Formation of 1-aza-2-silacyclopentanes and unexpected products from the insertion of phenylisocyanate into 2,2-dimethyl-1-(trimethylsilyl)-1-aza-2-silacyclopentane
-
Marcus Herbig



Abstract
Substances like 3-aminopropyltriethoxysilane are often used as adhesive promoters in various formulations for coatings or to adjust the properties of siloxanes and other polymers. Cyclisation of similar substances is also interesting because of the formation of the Si-N bond. 1-Aza-2-silacyclopentanes were synthesised from 3-aminopropylalkoxysilanes by intramolecular condensation reactions and substitution reactions at the silicon atom. The products tend to undergo ring-opening polymerisation. In contrast to literature reports, they can only be isolated as N-substituted derivatives. Phenylisocyanate inserts into the Si-N bonds of cyclic aminosilanes to form seven-membered heterocycles. Furthermore, phenylisocyanate reacts with N-H bonds in the same molecule. Two insertion products were isolated, and their crystal structures were determined.
Introduction
Substances like 3-aminopropyltriethoxysilane are often used as adhesive promoters in various formulations for coatings or to adjust the properties of siloxanes and other polymers. According to the literature, these substances undergo intramolecular condensations upon long time heating (Speier et al., 1971), which can be accelerated by the addition of hexamethyldisilazane (HMDS; Kirilin et al., 2009). The product was reported to be a cyclic N-aminosilane that is highly reactive to ring-opening polymerisation (Salikhov et al., 2014). However, the cyclic products have never been isolated (Scheme 1).
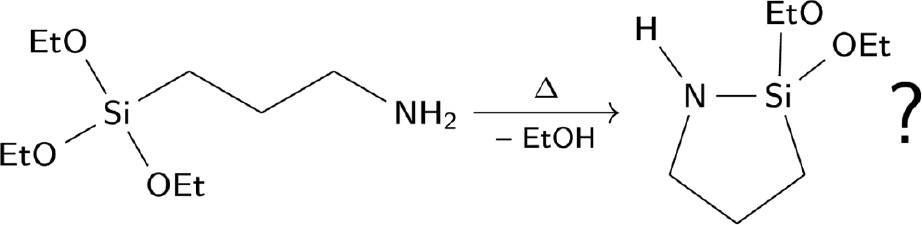
Cyclisation of 3-aminopropyltriethoxysilane by long-time heating.
The formation of 2,2-diethoxy-1-aza-2-silacyclopentane is dubious.
According to the patent literature, 1-aza-2-silacyclopentanes have been used to modify conjugated diene rubbers or surface properties similar to 3-aminopropyltriethoxysilane (Arkles et al., 2003; Ziche and Selbertinger, 2006). The reaction of isocyanates with azasilacyclopentanes has been used to manufacture thermoplastic rubbers by polymerisation with polyols (Pepe et al., 1994; Ziche and Selbertinger, 2006; Nyugaku et al., 2011; Tonomura et al., 2011). The Si-N bonds can react with heterocumulenes like CO2 and isocyanates to form carbamates and ureas (Kraushaar et al., 2014). The known reactions of primary N-silylamines are shown in Scheme 2, but many more insertion reactions are in principle possible (Scheme S1).
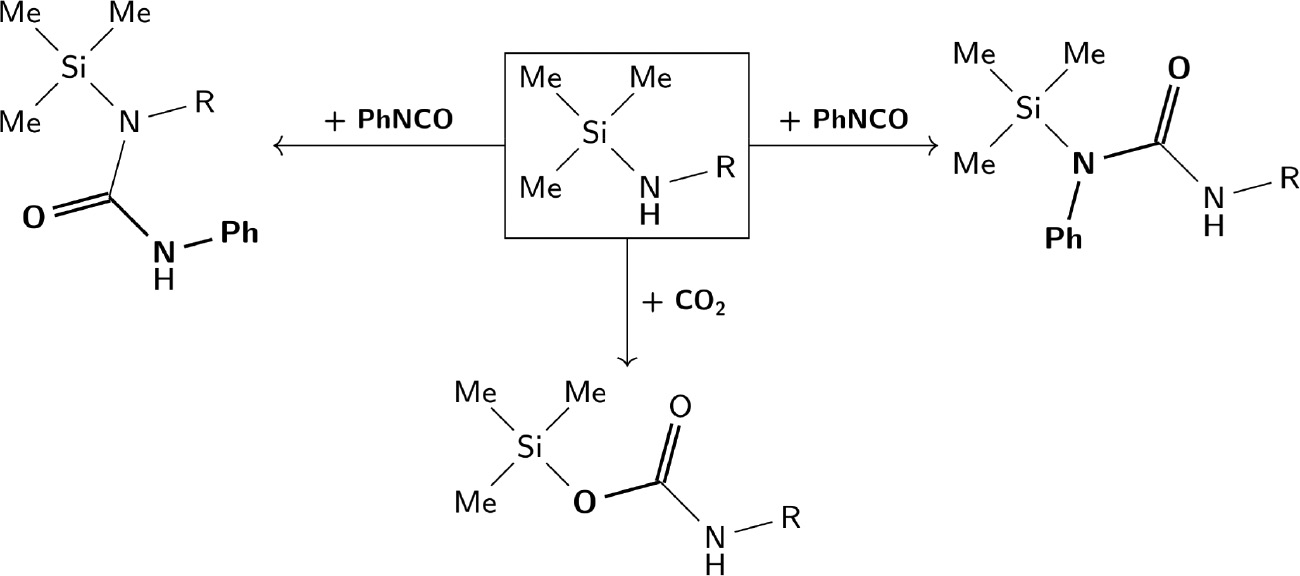
Reactions of primary N-silylamines with CO2 and isocyanates.
Both can insert into the Si-N- or the N-H bond, and twofold, threefold, or even fourfold insertion modes are possible (see also Scheme S1, supporting information).
We optimised the condensation reactions to synthesise cyclic N-aminosilanes with an exocyclic trimethylsilyl group, reducing the tendency to polymerisation. Furthermore, we investigated the insertion reaction with phenylisocyanate (PhNCO). Substitution reactions were performed to obtain a variety of cyclic products (2; Scheme 3). Having analysed the insertion products, we present the first molecular structure of a seven-membered silicon and urea moiety containing heterocycle and a five-membered cyclic N-aminosilane, which is not a silatrane or part of other oligocyclic moieties.
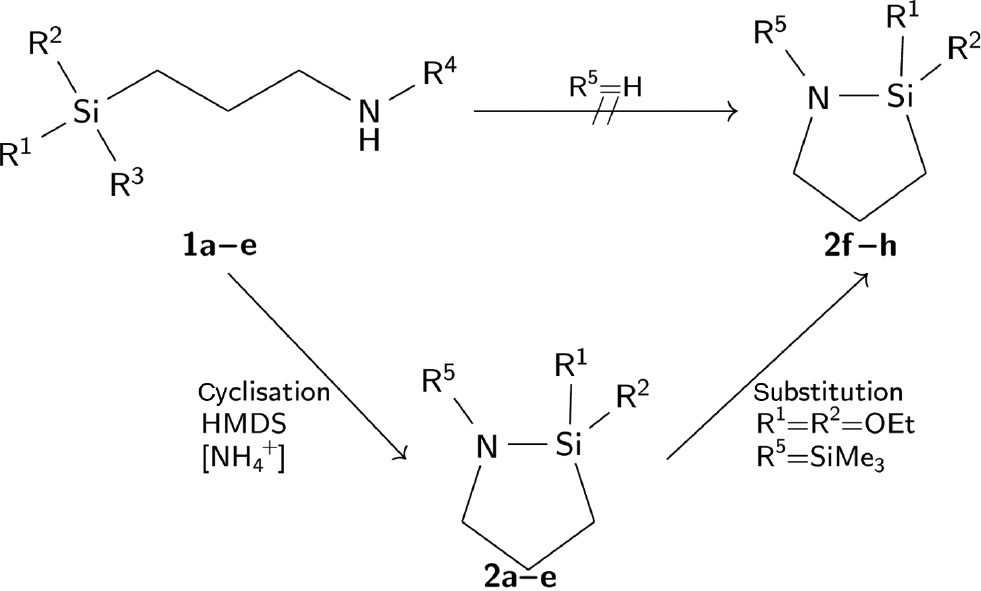
Synthesis of all cyclic N-aminosilanes by cyclisation or substitution reactions (R4=H or SiMe3).
N-unsubstituted 1-aza-2-silacyclopentasilanes could not be prepared.
Results and discussion
Synthesis of cyclic N-aminosilanes
All prepared cyclic N-aminosilanes are shown in Table 1. The starting materials can be heated at reflux and/or distilled without any evidence of the formation of cyclic products. Heating with sodium alcoholates (procedure E, similar to Ziche et al., 1996) is not as efficient as procedure D (see Table 3 in the Experimental section). According to the patent literature, the synthesis and isolation of unsubstituted cyclic N-aminosilanes is not possible (Pepe et al., 1994; Arkles et al., 2003). Nevertheless, Kirilin et al. (2009) reported the isolation of 2,2-diethoxy-1-aza-2-silacyclopentane with procedure B without mentioning the reaction time.
Cyclic N-aminosilanes prepared by cyclisation and substitution reactions according to procedures A, B, and D in Table 3.
| Substance no. | R1 | R2 | R5 | Yield (%) |
|---|---|---|---|---|
| 2a | OEt | OEt | SiMe3 | 61 |
| 2b | Me | OEt | SiMe3 | 73 |
| 2c | OMe | OMe | SiMe3 | 82 |
| 2d | Me | Me | SiMe3 | 90 |
| 2e | OMe | OMe | Me | 94a,b |
| 2f | OEt | Et | SiMe3 | 31a,b |
| 2g | OEt | nBu | SiMe3 | ~100a,b |
| 2h | nBu | nBu | SiMe3 | ~100a,b |
aAccording to 1H-NMR.
bProducts contain impurities, and yields were estimated via 1H-NMR spectra.
For the synthesis of the N-aminosilanes 2a–e in good yields and high purity, only procedure D is suitable. Heating without ammonium sulfate (procedures A and B) leaves the reactants mostly unchanged in the given time. Procedure C yields undefined polymers.
Ammonium sulfate is known as a catalyst for the transsilylation reaction of amines with HMDS. Therefore, it might be assumed that ammonium sulfate catalyses the silylation of the amine moiety. The lower boiling distillate was characterised to be the corresponding trimethylalkoxysilane that is formed by the intramolecular condensation of the N-trimethylsilylated 3-aminopropylsilane. The N-unsubstituted cyclic products could not be isolated. All attempts using procedure C yielded undefined polymers. Without a catalyst (procedure A), only small amounts have reacted in the given time, and no N-unsubstituted cyclic product could be found but N-trimethylsilylated ones. Also, no distillate is obtained by heating pure N-trimethylsilyl-3-aminopropyltrimethoxysilane with ammonium sulfate, but undefined polymers were found in the still pot showing similar 29Si nuclear magnetic resonance (NMR) signals like in experiments using procedure C with 1a. Excess of HMDS is needed for the synthesis of cyclic N-aminosilanes, which reduces the tendency of polymerisation, most likely because of steric effects by silylation of the remaining N-H bond. The presence of the SiMe3 moiety is confirmed by the NMR and infrared spectra, the elemental analysis (EA) of 2a, the determination of molar mass of 2a by ebullioscopy in cyclohexane, and the expected [M+H]+ peak in the mass spectrum of 2c. The N-trimethylsilylated products also tend to polymerise, forming high boiling macromolecules upon reflux but with much smaller reaction rates. Therefore, in all experiments performed with procedure D polymerisation, products can be found in the still pot after distillation lowering the yield of the pure cyclic product. On long time standing, the pure N-substituted cyclic N-aminosilane undergoes polymerisation, too. The polymerisation products can be converted into the starting materials by heating with excess of methanol or ethanol, respectively. The stabilisation of 1-aza-2-silacyclopentanes by the exocyclic SiMe3 moiety is confirmed by heating of N,N-bis(trimethylsilyl)-3-aminopropyltrimethoxysilane with catalytic amounts of ammonium sulfate. In this case, the formation of 2c and trimethylmethoxysilane is observed. N-Methyl-3-aminopropyltrimethoxysilane is less reactive and does not form 2e with a similar yield like the primary amines.
Further derivatives of 2 are accessible starting from 2a. Treatment of 2a with EtMgBr solution in a stoichiometry of 1:1 leads to 2f. A higher amount of EtMgBr per mole 2a does not react with the N-aminosilane to form the twofold substituted product (Scheme 4).
In contrast to this observation, the reaction with nBuLi leads to 2g and 2h depending on the stoichiometric ratio of the reactants. The removal of the byproducts by filtration and washing with n-pentane is not effective. Distillation should be avoided because of possible reactions at high temperature, i.e., polymerisation catalysed by the Grignard reagent or the corresponding ethanolate.
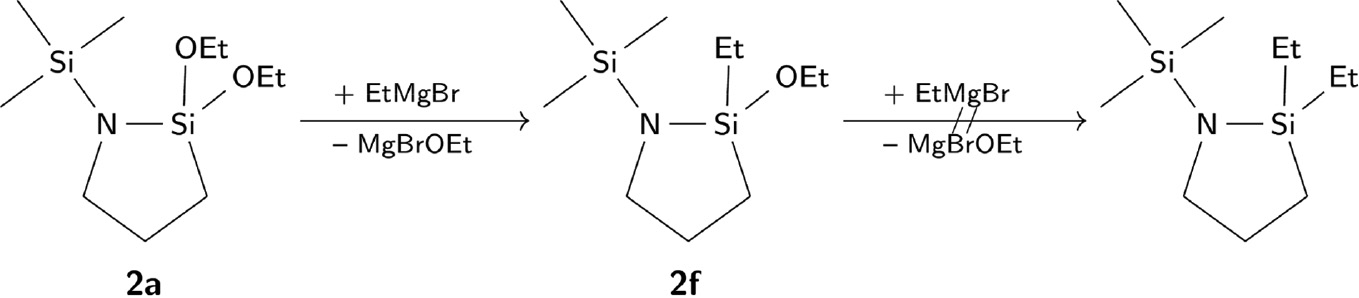
Substitution of the ethoxy group in 2a using EtMgBr-tetrahydrofuran solution.
Insertion reactions with PhNCO
The reaction of the cyclic N-aminosilanes with PhNCO results in a mixture of insertion and oligomerisation products of the isocyanate. Only in the reaction mixture obtained from 2d and PhNCO products 3 and 4 are crystallised and analysed (Scheme 5). In both molecules, the exocyclic trimethylsilane moiety is absent. There are two possibilities for this observation: (i) there were N-unsubstituted products in low concentration in the reaction mixture, not detected by NMR and having no influence on the other analyses; and (ii) partial hydrolysis took place. The Si-N bond is known to be moisture sensitive (see also section Alcoholysis reactions). Therefore, it is probable that this moiety is more prone to hydrolysis than the remaining groups in the cyclic N-aminosilanes 2. In view of 3 and 4, it is likely that the first reaction step is the insertion of PhNCO into the exocyclic Si-N bond. The urea moiety activates a second insertion into the endocyclic Si-N bond, probably due to steric reasons. The second molecule PhNCO inserts into the five-membered ring to form a seven-membered one. During this reaction step, the endocyclic Si-N bond is weakened and polymerisation as side reaction is more likely. Therefore, because of the evident partial hydrolysis, a mixture of different molecular species is obtained. Furthermore, isocyanates can undergo oligomerisation reactions, the products of which also are present in the final reaction mixture.
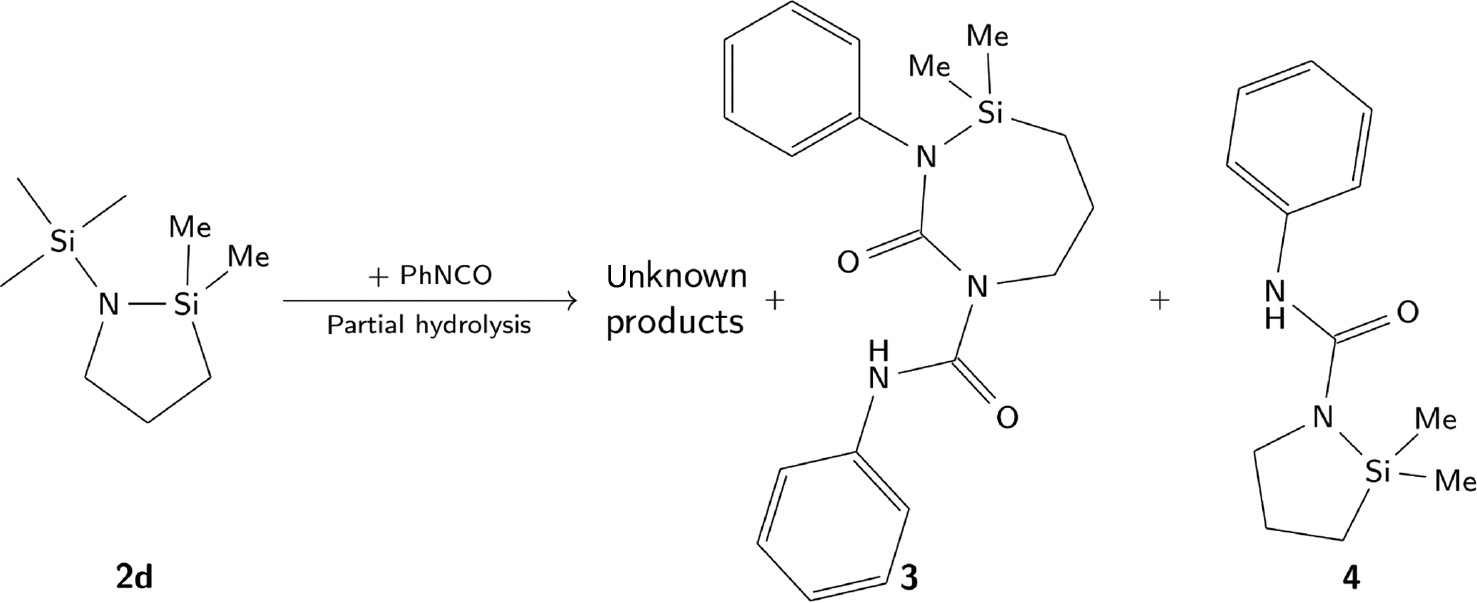
Reaction of 2d with PhNCO.
Two different products are crystallised from the reaction mixtures.
The observed insertion reaction could not be monitored by NMR spectroscopy upon melting solutions at −25°C of cyclic N-aminosilane and PhNCO in anhydrous nitromethane frozen separately in the NMR tube. Thus, after warming the mixture up to −25°C, the reaction was completed after seconds.
Alcoholysis reactions
In order to analyse the hydrolysis behaviour of 2a–d, we performed alcoholysis experiments with 1-pentanol and 2a. The results lead to the conclusion that the hydrolysis of both Si-N bonds is preferred compared with the hydrolysis of only one of them (Scheme 6). It is assumed that the first reaction step is the alcoholysis of the exocyclic Si-N bond, because of the steric hindrance of the endocyclic bond. Otherwise, the formation of 4 cannot be explained. The formed N-unsubstituted cyclic N-aminosilane undergoes ring-opening reaction with the alcohol but does not form polymers. In all alcoholysis experiments, 3-aminopropyldiethoxypentoxysilane was formed.
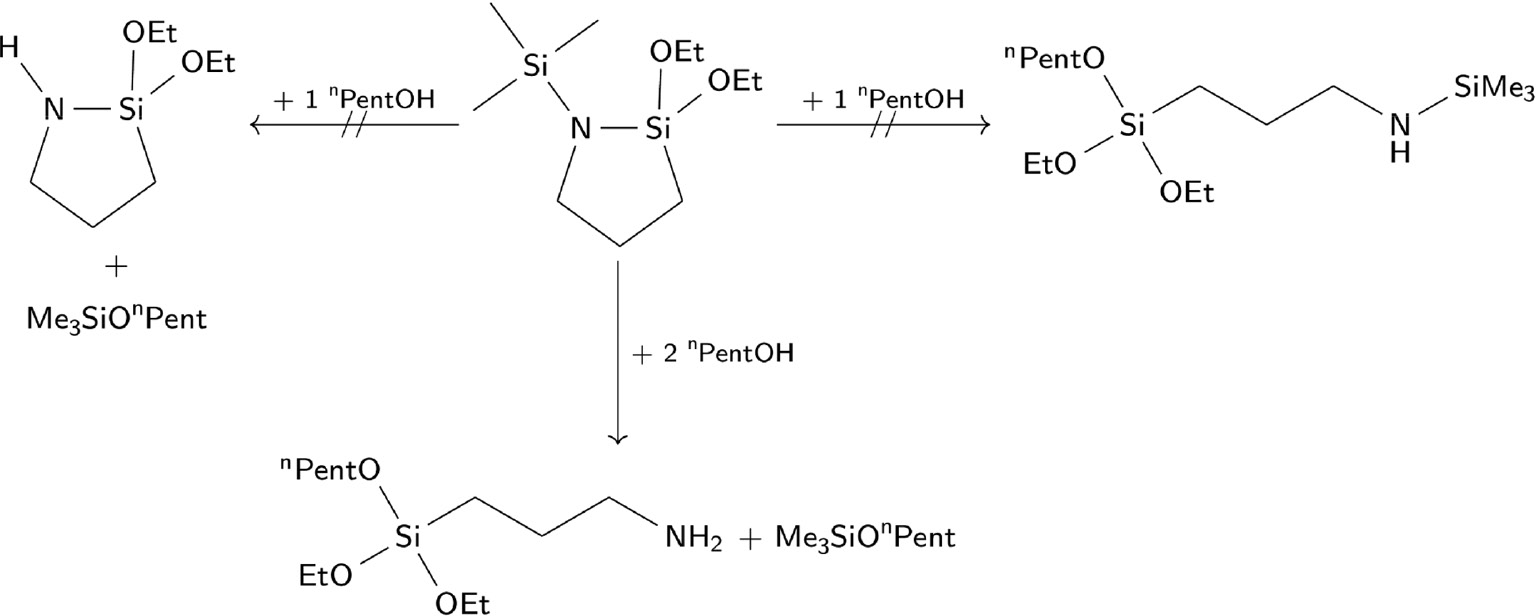
Alcoholysis of 2a with n-pentanol.
The cleavage of both Si-N bonds is preferred.
This is similar to the bis-insertion of CO2 into diaminosilanes of the type (CH3)2Si(NHR)2 to form bis-carbamates of the type (CH3)2Si(OCONHR)2. Mono-insertion products can neither be isolated nor detected (Kraushaar et al., 2012). Similar isocyanate insertions have also been reported (Schöne et al., 2010). Considering these results, we cannot determine if and when the loss of the SiMe3 moiety took place (via hydrolysis), leading to 3 and 4.
Single crystal X-ray diffraction analyses
Compound 3 crystallises in the orthorhombic space group Pbca with one crystallographically independent molecule in the asymmetric unit. Considering the outcome of the reaction leading to 3, one can state that one molecule of phenylisocyanate is inserted into the azasilacyclopentane ring of 2. This leads to a seven-membered ring with a 2,4-diazasilepane structure (Figure 1). Besides, a phenylcarbamic group replaces the trimethylsilyl group that was present in 2. Thus, a second insertion of an isocyanate molecule into the N-H bond has occurred.
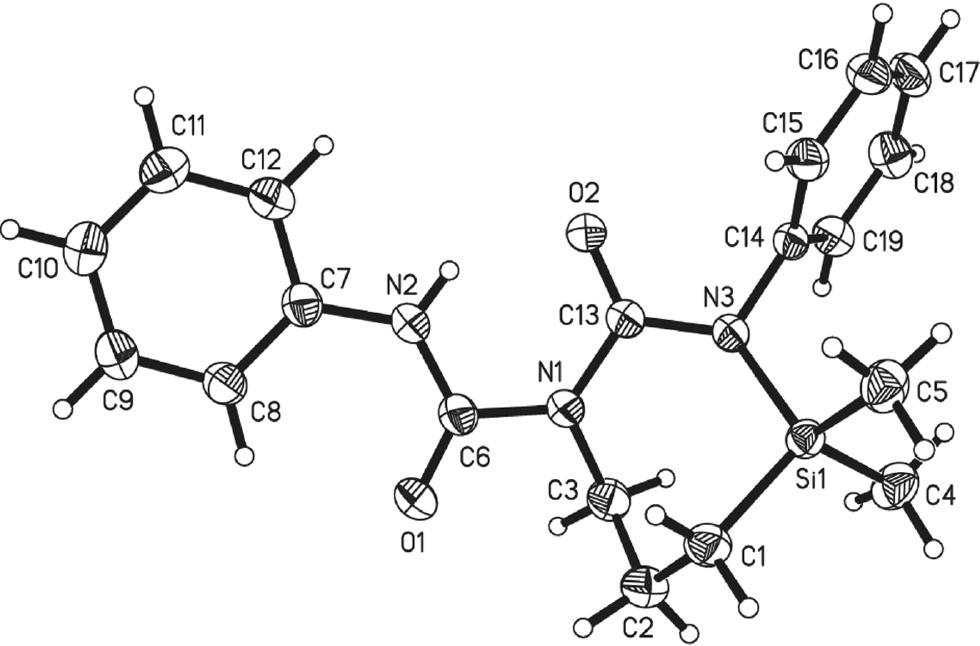
Molecular structure of 3 showing the atomic numbering scheme.
The thermal ellipsoids of the nonhydrogen atoms are drawn at the 50% probability level.
The ring pucker of the seven-membered ring (Si1-C1-C2-C3-N1-C13-N3) was analysed according to Evans and Boeyens (1989). The least-squares plane of these seven atoms in crystal coordinates is given by −8.404x−1.515y+14.142z=3.070. The out-of-plane displacements of the ring atoms in Å are 0.232 Si1, −0.595 C1, 0.130 C2, 0.602 C3, −0.391 N1, −0.278 C13, and 0.299 N3. These displacements lead to the conclusion that the seven-membered ring is in a boat conformation.
Besides, compound 3 contains a biuret unit (carbamylurea) given by the atoms N2-C6(=O1)-N1-C13(=O2)-N3. This unit is composed of two planar urea subunits with a dihedral angle between these subunits of 20.463(90)°. The phenyl ring C7-C12 is nearly coplanar with the urea subunit N2-C6(=O1)-N1 with a dihedral angle between these planes of 4.576(97)°. In contrast, the phenyl group C14-C19 shows a dihedral angle of 71.787(52)° with the urea subunit N1-C13(=O2)-N3. The angle between the phenyl units is 55.252(48)°. An intramolecular N2-H···O2 interaction (1.88Å, 142°) stabilises the molecular conformation.
The crystal packing is dominated by C-H···O interactions in the range of 2.4–2.7Å (Table 2). A bifurcated contact at O1 (C15-H15···O1, C5-H5···O1) leads to molecular chains along the crystallographic a axis. These chains are connected via C1-H1A···O2 along the crystallographic b axis. Further, C-H···π contacts of aliphatic and aromatic hydrogen atoms with both aryl moieties are in the range of 2.8–2.9Å.
Geometric parameters of selected intra- and intermolecular hydrogen bonds in 3 and 4.
| D-H···A | Symmetry | D-H (Å) | H···A (Å) | D···A (Å) | D-H···A (°) | |
|---|---|---|---|---|---|---|
| 3 | N2-H···O2 | x, y, z | 0.846(19) | 1.879(18) | 2.5983(15) | 141.9(17) |
| C15-H15···O1 | 1/2+x, y, 1/2−z | 0.95 | 2.44 | 3.3891(18) | 175.8 | |
| C5-H5C···O1 | 1/2+x, y, 1/2−z | 0.98 | 2.72 | 3.6442(18) | 158.4 | |
| C1-H1A···O2 | 1/2−x, 1/2+y, z | 0.99 | 2.67 | 3.4651(17) | 137.3 | |
| C5-H5B···Cg2a | 1/2−x, 1/2+y, z | 0.98 | 2.93 | 3.8117(17) | 150 | |
| C9-H9···Cg2a | x, 1/2−y, −1/2+z | 0.95 | 2.80 | 3.6607(15) | 151 | |
| C19-H19···Cg1a | −1/2+x, y, 1/2−z | 0.95 | 2.97 | 3.6899(15) | 134 | |
| 4 | N2A-H2NA···O1B | −x+1, y+1/2, −z+1/2 | 0.91(3) | 1.99(3) | 2.891(2) | 170(3) |
| N2B-H2NB···O1A | x+1, y, z | 0.90(3) | 2.20(3) | 3.058(2) | 159(2) | |
| C12B-H12B···O1A | x+1, y, z | 0.95 | 2.48 | 3.301(3) | 145.3 | |
| C3B-H3E···O1A | x+1, y, z | 0.99 | 2.49 | 3.417(3) | 156.2 | |
| C1B-H1C···Cg4 | −x, 1/2+y, 1/2−z | 0.99 | 2.71 | 3.684(6) | 170 | |
| C2A-H2B···Cg4 | −x, 1−y, −z | 0.99 | 2.71 | 3.655(3) | 159 | |
| C2C-H2E···Cg4 | −x, 1/2+y, 1/2−z | 0.99 | 2.56 | 3.43(3) | 146 |
aCg is defined as the centroid of the rings (centre of gravity): Cg1: C7-C12; Cg2: C14-C19; Cg4: C7B-C12B.
Compound 4 crystallises in the monoclinic space group P21/c with two crystallographically independent molecules in the asymmetric unit (Figure 2). The reaction from 2 to 4 involves replacement of the trimethylsilyl group by a phenylcarbamic group. The central urea unit of 4 is bound to a phenyl ring via N2 and to the 1,2-azasilolidine via N1. Both crystallographically independent molecules differ in the conformation of the phenyl ring with respect to the central urea unit formed by N1-C6(=O1)-N2. The dihedral angle between these planes is in molecule A 39.535(82)°, but in molecule B only 18.173(96)°. Intramolecular C8-H8···O1 distances differ from molecules A to B and influences the dihedral angle between the urea unit and the phenyl moiety (A: 2.45Å, 108°; B: 2.25Å, 121°). Five-membered rings may adopt an envelope or half-chair (twisted) conformation (Fuchs, 1978). Ring puckering analysis of 4 shows that the five-membered ring in molecule A adopts a twisted conformation with the atoms C1A and C2A −0.229 and 0.283Å out of the plane of the five-membered ring. The least-squares plane of the five atoms in crystal coordinates is given by 9.646x−2.079y+3.340z=2.250. Molecule B features a disorder at C1 and C2. Both five-membered rings formed by this disorder are in twisted conformation with the atoms C1B/C2B (−0.249/0.299Å) and C1C/C2C (0.282/−0.323Å) alternating above and below the plane formed by the five-membered rings. The least-squares planes of the five-membered rings in crystal coordinates are given by 1.135x−1.756y+18.192z=6.812 for Si1B-N1B-C1B-C2B-C3B and 2.865x+0.622y+ 17.546z=8.763 for Si1B-N1B-C1C-C2C-C3B.
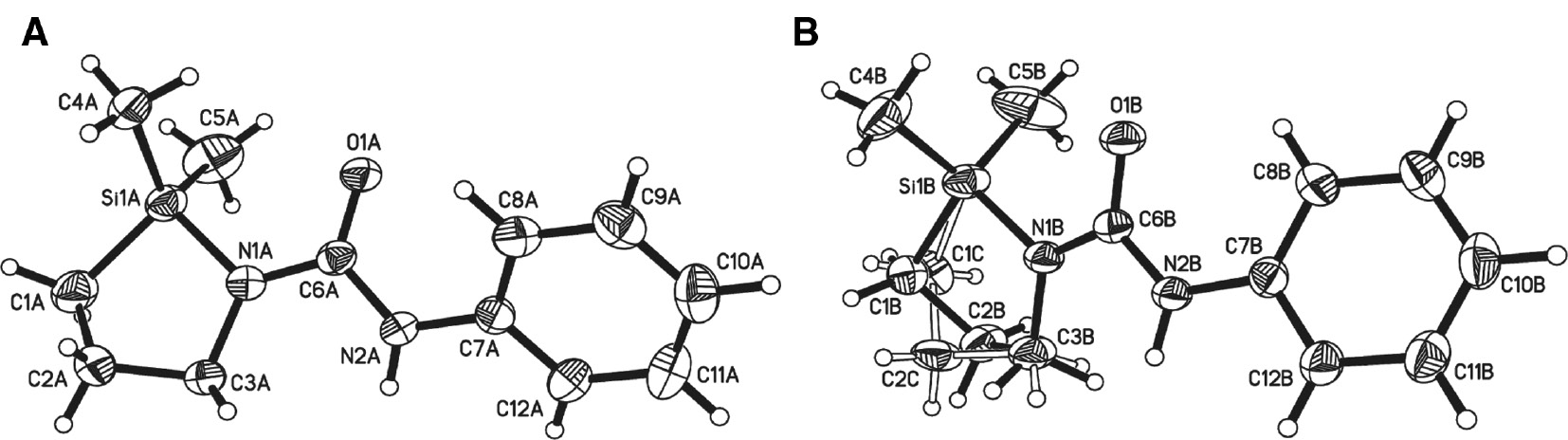
Molecular structures of the two crystallographically independent molecules of 4 showing the atomic numbering scheme.
Molecule A is labelled with A, molecule B with B. The thermal ellipsoids of the nonhydrogen atoms are drawn at the 50% probability level. Molecule B shows a disorder at the five-membered ring with s.o.f. of 0.85.
In contrast to 3, the crystal packing of 4 is dominated by C-H···π interactions (Nishio, 2004) in the range of 2.56 to 2.71Å. However, the π-system of molecule A is not involved in C-H···π contacts. Instead, the oxygen O1 atom interacts with the NH and two CH groups in strong N-H···O [2.20Å, 159(2)°] and weak C-H···O hydrogen bonding (2.5Å). These interactions occur in the same direction as a possible C-H···π can be localised (C3B-H3D···Cg3, with Cg3 as the centre of gravity of C7A-C12A) and form intermolecular dimers with molecule B. Furthermore, via N-H···O (Table 2) bridges of molecule B, the crystal packing is stabilised.
Conclusions
Intramolecular cyclisation reactions of 3-aminopropylalkoxysilanes to form 1-aza-2-silacyclopentane appear obvious and have been reported in literature. However, the corresponding five-membered cyclic condensation products have neither been isolated nor structurally analysed. Only in the presence of excess HMDS and (NH4)2SO4 as a catalyst it is possible that 1-aza-2-silacyclopentanes are formed and stabilised bearing SiMe3-substituents at the N atom.
Via this route, we obtained the derivatives 2a–2e in yields between 61% and 94%. Unsubstituted 1-aza-2-silacyclopentanes with N-H group are not obtainable. 2,2-Diethoxy-1-trimethylsilyl-1-aza-2-silacyclopentane (2a) can be treated with Grignard or alkyllithium reagents to form the monoalkylated or dialkylated products 2f–2g.
All 1-aza-2-silacyclopentanes tend to undergo ring-opening polymerisation reactions upon long-term standing at room temperature. Besides, hydrolysis and alcoholysis of the Si-N bond in 2a–2g can take place easily.
During our studies on PhNCO insertion reactions into the Si-N bonds of 2d, we isolated the partial hydrolysis and double-insertion product 3 and the mono-insertion product 4. The single crystal X-ray diffraction analyses indicate the unexpected formation of 2,4-diaza-1-silacycloheptane derivative 4 and an azasilacyclopentane 3 containing two and one urea units, respectively.
Experimental
All starting materials except 1d and N,N-bis(trimethylsilyl)-3-aminopropyl-trimethoxysilane were obtained from Sigma-Aldrich (St. Louis, MO, USA) and used as received. 1d and N,N-bis(trimethylsilyl)-3-amino-propyl-trimethoxysilane were obtained from ABCR (Karlsruhe, Germany) and used without further purification. All reactions were carried out under dry argon atmosphere using standard Schlenk technique.
NMR spectra were measured with a Bruker DPX400 (Bruker, Karlsruhe, Germany, frequencies: 1H, 400.13MHz; 13C, 100.61MHz; 29Si, 79.49MHz; all substitution reaction products) or a Bruker Avance III 500MHz (Bruker, Karlsruhe, Germany, frequencies: 1H, 500.13MHz; 13C, 125.76MHz; 29Si, 99.36MHz; all condensation and insertion products) spectrometer in CDCl3 (Armar Chemicals, Ahmedabad, Gujarat, India, purified by distillation from CaH2) with tetramethylsilane as internal standard.
Raman spectra were measured with Bruker Fourier Transform Raman Spectrometer RFS100/S (Bruker, Karlsruhe, Germany). The device works with an air-cooled Nd:YAG laser with a wavelength of 1064nm and a nitrogen-cooled detector. The samples were filled in glass tubes and sealed with PTFE-paste Triboflon MYN 503 (Spezialschmierstoffe Anja Heibach, Odenthal, Germany).
The EAs were conducted using a vario Micro cube (Elementar Analysensysteme GmbH, Hanau, Germany) with about 2mg samples in a tin capsule.
The ebullioscopic molecular mass determination was done with cyclohexane (VWR International GmbH, Darmstadt, Germany, purified by distillation from Na/benzophenone) as solvent and a four-wired Pt100 resistance thermometer.
The mass spectrum was measured with an Advion expression CMS mass spectrometer (Advion, Ithaca, NY, USA).
Synthesis of 2a–e by cyclisation reactions
The 3-aminopropylsilanes 1a–e were refluxed for 2 h with the reactants listed in Table 3. The reaction mixture was distilled under reduced pressure to isolate 2a–e. The catalyst and the amount of HMDS were varied in order to find optimised reaction conditions.
Parameters of different synthesis procedures.
| Procedure | Catalyst | Remarks | Yield (%) | ||||
|---|---|---|---|---|---|---|---|
| 2a | 2b | 2c | 2d | 2e | |||
| A | — | Stoichiometry HMDS:silane=1:1 | 9a | 5a | 2a | 0b | — |
| B | — | Excess HMDS | 4a | 9a | 5a | 6a | 0a |
| C | (NH4)2SO4 | Stoichiometry HMDS:silane=1:1 | 0c | 0c | 0c | 0c | 0c |
| D | (NH4)2SO4 | Excess HMDS | 61 | 73 | 82 | 90 | 94a |
| E | NaOMe | Without HMDS | 0a | 0a | 0a | 0a | 0a |
aAccording to 29Si-NMR, refer to 1a–e.
bMany impurities, not determined.
cUndefined polymer.
2,2-Diethoxy-1-trimethylsilyl-1-aza-2-silacyclopentane (2a):
1H-NMR: δ [ppm]=3.68 (q, 4H, CH3CH2O-, 3JH,H=7.0Hz), 2.85 (t, 2H, CH2-N, 3JH,H=5.9Hz), 1.72 (qu, 2H, CH2-CH2-CH2, 3JH,H=6.8Hz), 1.14 (t, 6H, CH3CH2O-, 3JH,H=7.0Hz), 0.50 (t, 2H, Si-CH2-CH2-, 3JH,H=7.6Hz), 0.33 (HMDS), 0.01 (s, 9H, Si(CH3)3); 13C-NMR: δ [ppm]=58.3 (CH3CH2O-), 45.4 (CH2-N), 25.4 (CH2-CH2-CH2), 18.3 (CH3CH2O-), 5.9 (Si-CH2-CH2-), −0.4 (Si(CH3)3); 29Si-NMR: δ [ppm]=2.6 (Si(CH3)3), −16.9 (Si1); see also Tsai and Marshall (1969); Raman ν [cm−1]=2958, 2927, 2900, 2839, 2763, 2715, 2671, 2620, 2485, 1478, 1452, 1409, 1290, 1247, 1193, 1144, 1089, 1020, 947, 888, 854, 771, 750, 679, 628, 573, 503, 416, 379, 194, 106; EA: calc for C10H25NO2Si2: C: 48.53%, H: 10.18%, N: 5.66%; found C: 48.23%, H: 10.29%, N: 5.47%; yield=6.82 g (61%, procedure D); M calc. for C10H25NO2Si2 247.5 g/mol, found M=(246±17) g/mol (ebullioscopically measured in cyclohexane).
2-Ethoxy-2-methyl-1-trimethylsilyl-1-aza-2-silacyclopentane (2b):
1H-NMR: δ [ppm]=3.61 (m, 2H, CH3CH2O-), 2.97 and 2.81 (m, each 1H, CH2-N), 1.70 (m, 2H, CH2-CH2-CH2), 1.14 (t, 3H, CH3CH2O-, 3JH,H=7.0Hz), 0.68 and 0.41 (m, each 1H, Si-CH2-CH2-), 0.14 (s, 3H, CH3Si1), 0.04 (s, 9H, Si(CH3)3); 13C-NMR: δ [ppm]=57.6 (CH3CH2O-), 47.1 (C3), 25.9 (CH2-CH2-CH2), 18.2 (CH3CH2O-), 10.6 (Si-CH2-CH2-), 2.4 (HMDS), −0.3 (Si(CH3)3), −1 (CH3Si1); 29Si-NMR: δ [ppm]=2.5 (Si(CH3)3), 11.1 (Si1); Raman ν [cm−1]=2956, 2925, 2899, 2834, 2714, 2669, 2618, 2487, 1477, 1452, 1409, 1287, 1246, 1137, 1106, 1061, 1013, 964, 890, 856, 750, 709, 684, 656, 626, 563, 485, 412, 232, 192; EA: calc for C9H23NOSi2: C: 49.71%, H: 10.66%, N: 6.44%; found C: 48.73%, H: 10.70%, N: 6.57%; yield=8.5 g (73%, procedure D).
2,2-Dimethoxy-1-trimethylsilyl-1-aza-2-silacyclopentane (2c):
1H-NMR: δ [ppm]=3.39 (m, 6H, CH3O-), 2.82 (q, 2H, CH2-N, 3JH,H=6.2Hz), 1.69 (m, 2H, CH2-CH2-CH2), 0.47 (q, 2H, Si-CH2-CH2-, 3JH,H=7.6Hz), 0.00 (m, 9H, Si(CH3)3); 13C-NMR: δ [ppm]=50.1 (CH3O-), 45.2 (CH2-N), 25.4 (CH2-CH2-CH2), 4.5 (Si-CH2-CH2-), −0.7 (Si(CH3)3); 29Si-NMR: δ [ppm]=2.9 (Si(CH3)3), −13.7 (Si1); see also Salikhov et al. (2014); Raman ν [cm−1]=2957, 2900, 2837, 2671, 2620, 2485, 1477, 1453, 1408, 1346, 1247, 1146, 1098, 1061, 1021, 987, 961, 888, 858, 784, 749, 676, 628, 565, 490, 412, 370, 219, 195, 108; yield=2.49 g (82%, procedure D); MS: [M+H]+m/z calc. for C8H21NO2Si2: 220 g/mol, found 220 g/mol.
2,2-Dimethyl-1-trimethylsilyl-1-aza-2-silacyclopentane (2d):
1H-NMR: δ [ppm]=2.91 (t, 2H, CH2-N, 3JH,H=6.0Hz), 1.73 (m, 2H, CH2-CH2-CH2), 0.63 (t, 2H, Si-CH2-CH2-, 3JH,H=7.4Hz), 0.31 (HMDS), 0.11 (s, 6H, CH3Si1), 0.05 (s, 9H, Si(CH3)3); 13C-NMR: δ [ppm]=48.3 (CH2-N), 26.6 (CH2-CH2-CH2), 13.23 (Si-CH2-CH2-), 1.6 (CH3Si1), 0.2 (Si(CH3)3); 29Si-NMR: δ [ppm]=22.8 (impurity), 18.2 (Si1), 2.1 (Si(CH3)3); Raman ν [cm−1]=2954, 2898, 2831, 2666, 2617, 2483, 1474, 1450, 1410, 1344, 1245, 1132, 1059, 1007, 889, 862, 832, 785, 746, 682, 645, 621, 554, 475, 368, 312, 213, 184, 109; yield=4.25 g (90%, procedure D).
2,2-Dimethoxy-1-methyl-1-aza-2-silacyclopentane (2e):
29Si-NMR: δ [ppm]=−20.8 (Si1), 2.4 (HMDS), −41.6 (N-methyl-3-aminopropyltrimethoxysilane). Because of impurities, no Raman spectrum was measured. Yield=94 mol% (according to 29Si-NMR, refer to 1e, procedure D).
Substitutions reactions with 2a
A solution of 2a and about 20 mL of n-pentane is cooled to −78°C by stirring in a bath of dry ice and isopropanol. Stoichiometric amounts of a solution of ethylmagnesiumbromide (1 m in tetrahydrofuran) or n-butyllithium (2.5 m in hexane) is added dropwise. After warming to room temperature, volatiles are removed in vacuo, and the residue is suspended in n-pentane. The suspension is filtered via a G4 frit, and the solvent is removed in vacuo. Some residues and byproducts remain in the liquid.
2-Ethoxy-2-ethyl-1-trimethylsilyl-1-aza-2-silacyclopentane (2f):
1H-NMR: δ [ppm]=3.67 (m, 2H, CH3CH2O-), 3.02 and 2.85 (m, each 1H, CH2-N), 1.75 (m, 2H, CH2-CH2-CH2), 1.18 (t, 3H, 3JH,H=7.0 Hz) and 0.94 (m, 3H, CH3CH2O- and CH3CH2Si1-), 0.63 (m, 4H, Si-CH2-CH2- and CH3CH2Si1-), 0.09 (s, 9H, Si(CH3)3); 13C-NMR: δ [ppm]=57.9 (CH3CH2O-), 47.6 (CH2-N), 26.4 (CH2-CH2-CH2), 18.5 (CH3CH2O-), 8.1 (CH3CH2Si1-), 7.2 (CH3CH2Si1-), 7.1 (Si-CH2-CH2-), −0.2 (Si(CH3)3); 29Si-NMR: δ [ppm]=12.9 (Si1), 2.2 (Si(CH3)3); yield=0.52 g (56%).
2-n-Butyl-2-ethoxy-1-trimethylsilyl-1-aza-2-silacyclopentane (2g):
1H-NMR: δ [ppm]=3.69 (m, 2H, CH3CH2O-), 3.03 and 2.85 (m, each 1H, CH2-N), 1.77 (m, 2H, CH2-CH2-CH2), 1.44 (m, 6H, CH2 of nBu); 1.22 (t, 3H, CH3CH2O-, 3JH,H=7.0Hz) and 0.98 (t, 3H, H4 and CH3 of nBu), 0.75 and 0.60 (m, 2H, Si-CH2-CH2-), 0.24 (s, 9H, Si(CH3)3); 13C-NMR: δ [ppm]=57.4 (CH3CH2O-), 47.2 (CH2-N), 26.4 and 26.3 and 26.0 (CH2-CH2-CH2 and CH2 of nBu), 18.3 (CH3CH2O-), 15.4 (CH2 of nBu); 13.8 (CH3 of nBu); 8.6 (Si-CH2-CH2-), −0.3 (Si(CH3)3); 29Si-NMR: δ [ppm]=11.2 (Si1), 2.2 (Si(CH3)3); Raman ν [cm−1]=2956, 2923, 2897, 2874, 2834, 2760, 2711, 2668, 2618, 2485, 1476, 1449, 1409, 1344, 1289, 1245, 1191, 1137, 1106, 1062, 1011, 963, 889, 852, 749, 683, 646, 629, 567, 499, 414, 224, 192, 108; yield=100% according to 1H-NMR, LiOEt not removed.
2,2-Di-n-butyl-1-trimethylsilyl-1-aza-2-silacyclopentane (2h):
1H-NMR: δ [ppm]=3.92 (m, 2H, LiOEt), 2.88 (t, 2H, CH2-N, 3JH,H=6.1 Hz), 1.69 (qu, 2H, CH2-CH2-CH2, 3JH,H=6.8 Hz), 1.33 (m, 12H, CH2 of nBu); 0.91 (m, 6H, CH3 of nBu), 0.65 (m, 2H, Si-CH2-CH2-), 0.60 (m, 3H, LiOEt); 0.12 (s, 9H, Si(CH3)3); 13C-NMR: δ [ppm]=58.0 (LiOEt), 48.6 (CH2-N), 26.9 and 26.6 and 26.2 (CH2-CH2-CH2 and CH2 of nBu), 22.8 (LiOEt), 16.6 (CH2 of nBu); 13.7 (CH3 of nBu); 9.5 (Si-CH2-CH2-), −0.4 (Si(CH3)3); 29Si-NMR: δ [ppm]=21.2 (Si1), 1.8 (Si(CH3)3); Raman ν [cm−1]=2955, 2934, 2898, 2874, 2859, 2734, 2705, 2668, 2616, 1447, 1410, 1343, 1289, 1245, 1188, 1133, 1052, 1006, 962, 888, 850, 747, 682, 639, 605, 564, 495, 417, 230, 188, 136; yield=100% according to 1H-NMR, LiOEt not removed.
Insertion reaction of PhNCO with 2d
The cyclic N-aminosilane 2d (one part) in n-pentane is cooled to 0°C, the stoichiometric amount (two parts) of PhNCO is carefully added via syringe, and the solution is stirred overnight. In the first experiment, 3 crystallised from this mixture overnight. In the second experiment, the solvent is removed in vacuo and replaced with dry CDCl3. After a few weeks, 4 crystallised from this solution.
3:
1H-NMR: δ [ppm]=10.85 (s, 1H, NH), 7.50–7.15 (m, 10H, Ar), 4.05 (t, 2H, CH2-N, 3JH,H=6.0Hz), 2.15 (qu, 2H, CH2-CH2-CH2, 3JH,H=7.0Hz), 1.08 (t, 2H, CH2-Si, 3JH,H=7.2Hz), 0.18 (s, 6H, CH3-Si ); 13C-NMR: δ [ppm]=163.0, 153.5 (C=O), 139.8, 138.4 (C(quart)), 130.1–.2 (Ar), 48.7 (CH2-N), 21.1 (CH2-CH2-CH2), 11.5 (CH2-Si), −0.7 (CH3-Si); 29Si-NMR: δ [ppm]=12.9.
4:
Crystallised from a CDCl3 solution after standing several weeks. NMR analysis of this solution showed that a mixture of insertion and oligomerisation products of phenylisocanate was formed. Therefore, unambiguous assignment of 1H- and 13C-NMR data of 4 is not possible in this case.
Alcoholysis of 2a
Stoichiometric as well as sub-stoichiometric amounts of dry 1-pentanol were added dropwise to a solution of 2a in n-pentane in an ice bath. The 29Si-NMR spectrum shows signals of 2a (δ=2.7, −17.0 ppm) and of the alcoholysis products (δ=16.7ppm (Me3SiOnPent) and −45.4 ppm (3-aminopropyldiethoxypenthoxysilane)).
Single crystal X-ray diffraction analysis
Data collection on 3 and 4 was performed on a STOE IPDS-2T image plate diffractometer (Stoe & Cie. GmbH, Darmstadt, Germany) equipped with a low-temperature device with Mo-Kα radiation (λ=0.71073 Å) using ω and φ scans. Software for data collection: X-AREA, cell refinement: X-AREA and data reduction: X-RED (Stoe and Cie, 2009). Preliminary structure models were derived by direct methods (Sheldrick, 2008), and the structures were refined by full-matrix least-squares calculations based on F2 for all reflections using SHELXL (Sheldrick, 2015). The hydrogen atoms at the amine nitrogen atoms N2 in both structures were localised from the differences in the Fourier maps and refined without restraints. All other hydrogen atoms were included in the models in calculated positions and were refined as constrained to the bonding atoms. Further crystallographic and structure refinement data are listed in Table 4.
Crystal data and structure refinement for 3 and 4.
| 3 | 4 | |
|---|---|---|
| Empirical formula | C19H23N3O2Si | C12H18N2OSi |
| Formula weight | 353.49 | 234.37 |
| Temperature (K) | 193(2) | 153(2) |
| Crystal system, space group | Orthorhombic, Pbca | Monoclinic, P21/c |
| a (Å) | 10.2679(3) | a=9.9834(4) |
| b (Å) | 14.4545(5) | b=14.3450(5) |
| c (Å) | 25.0332(7) | c=18.5345(9) |
| α (°) | 90 | 90 |
| β (°) | 90 | 91.947(4) |
| γ (°) | 90 | 90 |
| Volume (Å3) | 3715.4(2) | 2652.83(19) |
| Z | 8 | 8 |
| ρηοcalc (g/cm3) | 1.264 | 1.174 |
| Absorption coefficient (mm−1) | 0.144 | 0.160 |
| F(000) | 1504 | 1008 |
| Crystal size (mm) | 0.500×0.500×0.250 | 0.490×0.450×0.180 |
| Reflections collected/unique | 39 678/3638 [R(int)=0.0779] | 35 972/5697 [R(int)=0.0450] |
| Data/restraints/parameters | 3638/0/232 | 5697/3/320 |
| Final R indices [I>2σ(I)] | R1=0.0332, wR2=0.0865 | R1=0.0554, wR2=0.1227 |
| R indices (all data) | R1=0.0366, wR2=0.0897 | R1=0.0722, wR2=0.1343 |
Acknowledgements
The authors gratefully acknowledge help from Beate Kutzner (NMR), Regina Moßig (Raman), Konstantin Kraushaar (MS), and Ute Groß (EA). The German Research Foundation, DFG, is acknowledged for financial support. Part of this work was performed within the research group “Chemical utilization of carbon dioxide with aminosilanes (CO2-Sil)” that is financially supported by the European Union (European Regional Development Fund), the Ministry of Science and Art of Saxony (SMWK) and the Sächsische Aufbaubank (SAB).
References
Arkles, B. C.; Pan, Y.; Larson, G. L. WO2003091186A2, 2003.Search in Google Scholar
Evans, D. G.; Boeyens, J. C. A. Conformational analysis of ring pucker. Acta Cryst.1989, B45, 581–590.10.1107/S0108768189008190Search in Google Scholar
Fuchs, B. Conformations of Five-Membered Rings. In Topics in Stereochemistry. Eliel, E. L.; Allinger, N. L., Eds. John Wiley & Sons: New York, 1978; Vol. 10, pp 1–94.10.1002/9780470147191.ch1Search in Google Scholar
Kirilin, A. D.; Belova, L. O.; Korobova, E. A. Use of 3-(ethoxysilyl)-1-propylamine in the synthesis of silicon heterocycles. Russ. J. Gen. Chem.2009, 79, 2459–2460.10.1134/S1070363209110334Search in Google Scholar
Kraushaar, K.; Wiltzsch, C.; Wagler, J.; Boehme, U.; Schwarzer, A.; Roewer, G.; Kroke, E. From CO2 to polysiloxanes: di(carbamoyloxy)silanes Me2Si[(OCO)NRR’]2 as precursors for PDMS. Organometallics2012, 31, 4779–4785.10.1021/om300313fSearch in Google Scholar
Kraushaar, K.; Schmidt, D.; Schwarzer, A.; Kroke, E. Reactions of CO2 and CO2 Analogs (CXY with X, Y=O, S, NR) with Reagents Containing Si–H and Si–N Units. In Advances in Inorganic Chemistry: CO2 Chemistry. van Eldik, R.; Aresta, M., Eds. Academic Press: Waltham, MA, 2014; Vol. 66, pp 117–162.10.1016/B978-0-12-420221-4.00004-4Search in Google Scholar
Nishio, M. CH/π hydrogen bonds in crystals. Cryst. Eng. Commun.2004, 6, 130–158.10.1039/b313104aSearch in Google Scholar
Nyugaku, T.; Honma, T.; Tonomura, Y.; Kubota, T. JP201102267A, 2011.Search in Google Scholar
Pepe, E. J.; Schilling, C. L. Jr.; Bennett, E. W. WO9414820A1, 1994.Search in Google Scholar
Salikhov, T.; Kopylov, V.; Shragin, D. Silylated derivatives of azasilacyclopentanes. Russ. J. Gen. Chem.2014, 84, 875–882.10.1134/S1070363214050168Search in Google Scholar
Schöne, D.; Gerlach, D.; Wiltzsch, C.; Brendler, E.; Heine, T.; Kroke, E.; Wagler, J. A distorted trigonal antiprismatic cationic silicon complex with ureato ligands: syntheses, crystal structures and solid state 29Si NMR properties. Eur. J. Inorg. Chem.2010, 3, 461–467.10.1002/ejic.200900784Search in Google Scholar
Sheldrick, G. M. A short history of SHELX. Acta Cryst.2008, A64, 112–122.10.1107/S0108767307043930Search in Google Scholar PubMed
Sheldrick, G. M. Crystal structure refinement with SHELXL. Acta Cryst.2015, C71, 3–8.Search in Google Scholar
Speier, J. L.; Roth, C. A.; Ryan, J. Syntheses of (3-aminoalkyl) silicon compounds. W. J. Org. Chem.1971, 36, 3120–3126.10.1021/jo00820a011Search in Google Scholar
Stoe, Cie. X-RED and X-AREA. Stoe & Cie GmbH: Darmstadt, Germany, 2009.Search in Google Scholar
Tonomura, Y.; Kubota, T.; Honma, T. JP2001246392A, 2011.Search in Google Scholar
Tsai, T.-T.; Marshall, C. J. J. 1,1-Diethoxy-2-(trimethylsilyl)-1-sila- 2-azacyclopentane. J. Org. Chem.1969, 34, 3676–3679.10.1021/jo01263a119Search in Google Scholar
Ziche, W.; Selbertinger, E. WO 2006018143 A1, 2006.Search in Google Scholar
Ziche, W.; Ziemer, B.; John, P.; Weis, J.; Auner, N. Synthesis and structure of 1,6-diaza-2,2-dimethoxy-2-silacyclooctane. J. Organomet. Chem.1996, 521, 29–31.10.1016/0022-328X(96)06379-6Search in Google Scholar
Supplemental Material:
The online version of this article offers supplementary material (https://doi.org/10.1515/mgmc-2018-0005).
©2018 Walter de Gruyter GmbH, Berlin/Boston
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Articles in the same Issue
- Frontmatter
- Review
- Distortion isomers of PtM, Pt2M, PtM2 and PtMM′ (M = non-transition metals) derivatives-structural aspects
- Research Articles
- Formation of 1-aza-2-silacyclopentanes and unexpected products from the insertion of phenylisocyanate into 2,2-dimethyl-1-(trimethylsilyl)-1-aza-2-silacyclopentane
- ZnO-CeO2 nanocomposite: efficient catalyst for the preparation of thieno[2,3-d]pyrimidin-4(3H)-one derivatives
- Potential bioactive mononuclear diorganotin(IV) phenoxyacetohydroxamate complexes: synthesis, characterization and antimicrobial evaluation
- Geometrical and electronic properties of PdWSin (n=10–20) semiconductor materials
- Short Communication
- Synthesis and characterization of the coordination polymer [(THF)K(μ-OPri)2Al(μ-OPri)2]n
Articles in the same Issue
- Frontmatter
- Review
- Distortion isomers of PtM, Pt2M, PtM2 and PtMM′ (M = non-transition metals) derivatives-structural aspects
- Research Articles
- Formation of 1-aza-2-silacyclopentanes and unexpected products from the insertion of phenylisocyanate into 2,2-dimethyl-1-(trimethylsilyl)-1-aza-2-silacyclopentane
- ZnO-CeO2 nanocomposite: efficient catalyst for the preparation of thieno[2,3-d]pyrimidin-4(3H)-one derivatives
- Potential bioactive mononuclear diorganotin(IV) phenoxyacetohydroxamate complexes: synthesis, characterization and antimicrobial evaluation
- Geometrical and electronic properties of PdWSin (n=10–20) semiconductor materials
- Short Communication
- Synthesis and characterization of the coordination polymer [(THF)K(μ-OPri)2Al(μ-OPri)2]n