Genetic alterations in lymphoblastic leukaemia / lymphoma – a practical guide to WHO HAEM5
-
Doris Steinemann
Prof. Dr. rer. nat. Doris Steinemann

 ,
Małgorzata Dawidowska
,
Małgorzata Dawidowska
Małgorzata Dawidowska, Ph.D, DSc
,
Lisa J Russell
Dr. Lisa J Russell, Ph.D
,
Christine J Harrison
Prof. Christine J. Harrison, Ph.D
and
Gudrun Göhring
Prof. Dr. med. Gudrun Göhring

Abstract
We present a practical guide for analyzing the genetic aspects of lymphoblastic leukaemia/lymphoma according to the 5th edition of the World Health Organization (WHO) classification of haematolymphoid neoplasms (WHO-HAEM5) issued in 2024. The WHO-HAEM5 acknowledges the increasing importance of genetics in the diagnosis of lymphoid neoplasia. Classification is based on the established genetic subtypes according to cell lineage, with precursor cell neoplasms followed by mature malignancies. This guide describes those genetic abnormalities in acute precursor B- and T-cell neoplasms required for risk stratification, and for treatment, providing diagnostic algorithms under the headings of ‘essential’ and ‘desirable’ diagnostic criteria.
Keywords: WHO HAEM5 classification, Precursor B-cell neoplasms, B-ALL, T-ALL, LBL
Introduction
In the revised 5th edition of the World Health Organization (WHO) classification of haematolymphoid neoplasms (WHO-HAEM5), lymphoblastic leukaemia (ALL) and lymphoma (LBL) are grouped together into lymphoblastic neoplasms of B- and T-precursor cells [1]. The distinction between leukaemia and lymphoma depends on the primary involvement of blood and bone marrow (ALL) or lymph node and/or extra-nodal sites (LBL). The extensive development and integration of new genetic methods into routine diagnostics has greatly impacted the revised classification. While for the B-cell ALL/LBL, new genetic subgroups have been defined, in T-ALL/LBL, the classification remains based on immunophenotyping and histology.
B-ALL and lymphoma
ALL is the most frequent cancer in childhood, with B-ALL accounting for around 80 % of cases. The annual incidence rate of ALL is 3–4 cases per 100,000 children and young adults. The peak incidence occurs in young children [2], with long-term cure achieved in nearly 90 % of cases on most international contemporary protocols [3–6]. Adult ALL is rare and has inferior outcomes compared to children. Good-risk subtypes are more common in paediatric cases, while adverse-risk subtypes are more prevalent in adults [7]. Treatment is based on risk of relapse, predicted from a combination of clinical (e. g., age, white blood cell count), genetic, and morphological early response criteria. Monitoring of leukaemic blast clearance reflects treatment response and measurable residual disease (MRD), in association with genetics, are strong prognostic factors in ALL [4, 8]. B-LBL are morphologically identical to ALL, comprising around 10 % of LBL.
B-ALL/LBL genetic subclassification
A broad spectrum of established genetic aberrations with prognostic impact are used in risk stratification of B-ALL/LBL [9, 10]. These genetic markers may be numerical (aneuploidies) or structural (translocations, copy number variants) and stratify patients into low, standard, high or very high-risk arms, differing in treatment intensity. High hyperdiploidy (HHD) is the largest genetic subgroup, present in approximately 30 % of paediatric but only 1 % of adult B-ALL/LBL (Fig. 1). It is defined by a non-random pattern of chromosomal gains (X, 4, 6, 10, 14, 17, 18, and 21) resulting in a modal chromosome number of 51–67 chromosomes [11]. The overall prognosis is excellent. In contrast, hypodiploidy, defined by a modal number of 43 chromosomes or less, has a poor outcome. B-ALL/LBL with intrachromosomal amplification of chromosome 21 – iAMP21-ALL – was first described in 2003 as a distinct entity comprising around 2 % of paediatric B-ALL, with a median age of 9 years [12]. This subtype is also associated with a poor prognosis and benefits from treatment intensification [13]. Other markers of poor prognosis are the gene fusions: BCR::ABL1 resulting from the translocation t(9;22)(q34;q11), TCF3::HLF1/t(17;19)(q22;p13) and a range of KMT2A fusions including the most frequent partner genes: AFF1, MLLT1, MLLT3, MLLT10, AFDN, EPS15 and USP2 [14]. The rare fusion, TCF3::HLF, is a new entity within WHO-HAEM5. Whilst B-ALL/LBL harbouring this aberration is resistant to conventional chemotherapies, it has shown sensitivity to the BCL2-specific inhibitor venetoclax [15].
The ETV6::RUNX1 fusion, resulting from the translocation, t(12;21)(p13;q22) is associated with favorable outcome [16, 17]. B-ALL with a balanced t(1;19)(q23;p13) or its unbalanced form, der(19)t(1;19), giving rise to TCF3::PBX1 fusion, has an intermediate prognosis [18]. B-ALL/LBL with IG::IL3 fusion/t(5;14)(q31.1;q32.1) or other cryptic insertional rearrangements is a rare entity associated with accumulation of eosinophils. Herein, the IGH super-enhancers (14q32.1) are juxtaposed to the vicinity of the IL3 gene (5q31.1) leading to IL3 overexpression [19, 20].
B-ALL/LBL with BCR::ABL1-like features was introduced in WHO-HAEM4R, but ETV6::RUNX1-like-ALL is newly included in WHO-HAEM5. Although these entities lack the BCR::ABL1 and ETV6::RUNX1 fusions, respectively, they show similar gene expression profiles [21, 22]. ETV6::RUNX1-like ALL typically harbours deletions targeting ETV6 and IKZF1 [21].
B-ALL/LBL with other defined genetic abnormalities
Around 30 % of adult and 15 % of childhood B-ALL/LBL do not have established genetic abnormalities at diagnosis [23]. These cases were previously defined as B-other-ALL with highly variable prognosis and treatment response. Recently, whole genome- and transcriptome-sequencing have identified multiple new genomic subtypes of B-ALL. However, evidence for defining them as potential novel entities conferring distinct clinical, phenotypic and/or prognostic effects is limited. Thus, these new subtypes are listed under “B-ALL/LBL with other defined genetic abnormalities”. They include B-ALL/LBL with DUX4, MEF2D, ZNF384 or NUTM1 rearrangements, IG::MYC fusion, and cases with PAX5 alterations or PAX5 p.P80R. B-ALL/LBL that do not show a recurrent genetic alteration after comprehensive testing are summarized under NOS (not otherwise specified).
WHO Classification of B-lymphoblastic leukaemias/lymphomas (B-ALL/LBL) and of T-cell lymphoblastic leukaemia/lymphoma (T-ALL/LBL)From: The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms
|
5th edition of WHO classification |
|
B-ALL/LBL |
|
B-lymphoblastic leukaemia/lymphoma, NOS |
|
B-lymphoblastic leukaemia/lymphoma with high hyperdiploidy (HHD) |
|
B-lymphoblastic leukaemia/lymphoma with hypodiploidy |
|
B-lymphoblastic leukaemia/lymphoma with iAMP21 |
|
B-lymphoblastic leukaemia/lymphoma with BCR::ABL1 fusion |
|
B-lymphoblastic leukaemia/lymphoma with BCR::ABL1-like features |
|
B-lymphoblastic leukaemia/lymphoma with KMT2A rearrangement |
|
B-lymphoblastic leukaemia/lymphoma with ETV6::RUNX1 fusion |
|
B-lymphoblastic leukaemia/lymphoma with ETV6::RUNX1-like features |
|
B-lymphoblastic leukaemia/lymphoma with TCF3::PBX1 fusion |
|
B-lymphoblastic leukaemia/lymphoma with IGH::IL3 fusion |
|
B-lymphoblastic leukaemia/lymphoma with TCF3::HLF fusion |
|
B-lymphoblastic leukaemia/lymphoma with other defined genetic abnormalities |
|
T-ALL/LBL |
|
T-lymphoblastic leukaemia / lymphoma, NOS |
|
Early T-precursor lymphoblastic leukaemia / lymphoma |
NOS: not otherwise specified
DUX4-rearranged: Insertions of DUX4 close to the IGH super-enhancers and into intron 3 of the ERG locus have been described in about 4 % of B-ALL/LBL [21]. Frequent ERG deletions and a specific expression profile have been associated with this group [21, 24]. The detection of DUX4 rearrangements is challenging due to the small size of the rearrangement, the macrosatellite repeat nature of the DUX4 locus, as well as its subtelomeric localization. However, increased expression of DUX4 from RNA sequencing, immunohistochemical staining of CD2 [25], and CD371 expression by flow cytometry [26] facilitate identification of these cases. B-ALL/LBL with DUX4 rearrangements is associated with low relapse rates and high overall survival, despite persistent MRD early in the treatment course [27].
MEF2D-rearranged: MEF2D::BCL9 and MEF2D::HNRNPUL1 are the most common fusions, although other fusion partners have been rarely reported, including FOXJ2, CSF1R, HNRNPH1, PYGO2, BCL9L and SS18 [28]. A high risk of relapse has been associated with MEF2D::BCL9 fusions, but is not seen in patients with other MEF2D partners [28].
ZNF384-rearranged: Around 20 ZNF384 fusion partners have been identified so far, including EP300, TCF3, TAF15, CREBBP, EWSR1. The clinical significance of each fusion partner remains unclear due to the small number of reported cases. However, patients with EP300::ZNF384 ALL have been shown to have a lower cumulative relapse rate than other fusions [29].
NUTM1-rearranged: B-ALL/LBL with NUTM1 rearrangements, although rare, occur more frequently in infants without KMT2A rearrangements. They appear to have a favorable outcome [30].
IG::MYC: Although IG::MYC translocations are typical of Burkitt lymphoma (BL), WHO-HAEM5 reports infrequent cases of BL with a phenotype of precursor B-cells including expression of terminal deoxynucleotidyl transferase, sometimes CD34, and absence of CD20 and surface immunoglobulin expression. Not only their immunophenotype but also their molecular profiles, with frequent RAS-pathway mutations, and a distinct methylome indicate B-ALL/LBL rather than BL, where the IG::MYC translocation occurs at an early stage of B-cell maturation [31].
PAX5 alterations: Monoallelic deletions, ranging from focal to whole chromosome 9 deletions, are the most frequent PAX5 alterations in B-ALL/LBL. They act as cooperating events requiring other oncogenic lesions to induce overt malignant transformation. In contrast, PAX5 fusions – of which PAX5::ETV6 is the most recurrent – are founder lesions, displaying a relatively simple karyotype. PAX5 intragenic amplification (PAX5amp) has also been included in this subgroup based on similar RNA expression profiles [32].
PAX5 p.P80R: This subtype is characterized by biallelic alterations of PAX5, whereby one PAX5 allele acquires a deleterious mutation together with deletion or copy-neutral loss of heterozygosity of the WT allele. A reduced overall survival has been associated with this subtype [33].
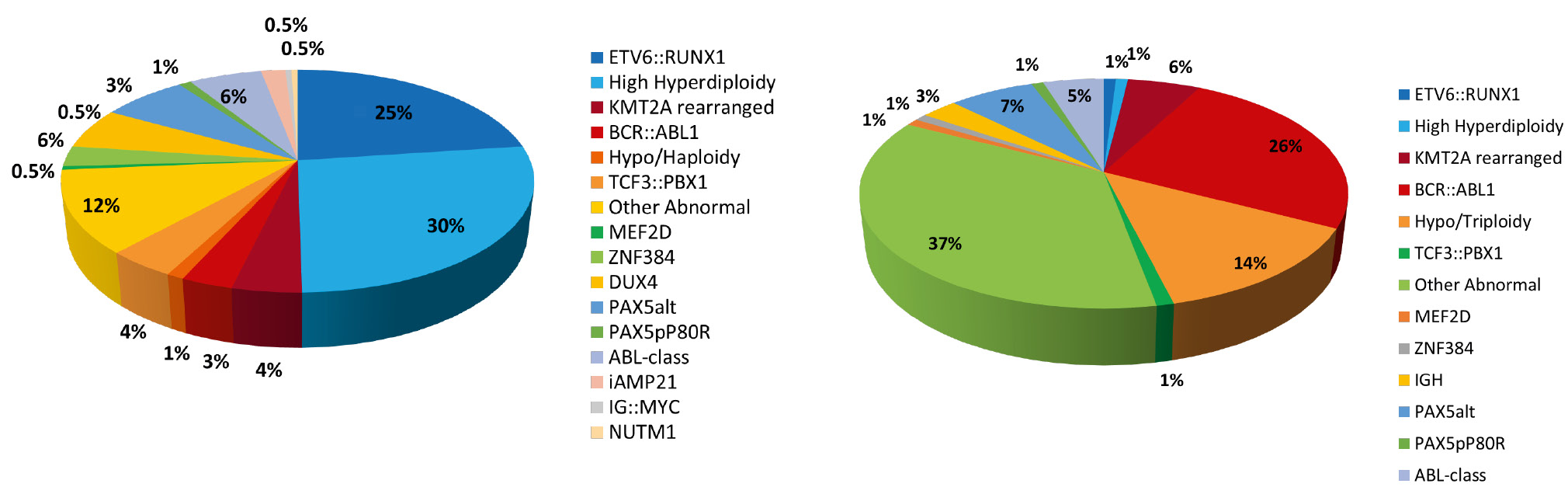
Frequency of B-ALL/LBL subtypes in children (left) and adults (right)
T-cell acute lymphoblastic leukaemia and lymphoma (T-ALL/LBL)
The classification of T-ALL/LBL, broadly used in the literature, is complex and based on the oncogenic activation of several transcription factors (TF), driving leukaemia development and defining major genetic subtypes: TAL1, TAL2, TLX1, TLX3, HOXA, LMO1/2, LMO2/LYL1, NKX2-1, SPI1 [34, 35]. Physiologically, these TF are involved in the development and maturation of T-cell precursors. In T-ALL/LBL they show ectopic expression due to rearrangements with TCR gene enhancers, structural variants or smaller mutations creating novel binding sites for factors enhancing the expression of these TF. Regardless of the mechanism, their oncogenic activation leads to a maturation arrest of T-cell precursors and to subtype-specific transcriptional reprograming. These genetic subtypes have been correlated with the maturation stages and with the presence of some genetic aberrations, e. g. TLX1 activation is frequently associated with a cortical immunophenotype; LMO2/LYL1 and HOXA subtypes are prevalent in ETP-ALL/LBL and are frequently associated with mutations activating JAK-STAT signaling; NUP214::ABL fusions are more prevalent in the TLX1/TLX3 subtype [36, 37] (Sin et al. 2021).
These proposed subtypes were defined by RNA-sequencing / gene expression profiles / RT-qPCR (to detect ectopic expression of TF) or genomic approaches (to detect the underlying genetic aberration, e. g. TCR::TF fusion). Increasing data from high throughput approaches, RNA-sequencing in particular, have shown that distinction between genetic subtypes is not clear-cut (some cases show expression of more than one TF driver oncogene). Despite improved understanding of the genetic landscape of T-ALL/LBL, evidence for the clinical relevance of the proposed genetic subtypes is lacking. WHO-HAEM5 includes only two entities: T-lymphoblastic leukaemia/lymphoma, NOS and Early T-precursor lymphoblastic leukaemia/lymphoma (ETP-ALL/LBL). The latter entity comprises approx. 5–17 % of paediatric and 7 % of adult T-ALL cases [37]. ETP-ALL/LBL is identified by similarity to earlier stages of T-cell precursors, based on gene expression profiling and detection of stem cell/myeloid markers by flow cytometry. The NK-lymphoblastic leukaemia/lymphoma was defined as a provisional entity in the WHO-HAEM4R, but it is no longer included in WHO-HAEM5 due to insufficient credibility of a distinct entity and the diagnostic criteria.
Diagnostic techniques for the detection of numerical and structural variants
A range of methods are currently used for the detection of genetic abnormalities in ALL/LBL. Most of the established abnormalities, defining the important genetic subtypes, can be recognized by cytogenetic testing. Karyotyping and fluorescence in-situ hybridization (FISH) have been used to identify good-risk (e. g. high hyperdiploidy and ETV6::RUNX1 fusion) and poor-risk (KMT2A rearrangements, BCR::ABL1, and hypodiploidy) subtypes of childhood B-ALL/LBL [38]. However, there is no clear agreement on the optimal method to define high hyperdiploidy. Karyotyping and/or DNA index (DI), a measure of the overall DNA content, were traditionally used. However, other genomic testing methods that allow identification of specific trisomies (e. g. WGS, SNP arrays, RNA-sequencing, FISH) are also being used. As increasing numbers of new and rare subtypes are being identified, it is becoming impossible to identify them all by karyotyping and FISH due to their limitations of resolution and time spent in analysis. For example, detection of ZNF384 and its fusion partners is challenging because they both are located close to telomeres. Identification of DUX4 rearrangements are impossible due to the small size of the insertions as well as the repetitive nature of its sequence, also challenging its detection by WGS. TCF3::HLF can be cryptic, thus RNA sequencing may be the best technique for its detection [39]. Optical genome mapping (OGM) has recently been introduced as an all-in-one high-resolution cytogenetic technique that is able to detect balanced and unbalanced translocations, the full range of copy number variations from a few kilobases to the chromosome level (aneuploidies), as well as genomic insertions and inversions [40]. However, it is unable to detect single nucleotide variants. High specificity and sensitivity of the chosen method(s) are important, as low tumour cell counts may further compromise detection.
Immunophenotyping
Immunophenotype characterization by flow cytometry is an essential tool for ALL/LBL diagnostics. WHO-HAEM5 continues to subclassify B-ALL/LBL and T-ALL/LBL by immunophenotype and no changes to flow cytometry criteria have been made. B-ALL/LBL typically expresses CD10, CD19, CD22, TdT, CD34, HLA-DR and CD45 (normal, diminished or negative) antigens, and is negative for surface immunoglobulin. Some cases have a more differentiated immunophenotype with slightly increased CD45 and diminished CD34 intensity, while expressing cytoplasmic immunoglobulin heavy chains (cµ). Interestingly, a correlation between phenotypic features and genetic subtypes has been observed. B-ALL/LBL with ETV6::RUNX1 shows intense expression of CD10 combined with decreased CD9 and CD20 expression [41], B-ALL/LBL associated with KMT2A rearrangements lacks expression of CD10 and CD24, but shows co-expression of myeloid markers CD15 and NG2 [42]. Identification of these phenotypic features by flow cytometry can provide the first clue to the presence of these significant genetic abnormalities.
Typically, blast cells in T-ALL/LBL express the following set of markers: CD45, CD7, cytoplasmic (cy) CD3, and nuclear TdT [36]. Additionally, variable expression of T-cell associated surface membrane antigens is seen: CD1a, CD2, CD3, CD4, CD5, CD8, CD99, TCRγδ, TCRαβ, dependent on the maturation stage of the blasts [36]. Although not included in the WHO-HAEM5, most flow cytometry laboratories routinely define T-ALL subtypes according to the European Group on Immunological Classification of Leukemia (EGIL): EGIL I–IV subtypes (Pro-T-ALL, Pre-T-ALL, Cortical T-ALL, Mature T-ALL) (Table 2) [37]. EGIL criteria provide additional information on the maturation stage of the lymphoblasts. In WHO-HAEM5, all of these subtypes fall into one entity – T-lymphoblastic leukaemia/lymphoma, NOS. The other entity – Early T-precursor lymphoblastic leukaemia/lymphoma (ETP-ALL/LBL) – is recognized by cyCD3+, CD7+, CD8-, CD1a-, CD5lo (less than 75 % of blasts positive), and co-expression of one or more stem cell or myeloid markers (in ≥ 25 % of blasts) including CD13, CD33, CD34, CD117 or HLA-DR [37]. The major challenge in the identification of this entity by flow cytometry is the availability of a broad panel of monoclonal antibodies relevant to all hematopoietic cell lineages to efficiently distinguish ETP-ALL/LBL from acute undifferentiated leukemia and acute leukemia with T / myeloid phenotype. ETP-ALL/LBL might also be identified by RNA-seq, but for the routine diagnostic laboratory setting, flow cytometry seems more feasible, despite its limitations.
Immunophenotypic criteria for the classification of T-cell lymphoblastic leukaemia/lymphoma (T-ALL/LBL) according to the European Group on Immunological Classification of Leukemia (EGIL)
|
|
Immunophenotypic markers |
|||||
|
Subtype |
cyCD3 |
CD7 |
CD2 |
CD5 |
CD1a |
smCD3 |
|
Pro-T-ALL |
+ |
+ |
– |
– |
– |
– |
|
Pre-T-ALL |
+ |
+ |
+/- |
+ |
– |
– |
|
Cortical T-ALL |
+ |
+ |
+/- |
+ |
+ |
+/- |
|
Mature T-ALL |
+ |
+ |
+/- |
+ |
– |
+ |
Measurable residual disease (MRD) in ALL
MRD, the detection of low levels of residual leukemic cells, is important in risk stratification as a strong predictor of survival [4]. Several methods are used to quantify the level of MRD, including flow cytometry, RT-PCR or Next Generation Sequencing, each with unique advantages and disadvantages with respect to specificity, sensitivity, applicability and reproducibility [43]. They rely on the identification of a blast population specific target (e. g. immunoglobulin heavy chain (IGH)/T cell receptor (TCR) gene rearrangement, fusion transcript). The EuroFlow Consortium, the European Study Group on MRD detection in ALL (EuroMRD) and the I-BFM-FLOW-Network are dedicated to the standardization of protocols to assure accurate and consistent MRD detection for clinical application across different laboratories [44–46].
Essential and desirable diagnostic criteria:
In B-ALL, flow cytomorphology is essential to quantify more than 20 % of B-lymphoblasts with B-cell lineage markers. In B-LBL, blasts must show a B-cell immunophenotype, markers of immaturity and be immunoglobulin negative. The identification of specific recurrent genetic abnormalities is essential in B-ALL/LBL diagnosis.
An immunophenotypic profile associated with specific genetic alterations is desirable in both disease subtypes. For T-ALL, flow cytomorphology is essential for the detection of immature T-cells. Their presence outside of the thymus – in peripheral blood, bone marrow or other tissues – is strongly indicative of a precursor T-cell neoplasm. It is desirable to identify maturation stage of the blasts by flow cytomorphology and to discriminate between T-lymphoblastic leukaemia/lymphoma, NOS vs. Early T-precursor lymphoblastic leukaemia/lymphoma.
Conclusion
In the new WHO-HAEM5, ALL and lymphoblastic lymphoma are taken together as lymphoblastic disease of B- and T-precursor cells. In B-ALL/LBL, new genetic subgroups have been defined, while in T-ALL/LBL, none have been identified, thus diagnosis is based on immunophenotyping and histology. For the first time, essential and desirable diagnostic criteria have been defined for ALL/LBL. The development and integration of many new genetic methods into routine diagnostics have greatly impacted the new classification. In the future, such new genetic methods will significantly impact further refinement of classification.
Affiliations
1Department of Human Genetics, Hannover Medical School, Hannover, Germany
2Institute of Human Genetics, Department of Molecular and Clinical Genetics, Poznan, Poland
3Biosciences Institute, Newcastle University Centre for Cancer, Newcastle upon Tyne, UK
4Leukaemia Research Cytogenetics Group, Translational and Clinical Research Institute, Newcastle University Centre for Cancer, Newcastle upon Tyne, UK
5Amedes genetics, MVZ wagnerstibbe für Laboratoriumsmedizin, Hämostaseologie, Humangenetik und Mikrobiologie, Hannover, Germany
About the authors

Prof. Dr. rer. nat. Doris Steinemann

Małgorzata Dawidowska, Ph.D, DSc

Dr. Lisa J Russell, Ph.D

Prof. Christine J. Harrison, Ph.D

Prof. Dr. med. Gudrun Göhring
Acknowledgments: Not applicable
Research funding: None declared
Author contributions: All authors have accepted responsibility for the entire content of this manuscript and approved its submission
Competing interests: Authors state no conflicts of interest.
Informed consent: Not applicable
Ethical approval: Not applicable
References
[1] WHO. WHO Classification of Tumours Editorial Board. Haematolymphoid tumours [Internet; beta version ahead of print]. WHO classification of tumours series. 2022. 5th ed.; vol. 11.Search in Google Scholar
[2] Maitra, A., et al., Precursor B-cell lymphoblastic lymphoma. A study of nine cases lacking blood and bone marrow involvement and review of the literature. Am J Clin Pathol, 2001. 115(6): p. 868–75.10.1309/Q5GV-3K00-WAC6-BBUBSearch in Google Scholar PubMed
[3] Moricke, A., et al., Dexamethasone vs prednisone in induction treatment of pediatric ALL: results of the randomized trial AIEOP-BFM ALL 2000. Blood, 2016. 127(17): p. 2101–12.10.1182/blood-2015-09-670729Search in Google Scholar PubMed
[4] Schrappe, M., et al., Late MRD response determines relapse risk overall and in subsets of childhood T-cell ALL: results of the AIEOP-BFM-ALL 2000 study. Blood, 2011. 118(8): p. 2077–2084.10.1182/blood-2011-03-338707Search in Google Scholar PubMed
[5] Hunger, S. P., et al., Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children’s oncology group. J Clin Oncol, 2012. 30(14): p. 1663–9.10.1200/JCO.2011.37.8018Search in Google Scholar PubMed PubMed Central
[6] Angiolillo, A. L., et al., Excellent Outcomes With Reduced Frequency of Vincristine and Dexamethasone Pulses in Standard-Risk B-Lymphoblastic Leukemia: Results From Children’s Oncology Group AALL0932. J Clin Oncol, 2021. 39(13): p. 1437–1447.10.1200/JCO.20.00494Search in Google Scholar PubMed PubMed Central
[7] Iacobucci, I. and C. G. Mullighan, Genetic Basis of Acute Lymphoblastic Leukemia. J Clin Oncol, 2017. 35(9): p. 975–983.10.1200/JCO.2016.70.7836Search in Google Scholar PubMed PubMed Central
[8] Borowitz, M. J., et al., Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children’s Oncology Group study. Blood, 2008. 111(12): p. 5477–85.10.1182/blood-2008-01-132837Search in Google Scholar PubMed PubMed Central
[9] Moorman, A. V., et al., Prognostic impact of chromosomal abnormalities and copy number alterations in adult B-cell precursor acute lymphoblastic leukaemia: a UKALL14 study. Leukemia, 2022. 36(3): p. 625–636.10.1038/s41375-021-01448-2Search in Google Scholar PubMed PubMed Central
[10] O’Connor, D., et al., Genotype-Specific Minimal Residual Disease Interpretation Improves Stratification in Pediatric Acute Lymphoblastic Leukemia. J Clin Oncol, 2018. 36(1): p. 34–43.10.1200/JCO.2017.74.0449Search in Google Scholar PubMed PubMed Central
[11] Paulsson, K. and B. Johansson, High hyperdiploid childhood acute lymphoblastic leukemia. Genes Chromosomes Cancer, 2009. 48(8): p. 637–60.10.1002/gcc.20671Search in Google Scholar PubMed
[12] Gao, Q., et al., The genomic landscape of acute lymphoblastic leukemia with intrachromosomal amplification of chromosome 21. Blood, 2023. 142(8): p. 711–723.10.1182/blood.2022019094Search in Google Scholar PubMed PubMed Central
[13] Moorman, A. V., et al., Risk-directed treatment intensification significantly reduces the risk of relapse among children and adolescents with acute lymphoblastic leukemia and intrachromosomal amplification of chromosome 21: a comparison of the MRC ALL97/99 and UKALL2003 trials. J Clin Oncol, 2013. 31(27): p. 3389–96.10.1200/JCO.2013.48.9377Search in Google Scholar PubMed
[14] Meyer, C., et al., The KMT2A recombinome of acute leukemias in 2023. Leukemia, 2023. 37(5): p. 988–1005.10.1038/s41375-023-01877-1Search in Google Scholar PubMed PubMed Central
[15] Fischer, U., et al., Genomics and drug profiling of fatal TCF3-HLF-positive acute lymphoblastic leukemia identifies recurrent mutation patterns and therapeutic options. Nat Genet, 2015. 47(9): p. 1020–1029.10.1038/ng.3362Search in Google Scholar PubMed PubMed Central
[16] Forestier, E., et al., Outcome of ETV6/RUNX1-positive childhood acute lymphoblastic leukaemia in the NOPHO-ALL–1992 protocol:: frequent late relapses but good overall survival. British Journal of Haematology, 2008. 140(6): p. 665–672.10.1111/j.1365-2141.2008.06980.xSearch in Google Scholar PubMed
[17] Bhojwani, D., et al., ETV6-RUNX1-positive childhood acute lymphoblastic leukemia: improved outcome with contemporary therapy. Leukemia, 2012. 26(2): p. 265–270.10.1038/leu.2011.227Search in Google Scholar PubMed PubMed Central
[18] Felice, M. S., et al., Prognostic impact of t(1;19) / TCF3-PBX1 in childhood acute lymphoblastic leukemia in the context of Berlin-Frankfurt-Munster-based protocols. Leuk Lymphoma, 2011. 52(7): p. 1215–21.10.3109/10428194.2011.565436Search in Google Scholar PubMed
[19] Fournier, B., et al., B-ALL With t(5;14)(q31;q32); IGH-IL3 Rearrangement and Eosinophilia: A Comprehensive Analysis of a Peculiar IGH-Rearranged B-ALL. Front Oncol, 2019. 9: p. 1374.10.3389/fonc.2019.01374Search in Google Scholar PubMed PubMed Central
[20] Bomken, S., et al., Cutaneous B-lymphoblastic lymphoma with IL3/IgH translocation presenting with hypereosinophilia and acute endocarditis. Pediatr Blood Cancer, 2015. 62(6): p. 1055–7.10.1002/pbc.25318Search in Google Scholar PubMed
[21] Lilljebjorn, H., et al., Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Nat Commun, 2016. 7: p. 11790.10.1038/ncomms11790Search in Google Scholar PubMed PubMed Central
[22] Den Boer, M. L., et al., A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol, 2009. 10(2): p. 125–34.10.1016/S1470-2045(08)70339-5Search in Google Scholar PubMed PubMed Central
[23] Paietta, E., et al., Molecular classification improves risk assessment in adult BCR-ABL1-negative B-ALL. Blood, 2021. 138(11): p. 948–958.10.1182/blood.2020010144Search in Google Scholar PubMed PubMed Central
[24] Zhang, J., et al., Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet, 2016. 48(12): p. 1481–1489.10.1038/ng.3691Search in Google Scholar PubMed PubMed Central
[25] Zaliova, M., et al., ERG deletion is associated with CD2 and attenuates the negative impact of deletion in childhood acute lymphoblastic leukemia. Leukemia, 2014. 28(1): p. 182–185.10.1038/leu.2013.282Search in Google Scholar PubMed
[26] Schinnerl, D., et al., CD371 cell surface expression: a unique feature of DUX4-rearranged acute lymphoblastic leukemia. Haematologica, 2019. 104(8): p. e352-e355.10.3324/haematol.2018.214353Search in Google Scholar PubMed PubMed Central
[27] Jeha, S., et al., Clinical significance of novel subtypes of acute lymphoblastic leukemia in the context of minimal residual disease-directed therapy. Blood Cancer Discov, 2021. 2(4): p. 326–337.10.1158/2643-3230.BCD-20-0229Search in Google Scholar PubMed PubMed Central
[28] Ohki, K., et al., Clinical characteristics and outcomes of B-cell precursor ALL with MEF2D rearrangements: a retrospective study by the Ponte di Legno Childhood ALL Working Group. Leukemia, 2023. 37(1): p. 212–216.10.1038/s41375-022-01737-4Search in Google Scholar PubMed PubMed Central
[29] Zhu, L., et al., ZNF384-Related Fusion Genes in Acute Lymphoblastic Leukemia. Cancer Control, 2023. 30: p. 10732748231182787.10.1177/10732748231182787Search in Google Scholar PubMed PubMed Central
[30] Boer, J. M., et al., Favorable outcome of NUTM1-rearranged infant and pediatric B cell precursor acute lymphoblastic leukemia in a collaborative international study. Leukemia, 2021. 35(10): p. 2978–2982.10.1038/s41375-021-01333-ySearch in Google Scholar PubMed PubMed Central
[31] Bomken, S., et al., Molecular characterization and clinical outcome of B-cell precursor acute lymphoblastic leukemia with IG-MYC rearrangement. Haematologica, 2023. 108(3): p. 717–731.10.3324/haematol.2021.280557Search in Google Scholar PubMed PubMed Central
[32] Jia, Z. and Z. Gu, PAX5 alterations in B-cell acute lymphoblastic leukemia. Front Oncol, 2022. 12: p. 1023606.10.3389/fonc.2022.1023606Search in Google Scholar PubMed PubMed Central
[33] Jung, M., et al., Frequency and prognostic impact of PAX5 p.P80R in pediatric acute lymphoblastic leukemia patients treated on an AIEOP-BFM acute lymphoblastic leukemia protocol. Genes Chromosomes Cancer, 2020. 59(11): p. 667–671.10.1002/gcc.22882Search in Google Scholar PubMed PubMed Central
[34] Inaba, H. and C. G. Mullighan, Pediatric acute lymphoblastic leukemia. Haematologica, 2020. 105(11): p. 2524–2539.10.3324/haematol.2020.247031Search in Google Scholar PubMed PubMed Central
[35] Seki, M., et al., Recurrent SPI1 (PU.1) fusions in high-risk pediatric T cell acute lymphoblastic leukemia. Nat Genet, 2017. 49(8): p. 1274–1281.10.1038/ng.3900Search in Google Scholar PubMed
[36] van Dongen, J. J., et al., EuroFlow antibody panels for standardized n-dimensional flow cytometric immunophenotyping of normal, reactive and malignant leukocytes. Leukemia, 2012. 26(9): p. 1908–75.10.1038/leu.2012.120Search in Google Scholar PubMed PubMed Central
[37] Sin, C. F. and P. M. Man, Early T-Cell Precursor Acute Lymphoblastic Leukemia: Diagnosis, Updates in Molecular Pathogenesis, Management, and Novel Therapies. Front Oncol, 2021. 11: p. 750789.10.3389/fonc.2021.750789Search in Google Scholar PubMed PubMed Central
[38] Harrison CJ, M. A., Schwab C, Iacobucci I, Mullighan C., Cytogenetics and Molecular Genetics In: Vora A, editor. Childhood Acute Lymphoblastic Leukemia, in Springer International Publishing. 2017. p. 61–98.10.1007/978-3-319-39708-5_4Search in Google Scholar
[39] Salim, M., et al., Cryptic TCF3 fusions in childhood leukemia: Detection by RNA sequencing. Genes Chromosomes Cancer, 2022. 61(1): p. 22–26.10.1002/gcc.22998Search in Google Scholar PubMed
[40] Lühmann, J. L. Z., M.; Hofmann, W.; Bergmann, A. K.; Möricke, A.; Cario, G.; Schrappe, M.; Schlegelberger, B.; Stanulla, M.; Steinemann, D., Deciphering the molecular complexity of the IKZF1plus genomic profile using Optical Genome Mapping. Haematologica, 2023.10.3324/haematol.2023.284115Search in Google Scholar PubMed PubMed Central
[41] De Zen, L., et al., Quantitative multiparametric immunophenotyping in acute lymphoblastic leukemia: correlation with specific genotype. I. ETV6/AML1 ALLs identification. Leukemia, 2000. 14(7): p. 1225–31.10.1038/sj.leu.2401824Search in Google Scholar PubMed
[42] Schwartz, S., et al., Expression of the human homologue of rat NG2 in adult acute lymphoblastic leukemia: close association with MLL rearrangement and a CD10(-)/CD24(-)/CD65s(+)/CD15(+) B-cell phenotype. Leukemia, 2003. 17(8): p. 1589–95.10.1038/sj.leu.2402989Search in Google Scholar PubMed
[43] Contreras Yametti, G. P., et al., Minimal Residual Disease in Acute Lymphoblastic Leukemia: Current Practice and Future Directions. Cancers (Basel), 2021. 13(8).10.3390/cancers13081847Search in Google Scholar PubMed PubMed Central
[44] Bruggemann, M., N. Gokbuget, and M. Kneba, Acute lymphoblastic leukemia: monitoring minimal residual disease as a therapeutic principle. Semin Oncol, 2012. 39(1): p. 47–57.10.1053/j.seminoncol.2011.11.009Search in Google Scholar PubMed
[45] Theunissen, P., et al., Standardized flow cytometry for highly sensitive MRD measurements in B-cell acute lymphoblastic leukemia. Blood, 2017. 129(3): p. 347–357.10.1182/blood-2016-07-726307Search in Google Scholar PubMed PubMed Central
[46] Maurer-Granofszky, M., et al., An Extensive Quality Control and Quality Assurance (QC/QA) Program Significantly Improves Inter-Laboratory Concordance Rates of Flow-Cytometric Minimal Residual Disease Assessment in Acute Lymphoblastic Leukemia: An I-BFM-FLOW-Network Report. Cancers (Basel), 2021. 13(23).10.3390/cancers13236148Search in Google Scholar PubMed PubMed Central
© 2024 the author(s), published by Walter de Gruyter GmbH, Berlin/Boston
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Frontmatter
- MAIN TOPIC Genetic Alterations of Haematolymphoid Tumours
- Genetic alterations in the classification of tumors of the haematopoietic system: From guiding diagnosis to instructing treatment in leukaemias and lymphomas
- Overview on WHO-HAEM5 and the diagnostic relevance of genetic alterations for the classification
- Appraisal of current technologies for the study of genetic alterations in hematologic malignancies with a focus on chromosome analysis and structural variants
- Genetic studies in clonal haematopoiesis, myelodysplastic neoplasms and acute myeloid leukaemia – a practical guide to WHO-HAEM5
- Genetic alterations in myeloproliferative and myelodysplastic/myeloproliferative neoplasms – a practical guide to WHO-HAEM5
- Genetic alterations in lymphoblastic leukaemia / lymphoma – a practical guide to WHO HAEM5
- Genetic alterations in chronic lymphocytic leukemia and plasma cell neoplasms – a practical guide to WHO HAEM5
- Genetic alterations in mature B- and T-cell lymphomas – a practical guide to WHO-HAEM5
- BERICHTE AUS DER HUMANGENETIK
- Personalia
- Ein Geburtstagsgruß für Ingo Hansmann zum 80. Geburtstag
- GfH-Verbandsmitteilungen
- Einladung zum Syndromtag 2024 in Göttingen vom 04.–05. Oktober
- Jahresberichte 2023 aus den GfH-Kommissionen und GfH-Arbeitskreisen
- BVDH-Verbandsmitteilungen
- Die neue HGQN-Variantendatenbank
- Laborqualität sichern – molekular- und zytogenetische Ringversuche des BVDH e. V.
Articles in the same Issue
- Frontmatter
- MAIN TOPIC Genetic Alterations of Haematolymphoid Tumours
- Genetic alterations in the classification of tumors of the haematopoietic system: From guiding diagnosis to instructing treatment in leukaemias and lymphomas
- Overview on WHO-HAEM5 and the diagnostic relevance of genetic alterations for the classification
- Appraisal of current technologies for the study of genetic alterations in hematologic malignancies with a focus on chromosome analysis and structural variants
- Genetic studies in clonal haematopoiesis, myelodysplastic neoplasms and acute myeloid leukaemia – a practical guide to WHO-HAEM5
- Genetic alterations in myeloproliferative and myelodysplastic/myeloproliferative neoplasms – a practical guide to WHO-HAEM5
- Genetic alterations in lymphoblastic leukaemia / lymphoma – a practical guide to WHO HAEM5
- Genetic alterations in chronic lymphocytic leukemia and plasma cell neoplasms – a practical guide to WHO HAEM5
- Genetic alterations in mature B- and T-cell lymphomas – a practical guide to WHO-HAEM5
- BERICHTE AUS DER HUMANGENETIK
- Personalia
- Ein Geburtstagsgruß für Ingo Hansmann zum 80. Geburtstag
- GfH-Verbandsmitteilungen
- Einladung zum Syndromtag 2024 in Göttingen vom 04.–05. Oktober
- Jahresberichte 2023 aus den GfH-Kommissionen und GfH-Arbeitskreisen
- BVDH-Verbandsmitteilungen
- Die neue HGQN-Variantendatenbank
- Laborqualität sichern – molekular- und zytogenetische Ringversuche des BVDH e. V.