
Clinical heterogeneity and molecular profile of triple A syndrome: a study of seven cases
-
Kanika Singh
 ,
Ratna Dua Puri
,
Ratna Dua Puri
 ,
Pratibha Bhai
,
Pratibha Bhai

Abstract
Background
Triple A syndrome is characterized by achalasia, alacrima and adrenal insufficiency with neurological manifestations occurring later in the course of the disease. It occurs due to biallelic mutations in the AAAS gene which codes for the nuclear pore protein ALADIN. A number of other features have been reported over time in this heterogeneous and multisystemic disorder. Unlike other autosomal recessive disorders, triple A syndrome patients show a wide phenotypic variability both among different patients and family members harboring the same mutation(s). A gene-environment interaction has been thought to be a plausible cause.
Methods
A retrospective analysis of six families and seven patients presenting with triple A syndrome was carried out. The clinical, biochemical and molecular testing data were collected and correlated. The results of treatment and follow-up and genetic counseling of the families were obtained wherever feasible.
Results
Our cohort consisted mostly of children and displayed a wide phenotypic variability in the presenting symptoms ranging from hypoglycemic seizures at the severe end of the spectrum to insidious hyperpigmentation and delayed development. Neurological and autonomic features were present in a few patients, suggesting requirement of prolonged follow-up for these patients. A significant gap between the onset of symptoms and confirmatory diagnosis was noted, suggesting that a high index of suspicion is required for diagnosing this disorder. Sudden unexplained death was observed in siblings, and early diagnosis and treatment could help in preventing early mortality and improving the quality of life for these patients.
Conclusion
High index of suspicion for a potentially treatable disorder allows early appropriate intervention.
Introduction
Triple A (AAA) syndrome or Allgrove syndrome (OMIM ID: 231550) was first described by Allgrove et al. in 1978 [1]. It is an autosomal recessive syndrome presenting with a clinical triad of adrenocorticotropic hormone (ACTH) resistant adrenal insufficiency, alacrima and achalasia. A number of other features have been found to be associated with this syndrome, such as xerostomia, dental caries, hyperkeratosis, gait disturbances and delayed puberty [2, 3]. In addition, some patients develop autonomic and neurological symptoms in adulthood [2, 4, 5]. In some cases, neurological symptoms have been noted earlier in childhood as well [2]. This suggests the multi-systemic nature of the disorder with a wide phenotypic variability. Mutations in the AAAS gene on chromosome 12q13, encoding the nuclear pore protein ALADIN, are known to cause this syndrome. Molecular testing of the AAAS gene allows confirmation of diagnosis and guides genetic counseling for the affected families. Isolated case reports of patients affected with this disorder have been described; however, there is limited molecular characterization and clinical profiling for this rare syndrome. The present study was carried out to describe the clinical profile, genotype and management of patients affected with triple A syndrome. A knowledge of the heterogeneous clinical presentation and high index of suspicion to diagnose this syndrome is essential to institute early treatment and improved outcomes.
Materials and methods
The case records of seven patients referred to the medical genetics and pediatric endocrinology clinic for the evaluation of triple A syndrome from January 2000 to December 2017 were reviewed. These seven patients had any two of the three features (alacrima, addisonianism, achalasia) suggestive of this syndrome and were thus included in this study. A detailed phenotype as per the proforma was compiled for all patients. Patients with incomplete data were recalled and re-examined as feasible.
The clinical data and relevant investigation information such as ACTH levels, serum cortisol, electrolytes and blood sugar were collected and correlated with the clinical presentation. The legal guardians gave permission for genetic testing of their child as per departmental protocol.
AAAS gene analysis
Genomic DNA was extracted from leukocytes isolated from peripheral blood samples of the patient using standard methods. Primers were designed to amplify all 16 coding exons and flanking introns of the AAAS gene (Accession No: NM_015665) using web primer software (primer sequences available on request). Amplified polymerase chain reaction (PCR) products were purified and sequenced using the BigDye Terminator sequencing kit and a 3500 genetic analyzer (Applied Biosystems, Foster city, CA, USA) according to the manufacturer’s protocol. Chromatograms obtained after sequencing were analyzed using the ChromasPro software (technelysium.com.au) and matched to the wild type AAAS gene sequence to identify the disease causing mutation.
In silico analysis of variants
In silico analysis tools like PolyPhen-2 [6], SIFT (sorting intolerant from tolerant) [7], Mutation Taster [8], Mutation Assessor [9], Provean [10] and BDGP (Berkeley Drosophila Genome Project) (for splice variant) [11] were used to predict the effect of variants.
An informed consent was obtained from all the patients/guardians for the molecular analysis of the AAAS gene. A gene study of the AAAS gene was carried out in six of the seven patients who provided consent for molecular testing.
Results
Clinical features
Patient characteristics are presented in Table 1. The prenatal and antenatal history were uneventful for all the patients. Consanguinity was present in three of the six families, the parents being first cousins. Five of the six families had more than one affected child. The total number of affected children in the five families were 11 (male=7, female=4). Molecular diagnosis was confirmed in six of the seven children who were brought for evaluation. Intrafamilial variability in the clinical symptoms was observed. Sudden death was reported in three of the siblings and a confirmatory diagnosis had not yet been established in them. In the case of patient 1 who presented chiefly with intellectual disability, her brother was more severely affected with a history of hyperpigmentation, adrenal insufficiency, poor growth and recurrent illnesses. Although he was on hydrocortisone replacement, the etiological diagnosis had not been established and he passed away during an illness at the age of 8 years. There was a history of sudden death of a sibling of patient 2 due to an unknown cause at 2 years of age. Patients 3 and 4 were siblings and were brought for the evaluation together and were similarly affected. However, an earlier sibling had passed away at 3 years of age and though there was a history of hyperpigmented skin, he had not been diagnosed. The sibling of patient 5 was more severely affected and diagnosed with impending adrenal failure and was under treatment at another facility. The sibling of patient 6 was reported to be healthy and the sibling of patient 7 had alacrima but was otherwise not significantly affected as per history. Thus, three of the 11 children had passed away without any definitive diagnosis.
Clinical and molecular details of the seven patients enrolled in the study.
| Patient no. | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| Gender | F | M | F | M | F | F | M |
| Consanguinity | + | + | – | – | – | + | – |
| Family no. | 1 | 2 | 3 | 3 | 4 | 5 | 6 |
| Family history | Affected sibling – died at 8 years | Asymptomatic sibling – died suddenly | Two affected siblings – one died | Two affected siblings – one died | Affected sibling+ | – | Affected sibling+ |
| Symptomatic age | 1 year | 2 years | 3 years | Birth | 1 year | 1 year | 1 year |
| Age at diagnosis | 12 years | 4.5 years | 3 years | 3 months | 5 years | 1 years 7 months | 8 years |
| Presenting symptom | Developmental delay | Hypoglycemic seizures | Developmental delay and hyperpigmentation | Developmental delay, hyperpigmentation | Hyperpigmentation and poor growth | Hyperpigmentation | Hyperpigmentation and gait imbalance |
| Alacrima | + | + | + | + | + | + | + |
| Adrenal insufficiency | + | + | + | + | + | + | + |
| Achalasia | + | – | – | – | – | + | + |
| Developmental delay | + | + | + | + | – | – | – |
| Poor growth | – | – | + | – | + | – | – |
| Hyperpigmentation of skin | + | + | + | + | + | + | + |
| Gait disturbance | + | – | – | – | – | – | + |
| Hyperreflexia | – | – | – | – | – | – | – |
| Distal weakness | – | – | – | – | – | – | – |
| Atrophy | – | – | – | – | – | – | – |
| Abnormal sweating | – | – | + | – | – | – | – |
| Postural hypotension | – | – | – | – | – | – | – |
| S. ACTH, pg/mL (n<28) | 271 | 1250 | 648 | N.A. | N.A. | >1250 | 3120 |
| S. cortisol 8 am, μg/dL (n>5) | <0.5 | 0.5 | 6.00 | N.A. | N.A. | 4.71 | 0.33 |
| Random blood sugar, mg/dL | N.A. | 30 | N.A. | N.A. | N.A. | 71 | 41.9 |
| S. aldosterone, pg/mL | N.A. | 519 | N.A. | N.A. | N.A. | ||
| S. Na/K | 145/4.8 | 136/4.7 | 146/3.9 | N.A. | N.A. | 132/4.4 | 138/4.7 |
| AAAS gene analysis | Homozygous c.762delC p.Ser255Valfs*35 | Homozygous c.899_901delinsTCT p.Ser300Ilefs*2 | Homozygous c.762delC p.Ser255Valfs*35 | Homozygous c.762delC p.Ser255Valfs*35 | Homozygous c.1432C>T p.Arg478* | Homozygous c.899_901delinsTCT p.Ser300Ilefs*2 | N.A. |
| Parents | Both heterozygous | Both heterozygous | Both heterozygous | Both heterozygous | N.A. | Both heterozygous | N.A. |
N.A., not available; M, male; F, female; N, normal value; S., serum.
The average age at recognition of symptoms by parents was 15 months and the average age at diagnosis was 5 years, indicating a delay in the recognition of symptoms by parents and/or physicians. The presenting complaint included developmental delay (patients 1, 3 and 4), hypoglycemia and seizures (patient 2), poor growth (patient 5), hyperpigmentation of skin (patients 3, 4, 5, 6, 7) and gait imbalance (patient 7).
Alacrima
Alacrima or the absence of tears was present in all the patients. The alacrima was noted since birth although it was not the presenting complaint for the patients.
Achalasia
Symptoms of achalasia were reported in three patients (patients 1, 6 and 7). It involved difficulty in swallowing foods and an aversion for food, along with increased time in completing meals, as perceived by the parents. These symptoms were seen to develop later in childhood (ages 3–15 years) as compared to the other symptoms. Patient 6 had severe symptoms and required a myotomy for the treatment of the achalasia.
Adrenal insufficiency/addisonianism
Adrenal insufficiency was biochemically confirmed in five of the seven patients. Hyperpigmentation of skin was seen in all patients at the time of presentation. Patient 2 presented with hypoglycemia, metabolic acidosis and seizures and patient 5 had poor growth which could be attributed to an underlying glucocorticoid deficiency.
Laboratory investigations (Table 1) showed high ACTH levels (normally up to 28 pg/mL in prepubertal children) and a low morning serum cortisol (cut-off <5 μg/dL) in five of the seven patients. Hypoglycemia as a consequence of adrenal insufficiency was seen in two patients (patients 2 and 7). The serum electrolytes were normal in patients 1, 2, 3, 6 and 7 reflecting a normal mineralocorticoid status.
Developmental delay
Developmental delay was present in four of the seven patients (patients 1, 2, 3 and 4). Although there was a mild delay in achieving the milestones in all the domains, there was a catch-up development in the motor domain. Intellectual disability was present in four patients (intelligence quotient [IQ]=50–70).
Skin abnormalities
All the patients had darkening of skin, which was noted prior to the onset of symptoms. In addition, patient 1 had hyperkeratosis of the palms and soles.
Poor growth
The weight of patients 3 and 5 was below the 3rd centile for age, while the height and head circumference were within the normal range for age. The other children did not show significantly altered growth parameters.
Neurological abnormalities
Unsteady gait and frequent falls were reported in two patients (patients 1 and 7). Other neurological findings that have been reported in the literature, albeit usually in adults like hyperreflexia, distal weakness and atrophy, were not seen in any of the children in our cohort.
Autonomic disturbances
One of the patients had a history of abnormal sweating (the elder sibling of the family, patient 3). No other patient had any symptom suggestive of postural hypotension or sweating disturbance to indicate autonomic involvement.
AAAS gene mutation study
Molecular testing of the AAAS gene was done in six of the seven patients. Three homozygous mutations were identified in the six patients: c.762delC in exon 8, c.899_901delinsTCT in exon 9 and c.1432C>T in exon 16. Two mutations (c.762delC and c.1432C>T) have been previously reported [12, 13]. Three patients, two being siblings (patients 1, 3 and 4), were homozygous for mutation c.762delC in exon 8. The mutation found in patients 2 and 6 (c.899_901delinsTCT) was a novel variant, and predicted to be disease causing by pathogenicity prediction software tool, Mutation Taster [8], (Table 2) and their parents were heterozygous carriers for the same variation (Figure 1).
AAAS pathogenic variants found in the study.
| Nucleotide change (zygosity) | Dbsnp ID | Exon no. | Amino acid change | Allele frequency in database | Novel/reported | In silico predictions | No of patients positive for mutation |
|---|---|---|---|---|---|---|---|
| c.762delC Homozygous | rs746057093 | 8 | p.Ser255Valfs*35 | ExAc: 0.00003 | Reported | – | 3 |
| c.899_901delinsTCT Homozygous | Not found in Dbsnp | 9 | Ser300Ilefs*2 | Neither found in ExAC nor 1000G | Novel | Pathogenica | 2 |
| c.1432C>T Homozygous | rs121918548 | 16 | p.Arg478* | ExAc: 0.00005 | Reported | – | 1 |
aAnalysis subjected to in silico tools like mutation Taster.
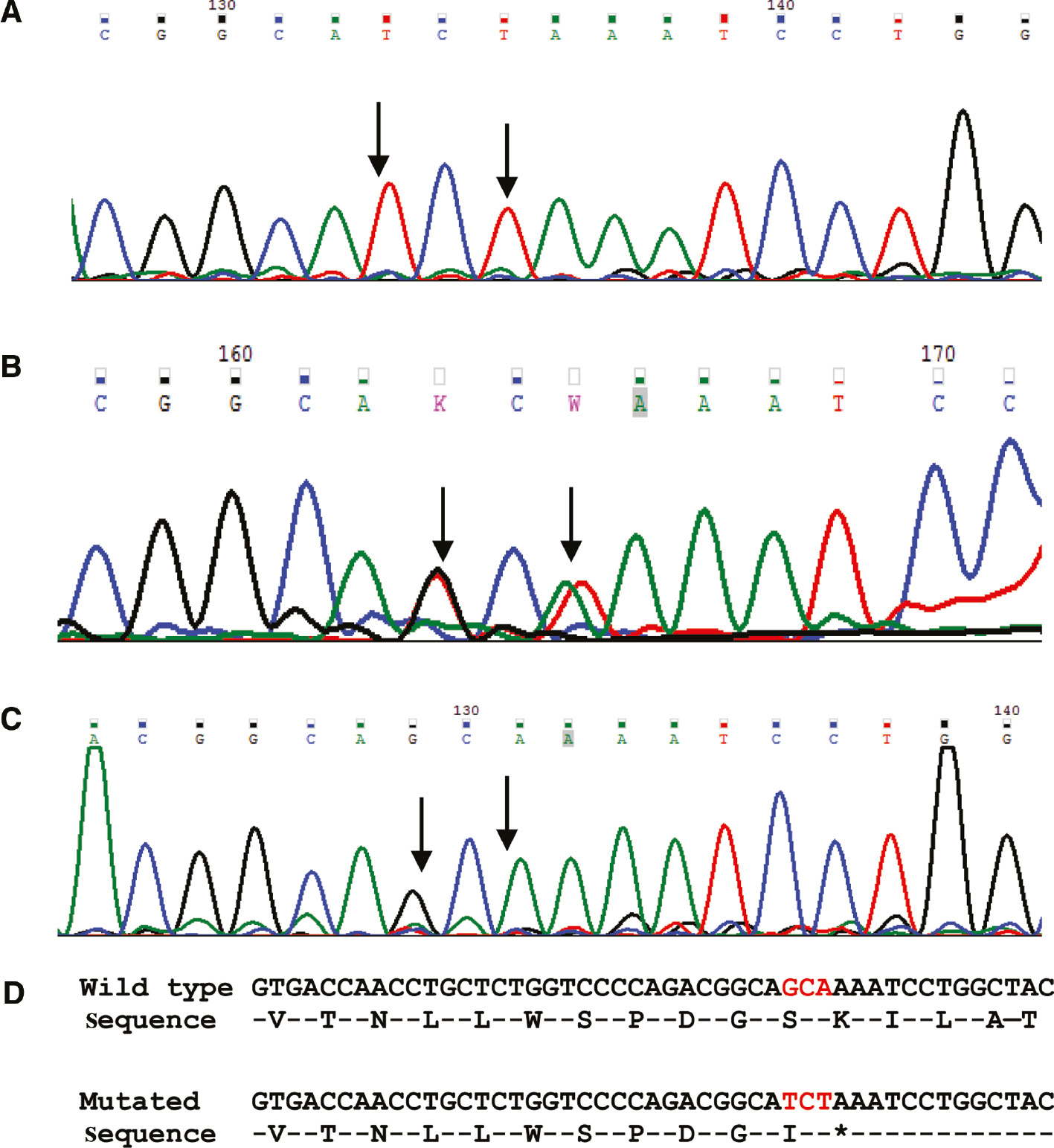
Electropherogram of the novel mutation identified in the AAAS gene (c.899_901delinsTCT).
(A) Electropherogram showing homozygous nucleotide changes in the affected child. (B) Electropherogram showing heterozygous nucleotide changes in the mother of affected child. (C) Electropherogram showing normal sequence in a negative control sample (normal individual). (D) Wild type and mutated DNA and amino acid sequence.  Arrows denote the position of nucleotide change.
Arrows denote the position of nucleotide change.
Treatment and course
All the patients were treated with oral hydrocortisone (10–15 mg/m2/day) and showed improvement in symptoms. The hyperpigmentation of skin decreased and the parents reported improvement in the general well-being and activity of their children. In addition, there was an upward trend in the weight centiles of patients 3 and 5. The patients were informed regarding the need to increase the dose during stress or infection and shift to intravenous (IV) hydrocortisone in case of an inability to accept it orally. They were advised lubricant eye drops to prevent dryness of eyes. The symptoms of achalasia were not severe and required a mild lifestyle adjustment, except for patient 6 who required a surgical myotomy. While the siblings (patients 3 and 4) had been lost to follow-up, all the other patients were doing well with treatment, highlighting the importance of establishing a definitive diagnosis and anticipating complications.
Genetic counseling and prenatal diagnosis
The families had been counseled of the autosomal recessive inheritance pattern of triple A syndrome and the 25% risk of an affected offspring in each conception. The availability of prenatal diagnosis was discussed with all couples planning another pregnancy. The carrier status of parents of all patients except patient 5 was confirmed. The parents of patient 2 desired a prenatal diagnosis in the subsequent pregnancy. A prenatal diagnosis was done by chorionic villus sampling at 11 weeks of pregnancy and the fetus was confirmed to be unaffected. The parents of patient 1 did not opt for a prenatal diagnosis in an ongoing pregnancy. The pregnancy resulted in a missed abortion. The family is now considering planning a future pregnancy using a donor egg or sperm to overcome the need for prenatal diagnosis as well as preimplantation genetic diagnosis.
The desire for prenatal diagnosis and termination in manageable and non-life threatening disorders in a developing country like India, where medical care is an out-of-pocket expenditure has been reported earlier [14]. A knowledge of differing attitudes toward prenatal diagnosis can help to guide genetic counseling in such disorders.
Discussion
We present here the clinical and molecular profile of six families with triple A syndrome. Triple A syndrome is typically diagnosed in children, with very few cases described in the literature as late onset or adult cases. The late onset cases are reported to present with neurological manifestations. Our cohort consisted of children who presented chiefly with features due to the underlying adrenal insufficiency.
Alacrima is the earliest and most consistent finding but is often overlooked by parents. This is possibly due to its negligible clinical relevance and morbidity. Although all the patients in this series had alacrima since birth it did not pre-empt medical attention. This was noted similarly by two other studies wherein all the patients had alacrima. Most patients come to attention due to the underlying adrenal insufficiency as observed in the present and earlier series [2, 15, 16, 17] (Table 3). The presentation of adrenal insufficiency was variable with severe manifestations of hypoglycemia and ketoacidosis (patient 2) at one end and insidious findings of hyperpigmentation of the skin (all patients) and poor growth (two of seven patients) at the other end of the spectrum. Presentation with hypoglycemia, metabolic acidosis and convulsions is reported earlier and should alert the physician to consider adrenal insufficiency as a potential cause [16, 18] (Table 3). The cause of death in three children might have been due to an event of adrenal crisis precipitated by stress or infection. It has been seen that in triple A syndrome, hypofunctioning of the adrenal glands is not present at birth but rather develops over time and is progressive. This seems plausible as all the patients in our cohort were reported to have alacrima since birth but the symptoms of adrenal insufficiency were noted only after 1 year of age. The adrenal impairment is generally restricted to glucocorticoid deficiency as was seen in this cohort, although mineralocorticoid deficiency has been reported in roughly 15% of patients in other studies [19]. Developmental delay was not present in all the patients, but when present it was a significant cause of concern for the parents. Though cognitive impairment may not be present in all cases, it has been reported in many children with triple A syndrome [2, 15, 16, 17] (Table 3). The reason for this is not clear, but is possibly a part of the central nervous system manifestations.
Comparison of clinical and molecular profile of patients with triple A syndrome previously reported.
| Milenković et al. [15] | Yassaee et al. [16] | Dumic et al. [2] | Kallabi et al. [17] | Our study | |
|---|---|---|---|---|---|
| Age group | 2–22 years | 5–21 years | 2–11 years | 1–10 years | 3 months–12 years |
| No of families, n | 18 | 5 | 5 | 25 | 6 |
| No of patients, n | 20 | 6 | 8 | 26 | 7 |
| Phenotype, n | 11 | 6 | 8 | 26 | 7 |
| Parental consanguinity present, n | N.A. | 5/5 | 0/5 | 17/25 | 3/6 |
| Family history positive, n | 1/18 | 2/5 | 3/5 | 1/25 | 5/6 |
| Presenting features | N.A. | Achalasia, hypoglycemia and seizures | Poor growth, hyperpigmentation, hypoglycemia, achalasia | Achalasia, hyperpigmentation, weakness | Developmental delay, hyperpigmentation, hypoglycemia and seizures |
| Alacrima, n | 9/11 | 6/6 | 8/8 | 26/26 | 7/7 |
| Achalasia, n | 10/11 | 6/6 | 6/8 | 23/26 | 3/7 |
| Glucocorticoid deficiency, n | 6/11 | 6/6 | 8/8 | 26/26 | 7/7 |
| Hyperpigmentation, n | N.A. | N.A. | N.A. | N.A. | 7/7 |
| Other skin findings, n | 4/11 | N.A. | 8/8 | N.A. | 1/7 |
| Poor growth, n | N.A. | N.A. | N.A. | N.A. | 2/7 |
| Neurological manifestations, n | 9/11 | 1/6 | 8/8 | 7/26 | 2/7 |
| Autonomic symptoms, n | 1/11 | N.A. | 6/8 | N.A. | 0/7 |
| Intellectual disability, n | N.A. | 2/6 | 3/8 | 4/26 | 4/7 |
| AAAS gene | Founder mutation S263Pa | S263P, Q15K, IVS14+ 1G>C, L356fs*, R312* | S263Pa, S296Y, G14fs*, Q387* | Founder mutation c.1331+1G>Ab | S300Ifs*c, S300Ifs*, R478* |
N.A., not available. aEuropean ethnic, bTunisian ethnic, cnovel.
The adrenal symptoms generally precede neurological symptoms which may not be seen at presentation in all the cases (Table 3). Rare cases have been described where the neurological symptoms were the presenting complaints with mild or no adrenal insufficiency [20, 21]. Other neurological symptoms that have been described include distal weakness and atrophy, mixed sensory motor demyelinating neuropathy, intention tremors, gait imbalance, motor neuron disease and optic atrophy [22, 23]. Postural hypotension and abnormal sweating were the most common manifestations of autonomic disturbance if present [2, 4]. Our cohort was largely devoid of significant neurological and autonomic findings except for gait imbalance in patients 2 and 7 and abnormal sweating in patient 3. This is in keeping with the late occurrence of neurological impairment in the course of the disease and the fact that our cohort consisted only of children. It is unclear whether the neurological and autonomic impairment is secondary to the effect of glucocorticoid deficiency on the postnatal development of the neurological system or is a primary manifestation. However, the former hypothesis has been suggested as an explanation for the delayed appearance of these symptoms in the lifetime [23]. A long-term follow-up of these patients would be required to monitor them for appearance of neurological symptoms and worsening.
This syndrome is an autosomal recessive disorder that occurs due to mutations in the AAAS gene consisting of 16 exons located on chromosome 12q13.13 [24]. The AAAS gene codes for a 546 amino acid long protein called ALADIN (59.6 kDa), localized to the cytoplasmic face of the nuclear pore complexes (NPCs). The NPCs are large multiprotein assemblies present on the nuclear envelope and are involved in nucleocytoplasmic transport. Functional studies have shown the inability of mutated ALADIN to localize to these NPCs and remain largely in the cytoplasm, signifying a functional rather than a morphological abnormality [25]. The AAAS gene is ubiquitously expressed in human tissues with abundant expression in the adrenal and pituitary glands, cerebellum, gastrointestinal organs and kidney [26]. This may explain the spectrum of symptoms and variability in clinical features seen in this syndrome.
We identified two reported and one novel variant in the AAAS gene, resulting in a truncated non-functional protein (Table 2). The c.899_901delinsTCT (p.Ser300Ilefs*2) has not been reported previously and is predicted to be disease causing by bioinformatics tools and noted to be conserved across different species. This mutation results in the substitution of serine at amino acid position 300 by isoleucine and a premature termination codon subsequently after amino acid position 300 and the formation of a truncated protein (300 amino acid long instead of 546 amino acid long wild type protein). Further segregation analysis showed the parents to be heterozygous for this variant. Mutations in patients with triple A syndrome are distributed through all 16 exons of the gene (Figure 2). No hot spots are identified in the gene and a comprehensive testing necessitates sequencing of the entire gene. Founder mutations have been reported in the European and Tunisian populations (Table 3).

Diagrammatic representation of pathogenic variants reported in the AAAS gene.
No clear-cut genotype-phenotype correlation could be deduced from our study, a lack of which has been seen in other studies as well [2, 27]. The late onset cases are often clinically ambiguous, highlighting the importance of molecular genetic diagnosis for confirmation of this disorder.
To conclude, this study emphasizes the variable interfamilial and intrafamilial clinical presentation of triple A syndrome. A high index of suspicion allows early diagnosis and management. Direct questioning to elucidate alacrima in a setting of adrenal insufficiency will enable appropriate clinical diagnosis. When suspected, molecular genetic testing offers definite confirmation of diagnosis, early management as well genetic counseling and reproductive options for the family.
Author contributions: All the authors have accepted responsibility for the entire content of this submitted manuscript and approved submission.
Research funding: None declared.
Employment or leadership: None declared.
Honorarium: None declared.
Competing interests: The funding organization(s) played no role in the study design; in the collection, analysis, and interpretation of data; in the writing of the report; or in the decision to submit the report for publication.
References
1. Allgrove J, Clayden GS, Grant DB, Macaulay JC. Familial glucocorticoid deficiency with achalasia of the cardia and deficient tear production. Lancet 1978;1:1284–6.10.1016/S0140-6736(78)91268-0Search in Google Scholar
2. Dumic M, Barišic N, Kusec V, Stingl K, Skegro M, et al. Long-term clinical follow-up and molecular genetic findings in eight patients with triple A syndrome. Eur J Pediatr 2012;171:1453–9.10.1007/s00431-012-1745-1Search in Google Scholar PubMed
3. Bustanji H, Sahar B, Huebner A, Ajlouni K, Landgraf D, et al. Triple A syndrome with a novel indel mutation in the AAAS gene and delayed puberty. J Pediatr Endocrinol Metab 2015;28:933–6.10.1515/jpem-2014-0401Search in Google Scholar PubMed
4. Vallet AE, Verschueren A, Petiot P, Vandenberghe N, Nicolino M, et al. Neurological features in adult Triple-A (Allgrove) syndrome. J Neurol 2012;259:39–46.10.1007/s00415-011-6115-9Search in Google Scholar PubMed
5. Jerie M, Vojtech Z, Malikova H, Prochazkova S, Vackova Z, et al. Allgrove syndrome with prominent neurological symptoms. Case Report. Neuro Endocrinol Lett 2016;37:184–8.Search in Google Scholar
6. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–9.10.1038/nmeth0410-248Search in Google Scholar PubMed PubMed Central
7. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009;4:1073–81.10.1038/nprot.2009.86Search in Google Scholar PubMed
8. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 2014;11:361–2.10.1038/nmeth.2890Search in Google Scholar PubMed
9. Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res 2011;39:e118.10.1093/nar/gkr407Search in Google Scholar PubMed PubMed Central
10. Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015;31:2745–7.10.1093/bioinformatics/btv195Search in Google Scholar PubMed PubMed Central
11. Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in genie. J Comp Biol 1997;4:311–23.10.1145/267521.267766Search in Google Scholar
12. Reimann J, Kohlschmidt N, Tolksdorf K, Weis J, Kuchelmeister K, et al. Muscle pathology as a diagnostic clue to Allgrove syndrome. J Neuropathol Exp Neurol 2017;76:337–41.10.1093/jnen/nlx016Search in Google Scholar PubMed
13. Yuksel B, Braun R, Topaloglu AK, Mungan NO, Ozer G, et al. Three children with triple A syndrome due to a mutation (R478X) in the AAAS gene. Horm Res 2004;61:3–6.10.1159/000075190Search in Google Scholar PubMed
14. Nahar R, Puri RD, Saxena R, Verma IC. Do parental perceptions and motivations towards genetic testing and prenatal diagnosis for deafness vary in different cultures? Am J Med Genet A 2013;161A:76–81.10.1002/ajmg.a.35692Search in Google Scholar PubMed
15. Milenković T, Koehler K, Krumbholz M, Zivanović S, Zdravković D, et al. Three siblings with triple A syndrome with a novel frameshift mutation in the AAAS gene and a review of 17 independent patients with the frequent p.Ser263Pro mutation. Eur J Pediatr 2008;167:1049–55.10.1007/s00431-007-0640-7Search in Google Scholar PubMed
16. Yassaee VR, Soltani Z, Ardakani BM. Mutation spectra of the AAAS gene in Iranian families with Allgrove syndrome. Arch Med Res 2011;42:163–8.10.1016/j.arcmed.2011.02.006Search in Google Scholar PubMed
17. Kallabi F, Belghuith N, Aloulou H, Kammoun T, Ghorbel S, et al. Clinical and genetic characterization of 26 Tunisian patients with Allgrove syndrome. Arch Med Res 2016;47:105–10.10.1016/j.arcmed.2016.04.004Search in Google Scholar PubMed
18. Ismail AF, Mohamed S, Ezz-Elregal M, Talat I, Muthurajan S, et al. Associations with hypoglycemia are useful: a case report of Allgrove syndrome. Turk J Gastroenterol 2012;23:608–9.10.4318/tjg.2012.0384Search in Google Scholar PubMed
19. Grant DB, Barnes ND, Dumic M, Ginalska-Malinowska M, Milla PJ, et al. Neurological and adrenal dysfunction in the adrenal insufficiency/alacrima/achalasia (3A) syndrome. Arch Dis Child 1993;68:779–82.10.1136/adc.68.6.779Search in Google Scholar PubMed PubMed Central
20. Barat P, Goizet C, Tullio-Pelet A, Puel O, Labessan C, et al. Phenotypic heterogeneity in AAAS gene mutation. Acta Paediatr 2004;93:1257–9.10.1111/j.1651-2227.2004.tb02760.xSearch in Google Scholar
21. Luigetti M, Pizzuti A, Bartoletti S, Houlden H, Pirro C, et al. Triple A syndrome: a novel compound heterozygous mutation in the AAAS gene in an Italian patient without adrenal insufficiency. J Neurol Sci 2010;290:150–2.10.1016/j.jns.2009.12.005Search in Google Scholar PubMed
22. Misgar RA, Pala NA, Ramzan M, Wani AI, Bashir MI, et al. Allgrove (Triple A) syndrome: a case report from the Kashmir Valley. Endocrinol Metab (Seoul) 2015;30:604–6.10.3803/EnM.2015.30.4.604Search in Google Scholar PubMed PubMed Central
23. Vishnu VY, Modi M, Prabhakar S, Bhansali A, Goyal MK. “A” motor neuron disease. J Neurol Sci 2014;336:251–3.10.1016/j.jns.2013.10.003Search in Google Scholar PubMed
24. Weber A, Wienker TF, Jung M, Easton D, Dean HJ, et al. Linkage of the gene for the triple A syndrome to chromosome 12q13 near the type II keratin gene cluster. Hum Mol Genet 1996;5:2061–6.10.1093/hmg/5.12.2061Search in Google Scholar PubMed
25. Cronshaw JM, Matunis MJ. The nuclear pore complex protein ALADIN is mislocalized in triple A syndrome. Proc Natl Acad Sci USA 2003;100:5823–7.10.1073/pnas.1031047100Search in Google Scholar PubMed PubMed Central
26. Handschug K, Sperling S, Yoon SJ, Hennig S, Clark AJ, et al. Triple A syndrome is caused by mutations in AAAS, a new WD-repeat protein gene. Hum Mol Genet 2001;10:283–90.10.1093/hmg/10.3.283Search in Google Scholar PubMed
27. Prpic I, Huebner A, Persic M, Handschug K, Pavletic M. Triple A syndrome:genotype-phenotype assessment. Clin Genet 2003;63:415–7.10.1034/j.1399-0004.2003.00070.xSearch in Google Scholar PubMed
©2018 Walter de Gruyter GmbH, Berlin/Boston
Articles in the same Issue
- Frontmatter
- Review
- Prevalence of cranial MRI findings in girls with central precocious puberty: a systematic review and meta-analysis
- Original Articles
- Can having a sibling with type 1 diabetes cause disordered eating behaviors?
- Wrist circumference as a novel predictor of obesity in children and adolescents: the CASPIAN-IV study
- Evaluation of Mn-superoxide dismutase and catalase gene expression in childhood obesity: its association with insulin resistance
- Impact of a group-based treatment program on adipocytokines, oxidative status, inflammatory cytokines and arterial stiffness in obese children and adolescents
- Clinical management of childhood hyperthyroidism with and without Down syndrome: a longitudinal study at a single center
- Detection of distant metastasis at the time of ablation in children with differentiated thyroid cancer: the value of pre-ablation stimulated thyroglobulin
- Random serum free cortisol and total cortisol measurements in pediatric septic shock
- Self-assessment of pubertal development in a puberty cohort
- Growth, the Mediterranean diet and the buying power of adolescents in Greece
- Clinical and molecular genetic characterization of two patients with mutations in the phosphoglucomutase 1 (PGM1) gene
- Genetic analysis of three families with X-linked dominant hypophosphatemic rickets
- Clinical heterogeneity and molecular profile of triple A syndrome: a study of seven cases
- No central adrenal insufficiency found in patients with Prader-Willi syndrome with an overnight metyrapone test
- Case Reports
- A novel mutation in the proopiomelanocortin (POMC) gene of a Hispanic child: metformin treatment shows a beneficial impact on the body mass index
- Hypertriglyceridemia thalassemia syndrome
- Severe consumptive hypothyroidism caused by multiple infantile hepatic haemangiomas
Articles in the same Issue
- Frontmatter
- Review
- Prevalence of cranial MRI findings in girls with central precocious puberty: a systematic review and meta-analysis
- Original Articles
- Can having a sibling with type 1 diabetes cause disordered eating behaviors?
- Wrist circumference as a novel predictor of obesity in children and adolescents: the CASPIAN-IV study
- Evaluation of Mn-superoxide dismutase and catalase gene expression in childhood obesity: its association with insulin resistance
- Impact of a group-based treatment program on adipocytokines, oxidative status, inflammatory cytokines and arterial stiffness in obese children and adolescents
- Clinical management of childhood hyperthyroidism with and without Down syndrome: a longitudinal study at a single center
- Detection of distant metastasis at the time of ablation in children with differentiated thyroid cancer: the value of pre-ablation stimulated thyroglobulin
- Random serum free cortisol and total cortisol measurements in pediatric septic shock
- Self-assessment of pubertal development in a puberty cohort
- Growth, the Mediterranean diet and the buying power of adolescents in Greece
- Clinical and molecular genetic characterization of two patients with mutations in the phosphoglucomutase 1 (PGM1) gene
- Genetic analysis of three families with X-linked dominant hypophosphatemic rickets
- Clinical heterogeneity and molecular profile of triple A syndrome: a study of seven cases
- No central adrenal insufficiency found in patients with Prader-Willi syndrome with an overnight metyrapone test
- Case Reports
- A novel mutation in the proopiomelanocortin (POMC) gene of a Hispanic child: metformin treatment shows a beneficial impact on the body mass index
- Hypertriglyceridemia thalassemia syndrome
- Severe consumptive hypothyroidism caused by multiple infantile hepatic haemangiomas