Clinical spectrum and management of imprinting disorders
-
Miriam Elbracht
 ,
Gerhard Binder
,
Gerhard Binder

Abstract
Imprinting disorders are exceptional within the group of monogenic syndromes. They are associated with molecular changes affecting imprinted regions and usually do not follow the rules of Mendelian inheritance. They account for a relevant proportion of congenital disorders, especially within the syndromal growth entities with endocrine, neurological, and skeletal characteristics. In patients with imprinting disorders and accelerated growth, significant tumor risks have to be considered. The number of known imprinting disorders increases with the identification of new regions in which parentally imprinted genes are located. Imprinting disorders are caused by genomic pathogenic variants affecting imprinted genes, as well as by aberrant imprinting marks (epimutations) in the patients themselves. Additionally, maternal effect mutations have recently been identified that trigger secondary epimutations in the offspring. These maternal effect mutations explain not only imprinting disorders in their children, but also recurrent reproductive failure in the families. This review aims to provide an overview of the recent findings in 13 well-known imprinting disorders relating to clinical diagnosis, management and counseling.
Introduction
Genomic imprinting is defined as the parent-specific expression of either the maternal or the paternal allele, and is regulated by different epigenetic processes (e. g., DNA methylation and histone modifications) (for review, see Prawitt and Haaf in this issue). Genomic imprinting has arisen in the evolution of higher mammals, and among different hypotheses about the evolutionary relevance of genomic imprinting, the “parental conflict hypothesis” has received greater attention [1]. It explains the functional inequality of maternal and paternal imprinted genes with the different “interests” of the parental genomes: Whereas the paternal interest lies in “promoting offspring,” it is essential for the mother to conserve resources for herself and for further offspring. Accordingly, many paternally expressed genes are growth-promoting, while maternally expressed genes are more growth-limiting. Disturbances of these imprinting patterns therefore often result in aberrant growth, and it is not astonishing that the majority of the so-called imprinting disorders are associated either with growth retardation or overgrowth (Table 1).
Clinical diagnostic criteria of the most prominent imprinting disorders. In case of SRS, BWS, and iPPSDs, clinical scoring systems have been suggested [4], [5], [37] which might help to decide on the molecular diagnostic testing procedure. (PTH, parathyroid hormone; TSH, thyroid-stimulating hormone).
| Imprinting disorder OMIM# | Prevalence | Chromosome | Molecular defects (frequency) | Reccurrence risk | MLID | Main clinical features |
| Transient neonatal diabetes mellitus (TNDM)601410 | 1/300,000 | 6q24 | upd(6)pat (41 %) | low | IUGR, transient diabetes mellitus, hyperglycemia without ketoacidosis, macroglossia, abdominal wall defects | |
| Paternal duplications (33 %): familial structural variants possible | familial structural variants possible | |||||
| PLAGL1:alt-TSS-DMR, LOM (26 %) | in case of MLID: autosomal recessive ZFP57 mutations | 50 % | ||||
| Silver–Russell syndrome (SRS)180860 | 1/75,000–1/100,000 | 7 | upd(7)mat (7–10 %) | only one familial translocation reported | 1 case | IUGR, PNGR, relative macrocephaly at birth, body asymmetry, prominent forehead, feeding difficulties |
| 11p15.15 | upd(11p15)mat (1 case) | not increased | ||||
| dup(11p15)mat (<1 %) | familial structural variants possible | |||||
| H19/IGF2:IG:DMR, LOM (40 %) | in case of MLID: maternal effect variants possible(familial) CNVs, SNVs in cis causing LOM reported in rare cases | 10 % | ||||
| CDKN1C, IGF2, HMGA2, PLAG1 point mutations | autosomal-dominant inheritance, with parental imprinting | |||||
| Birk–Barel syndrome 612292 | unknown | 8q24.3 | KCNK9 point mutations | autosomal-dominant inheritance with paternal imprinting | Intellectual disability, hypotonia, dysmorphism | |
| Beckwith–Wiedemann syndrome (BWS)130650 | 1/15,000 | 11p15.5 | upd(11p15)pat (20 %) | no | Macroglossia, exomphalos, lateralized overgrowth, Wilms tumor or nephroblastomatosis, hyperinsulinism, adrenal cortex cytomegaly, placental mesenchymal dysplasia, pancreatic adenomatosis | |
| dup(11p15)pat (2–4 %) | familial structural variants possible | |||||
| H19/IGF2:IG-DMR, GOM (4 %) | OCT4 binding site SNVs in 10 %, might be heritable | |||||
| KCNQ1OT1:TSS-DMR, LOM (50 %) | maternal effect variants in case of MLID | 25 % | ||||
| CDKN1C point mutations (5 % sporadic; familial 25–50 %) | autosomal-dominant inheritance with paternal imprinting | |||||
| Temple syndrome (TS14)616222 | unknown | 14q32 | upd(14)mat (70 %) | familial structural variants possible | IUGR, PNGR, neonatal hypotonia, feeding difficulties in infancy, truncal obesity, scoliosis, precocious puberty, small feet and hands | |
| del(14q32)pat (10 %) | familial structural variants possible | |||||
| MEG3/DLK1:IG-DMR-LOM (20 %) | low | single cases | ||||
| Kagami–Ogata syndrome (KOS14)608149 | unknown | 14q32 | upd(14)pat (70 %) | familial structural variants possible | IUGR, polyhydramnion, abdominal wall defects, bell-shaped thorax, coat-hanger ribs | |
| del(14q32)mat (20 %) | familial structural variants possible | |||||
| MEG3/DLK1:IG-DMR-GOM (10 %) | low | |||||
| (familial) Central precocious puberty (CPPB) | unknown | 14q32 | DLK1 point mutations | autosomal-dominant inheritance with paternal imprinting | Central precocious puberty | |
| Prader–Willi syndrome (PWS)176270 | 1/25,000–1/10,000 | 15q11q13 | del(15q11q13)pat (70–75 %) | familial structural variants possible | PNGR, intellectual disability, neonatal hypotonia, hypogenitalism, hypopigmentation, obesity, hyperphagia | |
| upd(15)mat (25–30 %) | familial structural variants possible | |||||
| SNURF:TSS-DMR, GOM (1 %) | 0–15 % due to an IC deletion | 1 case | ||||
| Angelman syndrome (AS)105830 | 1/20,000–1/12,000 | 15q11+C9q13 | del(15q11q13)mat (75 %) | familial structural variants possible | Severe intellectual disability, microcephaly, no speech, unmotivated laughing, ataxia, seizures, scoliosis | |
| upd(15)pat (1–2 %) | familial structural variants possible | |||||
| SNURF:TSS-DMR, LOM (1 %) | 0–15 % due to an IC deletion | |||||
| UBE3A point mutations (10 %) | autosomal-dominant inheritance with paternal imprinting | |||||
| Central precocious puberty 2 (CPPB2) 615356 | Unknown | 15q11.2 | MKRN3 point mutations | autosomal-dominant inheritance with paternal imprinting | Early activation of the hypothalamic–pituitary–gonadal axis resulting in gonadotropin-dependent precocious puberty | |
| Schaaf–Yang syndrome (SYS) 615547 | Unknown | 15q11.2 | MAGEL2 point mutations | autosomal-dominant inheritance with paternal imprinting | Delayed psychomotor development, intellectual disability, hypotonia | |
| Pseudohypoparathyroidism 1B (PHP1B) 603233 | Unknown | 20q13 | del(20q13)mat (single cases) | familial structural variants possible | Resistance to PTH and other hormones, albright hereditary osteodystrophy, subcutaneous ossifications, feeding behavior anomalies, abnormal growth patterns | |
| GNAS DMRs: LOM (>60 %) | familial (structural) variants possible,maternal effect variants in case of MLID | 12.5 % | ||||
| upd(20)pat (2–20 %) | familial structural variants possible | |||||
| GNAS point mutations | autosomal-dominant inheritance with paternal imprinting | |||||
| Mulchandani–Bhoj–Conlin syndrome (MBCS)617352 | Unknown | 20 | upd(20)mat | familial structural variants possible | IUGR, PNGR, feeding difficulties |
Imprinting disorders
Imprinting disorders belong to the group of rare diseases with a prevalence of less than 1:10,000 in newborns. Though some of them exhibit different and even opposite clinical features, they share the same types of molecular alterations affecting imprinted gene regions. So far, 13 syndromic clinical entities have been deeply defined (for overview, see Table 1) [2], [3]. In fact, the majority of the clinical findings are rather unspecific and manifest as impaired growth (pre- and/or postnatal growth retardation or acceleration), metabolic and endocrine disorders, and failure to thrive due to nutritional problems or abnormal eating habits. In addition, some imprinting disorders are accompanied by developmental delay, intellectual disability, and neurological symptoms. In imprinting disorders with accelerated growth, there is often an increased risk of tumors, with immediate consequences for the medical care of the patients.
![Figure 1 Pedigree of a family with a maternal effect NLRP7 variant (NM_001127255.1:c.2010_2011del, p.(Phe671Glnfs*18)) and reproductive histories of miscarriages affected by MLID (from [44], Oxford University Press). It should be noted that miscarriages only occur when the woman carries the variant, whereas paternal transmission is not associated with reproductive failure. It is assumed that the reproductive failure might be avoided by egg donation.](/document/doi/10.1515/medgen-2020-2044/asset/graphic/j_medgen-2020-2044_fig_001.jpg)
Pedigree of a family with a maternal effect NLRP7 variant (NM_001127255.1:c.2010_2011del, p.(Phe671Glnfs*18)) and reproductive histories of miscarriages affected by MLID (from [44], Oxford University Press). It should be noted that miscarriages only occur when the woman carries the variant, whereas paternal transmission is not associated with reproductive failure. It is assumed that the reproductive failure might be avoided by egg donation.
The clinical diagnosis of an imprinting disorder can be complicated by the variable expressivity of symptoms. In addition, some key features are transient or not obvious in adulthood (e. g., severe muscular hypotonia in neonates [“floppy infant”] with Prader–Willi syndrome [PWS] and dysmorphic signs in Silver–Russell syndrome [SRS]). As a result, an unknown number of patients with imprinting disorders remain either undiagnosed or misdiagnosed. To improve the clinical diagnoses, international consensus guidelines have been suggested for some imprinting disorders, including scoring systems to define a threshold for the clinical diagnosis and recommendations for the medical management (Tables 1, 2) [4], [5], [6], [7], [8], [9].
Multilocus imprinting disturbance (MLID)
A considerable number of individuals with imprinting disorders exhibit altered DNA methylation at additional imprinted loci compared with the disease-specific ones. This constitution is referred to as multilocus imprinting disturbance (MLID) (for review, see [10]; see also Prawitt and Haaf in this issue). Up to now, the majority of MLID carriers exhibit symptoms specific for one of the known imprinting disorders (i. e., Beckwith–Wiedemann syndrome [BWS] and SRS), and only single patients with overlapping phenotypes have been reported. However, it is conceivable that MLID patients currently escape detection as they do not show a special MLID phenotype and are therefore not tested. Furthermore, MLID often occurs as mosaicism, with a broad range of aberrant methylation patterns in different tissues (e. g., [11]). This mosaicism could escape detection and lead to a significant number of patients with apparent “isolated” loss of methylation (LOM) at a disease-specific locus in routinely tested lymphocyte DNA of patients who are effectively carriers of a MLID. As a result, the true prevalence of MLID is currently unknown.
MLID was first identified in patients with epimutations diagnosed as transient neonatal diabetes mellitus (TNDM) [12], and then in other classical imprinting syndromes (Table 1). The molecular cause of MLID is largely unknown, but the contribution of pathogenic variants in factors mediating the proper methylation at imprinted loci becomes obvious (for review, see [13] and Prawitt and Haaf in this issue). In some patients, factors encoded by the fetal genome harboring pathogenic variants are the cause of “inherited” MLID (i. e., autosomal recessive variants in ZFP57).
Additionally, these factors also comprise members of the subcortical maternal complex (SCMC) in the oocyte and early embryo. They are not expressed by the embryonal genome, but from the maternal genome and are hence named maternal effect genes (e. g., NLRP2, NLRP5, NLRP7, PADI6) [10]. The identification of MLID in context of maternal effect variants is currently conducted mainly in research context, but first studies show a high clinical relevance (Fig. 1). The involvement of single additional imprinted loci in case of MLID might be compatible with live-born children with an imprinting disorder, but unbalanced imprinting constitutions of a greater extent might be incompatible with life, and therefore lead to an increased rate of miscarriages. The most severe phenotypes of an imprinting disorder are familial or recurrent hydatidiform moles, defined as an aberrant human pregnancy with a placental overgrowth but severely abnormal or absent embryonic development.
Meanwhile, more than 110 pathogenic maternal effect variants affecting components of the SCMC have been identified in women suffering recurrent hydatidiform mole [14].
In summary, the pathogenic impact of genetic factors involved in the life cycle of imprinting is obvious. Therefore, the implementation of routine MLID testing in specific molecular imprinting disorders due to epimutations (TNDM, SRS, BWS, PHPIb; see Table 1) and a subsequent (next-generation sequencing-based) analysis of SCMC genes has to be discussed in the near future. And the other way round, MLID and SCMC gene testing in families with recurrent miscarriages might be discussed as well. In any case, the presence of maternal effect mutations should be considered in families with imprinting disorders or also unclear growth disorders and simultaneously frequent miscarriages.
Clinical diagnosis and clinical management – a brief overview
In the following, the most frequent imprinting disorders are briefly summarized in the order of their chromosomal localization, with regard to the characteristic symptoms and the common clinical management to allow a quick overview (see Table 1, which also summarizes the underlying molecular defects and their frequencies). The symptoms of imprinting disorders often overlap; therefore, a specific clinical diagnosis is not always possible. Rather, in the context of an intended molecular diagnostic confirmation of imprinting disorders, a broader molecular diagnostic approach is pursued with the simultaneous analysis of different imprinted loci. This approach is also the best way to identify patients with MLID.
Transient neonatal diabetes mellitus (TNDM; 6q24)
Clinical characteristics and diagnosis
The cardinal features of 6q24-associated TNDM comprise severe intrauterine growth retardation, hyperglycemia beginning in the neonatal period and often resolving within the first 18 months, dehydration, and absence of ketoacidosis (see also GeneReviews: NBK1534). Macroglossia and umbilical hernia can occur, in particular in cases with MLID. In the latter group, marked hypotonia, congenital heart disease, deafness, neurologic features including epilepsy, and renal malformations have been documented. Although insulin is usually required initially, the need for insulin gradually declines over time. Diabetes mellitus may recur in adolescence or later in adulthood; in particular women are at risk for relapse during pregnancy as gestational diabetes.
Management: treatment of manifestations, prevention of secondary complications
Rehydration and insulin are required at the time of diagnosis [15]. For further treatment and prevention we refer to the respective GeneReviews suggestions. For a personalized management, a rapid and comprehensive molecular characterization is required, and after exclusion of the 6q24 alterations (Table 1), multigene panel testing targeting the differential diagnosis genes of hyperinsulinism (i. e., KCNJ11, ABCC8, INS, GCK, and PDX1) should be considered.
Silver–Russell syndrome (SRS; 7p12, 7q32, 11p15.5)
Clinical characteristics and diagnosis
SRS is a clinically and genetically heterogeneous imprinting disorder characterized by moderate to severe intrauterine growth restriction, relative macrocephaly, failure to thrive, short stature, low muscle and fat mass (low BMI), body asymmetry, and a protruding forehead [4] (see also GeneReviews: NBK1324). Many other less frequent clinical characteristics have been described. The phenotype can be subtle. SRS is a clinical diagnosis: the recommended clinical scoring system (Table 2, Fig. 2) should lead the decision to perform genetic analysis, which is positive in approximately 60–70 % of the affected persons. A negative genetic analysis does not exclude SRS (see below). Mental development is normal in most of the children, while motor milestones may be delayed in infancy because of the large head and muscle weakness. Speech delay is apparent in a specific subgroup (upd[7]mat; see below). Gastro-esophageal reflux may occur in infancy and aggravate the feeding difficulties. Fasting tolerance is weak. There is no catch-up growth after birth. Untreated adult height is approximately 3 SD below target height [16]. Timing of puberty is early-normal. Premature adrenarche may occur, especially in children treated with rhGH binder [17]. Untreated body asymmetry may cause early arthrosis in adulthood. The small jaw frequently results in malposition of teeth.
Overview on the currently known imprinting disorders, their major molecular findings, and the main clinical features. Disorders are listed according to their chromosomal localization.
| Clinical diagnosis of Silver–Russell syndrome (SRS) (Netchine–Harbison score [NHS]) | |
| Clinical diagnosis is considered if patient scores at least four out of six from the following criteria. | |
| Clinical criterion | Definition |
| Small for gestational age (SGA) (birth weight and/or birth length) | ≤ −2 SDs for gestational age |
| Postnatal growth failure | Height at 24±1 months ≤ −2 SDs or height ≤ −2 SDs from mid-parental target height |
| Relative macrocephaly at birth | Head circumference at birth ≥ 1.5 SDs above average birth weight and/or length |
| Protruding forehead* (*equivalent to “prominent forehead”) | Forehead projecting beyond the facial plane on a side view as a toddler (1–3 years) |
| Body asymmetry | Leg length discrepancy (LLD) ≥ 0.5 cm or arm asymmetry or LLD < 0.5 cm with at least two other asymmetrical body parts (one not the face) |
| Feeding difficulties and/or low body mass index (BMI) | BMI ≤ −2 SDs at 24 months OR current use of a feeding tube or cyproheptadine for appetite stimulation |
| Clinical features of Beckwith–Wiedemann spectrum (BWSp) | |
| For a clinical diagnosis of classical BWS a score of ≥ 4 is required. Patients with a score of ≥ 2 merit genetic testing for investigation and diagnosis of BWS. | |
| Cardinal features (2 points per feature) | Suggestive features (1 point per feature) |
| Macroglossia | Birth weight > 2 SDs above the mean |
| Exomphalos | Facial nevus simplex |
| Lateralized overgrowth | Polyhydramnios and/or placentomegaly |
| Multifocal and/or bilateral Wilms tumor or nephroblastomatosis | Ear creases and/or pits |
| Hyperinsulinism (lasting beyond 1 week and requiring escalated treatment) | Transient hypoglycemia (lasting less than 1 week) |
| Pathology findings: adrenal cortex cytomegaly, placental mesenchymal dysplasia, or pancreatic adenomatosis | Typical BWSp tumors (neuroblastoma, rhabdomyosarcoma, unilateral Wilms tumor, hepatoblastoma, adrenocortical carcinoma, or phaeochromocytoma) |
| Nephromegaly and/or hepatomegaly | |
| Umbilical hernia and/or diastasis recti | |
| Clinical features of inactivating PTH/PTHrP signaling disorders (iPPSD) | |
| For a clinical diagnosis of iPPSD, the presence of one major criteria (either 1 or 2) or the presence of major criterion 3 and at least two minor criteria is required. The three major criteria have minimum or no overlap with other conditions due to different mechanisms. | |
| Major criteria | |
| 1 | PTH resistance |
| 2 | Ectopic ossification |
| 3 | Brachydactyly type E |
| Minor criteria | |
| 1 | TSH resistance |
| 2 | Other hormonal resistances |
| 3 | Motor and cognitive retardation or impairment |
| 4 | Intrauterine and postnatal growth retardation |
| 5 | Obesity/overweight |
| 6 | Flat nasal bridge and/or maxillary hypoplasia and/or round face |
GOM, gain of methylationIUGR, intrauterine growth restrictionLOM, loss of methylationPNGR, postnatal growth restrictionPTH, parathyroid hormoneUPD, uniparental disomy
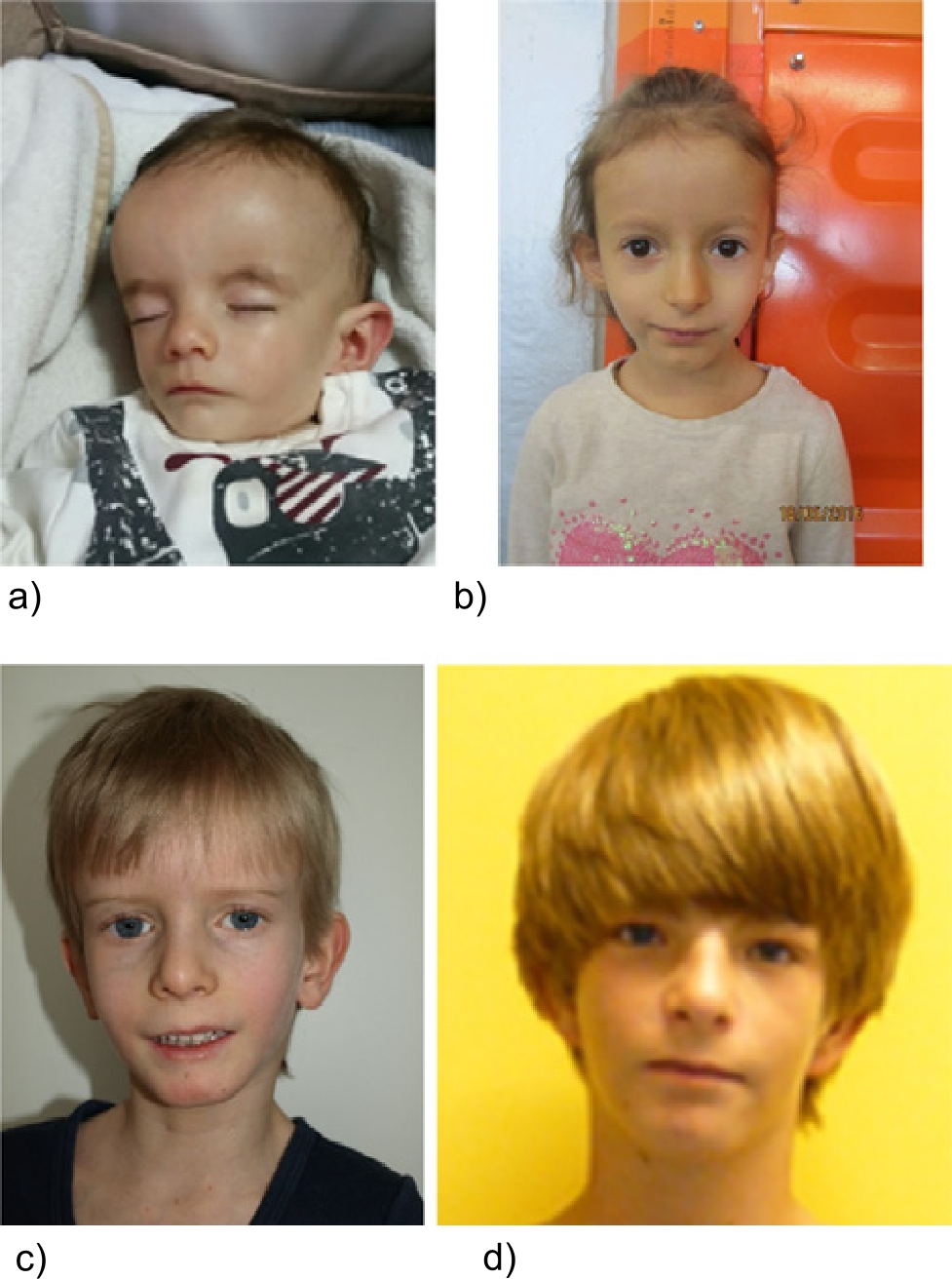
Patients referred for SRS testing and molecular diagnosis of IC2 LOM. Though the patients exhibit similar features, they illustrate the variable range of mosaicism as well as the possible occurrence of MLID. (a) One-year-old boy, IC2 LOM not detectable in blood, but in buccal swab DNA, NH-CSS: 5/5 criteria (by courtesy of V. Tasic). (b) Five-year-old girl, IC2 LOM in blood, NH-CSS: 3/3 criteria (only 3 criteria could be documented). (c) Eight-year-old boy, IC2 LOM in blood, NH-CSS: 6/6 criteria (by courtesy of C. Schröder). (d) Eighteen-year-old boy, MLID and IC2 LOM in blood, NH-CSS: 5/5 criteria.
Management: treatment of manifestations, prevention of secondary complications
The therapeutic approach is multidisciplinary and headed by a pediatric endocrinologist [4]. Treatment of feeding difficulties requires nutritionists, gastroenterologists, and speech therapists. The aim is an adequate nutritional status preserving normal growth for a child with SRS, adequate weight (always having in mind the constitutional low muscle and fat mass), vitality, and an adequate fasting tolerance. The parents should be educated in the avoidance, recognition, and treatment of hypoglycemia. Developmental delay of motor and speech skills should be diagnosed and treated by physiotherapy. Growth hormone treatment enables gain in muscle mass and strength (changing the body composition), enhances appetite, vitality. and the developmental progress, and promotes growth. Body asymmetry should be monitored by orthopedists and compensated if necessary. Teeth crowding should be treated by specialized dentists. Early puberty may compromise the achieved height gain after years of growth hormone treatment; pharmacological delay of central puberty by use of gonadotropin-releasing hormone analogues may preserve adult height potential [17]. Obesity should be avoided by early teaching of a healthy life style. Long-term follow-up data are missing and should be collected to optimize medical management in adulthood.
Birk–Barel syndrome (8q24)
Clinical characteristics and diagnosis
The main clinical features of Birk–Barel syndrome are moderate to severe intellectual disability, congenital central hypotonia, facial dysmorphism, and hyperactivity [18], [19]. Some of the common dysmorphic features are more prominent in childhood, and they comprise an elongated face and reduced facial movements. Eyebrows are flared, bushy, and arched upward, and ears are mildly protruding with a very prominent fold of the crux of the helix and a prominent antihelical fold. The nasal bridge is high and narrow with a broad nasal tip, and the philtrum is shortened. The maxillary and premaxillary regions are prominent with hypotonia of the mandible and micrognathia, leading to an open-mouthed appearance. The lips are thick, and a narrow, high-arched palate is reported.
Management: treatment of manifestations, prevention of secondary complications
In infancy, feeding difficulties might require tube feeding, followed by dysphagia of solid foods until puberty. For future therapeutic options, see also Horsthemke and Zechner in this issue.
Beckwith–Wiedemann syndrome (BWS; 11p15.5)
Clinical characteristics and diagnosis
According to recent international consensus guidelines [5], clinical and pathologic features of BWS have been grouped into two categories, macroglossia, exomphalos, lateralized overgrowth, multifocal and/or bilateral Wilms tumor or nephroblastomatosis, hyperinsulinism, adrenal cortex cytomegaly, and placental mesenchymal dysplasia or pancreatic adenomatosis being cardinal features (Table 2) (see also GeneReviews: NBK1394).
Genetic counseling and testing are recommended if an individual has at least two points in the scoring system, e. g., one of the cardinal features or at least two of the suggestive features. The newly defined term Beckwith–Wiedemann spectrum (BWSp) includes (a) patients with a clinical BWS diagnosis (at least four points when adding cardinal and suggestive features) independent of the presence of an (epi)genetic change at the BWS region, (b) patients with atypical BWS, defined by a clinical score of below 4 but with a proven (epi)genetic change at the BWS region, and (c) patients with isolated lateralized overgrowth with a proven (epi)genetic change at the BWS region.
Management: treatment of manifestations, prevention of secondary complications
Individuals with BWSp may face multiple clinical problems; therefore, a multidisciplinary approach is warranted. Relevant disciplines include, but are not limited to, neonatology, surgery, endocrinology, nephrology, neurology, cardiology, oral and maxillofacial surgery, pediatric orthopedics, and pediatric oncology. The following aspects are important. (1) Cancer surveillance with three monthly abdominal ultrasound examinations up to age 7 years. In children with the molecular diagnosis of IC2 LOM the cancer risk is lower and the recent guidelines do not recommend cancer surveillance in this group. (2) Annual measurement of body height and weight and lengths of both legs. In case of leg length difference, pediatric orthopedic evaluation is needed. (3) Monitoring of macroglossia and in case of secondary complications, surgery may be indicated. (4) Correction of abdominal wall defects. (5) Diagnosis and treatment of postnatal hypoglycemia. (6) Diagnosis and treatment of cardiac malformations. (7) Monitoring of neurological development. (8) Screening for renal complications. (8) Psychosocial support are further aims.
Temple syndrome (TS14; 14q32) (central precocious puberty [CPPB])
Clinical characteristics and diagnosis
Temple syndrome (TS14) is characterized by intrauterine and postnatal growth retardation, accompanied by hypotonia, feeding problems in early life with risk for obesity in later childhood and adolescents, early puberty, and recurrent otitis media [20], [21], [22]. As facial features, a broad forehead and a short nose with a wide nasal tip have been reported, and many patients have small hands and feet. In some patients (mild) intellectual delay has been described. In fact, the TS14 phenotype is nonspecific, and there is a broad clinical overlap with SRS and PWS. Therefore, many patients with molecular TS14 alterations are referred for SRS or PWS testing. Thus, TS14 testing should be considered in patients with negative SRS or PWS testing results (see Beygo et al. in this issue).
Recently, in patients of precocious puberty ([familial] central precocious puberty [CPPB]), pathogenic variants on the paternal allele have been identified of DLK1, a gene which is under the control of the TS14-associated MEG3:TSS-DMR [23].
Management: treatment of manifestations, prevention of secondary complications
Because of the clinical overlap with the more frequent imprinting disorders PWS and SRS, clinical care is symptomatic and oriented towards existing guidelines for these two disorders. It is essentially a multidisciplinary approach involving nutritional therapists, endocrinologists (growth hormone therapy, therapy of premature puberty), and orthopedic surgeons (scoliosis therapy). In case of developmental delay special education might be needed.
Kagami–Ogata syndrome (KOS14, 14q32)
Clinical characteristics and diagnosis
Kagami–Ogata syndrome (KOS14) is characterized by a unique constellation of features; in particular, the facial appearance with full cheeks and a protruding philtrum as well as the small bell-shaped thorax with a coat-hanger appearance of the ribs are prominent [24], [25]. Respiratory problems in the neonatal period can be life-threatening, but patients overcoming this vulnerable early phase often have a stable clinical course in the following childhood. Some clinical overlaps with BWS are obvious, i. e., abdominal wall defects, placentamegaly, and polyhydramnios. Moderate to severe developmental delay and/or intellectual disability are always present. In a small number of patients, hepatoblastomas have been identified.
Management: treatment of manifestations, prevention of secondary complications
Beside the possibly needed early intensive care intervention when severe pulmonary complications occur, an adequate support for developmental delay and periodical screening for hepatoblastoma in the affected patients are required.
Angelman syndrome (AS; 15q11q13)
Clinical characteristics and diagnosis
Major clinical signs of Angelman syndrome (AS) comprise severe developmental delay, unique behavioral features with frequent laughter and smiling, movement or balance disorder (ataxia, tremor), an easily excitable personality, and hypermotoric behavior, as well as speech impairment with none or minimal use of words [26] (see also GeneReviews: NBK1144). In about 90 % of individuals fulfilling the clinical criteria of AS a deficient expression or function of the maternally inherited UBE3A-allele confirms the diagnosis molecularly. Analysis of parent-specific DNA methylation imprints in the 15q11.2-q13 region reveals defects in approximately 80 % of individuals with AS, including deletions, uniparental disomy (UPD), or an imprinting defect; fewer than 1 % of individuals have a cytogenetically visible chromosome rearrangement (i. e., translocation or inversion). UBE3A-sequence analysis detects pathogenic variants in an additional approximately 11 % of patients.
Management: treatment of manifestations, prevention of secondary complications
The therapy of AS is symptom-oriented and therefore includes routine management of feeding difficulties, constipation, gastroesophageal reflux and strabismus, physical therapy, occupational therapy, and speech therapy with an emphasis on nonverbal methods of communication. Antiepileptic drugs should not include carbamezapine, vigabatrin, and tigabine as they may exacerbate seizures.
Some patients are at risk for medication overtreatment when movement abnormalities are mistaken for seizures and EEG abnormalities persist even when seizures are controlled. Sedating agents which might be helpful, e. g., for hypermotoric problems and nighttime wakefulness, can cause negative side effects, especially risperidone or other atypical antipsychotic drugs. Annual clinical examination for scoliosis and conservative or surgical treatment in symptomatic patients should be discussed. Evaluation of older children for obesity associated with an excessive appetite may be necessary (see GeneReviews).
Prader–Willi syndrome (PWS, 15q11q13)
Clinical characteristics and diagnosis
PWS is a complex multisystemic disorder (see also GeneReviews: NBK1330). It is characterized by severe neonatal muscular hypotonia and failure to thrive, early-onset extreme obesity due to insatiable appetite and hyperphagia, short stature, and hypogonadism, as well as variably impaired cognitive function, distinct behavioral problems, and psychiatric comorbidities (e. g., psychosis and autism spectrum disorders) [7], [27]. Hypothalamic dysfunction accounts for many clinical aspects of the PWS phenotype [28]. The prevalence of scoliosis in PWS is higher than in the general population [29].
Management: treatment of manifestations, prevention of secondary complications
The complexity of PWS requires a multidisciplinary therapeutic approach. Growth hormone therapy is approved for children with PWS and is recommended not only in order to improve adult height but also to ameliorate body composition by increasing lean body mass [30], [31]. Other endocrine manifestations requiring hormone replacement therapy are hypogonadism in almost all, as well as central hypothyroidism and adrenal insufficiency in some PWS individuals [32]. Morbid obesity and related complications must be avoided by dietary measures and strict control of food intake. Physiotherapy and psychological support are essential; some patients also need psychiatric treatment.
Schaaf–Yang syndrome (SYS; 15q11q13)
Clinical characteristics and diagnosis
Schaaf–Yang syndrome is a neurodevelopmental disorder which shares several symptoms with PWS including neonatal hypotonia, growth retardation and failure to thrive in the neonate period, hyperphagia, and obesity in later life, as well as impaired psychomotor and intellectual development. Joint contractures are typical findings, and a higher prevalence of autism disorders is reported. However, the phenotype is very variable [33], [34].
Management: treatment of manifestations, prevention of secondary complications
Due to the clinical overlap with PWS, the clinical management of SYS might lean on that for PWS but due to the rareness of the disease further data are needed as the basis for a more personalized therapy.
Central precocious puberty 2 (CPPB2; 15q11q13)
Clinical characteristics and diagnosis
CPPB is defined as a premature activation of the gonadotropic axis and development of secondary sexual characteristics before the age of 8 years in girls and 9 years in boys and is typically accompanied by a significant growth spurt. The timing of puberty is influenced by genetic, nutritional, and environmental factors and not all determinants involved are fully understood. Familial forms of GnRH-dependent precocious puberty have been reported in up to 27.5 % in some case series [35]. Central nervous system tumors as a cause of CPPB have to be ruled out.
Management: treatment of manifestations, prevention of secondary complications
CPPB can be treated with GnRH agonists. The decision for treatment has to be made individually. Age at onset, speed of progress of pubertal development, and extent of bone age acceleration have to be taken into consideration. Prevention of early epiphyseal fusion and short adult height is one therapeutic goal; postponing the emotional/psychological changes associated with puberty to a physiological age is another concern [36].
Pseudohypoparathyroidism/inactivating PTH/PTHrP signaling disorders (20q13.32)
Clinical characteristics and diagnosis
Pseudohypoparathyroidism (PHP) is historically a clinically heterogeneous hormone resistance syndrome mainly affecting the parathyroid hormone (PTH) pathway, described first by Albright in 1942. Nowadays all conditions of this group are summarized as inactivating PTH/PTHrP signaling disorders (iPPSD; Table 2) [37]. A clinical description is Albright’s hereditary osteodystrophy (AHO), which refers to the main features of short stature, round face, brachymetacarpia and -metatarsia, and ectopic ossification, as well as obesity and mild to moderate motor and cognitive disability. Laboratory findings may include the PTH resistance and also impairment of other Gs-alpha-related hormone functions, mainly TSH resistance. Underlying is a dysfunction of Gs-alpha-activity. PHPIA (nowadays iPPSD 2) refers to patients with AHO features and PTH resistance who have a diminished Gs-alpha-activity due to maternally inherited mutations of the GNAS gene. PPHP in this setting refers to patients with AHO, but without apparent endocrinopathies, despite diminished Gs-alpha-activity. The underlying cause are paternally transmitted inactivating mutations of the GNAS locus, leading to Gs-alpha deficiency in selected tissues due to tissue-specific imprinting. PHPIB (iPPSD3), in contrast, refers to patients with apparent PTH resistance, but no or little features of AHO. In these cases, different epigenetic aberrations affecting the GNAS gene locus can be detected.
Management: treatment of manifestations, prevention of secondary complications
As the clinical features of iPPSDs can be very heterogeneous and variable, an individualized approach for management has to be taken. PTH resistance is treated with activated Vitamin D metabolites, e. g., calcitriol, with the aim to reach normocalcemia without hypercalciuria. Ideally, PTH should then reach the usual reference interval. TSH resistance is treated with L-thyroxin in usual doses to reach normalized thyroid function tests. Management of AHO features is difficult. The recent consensus [6] recommends a multidisciplinary approach including a transition program, which should lead to a regular screen for secondary complications such as development of further endocrinopathies, ectopic bone formation, weight gain, development of diabetes, and support for cognitive and motor disabilities.
Mulchandani–Bhoj–Conlin syndrome (MBCS; 20q11q13)
Clinical characteristics and diagnosis
The phenotype of Mulchandani–Bhoj–Conlin syndrome (MBCS) is rather unspecific, with prenatal growth retardation, short stature with proportional head circumference, and feeding difficulties as the major features [38], [39]. Neurodevelopment seems to be normal. Due to the lack of specific features, upd(20)mat as the currently only molecular alteration of MBCS has mainly been identified in patients referred for SRS testing.
Management: treatment of manifestations, prevention of secondary complications
Data from growth hormone treatment in a small number of patients might indicate a favorable outcome; therefore, the clinical management might be leaned on that for SRS patients, but further studies are needed [4].
Genetic counseling
If the imprinting disorder is based on a UPD, there is virtually no increased recurrence risk for children of the UPD carrier. However, the situation is different for the parents of a UPD patient as some common familial chromosomal translocations can predispose to UPD formation. In particular, Robertsonian translocation carriers (i. e., translocations between the acrocentric chromosomes; frequency of 1/1,000 among newborns) have an increased risk for UPD formation. Thus, in imprinting disorder patients with UPDs of chromosomes 15 and 14, chromosomal analysis of the parents has to be considered [40]. Additionally, prenatal testing for UPD should be discussed with families transmitting structural variants affecting those chromosomes harboring clinically relevant imprinted regions [41].
(Familial) copy number variants (CNVs) and single nucleotide variants (SNVs) affecting genes causing imprinting disorders (e. g., UBE3A in AS and CDKN1C in BWS) are transmitted according to the autosomal dominant mode of inheritance. However, depending on the imprinting status of the affected gene, the clinical outcome depends on the sex of the parent contributing the affected allele. In rare familial cases, opposite clinical pictures can occur due to the transmission of the variant either from the mother or from the father [42]. Additionally, the size and gene content in case of chromosomal disturbances have to be determined as they can significantly alter and aggravate the clinical picture of its carrier and mask the imprinting disorder which is linked to the chromosomal region. In case of small pathogenic CNVs within differentially methylated regions (DMRs), the genotype–phenotype correlation might become ambiguous due to the complex interactions between the functional elements of imprinted loci (e. g., in 11p15.5 [43]). In these patients a precise molecular characterization of the alteration is needed.
For epimutation, it was assumed for a long time that they are not inherited, but arise sporadically. The recurrence risk for further children of the patients’ parents and also for children of the patients themselves was supposed to be very low, and in fact there is only a small number of reports on recurrent epimutations within the same families. However, it has now turned out that this assumption does not apply for so-called secondary epimutations, i. e., epimutations resulting from a genomic alteration indirectly affecting an imprinted locus. The number of genetic causes for secondary epimutations is growing, and they comprise the already mentioned maternal effect variants as well as small structural variants close to the locus of interest. Although probably not frequent, these possible inheritable causes of aberrant methylation have to be borne in mind in reporting and in genetic counseling.
Completely new aspects have to be considered in genetic counseling regarding the identification of maternal effect mutations. In these families the mother of a patient with an imprinting disorder mainly based on an MLID is carrier of a heterozygous, homozygous, or compound heterozygous variant in a maternal effect gene, which leads to imprinting disturbances in the growing fetus. A careful documentation of the family history of these mothers including miscarriages, pregnancy complications, and aneuploidies can help to identify them, because the clinical consequences of maternal effect mutations range from early abortions to late abortions and live-born children with or without different imprinting disorders in one and the same family. The children themselves do not have to carry the effect mutation to develop the clinical picture, so that there is no possibility of classical prenatal diagnosis. First descriptions of successful pregnancies following egg donation have been described in this context (for review, see [44]). Molecular testing for MLID has already been suggested on a research basis [4], [5] and its implementation in the routine diagnostic workup is currently in discussion. However, in families with MLID carriers and recurrent miscarriages it should be considered.
Prenatal testing for imprinting disorders might be offered to families with known genomic alterations predisposing for a child with imprinting disorders, i. e., in families with pathogenic CNVs and SNVs affecting imprinted regions. Additionally, prenatal molecular testing might be considered in case of prenatal ultrasound findings suggestive for an imprinting disorder, or prenatally detected chromosomal aberrations predisposing for a UPD (e. g., trisomy 15 in chorionic villous sampling). However, prior to molecular testing the methodical limitations, the reliability of the results, mosaicism, and ethical issues have to be discussed, preferably with the advice-seeking couple (see also Beygo et al. in this issue).
Patient support groups – international activities
Like for other rare diseases, patients and their families generally benefit from the exchange of information, so fortunately, national and international patient support groups have now been established for the more common imprinting disorders (for websites, see the list at the end of the manuscript).
Furthermore, in 2013 the European Network of Human Congenital Imprinting Disorders (EUCID.net) was founded to overcome the lacking standardization of diagnostics and clinical management strategies in the context of imprinting disorders. The formal COST-funding of EUCID.net ended in 2017, but it encouraged groups from European and non-European countries to collaborate (e. g., in the ENDO-ERN) and the exchange of knowledge and information is ongoing to further advance the scientific and clinical-diagnostic progress in the field of imprinting diseases (http://www.imprinting-disorders.eu/, https://endo-ern.eu/).
Conclusion and perspectives
The molecular and clinical overlaps between imprinting disorders are increasingly well understood and lead to reliable datasets which among other issues has resulted in consensus guidelines for the diagnosis and therapy of the more frequent imprinting disorders [4], [5], [6], [27]. In particular, the application of high-throughput sequencing and additional omic techniques provides new insights in basic mechanisms of imprinting and its disturbances. As an example, the list of possible maternal effect mutations leading to secondary epimutations in children is constantly increasing. Nevertheless, a lot of etiological questions and clinical observations are unanswered so far. Furthermore, a considerable number of patients with clinical symptoms indicating an imprinting disorder remain without molecular diagnostic confirmation. To decipher further molecular causes of imprinting disorders it is therefore helpful to include patients and their parents in (epi)genotype–phenotype studies. These studies provide the basis for future diagnostic and management recommendations, including the diagnosis of maternal effect mutations in women with frequent miscarriages. The extension from currently targeted diagnosis on specific imprinted loci to genome-wide approaches will increase the proportion of patients with hitherto unknown MLIDs. Additionally, the improvement of mosaic detection or the evaluation of tests on different cell systems will also reduce the proportion of imprinting disorder patients without molecular confirmation. It is desirable that a multidisciplinary exchange between the participating research groups and laboratories is maintained and embedded in national and international collaborations.
Website links (including patient organizations)
Angelman syndrome: https://www.angelman.de/
GeneReviews: https://www.ncbi.nlm.nih.gov/books/NBK1116/
Prader–Willi syndrome: https://prader-willi.de/
Silver–Russell syndrome: https://www.bkmf.de/arbeitsgruppen/srs-sga-whmangel/, https://silverrussellsyndrome.org/
References
[1] Haig D. Genomic imprinting and kinship: how good is the evidence? Annu Rev Genet. 2004;38:553–85.10.1146/annurev.genet.37.110801.142741Search in Google Scholar PubMed
[2] Soellner L, Begemann M, Mackay DJ, Gronskov K, Tumer Z, Maher ER et al. Recent Advances in Imprinting Disorders. Clin Genet. 2017;91(1):3–13.10.1111/cge.12827Search in Google Scholar PubMed
[3] Carli D, Riberi E, Ferrero GB, Syndromic MA. Disorders Caused by Disturbed Human Imprinting. J Clin Res Pediatr Endocrinol. 2020;12(1):1–16.10.4274/jcrpe.galenos.2019.2018.0249Search in Google Scholar PubMed PubMed Central
[4] Wakeling EL, Brioude F, Lokulo-Sodipe O, O’Connell SM Salem J, Bliek J et al. Diagnosis and management of Silver-Russell syndrome: first international consensus statement. Nat Rev Endocrinol. 2017;13(2):105–24.10.1038/nrendo.2016.138Search in Google Scholar PubMed
[5] Brioude F, Kalish JM, Mussa A, Foster AC, Bliek J, Ferrero GB et al. Expert consensus document: Clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement. Nat Rev Endocrinol. 2018;14(4):229–49.10.1038/nrendo.2017.166Search in Google Scholar PubMed PubMed Central
[6] Mantovani G, Bastepe M, Monk D, de Sanctis L, Thiele S, Usardi A et al. Diagnosis and management of pseudohypoparathyroidism and related disorders: first international Consensus Statement. Nat Rev Endocrinol. 2018;14(8):476–500.10.1038/s41574-018-0042-0Search in Google Scholar PubMed PubMed Central
[7] Goldstone AP, Holland AJ, Hauffa BP, Hokken-Koelega AC, Tauber M. speakers contributors at the Second Expert Meeting of the Comprehensive Care of Patients with PWS. Recommendations for the diagnosis and management of Prader-Willi syndrome. J Clin Endocrinol Metab. 2008;93(11):4183–97.10.1210/jc.2008-0649Search in Google Scholar PubMed
[8] Butler MG, Miller JL, Prader-Willi FJL. Syndrome – Clinical Genetics, Diagnosis and Treatment Approaches: An Update. Curr Pediatr Rev. 2019;15(4):207–44.10.2174/1573396315666190716120925Search in Google Scholar PubMed PubMed Central
[9] Bird LM. Angelman syndrome: review of clinical and molecular aspects. Appl Clin Genet. 2014;7:93–104.10.2147/TACG.S57386Search in Google Scholar PubMed PubMed Central
[10] Sanchez-Delgado M, Riccio A, Eggermann T, Maher ER, Lapunzina P, Mackay D et al. Causes and Consequences of Multi-Locus Imprinting Disturbances in Humans. Trends Genet. 2016;32(7):444–55.10.1016/j.tig.2016.05.001Search in Google Scholar PubMed
[11] Azzi S, Rossignol S, Steunou V, Sas T, Thibaud N, Danton F et al. Multilocus methylation analysis in a large cohort of 11p15-related foetal growth disorders (Russell Silver and Beckwith Wiedemann syndromes) reveals simultaneous loss of methylation at paternal and maternal imprinted loci. Hum Mol Genet. 2009;18(24):4724–33.10.1093/hmg/ddp435Search in Google Scholar PubMed
[12] Mackay DJ, Callaway JL, Marks SM, White HE, Acerini CL, Boonen SE et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet. 2008;40(8):949–51.10.1038/ng.187Search in Google Scholar PubMed
[13] Monk D, Mackay DJG, Eggermann T, Maher ER, Riccio A. Genomic imprinting disorders: lessons on how genome, epigenome and environment interact. Nat Rev Genet. 2019;20(4):235–48.10.1038/s41576-018-0092-0Search in Google Scholar PubMed
[14] Nguyen NMP, Khawajkie Y, Mechtouf N, Rezaei M, Breguet M, Kurvinen E et al. The genetics of recurrent hydatidiform moles: new insights and lessons from a comprehensive analysis of 113 patients. Mod Pathol. 2018;31(7):1116–30.10.1038/s41379-018-0031-9Search in Google Scholar PubMed
[15] Docherty LE, Kabwama S, Lehmann A, Hawke E, Harrison L, Flanagan SE et al. Clinical presentation of 6q24 transient neonatal diabetes mellitus (6q24 TNDM) and genotype-phenotype correlation in an international cohort of patients. Diabetologia. 2013;56(4):758–62.10.1007/s00125-013-2832-1Search in Google Scholar PubMed
[16] Binder G, Liebl M, Woelfle J, Eggermann T, Blumenstock G, Schweizer R. Adult height and epigenotype in children with Silver-Russell syndrome treated with GH. Horm Res Paediatr. 2013;80(3):193–200.10.1159/000354658Search in Google Scholar PubMed
[17] Binder G, Schweizer R, Blumenstock G, Ferrand N. Adrenarche in Silver-Russell Syndrome: Timing and Consequences. J Clin Endocrinol Metab. 2017;102(11):4100–8.10.1210/jc.2017-00874Search in Google Scholar PubMed
[18] Barel O, Shalev SA, Ofir R, Cohen A, Zlotogora J, Shorer Z et al. Maternally inherited Birk Barel mental retardation dysmorphism syndrome caused by a mutation in the genomically imprinted potassium channel KCNK9. Am J Hum Genet. 2008;83(2):193–9.10.1016/j.ajhg.2008.07.010Search in Google Scholar PubMed PubMed Central
[19] Graham JM Jr., Zadeh N, Kelley M, Tan ES, Liew W, Tan V et al. KCNK9 imprinting syndrome-further delineation of a possible treatable disorder. Am J Med Genet, Part A. 2016;170(10):2632–7.10.1002/ajmg.a.37740Search in Google Scholar PubMed
[20] Geoffron S, Abi Habib W, Chantot-Bastaraud S, Dubern B, Steunou V, Azzi S et al. Chromosome 14q32.2 Imprinted Region Disruption as an Alternative Molecular Diagnosis of Silver-Russell Syndrome. J Clin Endocrinol Metab. 2018;103(7):2436–46.10.1210/jc.2017-02152Search in Google Scholar PubMed
[21] Ioannides Y, Lokulo-Sodipe K, Mackay DJ, Davies JH, Temple IK. Temple syndrome: improving the recognition of an underdiagnosed chromosome 14 imprinting disorder: an analysis of 51 published cases. J Med Genet. 2014;51(8):495–501.10.1136/jmedgenet-2014-102396Search in Google Scholar PubMed
[22] Kagami M, Nagasaki K, Kosaki R, Horikawa R, Naiki Y, Saitoh S et al. Temple syndrome: comprehensive molecular and clinical findings in 32 Japanese patients. Genet Med. 2017;19(12):1356–66.10.1038/gim.2017.53Search in Google Scholar PubMed PubMed Central
[23] Gomes LG, Cunha-Silva M, Crespo RP, Ramos CO, Montenegro LR, Canton A et al. DLK1 Is a Novel Link Between Reproduction and Metabolism. J Clin Endocrinol Metab. 2019;104(6):2112–20.10.1210/jc.2018-02010Search in Google Scholar PubMed
[24] Kagami M, Kurosawa K, Miyazaki O, Ishino F, Matsuoka K, Ogata T. Comprehensive clinical studies in 34 patients with molecularly defined UPD(14) pat and related conditions (Kagami-Ogata syndrome). Eur J Hum Genet. 2015;23(11):1488–98.10.1038/ejhg.2015.13Search in Google Scholar PubMed PubMed Central
[25] Ogata T, Kagami M. Kagami-Ogata syndrome: a clinically recognizable upd(14) pat and related disorder affecting the chromosome 14q32.2 imprinted region. J Hum Genet. 2016;61(2):87–94.10.1038/jhg.2015.113Search in Google Scholar PubMed PubMed Central
[26] Williams CA, Beaudet AL, Clayton-Smith J, Knoll JH, Kyllerman M, Laan LA et al. Angelman syndrome 2005: updated consensus for diagnostic criteria. Am J Med Genet, Part A. 2006;140(5):413–8.10.1002/ajmg.a.31074Search in Google Scholar PubMed
[27] Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-Willi syndrome. Genet Med. 2012;14(1):10–26.10.1038/gim.0b013e31822bead0Search in Google Scholar PubMed
[28] Swaab DF. Prader-Willi syndrome and the hypothalamus. Acta Paediatr Suppl. 1997;423:50–4.10.1111/j.1651-2227.1997.tb18369.xSearch in Google Scholar PubMed
[29] van Bosse HJP, Butler MG. Clinical Observations and Treatment Approaches for Scoliosis in Prader-Willi Syndrome. Genes (Basel). 2020;11(3).10.3390/genes11030260Search in Google Scholar PubMed PubMed Central
[30] Festen DA, de Lind van Wijngaarden R, van Eekelen M, Otten BJ, Wit JM, Duivenvoorden HJ et al. Randomized controlled GH trial: effects on anthropometry, body composition and body proportions in a large group of children with Prader-Willi syndrome. Clin Endocrinol (Oxf). 2008;69(3):443–51.10.1111/j.1365-2265.2008.03228.xSearch in Google Scholar PubMed
[31] Lindgren AC, Lindberg A. Growth hormone treatment completely normalizes adult height and improves body composition in Prader-Willi syndrome: experience from KIGS (Pfizer International Growth Database). Horm Res. 2008;70(3):182–7.10.1159/000145019Search in Google Scholar PubMed
[32] Angulo MA, Butler MG, Cataletto ME. Prader-Willi syndrome: a review of clinical, genetic, and endocrine findings. J Endocrinol Invest. 2015;38(12):1249–63.10.1007/s40618-015-0312-9Search in Google Scholar PubMed PubMed Central
[33] McCarthy JM, McCann-Crosby BM, Rech ME, Yin J, Chen CA, Ali MA et al. Hormonal, metabolic and skeletal phenotype of Schaaf-Yang syndrome: a comparison to Prader-Willi syndrome. J Med Genet. 2018;55(5):307–15.10.1136/jmedgenet-2017-105024Search in Google Scholar PubMed
[34] McCarthy J, Lupo PJ, Kovar E, Rech M, Bostwick B, Scott D et al. Schaaf-Yang syndrome overview: Report of 78 individuals. Am J Med Genet, Part A. 2018;176(12):2564–74.10.1002/ajmg.a.40650Search in Google Scholar PubMed PubMed Central
[35] de Vries L, Kauschansky A, Shohat M, Phillip M. Familial central precocious puberty suggests autosomal dominant inheritance. J Clin Endocrinol Metab. 2004;89(4):1794–800.10.1210/jc.2003-030361Search in Google Scholar PubMed
[36] Aguirre RS, Eugster EA. Central precocious puberty: From genetics to treatment. Best Pract Res, Clin Endocrinol Metab. 2018;32(4):343–54.10.1016/j.beem.2018.05.008Search in Google Scholar PubMed
[37] Thiele S, Mantovani G, Barlier A, Boldrin V, Bordogna P, De Sanctis L et al. From pseudohypoparathyroidism to inactivating PTH/PTHrP signalling disorder (iPPSD), a novel classification proposed by the EuroPHP network. Eur J Endocrinol. 2016;175(6):P1–17.10.1530/EJE-16-0107Search in Google Scholar PubMed
[38] Mulchandani S, Bhoj EJ, Luo M, Powell-Hamilton N, Jenny K, Gripp KW et al. Maternal uniparental disomy of chromosome 20: a novel imprinting disorder of growth failure. Genet Med. 2016;18(4):309–15.10.1038/gim.2015.103Search in Google Scholar PubMed
[39] Hjortshoj TD, Sorensen AR, Yusibova M, Hansen BM, Duno M, Balslev-Harder M et al. upd(20)mat is a rare cause of the Silver-Russell-syndrome-like phenotype: Two unrelated cases and screening of large cohorts. Clin Genet. 2020;97(6):902–7.10.1111/cge.13727Search in Google Scholar PubMed
[40] Beygo J, Buiting K, Ramsden SC, Ellis R, Clayton-Smith J, Kanber D. Update of the EMQN/ACGS best practice guidelines for molecular analysis of Prader-Willi and Angelman syndromes. Eur J Hum Genet. 2019;27(9):1326–40.10.1038/s41431-019-0435-0Search in Google Scholar PubMed PubMed Central
[41] Shaffer LG, Agan N, Goldberg JD, Ledbetter DH, Longshore JW, American CSB. College of Medical Genetics statement of diagnostic testing for uniparental disomy. Genet Med. 2001;3(3):206–11.10.1097/00125817-200105000-00011Search in Google Scholar PubMed PubMed Central
[42] Jurkiewicz D, Kugaudo M, Skorka A, Smigiel R, Smyk M, Ciara E et al. A novel IGF2/H19 domain triplication in the 11p15.5 imprinting region causing either Beckwith-Wiedemann or Silver-Russell syndrome in a single family. Am J Med Genet, Part A. 2017;173(1):72–8.10.1002/ajmg.a.37964Search in Google Scholar PubMed
[43] Heide S, Chantot-Bastaraud S, Keren B, Harbison MD, Azzi S, Rossignol S et al. Chromosomal rearrangements in the 11p15 imprinted region: 17 new 11p15.5 duplications with associated phenotypes and putative functional consequences. J Med Genet. 2018;55(3):205–13.10.1136/jmedgenet-2017-104919Search in Google Scholar PubMed
[44] Elbracht M, Mackay D, Begemann M, Kagan KO, Eggermann T. Disturbed genomic imprinting and its relevance for human reproduction: causes and clinical consequences. Hum Reprod Update. 2020.10.1093/humupd/dmz045Search in Google Scholar PubMed
© 2020 Elbracht et al., published by De Gruyter
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Frontmatter
- MAIN TOPIC: IMPRINTING DISORDERS: NOVEL FINDINGS AND TRANSLATION INTO DIAGNOSTICS AND MANAGEMENT
- Imprinting disorders: novel findings and translation into diagnostics and management
- Basics and disturbances of genomic imprinting
- Molecular testing for imprinting disorders
- Clinical spectrum and management of imprinting disorders
- Novel strategies to cure imprinting disorders
- BERICHTE AUS DER HUMANGENETIK
- Personalia
- Personalia
- GfH-Verbandsmitteilungen
- Die GfH-JuniorAkademie 2020 – virtueller Vorreiter in der GfH
- ESHG warnt vor der Umsetzung der LDT-Anforderungen der IVDR
- BVDH-Verbandsmitteilungen
- Mitteilungen des Berufsverbandes Deutscher Humangenetiker e. V.
- Aktuelle Nachrichten
- Aktuelle Nachrichten
Articles in the same Issue
- Frontmatter
- MAIN TOPIC: IMPRINTING DISORDERS: NOVEL FINDINGS AND TRANSLATION INTO DIAGNOSTICS AND MANAGEMENT
- Imprinting disorders: novel findings and translation into diagnostics and management
- Basics and disturbances of genomic imprinting
- Molecular testing for imprinting disorders
- Clinical spectrum and management of imprinting disorders
- Novel strategies to cure imprinting disorders
- BERICHTE AUS DER HUMANGENETIK
- Personalia
- Personalia
- GfH-Verbandsmitteilungen
- Die GfH-JuniorAkademie 2020 – virtueller Vorreiter in der GfH
- ESHG warnt vor der Umsetzung der LDT-Anforderungen der IVDR
- BVDH-Verbandsmitteilungen
- Mitteilungen des Berufsverbandes Deutscher Humangenetiker e. V.
- Aktuelle Nachrichten
- Aktuelle Nachrichten