The clinical picture of symptomatic Rathke cleft cysts in children
-

 ,
,

Abstract
Objectives
Rathke cleft cysts (RCC) in childhood are rare, often asymptomatic, and thus discovered incidentally. We aimed to summarize clinical features and pituitary function of patients with symptomatic RCC.
Methods
This retrospective study included 14 patients (8 male) from the university hospital’s database (period 2005–2023).
Results
RCC diagnosis based on magnetic resonance imaging at 12.2 (1.8–17.6) years. Presenting symptoms were headaches (n=8), occurring alone (n=1) or with nausea/fatigue (n=2), polydipsia (n=2), or dizziness (n=3), followed by growth retardation (n=5), occurring alone (n=4) or with polydipsia (n=1). Two patients exhibited visual disturbances. Endocrinological evaluation revealed pituitary insufficiency in 10, including isolated or combined growth hormone (GH) deficiency (n=5), arginine-vasopressin deficiency (AVP-D; n=5), central hypothyroidism (n=2), and hypocortisolism (n=2). Three patients had hyperprolactinemia. Nine patients were monitored by regular imaging; five underwent surgery. In the observation group, cyst size remained unchanged in seven and decreased in two patients, while it increased in four patients treated surgically. At last presentation after 6.4 (0.33–14.8) years of follow-up, the endocrine status of the conservatively followed patients was normal in n=6, and pathological in n=3. Pituitary function did not normalize after surgery. Five patients developed hypogonadotropic hypogonadism, including two children who were followed conservatively.
Conclusions
We found a high incidence of pituitary insufficiency among symptomatic pediatric RCC patients. Pituitary function was not closely related to cyst size or location and did not improve after surgery. Regular clinical and radiological follow-up is mandatory for both conservatively and surgically treated patients.
Introduction
Rathke cleft cysts (RCC) are benign cystic remnants of Rathke’s pouch, typically located in the pars intermedia between the anterior and posterior pituitary lobes and usually regress during embryonic development [1]. They are often asymptomatic and incidentally discovered on cranial imaging. Diagnosing RCCs can be challenging due to the similarity of cystic lesions of the sellar region, including pituitary adenomas, craniopharyngiomas, and arachnoid cysts [2], 3]. RCCs are rare in the pediatric population [4]. The prevalence ranges from 1.2 % in asymptomatic children [5] to 13.5 % in children suffering from endocrine disorders [6]. Like adults, the most common presenting symptoms include headaches and endocrinological symptoms with varying frequencies across studies [6], [7], [8], [9].
The current literature contains limited information about the natural course and long-term outcomes, particularly in children who undergo conservative management. In the present study, we aim to analyze the clinical symptoms, endocrine dysfunction, and clinical course of pediatric RCC patients treated at a single institution.
Patients and methods
Patients
We retrospectively reviewed the medical records of 14 children with RCC diagnosed between 2005 and 2023 and treated in the Departments of Paediatrics and Adolescent Medicine and Neurosurgery at the Friedrich Alexander University Hospital, Erlangen, Germany. The patients were identified via the university hospital’s database. The study inclusion criteria included an RCC diagnosis confirmed by magnetic resonance imaging (MRI) and an age of 18 years or younger. Our institution’s Review Board approved the study.
Methods
The age at diagnosis, presenting symptoms, auxological data, the location and size of RCC on the first cranial MRI, the results of the ophthalmological examination, and laboratory results emphasizing endocrine dysfunction were extracted from medical files. The staging of papilledema was made according to Frisen [10]. The decision to perform a neurosurgical intervention based on clinical (refractory headaches resistant to appropriate analgesic treatment, ophthalmological disturbances) and radiological findings (signs of expanding or compressive lesions on the surrounding tissue on MRI) after discussion in our multidisciplinary board. The proposal for surgery was discussed with the parents/caregivers but was not accepted by all of them. In particular, the parents of the patient with papilledema Stage 1–2 according to Frisen (case No. 2) gave no consent to the operation. Finally, neurosurgery was performed in five patients. Overall, nine children were treated conservatively and followed by regular clinical examinations and yearly follow-up MRIs. Postoperatively, the first follow-up MRI was routinely performed at 3 months, thereafter at 6-to-12-month intervals or as clinically indicated.
Height standard deviation score (SDS) and body mass index (BMI)-SDS were calculated for chronological age (CA) and sex, using German references from Kromeyer and Hauschild [11]. Pubertal status was assessed according to Tanner [12], 13]. Growth retardation was defined as either a short stature (height≤2 SDS) or a height velocity <25th percentile, obesity as a BMI SDS>2, the onset of puberty as Tanner stage B2 in girls (both by inspection and palpation), and testicular sizes >3 mL (G2) in boys, respectively.
Hormonal assessment included measurement of the basal levels of serum thyroid stimulating hormone (TSH), free thyroxine (fT4), insulin-like growth factor-I (IGF-1), insulin-like growth factor binding protein-3 (IGF-BP3), luteinizing hormone (LH), follicle-stimulating hormone (FSH), estradiol/testosterone, plasma adrenocorticotropic hormone (ACTH), cortisol, and prolactin. A more detailed investigation via endocrine stimulation tests was undertaken if required. The definition of growth hormone deficiency (GHD) included a peak growth hormone (GH) level of <8 ng/mL in two different GH stimulation tests. Arginine vasopressin deficiency (AVP-D) was diagnosed based on serum electrolytes, serum and urine osmolality, and standardized water deprivation tests. Hyperprolactinemia was defined as a baseline prolactin level of >25 ng/mL. Central hypothyroidism was defined as subnormal free T4 levels in association with inappropriate normal or low TSH levels. ACTH deficiency was diagnosed in patients with low basal morning cortisol and ACTH levels, and lacking or inadequate cortisol increase after low-dose ACTH or metyrapone tests. Low basal gonadotropin levels and no response after gonadotrophic hormone-releasing hormone (GnRH) stimulation confirmed hypogonadotropic hypogonadism.
Statistical analysis
We performed descriptive statistical analysis. The median and range were determined for continuous variables, and the frequency and percentages were calculated for categorical data. All analyses were performed using IBM SPSS Statistical Software (version 29.0.0, SPSS Inc., Chicago, IL, USA).
Results
Presenting symptoms and baseline characteristics
We identified 14 patients (8 males, 6 females) with symptomatic RCC. The female-to-male ratio was 0.75. The age at diagnosis was 12.2 (1.8–17.6) years. Table 1 shows the clinical characteristics of the children.
Clinical data of the 14 patients with Rathke cleft cysts (RCC).
| Case, sex | At diagnosis | At last visit | ||||||
|---|---|---|---|---|---|---|---|---|
| Age, years | Clinical symptoms | Pituitary function | RCC location | RCC size max. diameter, mm | RCC therapy | Age, years | Pituitary function | |
| 1, F | 12.3 | Headache, dizziness | Normal | Intrasellar | 7 | Observation | 17.2 | Normal |
| 2, F | 17.6 | Syncope, papilledema | Normal | Intrasellar | 6 | Observation | 17.9 | Normal |
| 3, M | 10.8 | Height velocity <25th centile, headache, polydipsia | GHD, AVP-D, PRL↑ | Intrasellar/suprasellar | 14 | Surgery | 17.3 | GHD, ACTH↓, AVP-D, LH/FSH↓, PRL↑ |
| 4, M | 1.80 | Short stature | GHD, ACTH↓, TSH↓ | Intrasellar | 10 | Observation | 16.6 | GHD, ACTH↓, LH/FSH↓, TSH↓ |
| 5, F | 15.6 | Headache | Normal | Suprasellar | 4 | Observation | 16.7 | Normal |
| 6, M | 9.50 | Headache, polydipsia | AVP-D | Intrasellar | 2 | Observation | 16.7 | AVP-D |
| 7, M | 12.1 | Headache, papilledema, polydipsia/polyuria | AVP-D, PRL↑ | Intrasellar | 17 | Surgery | 15.8 | GHD, ACTH↓, AVP-D, LH/FSH↓, TSH↓ |
| 8, F | 14.6 | Headache, fatigue | AVP-D, ACTH↓, TSH↓, | Intrasellar/suprasellar | 18 | Surgery | 16.4 | ACTH↓, AVP-D, LH/FSH↓, TSH↓ |
| 9, F | 14.3 | Headache, weight gain, nausea | AVP-D, TSH↓, PRL↑ | Suprasellar | 18 | Surgery | 15.1 | AVP-D, PRL↑ |
| 10, M | 3.10 | Short stature | GHD | Intrasellar | 11 | Observation | 13.5 | Normal |
| 11, M | 3.60 | Short stature, micropenis | GHD, ACTH↓, TSH↓, | Intrasellar/suprasellar | 9 | Observation | 17.8 | GHD, ACTH↓, LH/FSH↓, TSH↓ |
| 12, F | 17.0 | Headache, dizziness | Normal | Suprasellar | 6 | Observation | 20.6 | Normal |
| 13, M | 16.4 | Headache, dizziness | TSH↓ | Intrasellar | 14 | Surgery | 20.9 | AVP-D, TSH↓ |
| 14, M | 5.70 | Short stature | GHD, TSH↓ | Intrasellar | 11 | Observation | 12.7 | GHD |
-
F, female; M, male; short stature, Height ≤-2 SDS; GHD, growth hormone deficiency; AVP-D, arginine vasopressin deficiency (diabetes insipidus centralis); PRL, prolactin; ACTH, adrenocorticotrophic hormone; TSH, thyroid-stimulating hormone; LH, luteinizing hormone; FSH, follicle-stimulating hormone.
The most common presenting symptom was headache (n=8). Headache was the only presenting symptom in one patient, appeared in combination with dizziness in three, with nausea/fatigue in two, and with polydipsia in two patients. Short stature was present in four patients; one patient presented with subnormal height velocity, combined with polydipsia and polyuria. Less frequently reported symptoms were fatigue (n=2), weight gain (n=1), and micropenis (n=1). Two patients exhibited visual disturbances in combination with papilledema (No. 2 and 7, Table 1). Seven patients were prepubertal, and seven patients (5 f, 2 m) were in puberty (Tanner stages 4–5). Mean height SDS was −0.43 (−4.1 to 1.3), and mean BMI-SDS was 0.88 (−1.26 to 2.72). Two patients (Patients No. 7 and 9; Table 1) had obesity, and one patient (No. 2) was overweight. Two of five female patients with Tanner stage V complained of irregular menstrual cycles.
Initial hormonal assessment
The endocrinological diagnostic work-up at presentation revealed pituitary insufficiency in 10 patients (71.4 %, Figure 1). Five patients had GHD. Three of them had a combination of GHD and central hypothyroidism or hypocortisolism; one patient had GHD and AVP-D, and another patient had isolated GHD. All five patients with GHD were prepubertal males. Five out of 14 patients had AVP-D, either alone (n=1) or in combination with central hypothyroidism (n=2) or hypocortisolism (n=2). Six patients had a TSH deficiency on diagnostic work-up, mostly in combination with other hormonal disturbances; only one patient exhibited isolated central hypothyroidism. Three patients (2 m, 1 f) had hyperprolactinaemia with a median level of 50.4 ng/mL (range 28.5–52.6). Two of the three patients with pituitary stalk dislocation (No. 7 and 11, Table 1) had multiple pituitary hormone deficiencies, whereas Patient No. 5 presented with no endocrine disturbances.
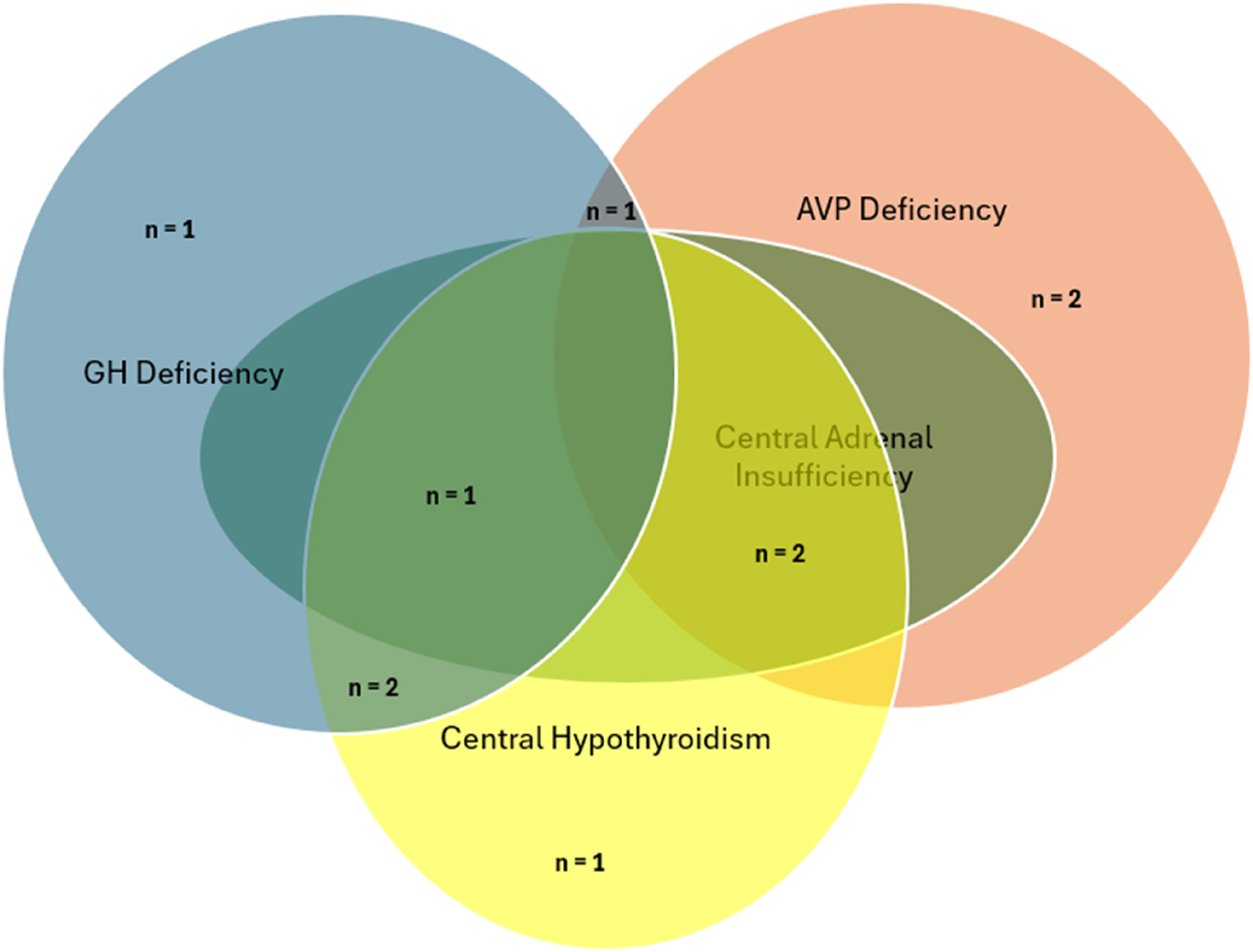
Prevalence of isolated or combined hormonal dysfunction in this cohort of 14 patients with Rathke cleft cysts. GH, growth hormone; AVP, arginine vasopressin.
Initial radiological findings
The median maximal axial cyst diameter on MRI was 10.5 mm (2–18). In nine patients, the cyst was located in the sellar region, while five patients had RCCs extending into the suprasellar region (Table 1). Three patients had a compression of the pituitary gland and/or optic chiasm, and three patients (Patients No. 5, 7, and 11, Table 1) had a dislocation of the pituitary stalk.
Although there was no correlation between the cyst size and endocrine dysfunction, all patients who underwent surgery had an RCC diameter of 14 mm or more (Table 1).
RCC treatment
Five patients underwent neurosurgery (transcranial n=1; transsphenoidal n=4, Table 1). The RCCs were removed entirely with total wall resection, and histology confirmed the diagnosis. Of the five patients treated surgically, the cyst size increased in four; three required a second operation (Patients No. 7, 9, and 13; Table 1). Nine children were observed by regular follow-ups (Table 1). In the observation group, cyst size remained unchanged in seven and decreased in two.
Endocrine treatment
Four patients with GHD were treated with rhGH; three of these from the conservatively followed group (No. 4, 10, and 11) and one (No. 3) from the surgically treated group. Patient No. 14 refused rhGH substitution therapy. All patients on rhGH were prepubertal males. Median duration of rhGH therapy was 8.5 years (4.0–13.9). Height-SDS improved from a median of −3.20 (−4.92 to −1.67) to a median of 0.65 (1.92–0.35).
Eight patients received l-thyroxine substitution, either alone (n=2), in combination with rhGH (n=3), or in combination with desmopressin and hydrocortisone (n=3). Six of them were still on l-thyroxine treatment during the follow-up period. Five patients initially received desmopressin: one alone (No. 6) and the remaining four combined with other hormone replacements. At the last visit, six patients received desmopressin: two alone (Patients No. 6 and 9) and four in combination with other hormones. Five of these six patients with AVP-D at the last visit belonged to the surgically treated group. Three patients with central adrenal insufficiency (CAI) after surgery received daily hydrocortisone (Patients No. 3, 7, and 8), and two patients (Patients No. 4 and 11) received hydrocortisone only as a stress medication. Hyperprolactinemia was not treated with dopamine agonists.
Outcomes
The follow-up period was 6.4 (0.33–14.8) years. Median age at the last presentation was 16.6 (12.7–20.9) years. Endocrinological evaluation at the last visit was normal in five of nine conservatively followed patients. However, two suffered from multiple pituitary hormone deficiencies, one with isolated GHD, and one patient exhibited isolated AVP-D (Table 1). Pituitary function did not normalize after surgery with the exception of hyperprolactinemia. During follow-up, five patients (four males and one female) developed hypogonadotropic hypogonadism, including two patients from the observation group. One of the two patients with initial visual disturbances Papilledema Stage 3 of Patient No. 7 (Table 1) disappeared after surgery. In contrast, Patient No. 2 who was followed conservatively exhibited still papilledema Stage 1–2 at the last visit, most probably associated with overweight-associated idiopathic intracranial hypertension.
Discussion
In the present study, we analyzed the clinical symptoms and endocrine disorders at presentation and during follow-up of 14 children and adolescents with symptomatic RCCs diagnosed over 18 years in our institution.
The diagnosis of RCC was made radiologically based on the characteristic appearance on MRI in all patients. It should be noted that a definitive diagnosis can only be made by histopathological examination of the cyst membrane [3]. It has been pointed out that in cases where membrane resection is not performed to preserve hypophyseal function, histological examination is incomplete, and the definitive diagnosis of the cyst remains uncertain [14]. The diagnosis was confirmed through microscopic pathological analysis of the membrane in five patients who had undergone surgery.
The number of our patients aligns with most published pediatric studies [8], [15], [16], [17], [18]. However, one study from South Korea reported the largest series of 93 children and adolescents from a single institution to date [19]. The authors found a slight female predominance (female to male ratio: 1.16) and a mean age of 10.9 years at diagnosis. The slight female-to-male predominance was also reported in a cohort of 24 children from a single institution [8] and found in 31 children with a histopathological diagnosis of RCC after surgery [20]. The smaller number of subjects studied could explain our lower female-to-male ratio. Our patients’ median age of 12 years at diagnosis matches well with data from the literature [8], 20].
Headaches have been reported as the most common symptom in pediatric studies, with prevalence ranging from 21 % [7] to 79 % [9]. In our study, eight of 14 patients (57 %) reported headaches as the presenting symptom, including all five surgically treated patients. The size of the cyst did not correlate with headaches. Even the patient with the smallest cyst size (Patient No. 6) in our cohort reported headaches and had signs of AVP-D. Recent data suggest that chronic adenohypophyseal inflammation may be the key to understanding why some patients with small RCCs experience severe headaches [21].
Our data confirm the low frequency of visual symptoms reported in other pediatric studies [7], 18]. In contrast, adult studies report visual impairments of up to 75 % [22].
Growth retardation and clinical signs of AVP-D were the most common endocrine abnormalities at presentation [15], 20]. Lee et al. report that endocrine abnormalities were more prevalent in the surgery group than in the observation group [19]. However, in the present study, we could not confirm these results. Patients with symptomatic RCC may display a variable degree of endocrine dysfunction. Our series revealed hormonal deficiencies in up to 71 % of the children at diagnosis. This study’s high frequency of GHD aligns with previous pediatric studies [7], 23], 24]. In contrast, AVP-D incidence was higher than previously reported in the literature [7], 8]. Arginine vasopressin deficiency was found in all five patients treated surgically (before surgery: n=4, after surgery: n=1), and only in one of the nine followed conservatively. Arginine vasopressin deficiency did not improve during the follow-up. Our results confirm the data of Lim HH et al., who stated that none of their 29 conservatively managed pediatric patients had AVP-D, whereas 40 % of surgically managed patients had AVP-D [23].
Hyperprolactinemia was reported as adults’ most common hormonal disturbance [25], whereas it seems to be rare in children [6], 20]. Hyperprolactinemia was found in three patients at diagnosis (2 m, 1 f) and in two patients at last visit (1 m, 1 f). None of the patients had galactorrhea, and the female patient (Pat. No. 9) had no menstrual irregularity. Our data do not confirm the reports of a high rate of precocious puberty, which has been reported between 4 and 37 % in other studies [7], 23]. This finding might be attributable to ethnic differences, the older age of the subjects, and the small number of female subjects in our study.
The data from the literature on complete resolution of prior hormone deficiencies after surgery range from 14.6 % [19] to 50 % [20]. In our cohort, endocrine deficits did not improve during our patients’ follow-up period. Moreover, at the last visit, five patients, including two patients of the observation group, developed hypogonadotropic hypogonadism during the clinical course as additional endocrine dysfunction.
The RCCs in childhood are usually small and typically located in the pars intermedia [26]. Previous studies reported that the median cyst diameter data varied between 5.5 mm [15] and 8 mm, ranging between 6 and 30 mm [8]. The median cyst size of our cohort was comparable to the literature. In five of our 14 patients, RCC showed a suprasellar extension on MRI. A former study suggested that suprasellar extension is associated with TSH, ACTH deficiencies, and AVP-D [23]. We did not observe a similar association; in fact, two of the patients with suprasellar RCC (Patients No. 5 and 12, Table 1) had a normal hormonal status at presentation.
The natural course of pediatric RCC remains unclear since most reported series do not provide long-term follow-up as in this study. In our observation group, all nine patients with RCC were diagnosed radiographically, differentiated from other cystic lesions by the location and lack of calcification [27]. The cyst size remained unchanged in seven and decreased in two. Of the five patients treated surgically, the cyst size increased in four; three required a second operation. Recurrence rates have been reported between 0 and 22 % [26]. Headache and hyperprolactinemia were the leading findings of cyst recurrence in our patients. In our cohort, two patients with recurrence had squamous metaplasia described in the histopathological examination. One of them had a postoperative cerebrospinal fluid leak. The other patient had a residual cyst demonstrated on postoperative MRI.
A recent systematic review and meta-analysis of 190 children with RCCs identified from 12 retrospective cohort studies compared surgical and conservative management approaches [28]. The authors reported that the conservative group demonstrated a broader age distribution than the surgical group, that headaches were more prevalent in surgical patients than in conservative patients, and that surgical patients exhibited higher rates of visual disturbances and endocrine dysfunction compared to conservative patients. Due to the relatively small number of patients in our cohort, we were unable to confirm these results.
Our data suggest that most pediatric RCC patients can be managed successfully. This finding is compatible with the study of Jung et al., in which only 7.5 % of patients were managed surgically, although 94.4 % were symptomatic [7]. While commonly accepted criteria for surgery include visual field deficits, radiographic evidence of chiasma compression, and medically refractory headaches, some authors suggest that the latter is a subjective surgical indication, as more than half of conservatively managed patients show spontaneous resolution of headaches [8]. None of our patients had headaches at the last visit. All five patients who underwent surgery had medically refractory headaches before surgery; we observed a complete resolution of headaches after the operation in all of them. However, the initial findings of endocrine deficiencies did not resolve; on the contrary, new hormonal deficiencies arose during follow-up in both observation and surgery groups. A recent study conducted on adult patients with RCC supports our findings [29]. The authors reported that the patients with presurgery pituitary dysfunction had the longest RCC diameter and that pituitary dysfunction did not improve after surgery. In our study, all five who had surgery had the largest RCC maximal dimension. The surgery led to an improvement of headache and hyperprolactinemia.
There are some limitations in the present study. We identified only 14 symptomatic patients with RCC over an 18-year period who fulfilled the criteria. Due to the retrospective design and the small number of subjects, detailed comparison analysis could not be performed, and thus, definitive conclusions cannot be drawn. Secondly, there might be selection bias, as we see the symptomatic patients referred to a tertiary university center due to further explanation of persistent headaches and hormonal disturbances. This might also be the explanation for the higher frequency of pituitary insufficiencies. Thirdly, the RCC diagnosis was based on the characteristic appearance of the MRI, and data on cyst size or location were extracted from the medical files.
The strengths of our study include its relatively long follow-up period. Only a few studies report the natural course of RCC during childhood. We demonstrated long-term outcomes and the factors associated with a recurrence. Secondly, we performed a detailed endocrine work-up in our cohort with stimulation tests. Lastly, a histopathological examination was performed on all surgically managed patients, allowing us to make a definitive diagnosis.
In conclusion, symptomatic RCCs are rare in children and are accompanied by a high frequency of endocrine disturbances, with GHD and AVP-D being the most common. Neither the size nor the location of RCC will help to define signs and symptoms. Surgery has benefits in improving headaches and visual disturbances, but not in improving endocrine deficiencies. Our findings suggest that most patients can be managed conservatively, and the cyst may shrink or even disappear entirely in the natural course. Residual cysts on postoperative MRI, recurrence of headache, and hyperprolactinemia might suggest a relapse.
-
Research ethics: This study protocol was reviewed and approved by our institution’s Review Board.
-
Informed consent: Not applicable.
-
Author contributions: ANC and HGD contributed to the study design, collected data, and prepared the final manuscript. ANC, MM, RT, MB, HGD, and JW were involved in data interpretation and manuscript drafting. All authors have accepted responsibility for the entire content of this manuscript and approved its submission.
-
Use of Large Language Models, AI and Machine Learning Tools: None declared.
-
Conflict of interest: The authors have no conflicts of interest to declare.
-
Research funding: None declared.
-
Data availability: The authors confirm that the data supporting the findings of this study are available within the article.
References
1. Teramoto, A, Hirakawa, K, Sanno, N, Osamura, Y. Incidental pituitary lesions in 1,000 unselected autopsy specimens. Radiology 1994;193:161–4. https://doi.org/10.1148/radiology.193.1.8090885.Search in Google Scholar PubMed
2. Gadelha, MR, Wildemberg, LE, Lamback, EB, Barbosa, MA, Kasuki, L, Ventura, N. Approach to the patient: differential diagnosis of cystic sellar lesions. J Clin Endocrinol Metab 2022;107:1751–8. https://doi.org/10.1210/clinem/dgac033.Search in Google Scholar PubMed
3. Nomikos, P, Buchfelder, M, Fahlbusch, R. Intra- and suprasellar colloid cysts. Pituitary 1999;2:123–6. https://doi.org/10.1023/a:1009983414014.10.1023/A:1009983414014Search in Google Scholar
4. Jahangiri, A, Molinaro, AM, Tarapore, PE, Blevins, L, Auguste, KI, Gupta, N, et al.. Rathke cleft cysts in pediatric patients: presentation, surgical management, and postoperative outcomes. Neurosurg Focus 2011;31:E3. https://doi.org/10.3171/2011.5.focus1178.Search in Google Scholar
5. Takanashi, J, Tada, H, Barkovich, AJ, Saeki, N, Kohno, Y. Pituitary cysts in childhood evaluated by MR imaging. Am J Neuroradiol 2005;26:2144–7.Search in Google Scholar
6. Güneş, A, Özbal Güneş, S. The neuroimaging features of Rathke’s cleft cysts in children with endocrine-related diseases. Diagn Interv Radiol (Ankara, Turkey) 2020;26:61–7. https://doi.org/10.5152/dir.2019.19352.Search in Google Scholar PubMed PubMed Central
7. Jung, JE, Jin, J, Jung, MK, Kwon, A, Chae, HW, Kim, DH, et al.. Clinical manifestations of Rathke’s cleft cysts and their natural progression during 2 years in children and adolescents. Ann Pediatr Endocrinol Metab 2017;22:164–9. https://doi.org/10.6065/apem.2017.22.3.164.Search in Google Scholar PubMed PubMed Central
8. Shepard, MJ, Elzoghby, MA, Kiehna, EN, Payne, SC, Jane, JA. Presentation and outcomes in surgically and conservatively managed pediatric Rathke cleft cysts. J Neurosurg Pediatr 2018;21:308–14. https://doi.org/10.3171/2017.9.Peds17400.Search in Google Scholar PubMed
9. Zada, G. Rathke cleft cysts: a review of clinical and surgical management. Neurosurg Focus 2011;31:E1. https://doi.org/10.3171/2011.5.Focus1183.Search in Google Scholar PubMed
10. Frisén, L. Swelling of the optic nerve head: a staging scheme. J Neurol Neurosurg Psychiatry 1982;45:13–18. https://doi.org/10.1136/jnnp.45.1.13.Search in Google Scholar PubMed PubMed Central
11. Kromeyer-Hauschild, K, Wabitsch, M, Kunze, D, Geller, F, Geiß, HC, Hesse, V, et al.. Perzentile für den Body-mass-Index für das Kindes- und Jugendalter unter Heranziehung verschiedener deutscher Stichproben. Monatsschr Kinderheilkd 2001;149:11.10.1007/s001120170107Search in Google Scholar
12. Marshall, WA, Tanner, JM. Variations in pattern of pubertal changes in girls. Arch Dis Child 1969;44:291–303. https://doi.org/10.1136/adc.44.235.291.Search in Google Scholar PubMed PubMed Central
13. Marshall, WA, Tanner, JM. Variations in the pattern of pubertal changes in boys. Arch Dis Child 1970;45:13–23. https://doi.org/10.1136/adc.45.239.13.Search in Google Scholar PubMed PubMed Central
14. Park, JK, Lee, EJ, Kim, SH. Optimal surgical approaches for Rathke cleft cyst with consideration of endocrine function. Neurosurgery 2012;70:250–6; discussion: 256–257. https://doi.org/10.1227/NEU.0b013e3182418034.Search in Google Scholar PubMed
15. Higuchi, Y, Hasegawa, K, Kubo, T, Tanaka, H, Tsukahara, H. The clinical course of Rathke’s cleft cysts in pediatric patients: impact on growth and pubertal development. Clin Pediatr Endocrinol 2022;31:38–43. https://doi.org/10.1297/cpe.2021-0034.Search in Google Scholar PubMed PubMed Central
16. Souteiro, P, Maia, R, Santos-Silva, R, Figueiredo, R, Costa, C, Belo, S, et al.. Pituitary incidentalomas in paediatric age are different from those described in adulthood. Pituitary 2019;22:124–8. https://doi.org/10.1007/s11102-019-00940-4.Search in Google Scholar PubMed
17. Iannelli, A, Martini, C, Cosottini, M, Castagna, M, Bogazzi, F, Muscatello, L. Rathke’s cleft cysts in children: clinical, diagnostic, and surgical features. Childs Nerv Syst 2012;28:297–303. https://doi.org/10.1007/s00381-011-1626-3.Search in Google Scholar PubMed
18. Müller, HL, Gebhardt, U, Faldum, A, Warmuth-Metz, M, Pietsch, T, Pohl, F, et al.. Xanthogranuloma, Rathke’s cyst, and childhood craniopharyngioma: results of prospective multinational studies of children and adolescents with rare sellar malformations. J Clin Endocrinol Metab 2012;97:3935–43. https://doi.org/10.1210/jc.2012-2069.Search in Google Scholar PubMed
19. Lee, JS, Kim, YH, Koh, EJ, Phi, JH, Kim, KH, et al.. Surgical indication of pediatric Rathke’s cleft cyst based on a 20-year retrospective cohort. J Neurosurg Pediatr 2023;32:729–38. https://doi.org/10.3171/2023.7.Peds23181.Search in Google Scholar PubMed
20. Brandel, MG, Lin, C, Rennert, RC, Plonsker, JH, Khan, UA, Crawford, JR, et al.. Surgical management of Rathke cleft cysts in pediatric patients: a single institution experience. Childs Nerv Syst 2024;40:1367–75. https://doi.org/10.1007/s00381-024-06277-z.Search in Google Scholar PubMed PubMed Central
21. Hayes, AG, Low, JP, Shoung, N, Fung, S, McCormack, AI. Inflammation of adenohypophysis is commonly associated with headache in surgically managed Rathke’s cleft cysts. Pituitary 2024;28:9. https://doi.org/10.1007/s11102-024-01486-w.Search in Google Scholar PubMed
22. Mukherjee, JJ, Islam, N, Kaltsas, G, Lowe, DG, Charlesworth, M, Afshar, F, et al.. Clinical, radiological and pathological features of patients with Rathke’s cleft cysts: tumors that may recur. J Clin Endocrinol Metab 1997;82:2357–62. https://doi.org/10.1210/jcem.82.7.4043.Search in Google Scholar PubMed
23. Lim, HH, Yang, SW. Risk factor for pituitary dysfunction in children and adolescents with Rathke’s cleft cysts. Korean J Pediatr 2010;53:759–65. https://doi.org/10.3345/kjp.2010.53.7.759.Search in Google Scholar PubMed PubMed Central
24. Oh, YJ, Park, HK, Yang, S, Song, JH, Hwang, IT. Clinical and radiological findings of incidental Rathke’s cleft cysts in children and adolescents. Ann Pediatr Endocrinol Metab 2014;19:20–6. https://doi.org/10.6065/apem.2014.19.1.20.Search in Google Scholar PubMed PubMed Central
25. Dadej, D, Skraba, K, Matyjaszek-Matuszek, B, Świrska, J, Ruchała, M, Ziemnicka, K. Presenting symptoms and endocrine dysfunction in Rathke cleft cysts - a two-centre experience. Endokrynol Pol 2021;72:505–11. https://doi.org/10.5603/EP.a2021.0091.Search in Google Scholar PubMed
26. Trifanescu, R, Ansorge, O, Wass, JA, Grossman, AB, Karavitaki, N. Rathke’s cleft cysts. Clin Endocrinol (Oxf) 2012;76:151–60. https://doi.org/10.1111/j.1365-2265.2011.04235.x.Search in Google Scholar PubMed
27. Shareef, M, Nasrallah, MP, AlArab, N, Atweh, LA, Zadeh, C, Hourani, R. Pituitary incidentalomas in paediatric population: incidence and characteristics. Clin Endocrinol (Oxf) 2021;94:269–76. https://doi.org/10.1111/cen.14353.Search in Google Scholar PubMed
28. Punukollu, A, Franklin, BA, Pineda, FG, Kuhar, K, Sayudo, IF, Chen, HC, et al.. Efficacy and safety of surgical management for Rathke’s cleft cysts in pediatric patients: a systematic review and meta-analysis. Neurosurg Rev 2024;48:13. https://doi.org/10.1007/s10143-024-03156-8.Search in Google Scholar PubMed
29. Alsavaf, MB, Gosal, JS, Wu, KC, Varthya, SB, Abouammo, MD, Prevedello, LM, et al.. Growth dynamics of Rathke’s Cleft cyst: a risk score system for surgical decision making. Acta Neurochir 2024;166:407. https://doi.org/10.1007/s00701-024-06299-1.Search in Google Scholar PubMed PubMed Central
© 2025 Walter de Gruyter GmbH, Berlin/Boston
Articles in the same Issue
- Frontmatter
- Review
- Subclinical but significant? Updated review of pediatric hypothyroidism
- Original Articles
- The differential impact of automated insulin delivery systems on body mass index in children with type 1 diabetes
- Maturity-onset diabetes of the young due to HNF1β variants (HNF1β-MODY): a 2-year follow-up study of six patients from a single diabetes center
- Investigating the kynurenine pathway in pediatric metabolic health
- Mucolipidosis type II and III: clinical spectrum, genetic landscape, and longitudinal outcomes in a pediatric cohort with six novel mutations
- Evaluation of the genetic alterations landscape of differentiated thyroid cancer in children
- Prognostic analysis of persistent disease in medium-to high-risk children and adolescents with differentiated thyroid carcinoma
- The clinical picture of symptomatic Rathke cleft cysts in children
- Pitfalls in the diagnosis of carnitine palmitoyltransferase 1 deficiency
- Effective treatment of hyperphosphatemia with denosumab in patients with loss of function of FGF23 and high bone density: case series
- Case Reports
- Dual molecular genetic diagnosis with combined malonic and methylmalonic aciduria (CMAMMA): implications of coexisting genetic disorders on clinical presentation
- Family experience with individuals of different ages and clinical presentations diagnosed with DI: do familial DI cases tolerate polyuria better?
- Atypical pediatric presentation of hyperparathyroidism: CDC73 gene mutation and parathyroid carcinoma
- Pseudohypertriglyceridemia as a clue: clinical and genetic spectrum of glycerol kinase deficiency in three pediatric cases
- Transient worsening of thyrotoxic myopathy following methimazole and metoprolol initiation in a 12-year-old girl: a case report and literature review
- Letter to the Editor
- Cognitive behavioral therapy (CBT) effect on diabetic youth depression, death anxiety and glycemic control
- Annual Reviewer Acknowledgment
- Reviewer Acknowledgment
Articles in the same Issue
- Frontmatter
- Review
- Subclinical but significant? Updated review of pediatric hypothyroidism
- Original Articles
- The differential impact of automated insulin delivery systems on body mass index in children with type 1 diabetes
- Maturity-onset diabetes of the young due to HNF1β variants (HNF1β-MODY): a 2-year follow-up study of six patients from a single diabetes center
- Investigating the kynurenine pathway in pediatric metabolic health
- Mucolipidosis type II and III: clinical spectrum, genetic landscape, and longitudinal outcomes in a pediatric cohort with six novel mutations
- Evaluation of the genetic alterations landscape of differentiated thyroid cancer in children
- Prognostic analysis of persistent disease in medium-to high-risk children and adolescents with differentiated thyroid carcinoma
- The clinical picture of symptomatic Rathke cleft cysts in children
- Pitfalls in the diagnosis of carnitine palmitoyltransferase 1 deficiency
- Effective treatment of hyperphosphatemia with denosumab in patients with loss of function of FGF23 and high bone density: case series
- Case Reports
- Dual molecular genetic diagnosis with combined malonic and methylmalonic aciduria (CMAMMA): implications of coexisting genetic disorders on clinical presentation
- Family experience with individuals of different ages and clinical presentations diagnosed with DI: do familial DI cases tolerate polyuria better?
- Atypical pediatric presentation of hyperparathyroidism: CDC73 gene mutation and parathyroid carcinoma
- Pseudohypertriglyceridemia as a clue: clinical and genetic spectrum of glycerol kinase deficiency in three pediatric cases
- Transient worsening of thyrotoxic myopathy following methimazole and metoprolol initiation in a 12-year-old girl: a case report and literature review
- Letter to the Editor
- Cognitive behavioral therapy (CBT) effect on diabetic youth depression, death anxiety and glycemic control
- Annual Reviewer Acknowledgment
- Reviewer Acknowledgment