supporting the concept that ApoC3 would be a promising target for HTG and atherosclerosis [88], [89], [90]. Indeed, several clinical trials with antisense nucleotide and monoclonal antibodies are now under development [91], [92], [93]. However, recent controversial findings are reported by other studies of human and experimental animals. Homozygotes lacking ApoC3 caused by a mutation called C19X display anti-atherogenic lipid profile, but their CHD proportion does not have any difference when compared to normal people or heterozygotes. Consistently, the beneficial effect of ApoC3 deficiency on atherosclerotic development in LDLR-deficient mice is not observed in our previous study [87], suggesting that ApoC3 plays a central role in triglyceride metabolism, but how it relates to ASCVD still remains elusive.
On this basis, we targeted ApoC3 in Syrian golden hamsters using CRISPR/Cas9 and fully characterized the lipoprotein metabolism and atherosclerosis of ApoC3 knockout hamsters. We found that plasma TG was significantly reduced in chow diet-fed ApoC3 knockout hamsters due to an increased clearance of circulating TRLs, which was consistent with the findings in ApoC3 knockout mice and hypertriglyceridemic patients receiving ASO of ApoC3. In addition, the key finding of less VLDLs and more LDLs uncovered by the lipoprotein disc electrophoresis was in agreement with the observation in carriers with ApoC3 R19X null mutation, indicating an increased conversion of VLDLs to LDLs, then leading to an elevation of LDLs [94, 95]. These elevated LDLs could be effectively cleared by liver through lipoprotein receptor-mediated uptake pathways. Although HDL-C levels did not alter in ApoC3 knockout hamsters on regular chow diet whereas homozygotes with ApoC3 R19X showed lower HDL-C levels, this difference in HDL-C between hamster and human could be attributed to the distinct diet ingredients. To our knowledge, unlike human diets that contain variable amounts of cholesterol and fat, the regular chow diets for rodent animals are generally poor in cholesterol and fat [96]. Moreover, HDL-C is susceptible to the environmental interventions, including high fat diets [97]. Therefore, the changes in HDL-C levels in WT and ApoC3 knockout hamsters were only observed upon high cholesterol/high fat diet feeding, but not regular chow diet, demonstrating that ApoC3 knockout hamsters also exhibited a trait of HDL metabolism similar to that observed in human with R19X mutation.
Consistently, ApoC3 knockout hamsters fed with HCHF diet for 4 months exhibited an anti-atherogenic profile with reduced TC and TG levels accompanied increased HDL-C level [98]. We speculated that quick clearance of large TRLs was still the major contributor to the reduction in the concentrations of TC and TG in ApoC3 knockout hamsters. As a consequence of the anti-atherosclerotic changes in lipid metabolism, the atherosclerotic plaque formation in vascular wall was obviously alleviated in ApoC3 knockout hamsters, suggesting that ApoC3 played a detrimental role in atherosclerosis. Importantly, the differences of aortic lesions between WT and ApoC3 knockout hamsters may be related to the distribution of lesions in hamsters because there are more lesions found in abdominal and thoracic aorta than aortic arch and sinus in hamsters than in mice [59]; therefore, the differences in these regions can be detected easily.
To our knowledge, inhibitors of ANGPTL3 and ApoC3 have been tested in patients with HTG in phase 3 clinical trials, whereas Evinacumab, a monoclonal antibody against ANGPTL3 was reported to reduce LDL levels in the patients with refractory hypercholesterolemia through endothelial lipase (EL) [99], but the beneficial effect of ApoC3 inhibition on FH has not been investigated yet. It will be tempting for us to elucidate the relationship between targeting ApoC3 and FH using LDLR and ApoC3 double knockout hamster and explore novel mechanisms by which ApoC3 inhibition lowers circulating lipid levels in the absence of LDLR in the near future.
HDL metabolism, hypoalphalipoproteinemia and Syrian golden hamsters with LCAT or ABCA1 deficiency
Under normal HDL metabolism condition, apolipoprotein A1 (ApoA1) is primarily synthesized and secreted by the liver and the intestine. Circulating ApoA1 serves as a receptor for phospholipid and unesterified cholesterol from phospholipid transfer protein (PLTP)-mediated lipid transfer and ATP-binding cassette transporter A1 (ABCA1)-mediated cholesterol efflux, respectively. This reaction is the initial step of reverse cholesterol transport (RCT), an important process to modulate cholesterol homeostasis and play beneficial roles in atherogenesis in vivo. Lecithin cholesterol acyltransferase (LCAT), a secretory protein that is mainly synthesized by the liver and the only cholesterol esterase in plasma [100] promotes the formation of αHDL, a mature form, by converting cholesterol and phosphatidylcholine into cholesteryl esters and lysolecithin, which is indispensable to maintain HDL stability and RCT [101] (Figure 3). Loss-of-function mutations in ABCA1 and LCAT cause hypoalphalipoproteinemia with extremely low HDL-C in patients and mice lacking these two genes also show undetectable HDL-C. However, unlike hypercholesterolemia and hypertriglyceridemia with elevated plasma cholesterol and triglyceride, respectively, which are positively associated with the increased risk of ASCVD, the relationship between low HDL cholesterol (HDL-C), so called “good cholesterol”, and ASCVD still needs to be investigated based on the inconsistent results from the studies of both human and experimental animal models, including mice and rabbits (Table 4). We will introduce two novel hamster models with LCAT and ABCA1 deficiency in this review.
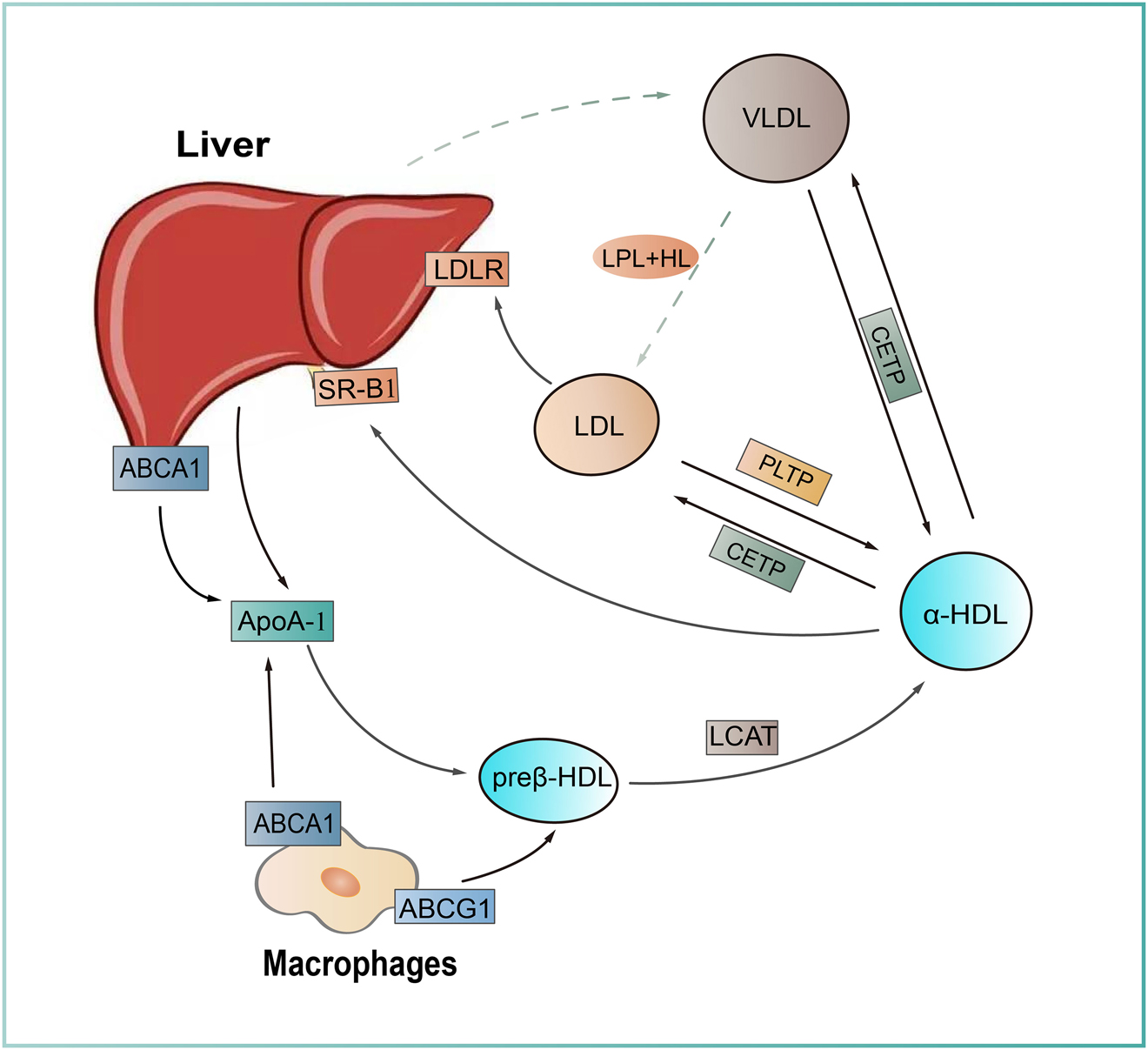
HDL metabolism and reverse cholesterol transport. Nascent high density lipoprotein (HDL) particles (preβ-HDL), originally formed from apolipoprotein A1 (ApoA1) secreted by the liver, become lipidated when cholesterols are loaded via ATP-binding cassette A1 (ABCA1) or ATP-binding cassette A1 (ABCG1) from different cell types. Free cholesterol within the HDL is transformed into CE by lecithin–cholesterol acyltransferase (LCAT). Lipid-rich HDL particles (α-HDL) undergo lipid exchange with ApoB-containing particles (VLDL, LDL) through cholesteryl ester transfer protein (CETP) and phospholipid transfer protein (PLTP), followed by a hepatic degradation through scavenger receptor class B type 1 (SR-B1) mediated pathway, which is called reverse cholesterol transport (RCT) that is important for lipid homeostasis in vivo.
LCAT, ABCA1, low HDL-C and ASCVD.
| Genes | Species | Gene loci | Intermediate phenotypes | ASCVD risk | Refs |
|---|---|---|---|---|---|
| LCAT | Human | LCAT mutant | Extremely low HDL-C | Unchanged | [125, 126] |
 Increased Increased |
[127, 128] | ||||
| Heterozygous LCAT deficiency | Extremely low HDL-C |
 Increased Increased |
[127, 129, 130] | ||
| Homozygous LCAT deficiency | Unchanged | [129] | |||
| Mouse | LCAT overexpression | Increased HDL-C |
 Increased Increased |
[131] | |
| LCAT deficiency | Decreased HDL-C | Unchanged | [132] | ||
 Decreased Decreased |
[107, 133] | ||||
| Decreased HDL-C, increased cholesteryl esters in apoB-containing lipoproteins |
 Increased Increased |
[106] | |||
| Rabbit | LCAT overexpression | Increased HDL-C |
 Decreased Decreased |
[134], [135], [136] | |
| ABCA1 | Human | SNPs located near the ABCA1 gene | Small changes in the levels of HDL-C | Unchanged | [137, 138] |
| Missense variants in ABCA1 | Decreases in cholesterol efflux |
 Increased Increased |
[139], [140], [141], [142] | ||
| Lower plasma levels of HDL cholesterol | Unchanged | [142, 143] | |||
| Mouse | Macrophage ABCA1 deficiency | Low HDL-C |
 Increased Increased |
[144] | |
| Hepatic ABCA1 deficiency |
 Increased Increased |
[144, 145] | |||
| Leukocyte ABCA1 deficiency | Unchanged HDL-C |
 Increased Increased |
[118] | ||
| ABCA1 global knockout | Low HDL-C | Unchanged | [144] |
-
HDL-C: high density lipoprotein-cholesterol; ASCVD: atherosclerosis-related cardiovascular disease.
LCAT, low HDL and LCAT knockout hamsters
As an enzyme, LCAT has two different catalytic activities. On one hand, it exerts the function of phospholipase A2 activity to release fatty acids from the sn-2 position of phosphatidylcholine; on the other hand, it possesses transesterification activity, which transfers fatty acids to hydroxyl group of cholesterol. In humans, the symptoms of LCAT deficiency are mainly characterized by extremely low HDL-C levels, elevated free cholesterol and triglyceride levels. Basically, LCAT deficiency is divided into two types: familial LCAT deficiency (FLD) with complete loss of both α and β activities required for the esterification of free cholesterol in HDL and LDL, respectively, whereas fish eye disease (FED) with partial loss of LCAT activity with preserved β activity. Anemia and renal dysfunction are the most common manifestations in FLD patients, while corneal turbidity is always observed in FED patients. Although LCAT has been proposed to have α and β activities, which have different impacts on atherosclerosis [102]. However, there is no direct evidence elucidating this unsolved problem to date since LCAT was discovered in 1967.
It has been reported that HDL plays multiple favorable roles of RCT, anti-inflammation and anti-oxidation in atheroprevention. Low HDL is closely associated with hypertriglyceridemia and diabetes mellitus, contributing to accelerated atherosclerosis. Although deficit of LCAT activity leads to a significant reduction in HDL levels, which might theoretically exacerbate atherosclerosis, clinical findings are contradictory, even showing that low HDL is not associated with atherosclerotic development [103]. Recently, Oldoni et al. [102] reported that preserved β activity of LCAT to maintain cholesterol esterification in ApoB-containing lipoproteins, such as LDLs, was a determinant of atherosclerosis in the patients with LCAT mutations when using carotid intimal-media thickness (cIMT); however, the application of cIMT for the evaluation of atherosclerosis in vivo may be revisited because it cannot accurately measure the atherosclerotic lesions in vascular wall [104, 105]. In the studies using experimental animals, LCAT knockout mice crossed with atherosclerosis prone mouse models, LDLR or ApoE-deficient mice, showed either pro-atherogenic or antiatherogenic effects, depending on lipoprotein profiles modulated by dietary interventions, but not LCAT per se [106, 107].
To answer this question, LCAT knockout hamsters were created using CRISPR/Cas9 technique. Maintained on normal chow diet, LCAT knockout hamsters displayed extremely low HDL and markedly increased free cholesterol in plasma, accompanied by elevated TG, a phenotype similar to that observed in humans with LCAT deficiency. Lipidomics analysis revealed that circulating phosphatidylcholine and phosphatidylethanolamine, two substrates of LCAT-mediated cholesteryl esterification were significantly increased, whereas cholesteryl ester and lysolecithin, two products of LCAT-mediated reaction, were largely reduced [108]. Moreover, TG (54:2), one of the 3 top lipid species used to predict the risk of CVD in the prospective population-based Bruneck [108], was also increased by 2.5 folds in LCAT knockout hamsters. Accumulation of free cholesterol in plasma inhibited LPL activity, leading to elevated TG levels. Upon high cholesterol/high fat diet feeding, LCAT knockout hamsters developed more atherosclerotic lesions relative to wild type controls [109]. In the following study, Guo et al. [110] investigated the influence of LCAT deficiency on spontaneous atherosclerosis. They found that although male and female LCAT exhibited different blood lipid profiles, especially total cholesterol, aged LCAT knockout hamsters (>16 months) present accelerated atherosclerotic lesions, which was only positively correlated with plasma malondialdehyde (MDA) independent of total cholesterol levels, indicating that the ratio of HDL-C and TG or lipid status could be considered as a reliable predictor for atherosclerosis in FLD patients. Of note, these observations confirm that LCAT plays a protective role in atherosclerosis, suggesting that LCAT will be a promising target for ASCVD and the hamster model with LCAT deficiency will provide a platform to develop therapeutic approaches to low HDL and ASCVD caused by FLD.
ABCA1, hypolipoproteinemia and ABCA1 knockout hamsters
Since ABCA1 participates in the initial step of RCT and HDL maturation, it is referred as RCT gatekeeper [111]. This ABCA1 is a key modulator of cholesterol efflux to reduce foam cell formation and thus inflammatory response to lipid accumulation in vascular wall [112]. Previous work demonstrate that an increase in cholesterol efflux by 50% and plasma HDL-C levels by 30%, both of which are mediated by ABCA, is associated with reduced incidence of CAD by up to 50% [113]. Recently, ABCA1 expression at the transcript levels is inversely associated with plasma hs-CRP concentration by Li et al. [114]. In vitro experiments showed that could directly inhibit the expression of inflammatory markers, such as ABCA1 interleukin-1β (IL-1β), interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) [115]. This evidence demonstrates that ABCA1 also possesses anti-inflammatory property [113, 114].
In 1999, ABCA1 gene was firstly linked to Tangier’s disease (TD) in humans, a rare genetic disorder with complete loss of HDL in circulation, abnormal intracellular cholesterol accumulation and impaired efflux of cholesterol and phospholipid together with obvious systemic inflammation, which was originally reported in 1959. Today, there are approximately 100 cases identified worldwide. Patients with TD have been reported to have an increased risk of atherosclerosis [116] and kidney glomerulonephritis [117]. Surprisingly, ABCA1 knockout mice with very low HDL-C and normal TG levels, had no spontaneous atherosclerosis nor significant renal damage on normal chow diet. After crossed with ApoE- or LDLR-deficient mice, atherosclerotic lesions were not significantly aggravated by ABCA1 deficiency, which was attributable to a marked reduction in plasma total cholesterol. A significant difference in atherosclerotic lesion was observed only when ABCA1 was specifically deleted from macrophages without affecting total cholesterol levels compared to control mice [118]. Unfortunately, such inherited tissue-specific gene mutations do not occur in humans, implying that conditional knockout mice cannot mimic TD for future translational study.
Therefore, it is rational to establish another ABCA1 knockout animal model with different species to study the relationship between ABCA1 and atherosclerosis. Using CRISPR/Cas9, we and Qin generated a hamster model lacking ABCA1 in China. As expected, ABCA1 knockout hamsters showed similar characteristics in terms of dyslipidemia to TD patients with loss-of-function mutations in ABCA1 gene: (1) undetectable HDL and ApoA1; (2) increased free cholesterol levels; (3) no significant change in total cholesterol; (4) elevated TG concentration in VLDL fractions; (5) decreased CETP activity (unpublished data). Importantly, while ABCA1 knockout hamsters present a significantly shorter lifespan of 6–8 months with reduced body weight and abnormal blood biochemical parameters, while wild type hamsters could survive up to 3 years. Spontaneous atherosclerosis was observed in normal chow-fed ABCA1 knockout hamsters before they died (unpublished data).
Vascular dementia not only triggers atherosclerotic plaques, but also brain dysfunction. As ABCA1 plays an important role in vascular disease, the same gene definitely affects central nerve system because of its high expression in brain. Previously, Nordestgaard et al. [119] found that loss-of-function ABCA1 gene mutations predisposed individuals to a high risk of Alzheimer’s disease, suggesting that disrupting normal ABCA function will cause impairment of recognition and memory. In our study of Y-maze and novelty recognition, ABCA1 knockout hamsters showed impaired ability of learning and memory compared with WT hamsters, suggesting that loss of ABCA1 had a detrimental effect on brain function. Further study demonstrated that this impairment in brain was largely attributed to serious inflammation, leading to the disappearance of cytoplasmic Nissl bodies, neuronal swelling.
These results described above suggest that the ABCA1 knockout hamsters resemble TD patients in clinics caused by loss-of-function mutations in ABCA1 gene, providing the encouraging experimental evidence for clinical practice and will be a reliable tool to study ABCA1, hypoalphalipoproteinemia and related diseases.
Limitations and perspective of hamsters as animal models of dyslipidemia and atherosclerosis
Although hamsters have many advantages over other species as a rodent model similar to human in the traits of lipid metabolism and the distribution of atherosclerotic lesions, the limitations cannot be ignored summarized below, which need to be improved in future study.
The establishment of standardized experimental methods. Small rodent species, such as mice and rats, are widely used for basic research with standard protocols that has not been established and optimized in hamsters due to the small application range of hamsters. For example, blood collection. As we know, the tail of hamster is short, making it impossible to use regular tail vein injection for some experiments, but intravenous administration should be considered. Herein, we suggest that a route of administration through the external jugular vein is the best method. Although technically, it is more complex, the reliability of intravenous administration can be ensured.
Lack of dedicated equipment and tools. Recently, the existing experimental equipment and reagents are mostly designed for humans and mice. Because there is no equipment specific for hamsters, sometime it is inconvenient to set up experiments. To our knowledge, a common sphygmomanometer used for mice and rats cannot be applied for hamsters due to differences in body size between hamsters and other small rodent animals. The probes of B-ultrasonography are also designed for mice and rats, but not hamsters. Moreover, various commercially available antibodies against different hamster antigens are still lacking, indicating that only polyclonal antibodies with cross reaction among different species can be used.
Lack of inbred strains. Currently, most hamsters for laboratory use belong to the close colony, but inbred strains, leading to a relatively large variation in the individuals from intra-group sample and difficulty to make a clear conclusion when the sample size is small in some experiments. Therefore, the sample size required for the experiment is large to make the experiment more difficult. However, given that human populations over the world are diverse, this trait of hamsters is highly similar to that of human, giving hamsters a lead in precision medicine and translational research of human diseases. Eventually, the conclusions drawn from the experiments using hamsters as experimental animal models may be closer to the discoveries in humans when the sample size is appropriately increased.
In summary, although there still are those limitations, hamster as a valuable experimental model will gain more attention in the fields of metabolic disorders and ASCVD because of its high similarity to human in terms of lipid metabolism and the success of more genetically engineered models. With the development of gene editing technology, the standardization of experimental protocols and new experimental products, and the generation of inbred strains, we believe that the application perspective of genetically engineered hamster models in the field of lipid metabolism and ASCVD will be brilliant.
Funding source: National Natural Science Foundation of China 10.13039/501100001809
Award Identifier / Grant number: 31520103909
Award Identifier / Grant number: 91739105
Award Identifier / Grant number: 81770449
Award Identifier / Grant number: 82070460
Funding source: Peking University 10.13039/501100007937
Award Identifier / Grant number: BMU2020MX001
Award Identifier / Grant number: BMU2020XY011
-
Author contributions: All authors have accepted responsibility for the entire content of this manuscript and approved its submission.
-
Research funding: This work is supported by National Natural Science Foundation of China NSFC 31520103909 and 91739105 to G.L., 81770449 to Y.W. and 82070460 to X.X. Peking University BMU2020MX001 and BMU2020XY011 to X. X. G.L. is a fellow at the Collaborative Innovation Center for Cardiovascular Disease Translational Medicine, Nanjing Medical University.
-
Competing interests: Authors state no conflict of interest.
-
Informed consent: Informed consent was obtained from all individuals included in this study.
-
Ethical approval: The local Institutional Review Board deemed the study exempt from review.
References
1. Ma, LY, Chen, WW, Gao, RL, Liu, LS, Zhu, ML, Wang, YJ, et al.. China cardiovascular diseases report 2018: an updated summary. J Geriatr Cardiol 2020;17:1–8. https://doi.org/10.11909/j.issn.1671-5411.2020.01.001.Search in Google Scholar
2. Sandesara, PB, Virani, SS, Fazio, S, Shapiro, MD. The forgotten lipids: triglycerides, remnant cholesterol, and atherosclerotic cardiovascular disease risk. Endocr Rev 2019;40:537–57. https://doi.org/10.1210/er.2018-00184.Search in Google Scholar
3. Getz, GS, Reardon, CA. Animal models of atherosclerosis. Arterioscler Thromb Vasc Biol 2012;32:1104–15. https://doi.org/10.1161/atvbaha.111.237693.Search in Google Scholar
4. Iwatsuki-Horimoto, K, Nakajima, N, Ichiko, Y, Sakai-Tagawa, Y, Noda, T, Hasegawa, H, et al.. Syrian hamster as an animal model for the study of human influenza virus infection. J Virol 2018;92:e01693–17. https://doi.org/10.1128/JVI.01693-17.Search in Google Scholar
5. Wattenberg, LW, Wiedmann, TS, Estensen, RD. Chemoprevention of cancer of the upper respiratory tract of the Syrian golden hamster by aerosol administration of difluoromethylornithine and 5-fluorouracil. Cancer Res 2004;64:2347–9. https://doi.org/10.1158/0008-5472.can-03-4032.Search in Google Scholar
6. Rogers, TF, Zhao, F, Huang, D, Beutler, N, Burns, A, He, WT, et al.. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science 2020;369:956–63. https://doi.org/10.1126/science.abc7520.Search in Google Scholar
7. Sia, SF, Yan, LM, Chin, AWH, Fung, K, Choy, KT, Wong, AYL, et al.. Pathogenesis and transmission of SARS-CoV-2 in golden hamsters. Nature 2020;583:834–8. https://doi.org/10.1038/s41586-020-2342-5.Search in Google Scholar
8. Hou, YJ, Chiba, S, Halfmann, P, Ehre, C, Kuroda, M, Dinnon, KH3rd, et al.. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020;370:1464–8. https://doi.org/10.1126/science.abe8499.Search in Google Scholar
9. Sanchez-Felipe, L, Vercruysse, T, Sharma, S, Ma, J, Lemmens, V, Van Looveren, D, et al.. A single-dose live-attenuated YF17D-vectored SARS-CoV-2 vaccine candidate. Nature 2021;590:320–5. https://doi.org/10.1038/s41586-020-3035-9.Search in Google Scholar
10. Altschul, R. Experimental cholesterol arteriosclerosis. II. Changes produced in golden hamsters and in Guinea pigs. Am Heart J 1950;40:401–9. https://doi.org/10.1016/0002-8703(50)90323-1.Search in Google Scholar
11. Coyne, MJ, Bonorris, GG, Chung, A, Cove, H, Schoenfield, LJ. Dietary cholesterol affects chenodeoxycholic acid action on biliary lipids. Gastroenterology 1977;72:927–31. https://doi.org/10.1016/s0016-5085(77)80213-8.Search in Google Scholar
12. Filip, DA, Nistor, A, Bulla, A, Radu, A, Lupu, F, Simionescu, M. Cellular events in the development of valvular atherosclerotic lesions induced by experimental hypercholesterolemia. Atherosclerosis 1987;67:199–214. https://doi.org/10.1016/0021-9150(87)90280-2.Search in Google Scholar
13. Cincotta, AH, Meier, AH. Circadian rhythms of lipogenic and hypoglycaemic responses to insulin in the golden hamster (Mesocricetus auratus). J Endocrinol 1984;103:141–6. https://doi.org/10.1677/joe.0.1030141.Search in Google Scholar
14. Nistor, A, Bulla, A, Filip, DA, Radu, A. The hyperlipidemic hamster as a model of experimental atherosclerosis. Atherosclerosis 1987;68:159–73. https://doi.org/10.1016/0021-9150(87)90106-7.Search in Google Scholar
15. Chen, J, Song, W, Redinger, RN. Effects of dietary cholesterol on hepatic production of lipids and lipoproteins in isolated hamster liver. Hepatology 1996;24:424–34. https://doi.org/10.1002/hep.510240222.Search in Google Scholar
16. Woollett, LA, Kearney, DM, Spady, DK. Diet modification alters plasma HDL cholesterol concentrations but not the transport of HDL cholesteryl esters to the liver in the hamster. J Lipid Res 1997;38:2289–302. https://doi.org/10.1016/s0022-2275(20)34943-9.Search in Google Scholar
17. Otto, J, Ordovas, JM, Smith, D, van Dongen, D, Nicolosi, RJ, Schaefer, EJ. Lovastatin inhibits diet induced atherosclerosis in F1B golden Syrian hamsters. Atherosclerosis 1995;114:19–28. https://doi.org/10.1016/0021-9150(94)05457-t.Search in Google Scholar
18. Taghibiglou, C, Carpentier, A, Van Iderstine, SC, Chen, B, Rudy, D, Aiton, A, et al.. Mechanisms of hepatic very low density lipoprotein overproduction in insulin resistance. Evidence for enhanced lipoprotein assembly, reduced intracellular ApoB degradation, and increased microsomal triglyceride transfer protein in a fructose-fed hamster model. J Biol Chem 2000;275:8416–25. https://doi.org/10.1074/jbc.275.12.8416.Search in Google Scholar
19. Taghibiglou, C, Rudy, D, Van Iderstine, SC, Aiton, A, Cavallo, D, Cheung, R, et al.. Intracellular mechanisms regulating ApoB-containing lipoprotein assembly and secretion in primary hamster hepatocytes. J Lipid Res 2000;41:499–513. https://doi.org/10.1016/s0022-2275(20)32397-x.Search in Google Scholar
20. Adeli, K, Taghibiglou, C, Van Iderstine, SC, Lewis, GF. Mechanisms of hepatic very low-density lipoprotein overproduction in insulin resistance. Trends Cardiovasc Med 2001;11:170–6. https://doi.org/10.1016/s1050-1738(01)00084-6.Search in Google Scholar
21. Mangaloglu, L, Cheung, RC, Van Iderstine, SC, Taghibiglou, C, Pontrelli, L, Adeli, K. Treatment with atorvastatin ameliorates hepatic very-low-density lipoprotein overproduction in an animal model of insulin resistance, the fructose-fed Syrian golden hamster: evidence that reduced hypertriglyceridemia is accompanied by improved hepatic insulin sensitivity. Metabolism 2002;51:409–18. https://doi.org/10.1053/meta.2002.30954.Search in Google Scholar PubMed
22. Au, CS, Wagner, A, Chong, T, Qiu, W, Sparks, JD, Adeli, K. Insulin regulates hepatic apolipoprotein B production independent of the mass or activity of Akt1/PKBalpha. Metabolism 2004;53:228–35. https://doi.org/10.1016/j.metabol.2003.09.011.Search in Google Scholar PubMed
23. Qiu, W, Kohen-Avramoglu, R, Mhapsekar, S, Tsai, J, Austin, RC, Adeli, K. Glucosamine-induced endoplasmic reticulum stress promotes ApoB100 degradation: evidence for Grp78-mediated targeting to proteasomal degradation. Arterioscler Thromb Vasc Biol 2005;25:571–7. https://doi.org/10.1161/01.atv.0000154142.61859.94.Search in Google Scholar
24. Su, Q, Tsai, J, Xu, E, Qiu, W, Bereczki, E, Santha, M, et al.. Apolipoprotein B100 acts as a molecular link between lipid-induced endoplasmic reticulum stress and hepatic insulin resistance. Hepatology 2009;50:77–84. https://doi.org/10.1002/hep.22960.Search in Google Scholar PubMed
25. Qiu, W, Zhang, J, Dekker, MJ, Wang, H, Huang, J, Brumell, JH, et al.. Hepatic autophagy mediates endoplasmic reticulum stress-induced degradation of misfolded apolipoprotein B. Hepatology 2011;53:1515–25. https://doi.org/10.1002/hep.24269.Search in Google Scholar PubMed
26. Hsieh, J, Longuet, C, Baker, CL, Qin, B, Federico, LM, Drucker, DJ, et al.. The glucagon-like peptide 1 receptor is essential for postprandial lipoprotein synthesis and secretion in hamsters and mice. Diabetologia 2010;53:552–61. https://doi.org/10.1007/s00125-009-1611-5.Search in Google Scholar PubMed
27. Hsieh, J, Longuet, C, Maida, A, Bahrami, J, Xu, E, Baker, CL, et al.. Glucagon-like peptide-2 increases intestinal lipid absorption and chylomicron production via CD36. Gastroenterology 2009;137:997–1005.e1–4. https://doi.org/10.1053/j.gastro.2009.05.051.Search in Google Scholar PubMed
28. Hein, GJ, Baker, C, Hsieh, J, Farr, S, Adeli, K. GLP-1 and GLP-2 as yin and yang of intestinal lipoprotein production: evidence for predominance of GLP-2-stimulated postprandial lipemia in normal and insulin-resistant states. Diabetes 2013;62:373–81. https://doi.org/10.2337/db12-0202.Search in Google Scholar PubMed PubMed Central
29. Taher, J, Baker, CL, Cuizon, C, Masoudpour, H, Zhang, R, Farr, S, et al.. GLP-1 receptor agonism ameliorates hepatic VLDL overproduction and de novo lipogenesis in insulin resistance. Mol Metab 2014;3:823–33. https://doi.org/10.1016/j.molmet.2014.09.005.Search in Google Scholar PubMed PubMed Central
30. Hsieh, J, Trajcevski, KE, Farr, SL, Baker, CL, Lake, EJ, Taher, J, et al.. Glucagon-like peptide 2 (GLP-2) stimulates postprandial chylomicron production and postabsorptive release of intestinal triglyceride storage pools via induction of nitric oxide signaling in male hamsters and mice. Endocrinology 2015;156:3538–47. https://doi.org/10.1210/en.2015-1110.Search in Google Scholar PubMed
31. Farr, S, Baker, C, Naples, M, Taher, J, Iqbal, J, Hussain, M, et al.. Central nervous system regulation of intestinal lipoprotein metabolism by glucagon-like peptide-1 via a brain-gut axis. Arterioscler Thromb Vasc Biol 2015;35:1092–100. https://doi.org/10.1161/atvbaha.114.304873.Search in Google Scholar
32. Breslow, JL. Transgenic mouse models of lipoprotein metabolism and atherosclerosis. Proc Natl Acad Sci USA 1993;90:8314–8. https://doi.org/10.1073/pnas.90.18.8314.Search in Google Scholar
33. Zhang, SH, Reddick, RL, Piedrahita, JA, Maeda, N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science 1992;258:468–71. https://doi.org/10.1126/science.1411543.Search in Google Scholar
34. Ishibashi, S, Brown, MS, Goldstein, JL, Gerard, RD, Hammer, RE, Herz, J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J Clin Invest 1993;92:883–93. https://doi.org/10.1172/jci116663.Search in Google Scholar
35. Castro-Perez, J, Briand, F, Gagen, K, Wang, SP, Chen, Y, McLaren, DG, et al.. Anacetrapib promotes reverse cholesterol transport and bulk cholesterol excretion in Syrian golden hamsters. J Lipid Res 2011;52:1965–73. https://doi.org/10.1194/jlr.m016410.Search in Google Scholar
36. Dietschy, JM, Turley, SD, Spady, DK. Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J Lipid Res 1993;34:1637–59. https://doi.org/10.1016/s0022-2275(20)35728-x.Search in Google Scholar
37. Fernandez, ML, Wilson, TA, Conde, K, Vergara-Jimenez, M, Nicolosi, RJ. Hamsters and Guinea pigs differ in their plasma lipoprotein cholesterol distribution when fed diets varying in animal protein, soluble fiber, or cholesterol content. J Nutr 1999;129:1323–32. https://doi.org/10.1093/jn/129.7.1323.Search in Google Scholar PubMed
38. Spady, DK, Dietschy, JM. Dietary saturated triacylglycerols suppress hepatic low density lipoprotein receptor activity in the hamster. Proc Natl Acad Sci U S A 1985;82:4526–30. https://doi.org/10.1073/pnas.82.13.4526.Search in Google Scholar PubMed PubMed Central
39. Sima, A, Bulla, A, Simionescu, N. Experimental obstructive coronary atherosclerosis in the hyperlipidemic hamster. J Submicrosc Cytol Pathol 1990;22:1–16.Search in Google Scholar
40. Dekker, MJ, Baker, C, Naples, M, Samsoondar, J, Zhang, R, Qiu, W, et al.. Inhibition of sphingolipid synthesis improves dyslipidemia in the diet-induced hamster model of insulin resistance: evidence for the role of sphingosine and sphinganine in hepatic VLDL-apoB100 overproduction. Atherosclerosis 2013;228:98–109. https://doi.org/10.1016/j.atherosclerosis.2013.01.041.Search in Google Scholar PubMed
41. Dillard, A, Matthan, NR, Lichtenstein, AH. Use of hamster as a model to study diet-induced atherosclerosis. Nutr Metab (Lond) 2010;7:89. https://doi.org/10.1186/1743-7075-7-89.Search in Google Scholar PubMed PubMed Central
42. Gofman, JW, Lindgren, F. The role of lipids and lipoproteins in atherosclerosis. Science 1950;111:166–71. https://doi.org/10.1126/science.111.2877.166.Search in Google Scholar
43. Brown, MS, Goldstein, JL. How LDL receptors influence cholesterol and atherosclerosis. Sci Am 1984;251:58–66. https://doi.org/10.1038/scientificamerican1184-58.Search in Google Scholar
44. Di Angelantonio, E, Sarwar, N, Perry, P, Kaptoge, S, Ray, KK, Thompson, A, et al.. Major lipids, apolipoproteins, and risk of vascular disease. JAMA 2009;302:1993–2000. https://doi.org/10.1001/jama.2009.1619.Search in Google Scholar
45. Gerald, H, Tomkin, Owens, D. LDL as a cause of atherosclerosis. Open Atherosclerosis Thromb J 2012;5:13–21.10.2174/1876506801205010013Search in Google Scholar
46. Linton, MF, Yancey, PG, Davies, SS, Jerome, WG, Linton, EF, Song, WL, et al.. The role of lipids and lipoproteins in atherosclerosis. In: Feingold, KR, Anawalt, B, Boyce, A, Chrousos, G, de Herder, WW, Dungan, K, et al.., editors. Endotext. South Dartmouth (MA): MDText.com, Inc. Copyright © 2000–2021, MDText.com, Inc.; 2000.Search in Google Scholar
47. Brown, MS, Goldstein, JL. Analysis of a mutant strain of human fibroblasts with a defect in the internalization of receptor-bound low density lipoprotein. Cell 1976;9:663–74. https://doi.org/10.1016/0092-8674(76)90130-6.Search in Google Scholar
48. Defesche, JC, Gidding, SS, Harada-Shiba, M, Hegele, RA, Santos, RD, Wierzbicki, AS. Familial hypercholesterolaemia. Nat Rev Dis Primers 2017;3:17093. https://doi.org/10.1038/nrdp.2017.93.Search in Google Scholar PubMed
49. Benn, M, Watts, GF, Tybjaerg-Hansen, A, Nordestgaard, BG. Familial hypercholesterolemia in the Danish general population: prevalence, coronary artery disease, and cholesterol-lowering medication. J Clin Endocrinol Metab 2012;97:3956–64. https://doi.org/10.1210/jc.2012-1563.Search in Google Scholar PubMed
50. Gidding, SS, Champagne, MA, de Ferranti, SD, Defesche, J, Ito, MK, Knowles, JW, et al.. The agenda for familial hypercholesterolemia: a scientific statement from the American heart association. Circulation 2015;132:2167–92. https://doi.org/10.1161/cir.0000000000000297.Search in Google Scholar
51. Meshkov, A, Ershova, A, Kiseleva, A, Zotova, E, Sotnikova, E, Petukhova, A, et al.. The LDLR, APOB, and PCSK9 variants of index patients with familial hypercholesterolemia in Russia. Genes (Basel). 2021;12:66. https://doi.org/10.3390/genes12010066.Search in Google Scholar PubMed PubMed Central
52. Powell-Braxton, L, Véniant, M, Latvala, RD, Hirano, KI, Won, WB, Ross, J, et al.. A mouse model of human familial hypercholesterolemia: markedly elevated low density lipoprotein cholesterol levels and severe atherosclerosis on a low-fat chow diet. Nat Med 1998;4:934–8. https://doi.org/10.1038/nm0898-934.Search in Google Scholar PubMed
53. Sithu, SD, Malovichko, MV, Riggs, KA, Wickramasinghe, NS, Winner, MG, Agarwal, A, et al.. Atherogenesis and metabolic dysregulation in LDL receptor-knockout rats. JCI Insight 2017;2:e86442. https://doi.org/10.1172/jci.insight.86442.Search in Google Scholar PubMed PubMed Central
54. Shiomi, M. The history of the WHHL rabbit, an animal model of familial hypercholesterolemia (II) – contribution to the development and validation of the therapeutics for hypercholesterolemia and atherosclerosis. J Atheroscler Thromb 2020;27:119–31. https://doi.org/10.5551/jat.rv17038-2.Search in Google Scholar PubMed PubMed Central
55. Lu, R, Yuan, T, Wang, Y, Zhang, T, Yuan, Y, Wu, D, et al.. Spontaneous severe hypercholesterolemia and atherosclerosis lesions in rabbits with deficiency of low-density lipoprotein receptor (LDLR) on exon 7. EBioMedicine 2018;36:29–38. https://doi.org/10.1016/j.ebiom.2018.09.020.Search in Google Scholar PubMed PubMed Central
56. Davis, BT, Wang, XJ, Rohret, JA, Struzynski, JT, Merricks, EP, Bellinger, DA, et al.. Targeted disruption of LDLR causes hypercholesterolemia and atherosclerosis in Yucatan miniature pigs. PLoS One 2014;9:e93457. https://doi.org/10.1371/journal.pone.0093457.Search in Google Scholar PubMed PubMed Central
57. Li, Y, Fuchimoto, D, Sudo, M, Haruta, H, Lin, QF, Takayama, T, et al.. Development of human-like advanced coronary plaques in low-density lipoprotein receptor knockout pigs and justification for statin treatment before formation of atherosclerotic plaques. J Am Heart Assoc 2016;5:e002779. https://doi.org/10.1161/JAHA.115.002779.Search in Google Scholar PubMed PubMed Central
58. Guo, X, Gao, M, Wang, Y, Lin, X, Yang, L, Cong, N, et al.. LDL receptor gene-ablated hamsters: a rodent model of familial hypercholesterolemia with dominant inheritance and diet-induced coronary atherosclerosis. EBioMedicine 2018;27:214–24. https://doi.org/10.1016/j.ebiom.2017.12.013.Search in Google Scholar PubMed PubMed Central
59. He, K, Wang, J, Shi, H, Yu, Q, Zhang, X, Guo, M, et al.. An interspecies study of lipid profiles and atherosclerosis in familial hypercholesterolemia animal models with low-density lipoprotein receptor deficiency. Am J Transl Res 2019;11:3116–27.Search in Google Scholar
60. Wang, J, He, K, Yang, C, Lin, X, Zhang, X, Wang, Y, et al.. Dietary cholesterol is highly associated with severity of hyperlipidemia and atherosclerotic lesions in heterozygous LDLR-deficient hamsters. Int J Mol Sci 2019;20:3515. https://doi.org/10.3390/ijms20143515.Search in Google Scholar PubMed PubMed Central
61. Getz, GS, Reardon, CA. Do the apoe–/– and Ldlr–/– mice yield the same insight on atherogenesis? Arterioscler Thromb Vasc Biol 2016;36:1734–41. https://doi.org/10.1161/atvbaha.116.306874.Search in Google Scholar PubMed PubMed Central
62. Wu, Y, Xu, MJ, Cao, Z, Yang, C, Wang, J, Wang, B, et al.. Heterozygous Ldlr-deficient hamster as a model to evaluate the efficacy of PCSK9 antibody in hyperlipidemia and atherosclerosis. Int J Mol Sci 2019;20:5936. https://doi.org/10.3390/ijms20235936.Search in Google Scholar PubMed PubMed Central
63. Sun, Z, Zhang, L, Li, L, Shao, C, Liu, J, Zhou, M, et al.. Galectin-3 mediates cardiac remodeling caused by impaired glucose and lipid metabolism through inhibiting two pathways of activating Akt. Am J Physiol Heart Circ Physiol 2021;320:H364–80. https://doi.org/10.1152/ajpheart.00523.2020.Search in Google Scholar PubMed
64. Peng, J, Luo, F, Ruan, G, Peng, R, Li, X. Hypertriglyceridemia and atherosclerosis. Lipids Health Dis 2017;16:233. https://doi.org/10.1186/s12944-017-0625-0.Search in Google Scholar PubMed PubMed Central
65. Cobbaert, CM, Althaus, H, Begcevic Brkovic, I, Ceglarek, U, Coassin, S, Delatour, V, et al.. Towards an SI-traceable reference measurement system for seven serum apolipoproteins using bottom-up quantitative proteomics: conceptual approach enabled by cross-disciplinary/cross-sector collaboration. Clin Chem 2021;67:478–89. https://doi.org/10.1093/clinchem/hvaa239.Search in Google Scholar PubMed
66. Ku, EJ, Cho, KC, Lim, C, Kang, JW, Oh, JW, Choi, YR, et al.. Discovery of plasma biomarkers for predicting the severity of coronary artery atherosclerosis by quantitative proteomics. BMJ Open Diabetes Res Care 2020;8:e001152. https://doi.org/10.1136/bmjdrc-2019-001152.Search in Google Scholar PubMed PubMed Central
67. Do, R, Willer, CJ, Schmidt, EM, Sengupta, S, Gao, C, Peloso, GM, et al.. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet 2013;45:1345–52. https://doi.org/10.1038/ng.2795.Search in Google Scholar PubMed PubMed Central
68. Wolska, A, Dunbar, RL, Freeman, LA, Ueda, M, Amar, MJ, Sviridov, DO, et al.. Apolipoprotein C-II: new findings related to genetics, biochemistry, and role in triglyceride metabolism. Atherosclerosis 2017;267:49–60. https://doi.org/10.1016/j.atherosclerosis.2017.10.025.Search in Google Scholar PubMed PubMed Central
69. Wang, H, Eckel, RH. Lipoprotein lipase: from gene to obesity. Am J Physiol Endocrinol Metab 2009;297:E271–88. https://doi.org/10.1152/ajpendo.90920.2008.Search in Google Scholar PubMed
70. Shachter, NS, Hayek, T, Leff, T, Smith, JD, Rosenberg, DW, Walsh, A, et al.. Overexpression of apolipoprotein CII causes hypertriglyceridemia in transgenic mice. J Clin Invest 1994;93:1683–90. https://doi.org/10.1172/jci117151.Search in Google Scholar
71. Sakurai, T, Sakurai, A, Vaisman, BL, Amar, MJ, Liu, C, Gordon, SM, et al.. Creation of apolipoprotein C-II (ApoC-II) mutant mice and correction of their hypertriglyceridemia with an ApoC-II mimetic peptide. J Pharmacol Exp Therapeut 2016;356:341–53. https://doi.org/10.1124/jpet.115.229740.Search in Google Scholar PubMed PubMed Central
72. Liu, C, Gates, KP, Fang, L, Amar, MJ, Schneider, DA, Geng, H, et al.. Apoc2 loss-of-function zebrafish mutant as a genetic model of hyperlipidemia. Dis Model Mech 2015;8:989–98. https://doi.org/10.1242/dmm.019836.Search in Google Scholar PubMed PubMed Central
73. Gao, M, Yang, C, Wang, X, Guo, M, Yang, L, Gao, S, et al.. ApoC2 deficiency elicits severe hypertriglyceridemia and spontaneous atherosclerosis: a rodent model rescued from neonatal death. Metabolism 2020;109:154296. https://doi.org/10.1016/j.metabol.2020.154296.Search in Google Scholar PubMed
74. Yang, C, Tian, W, Ma, S, Guo, M, Lin, X, Gao, F, et al.. AAV-mediated ApoC2 gene therapy: reversal of severe hypertriglyceridemia and rescue of neonatal death in ApoC2-deficient hamsters. Mol Ther Methods Clin Dev 2020;18:692–701. https://doi.org/10.1016/j.omtm.2020.07.011.Search in Google Scholar
75. Pulawa, LK, Jensen, DR, Coates, A, Eckel, RH. Reduction of plasma triglycerides in apolipoprotein C-II transgenic mice overexpressing lipoprotein lipase in muscle. J Lipid Res 2007;48:145–51. https://doi.org/10.1194/jlr.m600384-jlr200.Search in Google Scholar
76. Suárez-Sánchez, F, Klunder-Klunder, M, Valladares-Salgado, A, Gómez-Zamudio, J, Peralta-Romero, J, Meyre, D, et al.. APOA5 and APOA1 polymorphisms are associated with triglyceride levels in Mexican children. Pediatr Obes 2017;12:330–6. https://doi.org/10.1111/ijpo.12147.Search in Google Scholar
77. Chen, H, Ding, S, Zhou, M, Wu, X, Liu, X, Wu, Y, et al.. Association of rs662799 in APOA5 with CAD in Chinese Han population. BMC Cardiovasc Disord 2018;18:2. https://doi.org/10.1186/s12872-017-0735-7.Search in Google Scholar
78. Sánchez-Moreno, C, Ordovás, JM, Smith, CE, Baraza, JC, Lee, YC, Garaulet, M. APOA5 gene variation interacts with dietary fat intake to modulate obesity and circulating triglycerides in a Mediterranean population. J Nutr 2011;141:380–5. https://doi.org/10.3945/jn.110.130344.Search in Google Scholar
79. Mansouri, RM, Baugé, E, Gervois, P, Fruchart-Najib, J, Fiévet, C, Staels, B, et al.. Atheroprotective effect of human apolipoprotein A5 in a mouse model of mixed dyslipidemia. Circ Res 2008;103:450–3. https://doi.org/10.1161/circresaha.108.179861.Search in Google Scholar
80. Shu, X, Nelbach, L, Ryan, RO, Forte, TM. Apolipoprotein A-V associates with intrahepatic lipid droplets and influences triglyceride accumulation. Biochim Biophys Acta 2010;1801:605–8. https://doi.org/10.1016/j.bbalip.2010.02.004.Search in Google Scholar
81. van den Berg, SA, Heemskerk, MM, Geerling, JJ, van Klinken, JB, Schaap, FG, Bijland, S, et al.. Apolipoprotein A5 deficiency aggravates high-fat diet-induced obesity due to impaired central regulation of food intake. FASEB J 2013;27:3354–62. https://doi.org/10.1096/fj.12-225367.Search in Google Scholar
82. Camporez, JP, Kanda, S, Petersen, MC, Jornayvaz, FR, Samuel, VT, Bhanot, S, et al.. ApoA5 knockdown improves whole-body insulin sensitivity in high-fat-fed mice by reducing ectopic lipid content. J Lipid Res 2015;56:526–36. https://doi.org/10.1194/jlr.m054080.Search in Google Scholar
83. Brown, WV, Baginsky, ML. Inhibition of lipoprotein lipase by an apoprotein of human very low density lipoprotein. Biochem Biophys Res Commun 1972;46:375–82. https://doi.org/10.1016/s0006-291x(72)80149-9.Search in Google Scholar
84. Shelburne, F, Hanks, J, Meyers, W, Quarfordt, S. Effect of apoproteins on hepatic uptake of triglyceride emulsions in the rat. J Clin Invest 1980;65:652–8. https://doi.org/10.1172/jci109710.Search in Google Scholar PubMed PubMed Central
85. Pechlaner, R, Tsimikas, S, Yin, X, Willeit, P, Baig, F, Santer, P, et al.. Very-low-density lipoprotein-associated apolipoproteins predict cardiovascular events and are lowered by inhibition of APOC-III. J Am Coll Cardiol 2017;69:789–800. https://doi.org/10.1016/j.jacc.2016.11.065.Search in Google Scholar PubMed PubMed Central
86. Ebara, T, Ramakrishnan, R, Steiner, G, Shachter, NS. Chylomicronemia due to apolipoprotein CIII overexpression in apolipoprotein E-null mice. Apolipoprotein CIII-induced hypertriglyceridemia is not mediated by effects on apolipoprotein E. J Clin Invest 1997;99:2672–81. https://doi.org/10.1172/jci119456.Search in Google Scholar
87. Li, H, Han, Y, Qi, R, Wang, Y, Zhang, X, Yu, M, et al.. Aggravated restenosis and atherogenesis in ApoCIII transgenic mice but lack of protection in ApoCIII knockouts: the effect of authentic triglyceride-rich lipoproteins with and without ApoCIII. Cardiovasc Res 2015;107:579–89. https://doi.org/10.1093/cvr/cvv192.Search in Google Scholar PubMed
88. Pollin, TI, Damcott, CM, Shen, H, Ott, SH, Shelton, J, Horenstein, RB, et al.. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science 2008;322:1702–5. https://doi.org/10.1126/science.1161524.Search in Google Scholar PubMed PubMed Central
89. Jørgensen, AB, Frikke-Schmidt, R, Nordestgaard, BG, Tybjærg-Hansen, A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med 2014;371:32–41.10.1056/NEJMoa1308027Search in Google Scholar PubMed
90. Timpson, NJ, Walter, K, Min, JL, Tachmazidou, I, Malerba, G, Shin, SY, et al.. A rare variant in APOC3 is associated with plasma triglyceride and VLDL levels in Europeans. Nat Commun 2014;5:4871. https://doi.org/10.1038/ncomms5871.Search in Google Scholar PubMed PubMed Central
91. Gaudet, D, Brisson, D, Tremblay, K, Alexander, VJ, Singleton, W, Hughes, SG, et al.. Targeting APOC3 in the familial chylomicronemia syndrome. N Engl J Med 2014;371:2200–6. https://doi.org/10.1056/nejmoa1400284.Search in Google Scholar PubMed
92. Alexander, VJ, Xia, S, Hurh, E, Hughes, SG, O’Dea, L, Geary, RS, et al.. N-acetyl galactosamine-conjugated antisense drug to APOC3 mRNA, triglycerides and atherogenic lipoprotein levels. Eur Heart J 2019;40:2785–96. https://doi.org/10.1093/eurheartj/ehz209.Search in Google Scholar PubMed PubMed Central
93. Khetarpal, SA, Zeng, X, Millar, JS, Vitali, C, Somasundara, AVH, Zanoni, P, et al.. A human APOC3 missense variant and monoclonal antibody accelerate apoC-III clearance and lower triglyceride-rich lipoprotein levels. Nat Med 2017;23:1086–94. https://doi.org/10.1038/nm.4390.Search in Google Scholar PubMed PubMed Central
94. Saleheen, D, Natarajan, P, Armean, IM, Zhao, W, Rasheed, A, Khetarpal, SA, et al.. Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature 2017;544:235–9. https://doi.org/10.1038/nature22034.Search in Google Scholar PubMed PubMed Central
95. Reyes-Soffer, G, Sztalryd, C, Horenstein, RB, Holleran, S, Matveyenko, A, Thomas, T, et al.. Effects of APOC3 heterozygous deficiency on plasma lipid and lipoprotein metabolism. Arterioscler Thromb Vasc Biol 2019;39:63–72. https://doi.org/10.1161/atvbaha.118.311476.Search in Google Scholar
96. Getz, GS, Reardon, CA. Diet and murine atherosclerosis. Arterioscler Thromb Vasc Biol 2006;26:242–9. https://doi.org/10.1161/01.atv.0000201071.49029.17.Search in Google Scholar PubMed
97. McGillicuddy, FC, Roche, HM. Nutritional status, genetic susceptibility, and insulin resistance – important precedents to atherosclerosis. Mol Nutr Food Res 2012;56:1173–84. https://doi.org/10.1002/mnfr.201100785.Search in Google Scholar PubMed
98. Guo, M, Xu, Y, Dong, Z, Zhou, Z, Cong, N, Gao, M, et al.. Inactivation of ApoC3 by CRISPR/Cas9 protects against atherosclerosis in hamsters. Circ Res 2020;127:1456–8. https://doi.org/10.1161/circresaha.120.317686.Search in Google Scholar
99. Rosenson, RS, Burgess, LJ, Ebenbichler, CF, Baum, SJ, Stroes, ESG, Ali, S, et al.. Evinacumab in patients with refractory hypercholesterolemia. N Engl J Med 2020;383:2307–19. https://doi.org/10.1056/nejmoa2031049.Search in Google Scholar PubMed
100. Kunnen, S, Van Eck, M. Lecithin:cholesterol acyltransferase: old friend or foe in atherosclerosis? J Lipid Res 2012;53:1783–99. https://doi.org/10.1194/jlr.r024513.Search in Google Scholar PubMed PubMed Central
101. Asztalos, BF, Schaefer, EJ, Horvath, KV, Yamashita, S, Miller, M, Franceschini, G, et al.. Role of LCAT in HDL remodeling: investigation of LCAT deficiency states. J Lipid Res 2007;48:592–9. https://doi.org/10.1194/jlr.m600403-jlr200.Search in Google Scholar
102. Oldoni, F, Baldassarre, D, Castelnuovo, S, Ossoli, A, Amato, M, van Capelleveen, J, et al.. Complete and partial lecithin:cholesterol acyltransferase deficiency is differentially associated with atherosclerosis. Circulation 2018;138:1000–7. https://doi.org/10.1161/circulationaha.118.034706.Search in Google Scholar
103. Rousset, X, Shamburek, R, Vaisman, B, Amar, M, Remaley, AT. Lecithin cholesterol acyltransferase: an anti- or pro-atherogenic factor? Curr Atherosclerosis Rep 2011;13:249–56. https://doi.org/10.1007/s11883-011-0171-6.Search in Google Scholar PubMed PubMed Central
104. Johri, AM, Nambi, V, Naqvi, TZ, Feinstein, SB, Kim, ESH, Park, MM, et al.. Recommendations for the assessment of carotid arterial plaque by ultrasound for the characterization of atherosclerosis and evaluation of cardiovascular risk: from the American Society of Echocardiography. J Am Soc Echocardiogr 2020;33:917–33. https://doi.org/10.1016/j.echo.2020.04.021.Search in Google Scholar
105. Inaba, Y, Chen, JA, Bergmann, SR. Carotid plaque, compared with carotid intima-media thickness, more accurately predicts coronary artery disease events: a meta-analysis. Atherosclerosis 2012;220:128–33. https://doi.org/10.1016/j.atherosclerosis.2011.06.044.Search in Google Scholar
106. Furbee, JWJr, Sawyer, JK, Parks, JS. Lecithin:cholesterol acyltransferase deficiency increases atherosclerosis in the low density lipoprotein receptor and apolipoprotein E knockout mice. J Biol Chem 2002;277:3511–9. https://doi.org/10.1074/jbc.m109883200.Search in Google Scholar
107. Ng, DS, Maguire, GF, Wylie, J, Ravandi, A, Xuan, W, Ahmed, Z, et al.. Oxidative stress is markedly elevated in lecithin:cholesterol acyltransferase-deficient mice and is paradoxically reversed in the apolipoprotein E knockout background in association with a reduction in atherosclerosis. J Biol Chem 2002;277:11715–20. https://doi.org/10.1074/jbc.m112320200.Search in Google Scholar
108. Stegemann, C, Pechlaner, R, Willeit, P, Langley, SR, Mangino, M, Mayr, U, et al.. Lipidomics profiling and risk of cardiovascular disease in the prospective population-based Bruneck study. Circulation 2014;129:1821–31. https://doi.org/10.1161/circulationaha.113.002500.Search in Google Scholar
109. Dong, Z, Shi, H, Zhao, M, Zhang, X, Huang, W, Wang, Y, et al.. Loss of LCAT activity in the golden Syrian hamster elicits pro-atherogenic dyslipidemia and enhanced atherosclerosis. Metabolism 2018;83:245–55. https://doi.org/10.1016/j.metabol.2018.03.003.Search in Google Scholar
110. Guo, M, Liu, Z, Xu, Y, Ma, P, Huang, W, Gao, M, et al.. Spontaneous atherosclerosis in aged LCAT-deficient hamsters with enhanced oxidative stress-brief report. Arterioscler Thromb Vasc Biol 2020;40:2829–36. https://doi.org/10.1161/atvbaha.120.315265.Search in Google Scholar
111. Oram, JF, Lawn, RM. ABCA1. The gatekeeper for eliminating excess tissue cholesterol. J Lipid Res 2001;42:1173–9. https://doi.org/10.1016/s0022-2275(20)31566-2.Search in Google Scholar
112. Nagappa, M, Taly, AB, Mahadevan, A, Pooja, M, Bindu, PS, Chickabasaviah, YT, et al.. Tangier’s disease: an uncommon cause of facial weakness and non-length dependent demyelinating neuropathy. Ann Indian Acad Neurol 2016;19:137–9. https://doi.org/10.4103/0972-2327.175436.Search in Google Scholar PubMed PubMed Central
113. Singaraja, RR, Brunham, LR, Visscher, H, Kastelein, JJ, Hayden, MR. Efflux and atherosclerosis: the clinical and biochemical impact of variations in the ABCA1 gene. Arterioscler Thromb Vasc Biol 2003;23:1322–32. https://doi.org/10.1161/01.atv.0000078520.89539.77.Search in Google Scholar PubMed
114. Li, C, Guo, R, Lou, J, Zhou, H. The transcription levels of ABCA1, ABCG1 and SR-BI are negatively associated with plasma CRP in Chinese populations with various risk factors for atherosclerosis. Inflammation 2012;35:1641–8. https://doi.org/10.1007/s10753-012-9479-9.Search in Google Scholar
115. Tang, C, Liu, Y, Kessler, PS, Vaughan, AM, Oram, JF. The macrophage cholesterol exporter ABCA1 functions as an anti-inflammatory receptor. J Biol Chem 2009;284:32336–43. https://doi.org/10.1074/jbc.m109.047472.Search in Google Scholar
116. Brunham, LR, Singaraja, RR, Hayden, MR. Variations on a gene: rare and common variants in ABCA1 and their impact on HDL cholesterol levels and atherosclerosis. Annu Rev Nutr 2006;26:105–29. https://doi.org/10.1146/annurev.nutr.26.061505.111214.Search in Google Scholar
117. Christiansen-Weber, TA, Voland, JR, Wu, Y, Ngo, K, Roland, BL, Nguyen, S, et al.. Functional loss of ABCA1 in mice causes severe placental malformation, aberrant lipid distribution, and kidney glomerulonephritis as well as high-density lipoprotein cholesterol deficiency. Am J Pathol 2000;157:1017–29. https://doi.org/10.1016/s0002-9440(10)64614-7.Search in Google Scholar
118. van Eck, M, Bos, IS, Kaminski, WE, Orsó, E, Rothe, G, Twisk, J, et al.. Leukocyte ABCA1 controls susceptibility to atherosclerosis and macrophage recruitment into tissues. Proc Natl Acad Sci U S A 2002;99:6298–303. https://doi.org/10.1073/pnas.092327399.Search in Google Scholar
119. Nordestgaard, LT, Tybjærg-Hansen, A, Nordestgaard, BG, Frikke-Schmidt, R. Loss-of-function mutation in ABCA1 and risk of Alzheimer’s disease and cerebrovascular disease. Alzheimers Dement 2015;11:1430–8. https://doi.org/10.1016/j.jalz.2015.04.006.Search in Google Scholar
120. Liu, GL, Fan, LM, Redinger, RN. The association of hepatic apoprotein and lipid metabolism in hamsters and rats. Comp Biochem Physiol A Comp Physiol 1991;99:223–8. https://doi.org/10.1016/0300-9629(91)90263-c.Search in Google Scholar
121. Safronetz, D, Ebihara, H, Feldmann, H, Hooper, JW. The Syrian hamster model of hantavirus pulmonary syndrome. Antivir Res 2012;95:282–92. https://doi.org/10.1016/j.antiviral.2012.06.002.Search in Google Scholar PubMed PubMed Central
122. Dondji, B, Bungiro, RD, Harrison, LM, Vermeire, JJ, Bifulco, C, McMahon-Pratt, D, et al.. Role for nitric oxide in hookworm-associated immune suppression. Infect Immun 2008;76:2560–7. https://doi.org/10.1128/iai.00094-08.Search in Google Scholar PubMed PubMed Central
123. da Silva-Couto, L, Ribeiro-Romão, RP, Saavedra, AF, da Silva Costa Souza, BL, Moreira, OC, Gomes-Silva, A, et al.. Intranasal vaccination with leishmanial antigens protects golden hamsters (Mesocricetus auratus) against Leishmania (Viannia) Braziliensis infection. PLoS Neglected Trop Dis 2015;9:e3439. https://doi.org/10.1371/journal.pntd.0003439.Search in Google Scholar PubMed PubMed Central
124. Kuehne, SA, Collery, MM, Kelly, ML, Cartman, ST, Cockayne, A, Minton, NP. Importance of toxin A, toxin B, and CDT in virulence of an epidemic Clostridium difficile strain. J Infect Dis 2014;209:83–6. https://doi.org/10.1093/infdis/jit426.Search in Google Scholar PubMed PubMed Central
125. Calabresi, L, Baldassarre, D, Castelnuovo, S, Conca, P, Bocchi, L, Candini, C, et al.. Functional lecithin:cholesterol acyltransferase is not required for efficient atheroprotection in humans. Circulation 2009;120:628–35. https://doi.org/10.1161/CIRCULATIONAHA.108.818143.Search in Google Scholar PubMed
126. Haase, CL, Tybjærg-Hansen, A, Qayyum, AA, Schou, J, Nordestgaard, BG, Frikke-Schmidt, R. LCAT, HDL cholesterol and ischemic cardiovascular disease: a Mendelian randomization study of HDL cholesterol in 54,500 individuals. J Clin Endocrinol Metab 2012;97:E248–56. https://doi.org/10.1210/jc.2011-1846.Search in Google Scholar PubMed
127. Duivenvoorden, R, Holleboom, AG, van den Bogaard, B, Nederveen, AJ, de Groot, E, Hutten, BA, et al.. Carriers of lecithin cholesterol acyltransferase gene mutations have accelerated atherogenesis as assessed by carotid 3.0-T magnetic resonance imaging [corrected]. J Am Coll Cardiol 2011;58:2481–7. https://doi.org/10.1016/j.jacc.2010.11.092.Search in Google Scholar PubMed
128. Hovingh, GK, Hutten, BA, Holleboom, AG, Petersen, W, Rol, P, Stalenhoef, A, et al.. Compromised LCAT function is associated with increased atherosclerosis. Circulation 2005;112:879–84. https://doi.org/10.1161/circulationaha.105.540427.Search in Google Scholar
129. Ayyobi, AF, McGladdery, SH, Chan, S, John Mancini, GB, Hill, JS, Frohlich, JJ. Lecithin:cholesterol acyltransferase (LCAT) deficiency and risk of vascular disease: 25 year follow-up. Atherosclerosis 2004;177:361–6. https://doi.org/10.1016/j.atherosclerosis.2004.07.018.Search in Google Scholar PubMed
130. van den Bogaard, B, Holleboom, AG, Duivenvoorden, R, Hutten, BA, Kastelein, JJ, Hovingh, GK, et al.. Patients with low HDL-cholesterol caused by mutations in LCAT have increased arterial stiffness. Atherosclerosis 2012;225:481–5. https://doi.org/10.1016/j.atherosclerosis.2012.09.022.Search in Google Scholar PubMed
131. Vaisman, BL, Klein, HG, Rouis, M, Bérard, AM, Kindt, MR, Talley, GD, et al.. Overexpression of human lecithin cholesterol acyltransferase leads to hyperalphalipoproteinemia in transgenic mice. J Biol Chem 1995;270:12269–75. https://doi.org/10.1074/jbc.270.20.12269.Search in Google Scholar PubMed
132. Calabresi, L, Franceschini, G. Lecithin:cholesterol acyltransferase, high-density lipoproteins, and atheroprotection in humans. Trends Cardiovasc Med 2010;20:50–3. https://doi.org/10.1016/j.tcm.2010.03.007.Search in Google Scholar PubMed
133. Lambert, G, Sakai, N, Vaisman, BL, Neufeld, EB, Marteyn, B, Chan, CC, et al.. Analysis of glomerulosclerosis and atherosclerosis in lecithin cholesterol acyltransferase-deficient mice. J Biol Chem 2001;276:15090–8. https://doi.org/10.1074/jbc.m008466200.Search in Google Scholar PubMed
134. Hoeg, JM, Vaisman, BL, Demosky, SJJr, Meyn, SM, Talley, GD, Hoyt, RFJr, et al.. Lecithin:cholesterol acyltransferase overexpression generates hyperalpha-lipoproteinemia and a nonatherogenic lipoprotein pattern in transgenic rabbits. J Biol Chem 1996;271:4396–402. https://doi.org/10.1074/jbc.271.8.4396.Search in Google Scholar
135. Brousseau, ME, Hoeg, JM. Transgenic rabbits as models for atherosclerosis research. J Lipid Res 1999;40:365–75. https://doi.org/10.1016/s0022-2275(20)32440-8.Search in Google Scholar
136. Brousseau, ME, Santamarina-Fojo, S, Vaisman, BL, Applebaum-Bowden, D, Bérard, AM, Talley, GD, et al.. Overexpression of human lecithin:cholesterol acyltransferase in cholesterol-fed rabbits: LDL metabolism and HDL metabolism are affected in a gene dose-dependent manner. J Lipid Res 1997;38:2537–47. https://doi.org/10.1016/s0022-2275(20)30038-9.Search in Google Scholar
137. Willer, CJ, Schmidt, EM, Sengupta, S, Peloso, GM, Gustafsson, S, Kanoni, S, et al.. Discovery and refinement of loci associated with lipid levels. Nat Genet 2013;45:1274–83. https://doi.org/10.1038/ng.2797.Search in Google Scholar PubMed PubMed Central
138. Deloukas, P, Kanoni, S, Willenborg, C, Farrall, M, Assimes, TL, Thompson, JR, et al.. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet 2013;45:25–33. https://doi.org/10.1038/ng.2480.Search in Google Scholar PubMed PubMed Central
139. Bochem, AE, van Wijk, DF, Holleboom, AG, Duivenvoorden, R, Motazacker, MM, Dallinga-Thie, GM, et al.. ABCA1 mutation carriers with low high-density lipoprotein cholesterol are characterized by a larger atherosclerotic burden. Eur Heart J 2013;34:286–91. https://doi.org/10.1093/eurheartj/ehs376.Search in Google Scholar PubMed
140. Frikke-Schmidt, R, Nordestgaard, BG, Schnohr, P, Steffensen, R, Tybjaerg-Hansen, A. Mutation in ABCA1 predicted risk of ischemic heart disease in the copenhagen city heart study population. J Am Coll Cardiol 2005;46:1516–20. https://doi.org/10.1016/j.jacc.2005.06.066.Search in Google Scholar PubMed
141. Frikke-Schmidt, R, Nordestgaard, BG, Jensen, GB, Steffensen, R, Tybjaerg-Hansen, A. Genetic variation in ABCA1 predicts ischemic heart disease in the general population. Arterioscler Thromb Vasc Biol 2008;28:180–6. https://doi.org/10.1161/atvbaha.107.153858.Search in Google Scholar
142. Hovingh, GK, Kuivenhoven, JA, Bisoendial, RJ, Groen, AK, van Dam, M, van Tol, A, et al.. HDL deficiency and atherosclerosis: lessons from Tangier disease. J Intern Med 2004;255:299–301. https://doi.org/10.1046/j.0954-6820.2003.01256.x.Search in Google Scholar PubMed
143. Frikke-Schmidt, R, Nordestgaard, BG, Stene, MC, Sethi, AA, Remaley, AT, Schnohr, P, et al.. Association of loss-of-function mutations in the ABCA1 gene with high-density lipoprotein cholesterol levels and risk of ischemic heart disease. JAMA 2008;299:2524–32. https://doi.org/10.1001/jama.299.21.2524.Search in Google Scholar PubMed
144. Aiello, RJ, Brees, D, Bourassa, PA, Royer, L, Lindsey, S, Coskran, T, et al.. Increased atherosclerosis in hyperlipidemic mice with inactivation of ABCA1 in macrophages. Arterioscler Thromb Vasc Biol 2002;22:630–7. https://doi.org/10.1161/01.atv.0000014804.35824.da.Search in Google Scholar PubMed
145. Brunham, LR, Singaraja, RR, Duong, M, Timmins, JM, Fievet, C, Bissada, N, et al.. Tissue-specific roles of ABCA1 influence susceptibility to atherosclerosis. Arterioscler Thromb Vasc Biol 2009;29:548–54. https://doi.org/10.1161/atvbaha.108.182303.Search in Google Scholar
© 2021 George Liu et al., published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
Articles in the same Issue
- Frontmatter
- Preface
- New challenges in medicine
- Inaugural Editorial
- Welcome to your new journal: Medical Review
- Editorial
- Cell–cell crosstalk in the heart
- Perspectives
- Gene editing therapy ready for cardiovascular diseases: opportunities, challenges, and perspectives
- Resident stem cells in the heart
- “Know Diabetes by Heart”: role of adipocyte-cardiomyocyte communications
- Reviews
- A novel function of CREG in metabolic disorders
- The gut-cardiovascular connection: new era for cardiovascular therapy
- Sympatho-adrenergic mechanisms in heart failure: new insights into pathophysiology
- The role of caveolae in endothelial dysfunction
- Genetically-engineered hamster models: applications and perspective in dyslipidemia and atherosclerosis-related cardiovascular disease
Articles in the same Issue
- Frontmatter
- Preface
- New challenges in medicine
- Inaugural Editorial
- Welcome to your new journal: Medical Review
- Editorial
- Cell–cell crosstalk in the heart
- Perspectives
- Gene editing therapy ready for cardiovascular diseases: opportunities, challenges, and perspectives
- Resident stem cells in the heart
- “Know Diabetes by Heart”: role of adipocyte-cardiomyocyte communications
- Reviews
- A novel function of CREG in metabolic disorders
- The gut-cardiovascular connection: new era for cardiovascular therapy
- Sympatho-adrenergic mechanisms in heart failure: new insights into pathophysiology
- The role of caveolae in endothelial dysfunction
- Genetically-engineered hamster models: applications and perspective in dyslipidemia and atherosclerosis-related cardiovascular disease