A germline mutation of HRPT2/CDC73 (70 G>T) in an adolescent female with parathyroid carcinoma: first case report and a review of the literature
-
Monica Serrano-Gonzalez


Abstract
Parathyroid carcinoma is a rare cause of primary hyperparathyroidism amongst children, with only nine previously reported cases. The objective of the study was to present the first pediatric case with a germline CDC73 (formerly known as HRPT2) mutation, and to review the literature. A 14-year-old girl presented with pathologic slipped capital femoral epiphysis (SCFE). The patient was noted to have an elevated calcium level of 3.4 mmol/L (13.4 mg/dL), a parathyroid hormone (PTH) level of 1013 ng/L (1013 pg/mL), and a 3-cm palpable neck mass. Ultrasound and 99mTc-Sestamibi confirmed the suspicion of a parathyroid mass. Intraoperative findings and pathology confirmed the diagnosis of parathyroid carcinoma. Post-operative PTH decreased to 14 ng/L (14 pg/mL). Genetic testing showed a germline 70 G>T HRPT2/CDC73 mutation. This is the first case documenting a germline 70 G>T HRPT2/CDC73 gene mutation in a pediatric parathyroid carcinoma. Patients with sporadic parathyroid carcinoma may benefit from HRPT2/CDC73 gene mutation screening.
Introduction
Primary hyperparathyroidism is very rare in children and adolescents, with an estimated incidence of 2–5 in 100,000 [1]. Parathyroid carcinoma is a rare cause of primary hyperparathyroidism, with a reported incidence of 0.4–5% of hyperparathyroid-related hypercalcemia [2], [3], [4], [5], [6]. Additionally, parathyroid carcinoma only accounts for 0.005% of all malignancies [7]. As a result, a relative paucity of literature exists on parathyroid carcinoma. Furthermore, parathyroid carcinoma amongst children and adolescents is exceedingly rare, with only nine previously reported cases in the English literature occurring in children under the age of 16 [2], [8], [9], [10], [11], [12], [13], [14], [15]. None of these cases has had documented HRPT2/CDC73 gene mutation analysis. We report the tenth case, which is also the first pediatric case to have a reported HRPT2/CDC73 mutation.
Materials and methods
Clinical
Routine laboratory studies on serum were performed at Children’s Hospital Los Angeles laboratory, Los Angeles, CA, USA. Calcium and serum phosphorus were measured by colorimetric spectrophotometry (Ortho Clinical Diagnostics, Rochester, NY, USA). Intact PTH (iPTH), 25-OH vitamin D and 1,25-OH vitamin D were measured at Quest Diagnostics Nichols Institute, San Juan Capistrano, CA, USA. iPTH was measured by an immunoassay method (UniCel Dxl; Beckman Coulter, Brea, CA, USA); 25-OH vitamin D and 1,25-OH vitamin D were measured by liquid chromatography-mass spectrometry (LC/MS/MS; Thermo Instrument Systems, Waltham, MA, USA). Tumor histologic evaluation was done at the pathologic laboratory at Children’s Hospital Los Angeles.
Genetic analysis
Peripheral blood samples were obtained for genetic analysis, which was performed at GeneDx, Gaithersburg, MD, USA. Genomic DNA was PCR-amplified for analysis of the coding region (exons 2–10) of the MEN1 gene as well as exons 1–17 of the HRPT2/CDC73 gene and their flanking splice sites. Bi-directional sequence was obtained and DNA sequence was analyzed and compared to the published gene sequences.
Results
Case description
A previously healthy 14-year-old Hispanic girl suffered from chronic leg pain since 6 years of age and her pediatrician repeatedly reassured the parents that this was likely due to ‘growing pains’. At 7 years old she was diagnosed with severe thoracolumbar scoliosis for which she was seen by an orthopedist and was prescribed a brace, to which she had poor treatment adherence. She developed progressive bowing of her legs together with worsening leg pain as well as bilateral groin pain that gradually increased in severity. She started to limp, and her symptoms worsened to the point where she could only walk very short distances and otherwise had to be carried. At that time, she presented to an orthopedist and was found to have a right-sided slipped capital femoral epiphysis (SCFE). The patient underwent in situ fixation of the right femur. The patient was found to have hypercalcemia of 3.05 mmol/L (12.2 mg/dL) at the time of orthopedic intervention, which warranted further work up and referral to endocrinology. Several months later, upon our initial evaluation, the patient reported diffuse bone pain most notably in her knees, increased thirst, insomnia, fatigue and difficulty concentrating at school. She also reported mild globus sensation when swallowing solid foods but denied abdominal pain. She had no history of renal disease or thyroid dysfunction. There was no family history of thyroid, parathyroid, or renal disease. On physical examination, the patient had a left anterior 3×3 cm neck mass. Pertinent preoperative laboratory data included serum total calcium of 3.4 mmol/L (13.4 mg/dL, reference range, 8.4–10.2 mg/dL), phosphorus 1.1 mmol/L (3.5 mg/dL, reference range 2.5–4.5 mg/dL), intact parathyroid hormone (iPTH) of 1013 ng/L (1013 pg/mL, reference range, 9–69 pg/mL), 25-OH vitamin D of <12.5 nmol/L (<5 ng/mL, sufficient >30 ng/mL), 1,25-OH vitamin D of 369.2 pmol/L (142 pg/mL, reference range 19–83 pg/mL), serum albumin of 43 g/L (4.3 g/dL, reference range, 3.2–4.5 g/dL), spot urine calcium of 5.6 mmol/L (22.4 mg/dL), spot urine creatinine of 6895 mmol/L (78 mg/dL), urine calcium:creatinine ratio of 0.29. All other laboratory values were within normal limits.
A 99mTechnetium (Tc)-Sestamibi scan demonstrated a large extrathyroidal 99mTc uptake at the left inferior thyroid pole that persisted at the 3-h mark, suspicious for parathyroid adenoma. A high-resolution ultrasound of the neck found a well-defined extrathyroidal mass at the inferior aspect of the left thyroid lobe that correlated with the 99mTc-Sestamibi scan. The mass measured 3.3×2.3×2.9 cm with central heterogeneous echogenicity, soft tissue echogenicity, and some cystic portions, giving it an unusual appearance for a parathyroid adenoma.
The patient underwent left neck exploration and parathyroidectomy with recurrent laryngeal nerve monitoring. At the time of surgery, a firm, 3.5×3×1.3 cm left inferior parathyroid mass was encountered, and found to be adherent to surrounding tissues, including the anterior tracheal wall fascia, the left inferior thyroid, and the left thymic tongue; thus, a wide local excision was performed. There was no cervical adenopathy identified in the tracheoesophageal groove, but the left recurrent laryngeal nerve was significantly displaced laterally by the tumor. En-bloc resection of the tumor along with a left inferior thyroid lobectomy was performed. The performed neck dissection included an ipsilateral selective neck dissection of levels III and IV from the left common carotid artery laterally to the anterior tracheal wall fascia medially and a complete level VI lymph node dissection down to the brachiocephalic artery including the thymic tongue. The neck exploration was limited to the ipsilateral and central neck compartments as the tumor did not cross midline, intraoperative frozen sections on all surgical margins were negative for tumor, and there was no clinical lymphadenopathy observed. The surgical specimen was dark red-purple in color and weighed 14 g. On histologic evaluation, the tumor demonstrated focal vascular invasion, consistent with parathyroid carcinoma (Figure 1). Otherwise, the tumor capsule was intact, all margins were negative including the inferior portion of the left thyroid lobe. The left superior parathyroid gland was biopsied and reported to be normal parathyroid tissue on histological analysis. Six level VI lymph nodes were analyzed and negative for tumor. Intra-operatively, iPTH fell to 41 ng/L (41 pg/mL) and post-operatively it fell to 14 ng/L (14 pg/mL) and she had a calcium nadir of 1.7 nmol/L (6.8 mg/dL). She was started on cholecalciferol, calcitriol and calcium supplementation in the immediate post-operative period. She was persistently hypocalcemic despite calcium supplementation until postoperative day 4 when her calcium normalized. Renal ultrasound was negative for nephrocalcinosis, calculi or hydroureteronephrosis, and pelvic ultrasound was negative for uterine tumors. The patient underwent a skeletal survey that demonstrated cystic changes to the left femoral neck requiring further evaluation. There was no suspicion for jaw tumors. MRI of the left hip without contrast demonstrated a multi-lobulated cystic lesion within the proximal left femoral neck involving nearly 50% of the diameter of the femoral neck consistent with a brown tumor (Figure 2). The patient was made non-weight bearing to the left lower extremity, and was discharged on postoperative day 5 in good condition.
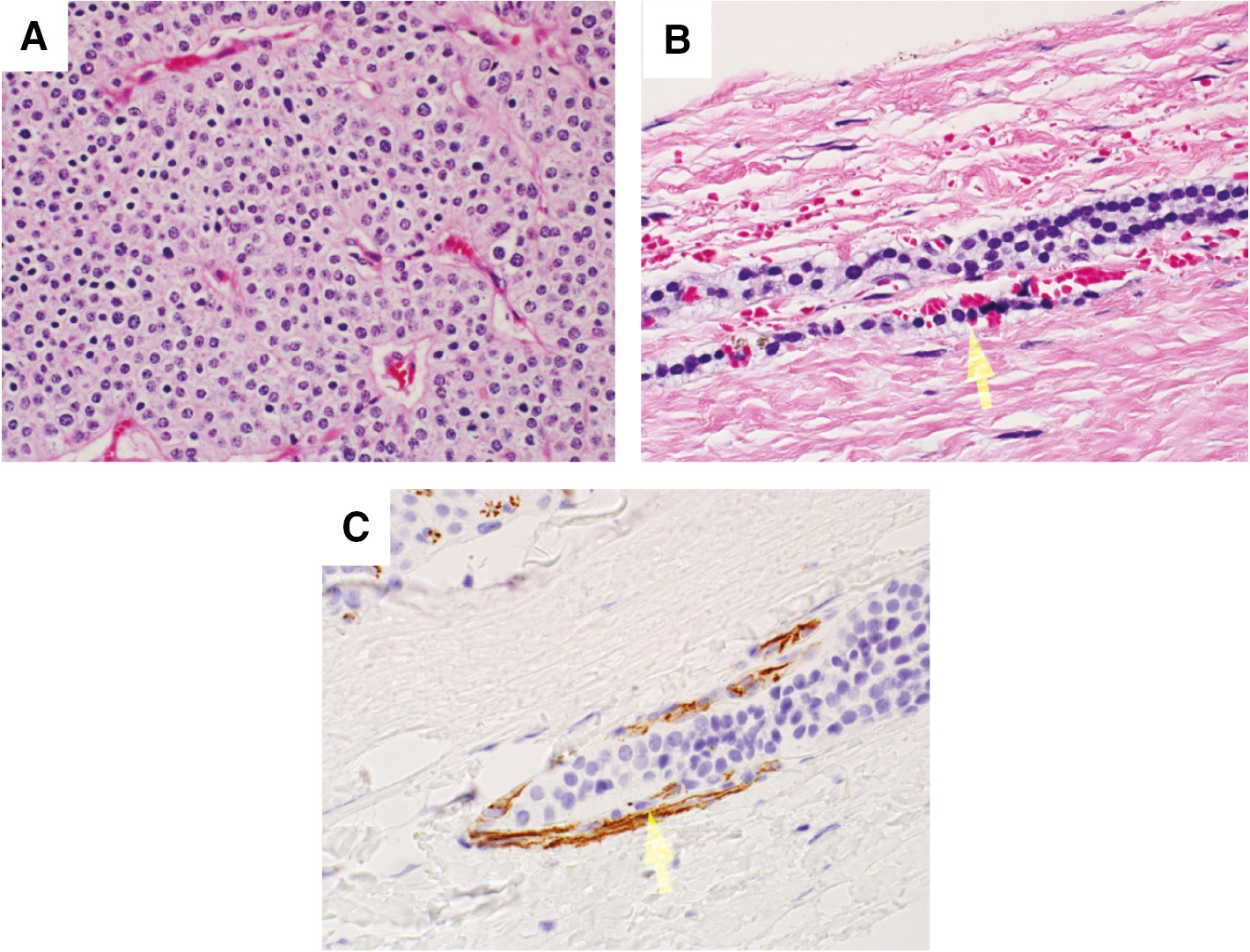
H&E and immunohistochemistry stains.
(A) Photomicrograph of parathyroid carcinoma showing scattered atypical cells and focal intervening fibrous bands (H&E). (B) Photomicrograph showing vascular invasion (H&E). Arrow points to vascular invasion. (C) CDC31 immunohistochemistry showing vascular invasion. Arrow points to vascular invasion.
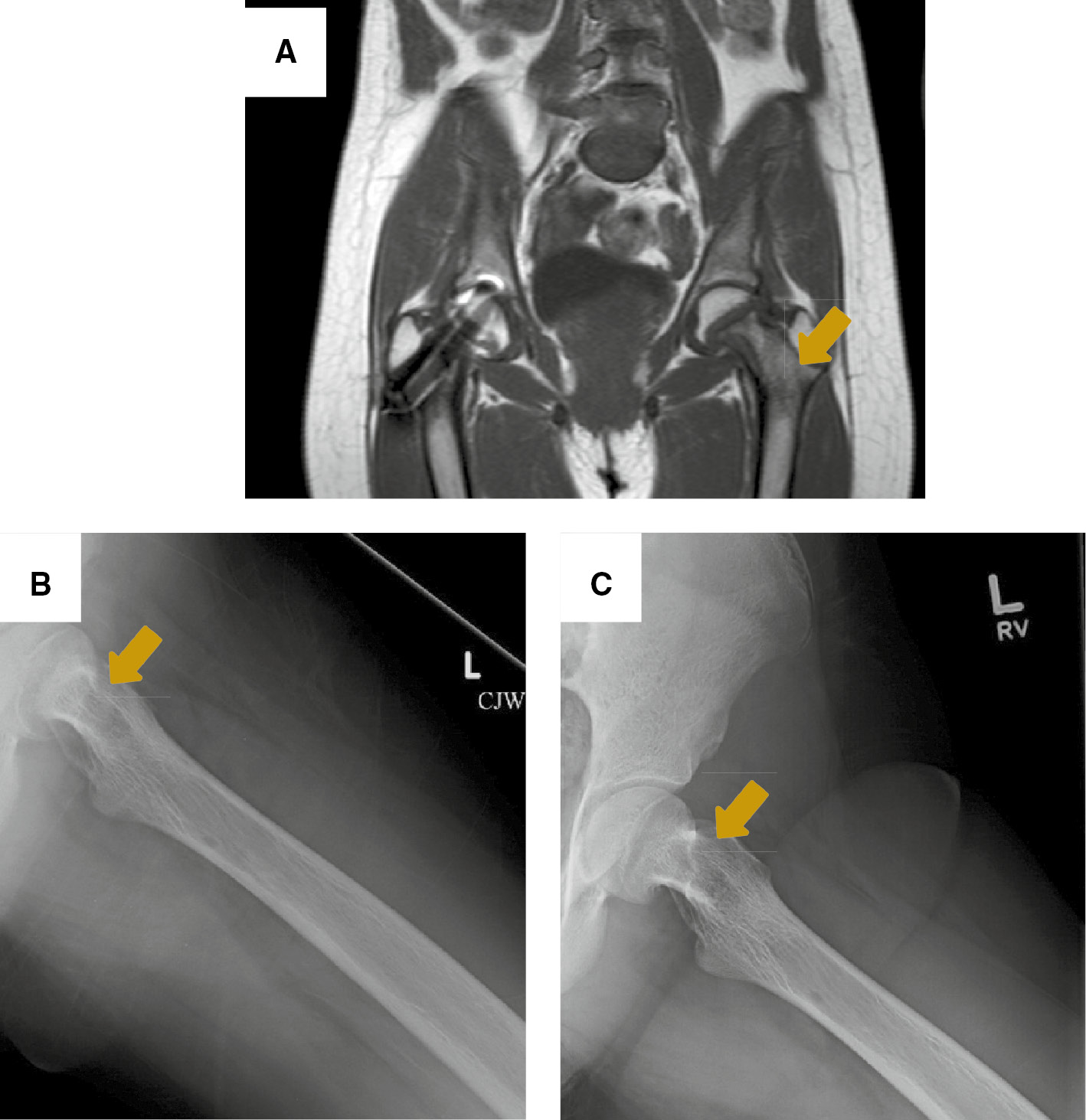
Brown tumor to the left femoral neck.
(A) MRI of the hip on coronal plane showing a multilobulated cystic lesion occupying more than 50% of the left femoral neck. Arrow points to a brown tumor. X-ray views showing thickening of the trabeculae and a well-defined lucency to the medial aspect of femoral neck on post-operative week 1 (B), and 6 months later (C). Arrows point to a brown tumor.
The patient has been followed in the otolaryngology, endocrinology and orthopedic surgery clinics postoperatively and is doing well. Calcium levels normalized to 2.15 mmol/L (8.6 mg/dL) when checked on postoperative day 16; calcitriol was discontinued by 2 months’ post-operatively once serum calcium levels improved, iPTH remains in the normal range and there is no hypercalcemia on calcium and cholecalciferol supplementation at 6 months’ post-operatively. Her left femoral brown tumor has significantly improved on X-rays (Figure 2) and her weight bearing has progressed to crutch use only outside of the home. Unfortunately, her scoliosis has progressed from 59 degrees at time of presentation to 63 degrees 6 months later, and thus she underwent posterior spinal fusion from T4 to L2 per orthopedics recommendation. Otherwise, her bone pain has essentially resolved and she remains disease-free.
Genetic analysis
MEN1 gene mutation analysis from a peripheral blood sample was negative for disease-causing mutations. However, HRPT2/CDC73 gene analysis from peripheral blood leukocytes was positive for a germline, heterozygous mutation, c.70 G>T, p. Glu24Ter, in exon 1 of the HRPT2/CDC73 gene, which codes for an early stop codon. Parental genetic testing has not been done due to financial constraints.
Discussion
We describe the case of a 14-year-old female with parathyroid carcinoma that constitutes the first pediatric case with a reported HRPT2/CDC73 mutation, c.70 G>T, which has only been described as a somatic mutation in two adult cases before [16], [17]. Parathyroid carcinoma in the pediatric population is a rare entity; to date, only nine previous cases have been reported in children under 16 years of age [2], [8], [9], [10], [11], [12], [13], [14], [15], affecting children ranging from 8 to 15 years of age (Table 1). Most previous cases were sporadic parathyroid carcinomas, except for two. The first one was a 14-year-old male reported by McHenry et al. that presented with a hypercalcemic crisis in the context of familial hyperparathyroidism. The second case was reported by Hamill et al., of an 8-year-old female with family history of primary hyperparathyroidism suggesting a genetic predisposition [2], [8], [9], [10], [11], [12], [13], [14], [15].
Clinical characteristics in pediatric patients with parathyroid carcinoma.
| Author, year | Age, year | Sex | Family history | Presentation | Renal US | Palpable neck mass | Pre-operative total calcium, mmol/L (mg/dL) | Location | Size, cm | Menin gene | HRPT2 gene |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Hamill et al., 2002 [9] | 8 | F | Yes | Lethargy, nocturia, gross hematuria | NS | Yes | 3.6 (14.24) | Left inferior | 1.5×1×1 | (−) | NS |
| Fiedler et al., 2009 [13] | 10 | M | Noa | Anorexia, fatigue, knee pain | NS | No | 3.9 (15.5) | Ectopic (mediastinum) | NS | NS | NS |
| Vinodh and Rajeshari, 2012 [14] | 11 | M | No | Genu valgum | Cystitis | Yes | 1.62 (6.48)c | Right inferior | 2.5×1×1.5 | NS | NS |
| Schantz and Castleman, 1973 [8] | 13 | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS |
| Fujimoto et al., 1984 [2] | 13 | F | NS | Bone pain, malaise | NS | Yes | 4.0 (15.9) | Right superior | 15 g | NS | NS |
| Kim, 2012 [15] | 13 | F | No | Neck mass | NS | Yes | 3 (12.0) | Right | 22 g | NS | NS |
| McHenry et al., 1993 [10] | 14 | M | Nob | Bone pain, malaise, polyuria, polydipsia | NS | Yes | 3.3 (13.16) | Right inferior | 2.5×3 | NS | NS |
| Young et al., 1984 [11] | 15 | M | NS | Pancreatitis, seizures | NS | No | 4 (16) | Left inferior | 2×2 | NS | NS |
| Meier et al., 1999 [12] | 15 | M | NS | Fatigue, weight loss | NS | Yes | 5.2 (20.7) | Left inferior | 3×2×2 | NS | NS |
| Our case | 14 | F | No | SCFE, scoliosis | Negative | Yes | 3.4 (13.4) | Left inferior | 3.5×3×1.3, 14 g | (−) | (+) |
aDespite negative family history of parathyroid carcinoma, there was positive family history of thyroid adenoma (maternal grandmother) and parathyroid adenoma (maternal cousin). bPositive family history of parathyroid adenoma (mother). cIn this case report, only ionized calcium was reported. NS, not specified.
Patients with parathyroid carcinoma have markedly elevated calcium levels above 3.5 mmol/L (14 mg/dL), or 3–4 mg/dL (0.85–1 mmol/L) above the upper limit of normal, in contrast to patients with parathyroid adenoma. For the reported pediatric cases, including ours, average total calcium at presentation is 3.8 mmol/L (15.1 mg/dL, reference range, 8.4–10.2 mg/dL) [2], [8], [9], [10], [11], [12], [13], [14], [15]. This severe hypercalcemia is generally associated with presenting symptoms of fatigue, weakness, weight loss, anorexia, polyuria, polydipsia, renal colic, and bone pain. Additionally, patients with parathyroid carcinoma present with extremely high iPTH levels, generally 3–10 times above the upper limit of normal.
A palpable neck mass is a remarkably rare finding in parathyroid adenoma, but has been associated with up to 76% of cases of parathyroid carcinoma [18], [19]. Of the nine reported cases of pediatric parathyroid carcinoma with a reported physical exam (one of them did not report it), a clinically palpable neck mass was found in seven patients, equivalent to an incidence of 77.8% in children <16 years old [2], [8], [9], [10], [11], [12], [13], [14], [15]. Higher-resolution anatomical studies such as computed tomography (CT) or magnetic resonance imaging (MRI) are of significant value in the preoperative evaluation when malignancy is suspected, as they provide details on the location of the lesion and its relation to other structures, as well as reveal invasion of surrounding structures and enlarged lymph nodes [19].
Our patient became significantly hypocalcemic down to 1.7 mmol/L (6.8 mg/dL) on post-operative day 1, and was symptomatic, with numbness and tingling to her face and legs as well as a positive Chvostek sign. She required intravenous calcium and calcitriol supplementation. Her phosphorus was as low as 0.7 mmol/L (2.0 mg/dL) the first day post-operatively, and then remained normal until 1 month after surgery when it started to rise, to a peak of 2.0 mmol/L (6.1 mg/dL) 2 months’ post-operatively and was still elevated at 5 months. She required calcitriol supplementation for 2 months after tumor removal and calcium supplementation still at 6 months after surgery. Her post-operative hypocalcemia, even though confounded by hungry bone syndrome and vitamin D deficiency, is most likely explained by chronic suppression of the three remaining parathyroid glands in the face of chronic hyperparathyroidism.
To our knowledge, this is the first case documenting parathyroid carcinoma in a child presenting with SCFE. While the link between primary hyperparathyroidism and reduced bone mineral density with cortical bone disease is well established in adults [20], there have only been 10 pediatric case reports worldwide linking primary hyperparathyroidism and SCFE [21], [22], [23], [24], [25], [26], [27], [28], [29], [30]. Our patient presented initially with SCFE and was found after full skeletal survey to have a cystic lesion to the left femoral neck osteitis fibrosa cystica. With an incidence of 10.8 per 100,000 children, SCFE is the most common hip disorder affecting children and adolescents, usually in obese patients between the ages of 8 and 15 years [31], [32], [33], [34]. However, the vast majority of children with SCFE do not have hyperparathyroidism or parathyroid carcinoma.
Even with early consideration of parathyroid carcinoma, the diagnosis is a difficult one to make, and often is not suspected until the tumor is visualized by the surgeon. In about 50% of cases, parathyroid carcinomas are surrounded by a dense, fibrous, grayish-white capsule that adheres tenaciously to adjacent tissues and makes the tumor difficult to separate from contiguous structures [18]. Adhesions are partly due to a desmoplastic reaction, which is a reactive stromal response that creates a dense fibrotic tissue surrounding the tumor [35]. Thus, a firm, lobulated, or fixed tumor that is adherent to surrounding structures should alert the surgeon to a possible malignant diagnosis [18].
Once the diagnosis is suspected, complete en bloc resection of the tumor and surrounding neck structures should be performed [2], [36]. In adults, successful en bloc resection of the tumor results in 90% long-term survival, while incomplete tumor resection has been shown to have a local recurrence rate of 50% with a mortality rate of 46% [37], [38]. Due to its short half-life, intra-operative PTH measurement accurately predicts the outcome of parathyroidectomy [39].
In 2002, the gene CDC73 (formerly known as HRPT2), encoded in chromosome 1q25, and its corresponding protein parafibromin were identified [40], and mutations in this tumor suppressor gene have been found to be associated with cases of sporadic parathyroid carcinoma, autosomal dominant hyperparathyroidism-jaw tumor syndrome (HPT-JT), and familial isolated hyperparathyroidism (FIHP). Up to 70% of sporadic cases of parathyroid carcinoma are found to have HRPT2/CDC73 mutations [17], [40], [41], [42], [43], as opposed to <2% of cases of parathyroid adenoma [44]. HRPT2/CDC73 encodes the 531 amino acid protein parafibromin (parathyroid disease and fibro-osseous lesions), which is the human homologue of yeast CDC73. Parafibromin is located in the nucleus and is part of the complex that contains the human homologues of the yeast Paf1 complex (hPaf1). Parafibromin acts as a tumor suppressor and is involved in transcription regulation, histone modification, RNA processing and cell proliferation [16], [45]. It has been shown that parafibromin down-regulates the expression of the oncogene cyclin D1 [46] and that increased expression of cyclin D1 is associated with the formation of parathyroid adenomas and carcinomas [42].
Our patient was found to be heterozygous for a germline mutation in HRPT2/CDC73 (c.70 G>T, p. Glu24Ter), which is located in exon 1, and is predicted to cause loss of normal protein function by creating an early stop codon, E24X. In a review by Newey et al., among patients with HPT-JT syndrome, FIHP, parathyroid carcinoma, parathyroid adenoma and sporadic ossifying fibromas of the jaw, mutations in exon 1, 2 and 7 were overrepresented, even after adjusting for exon size, accounting for 34, 17 and 21%, respectively [47]. The author also reviewed that there had been 111 mutations to HRPT2/CDC73 reported as of 2008, 68 germline and 38 somatic [47]. Most mutations to HRPT2/CDC73 are frameshift, nonsense, and missense; however, as many as 35% of patients with HPT-JT have deletions of HRPT2/CDC73 and it is recommended that deletion analysis of the HRPT2/CDC73 gene via MPLA (multiplex ligation-dependent probe amplification) be performed in patients with parathyroid carcinoma who do not have HRPT2/CDC73 point mutations detected by sequencing [48], [49]. The same patient may have both germline and somatic mutations or two different somatic mutations to HRPT2/CDC73, and this explains why patients with a germline mutation can have asynchronous parathyroid neoplasms, consistent with the Knudson ‘two hit’ concept of biallelic inactivation of tumor suppressor genes, such as HRPT2/CDC73 [17], [50]. The 70 G>T mutation has been reported previously as a somatic mutation in two adult patients with sporadic parathyroid carcinoma [16], [17]; however, to our knowledge it has not been reported as a germline mutation and it is also the first HRPT2/CDC73 mutation reported in a pediatric case of parathyroid carcinoma.
The finding of unexpected germline mutations in apparently sporadic parathyroid carcinomas has already been reported by other authors, and as these cancers may represent an occult form of HPT-JT, clinical monitoring for jaw tumor and kidney lesions is advised and gene testing of family members is suggested, with monitoring of serum calcium in gene carriers [17], [43]. The HRPT2 protein product parafibromin is being investigated as an immunohistochemical marker for parathyroid carcinoma [41]. Cetani et al. [16] found that loss of parafibromin staining had a sensitivity of 100% and a specificity of 88%, compared to 82% sensitivity and 96% specificity for HRPT2/CDC73 mutation. The author recommends that both HRPT2/CDC73 gene testing and parafibromin staining should be done in the context of parathyroid tumors with atypical features. Wang et al., in turn, reported that combination of both markers had a sensitivity of 92.3% without reduction in specificity, which was 100% for HRPT2/CDC73 mutations and 95% for loss of parafibromin expression [51]. Using genetic testing for HRPT2 mutations and staining for parafibromin is especially helpful considering that parathyroid carcinoma is particularly difficult to diagnose pathologically: in a large study of 40 parathyroid carcinomas defined based on recurrence and metastasis rather than by pathological criteria alone, less than half were correctly diagnosed as malignant at presentation based on the combined clinical and pathological findings [52].
While parathyroid carcinoma is an exceedingly rare entity amongst the pediatric population, it is an important diagnosis that should be considered by healthcare providers when a patient presents with hyperparathyroidism and a palpable neck mass. Serum calcium level is the most reliable indicator of disease recurrence [39]. Close follow-up is required during the first 2 years after surgery because of the higher rate of recurrence during this period; however, long-term follow-up is recommended because recurrence can happen up to 15 years after surgery [52]. In patients with HRPT2/CDC73 mutations, Witteveen et al. reported a seven-fold increased risk of developing local or distant metastasis. All four patients with the gene mutation developed local recurrence and/or distant metastasis, which occurred only in 10 of 19 patients without mutations [53]. Wang et al. did not find a significant correlation between a mutation in HRPT2/CDC73 and distant metastases but did find a higher incidence of recurrence in patients with HRPT2/CDC73 mutations (100% vs. 50%, p=0.031), after adjusting for gender, age, initial serum calcium and duration of follow-up [51].
In conclusion, parathyroid carcinoma is rare in pediatrics but should be considered in a patient with hypercalcemia and a neck mass. We report the first pediatric patient with a HRPT2/CDC73 mutation in the context of parathyroid carcinoma. Genetic screening for HRPT2/CDC73 mutations is prudent in pediatric patients with parathyroid carcinoma. Close follow-up is required for patients who carry this mutation due to a higher risk of metastasis and recurrence. Screening of family members for this mutation may be necessary.
Acknowledgments
The authors would like to thank David M. Parham for his help in interpreting the pathology and preparing the photomicrograph, as well as Dr. Richard Boles and Dr. Susanna Sorrentino for their assistance with interpretation of results.
Author contributions: All the authors have accepted responsibility for the entire content of this submitted manuscript and approved submission.
Research funding: None declared.
Employment or leadership: None declared.
Honorarium: None declared.
Competing interests: The funding organization(s) played no role in the study design; in the collection, analysis, and interpretation of data; in the writing of the report; or in the decision to submit the report for publication.
References
1. Menon P, Dayal D, Rao SG, Bhattacharya A, Narasimha Rao KL. Childhood parathyroid adenoma: a rare but important cause of nephrolithiasis. J Pediatr Endocrinol Metab 2016;29:853–6.10.1515/jpem-2015-0369Search in Google Scholar
2. Fujimoto Y, Obara T, Ito Y. Surgical treatment of ten cases of parathyroid carcinoma: importance of an initial en bloc tumor resection. World J Surg 1984;8:392–400.10.1007/BF01655086Search in Google Scholar
3. Wang CA, Gaz RD. Natural history of parathyroid carcinoma. Diagnosis, treatment, and results. Am J Surg 1985;149:522–7.10.1016/S0002-9610(85)80050-7Search in Google Scholar
4. Pelizzo MR, Piotto A, Bergamasco A, Rubello D, Casara D. [Parathyroid carcinoma. Therapeutic strategies derived from 20 years of experience]. Minerva Endocrinol 2001;26:23–9.Search in Google Scholar
5. van Heerden JA, Weiland LH, ReMine WH, Walls JT, Purnell DC. Cancer of the parathyroid glands. Arch Surg 1979;114:475–80.10.1001/archsurg.1979.01370280129019Search in Google Scholar
6. Dotzenrath C, Goretzki PE, Sarbia M, Cupisti K, Feldkamp J, et al. Parathyroid carcinoma: problems in diagnosis and the need for radical surgery even in recurrent disease. Eur J Surg Oncol 2001;27:383–9.10.1053/ejso.2001.1122Search in Google Scholar
7. Dudney WC, Bodenner D, Stack BC. Parathyroid carcinoma. Otolaryng Clin N Am 2010;43:441–53.10.1016/j.otc.2010.01.011Search in Google Scholar
8. Schantz A, Castleman B. Parathyroid carcinoma. A study of 70 cases. Cancer 1973;31:600–5.10.1002/1097-0142(197303)31:3<600::AID-CNCR2820310316>3.0.CO;2-0Search in Google Scholar
9. Hamill J, Maoate K, Beasley SW, Corbett R, Evans J. Familial parathyroid carcinoma in a child. J Paediatr Child Health 2002;38:314–7.10.1046/j.1440-1754.2002.00802.xSearch in Google Scholar
10. McHenry CR, Rosen IB, Walfish PG, Cooter N. Parathyroid crisis of unusual features in a child. Cancer 1993;71:1923–7.10.1002/1097-0142(19930301)71:5<1923::AID-CNCR2820710531>3.0.CO;2-VSearch in Google Scholar
11. Young TO, Saltzstein EC, Boman DA. Parathyroid carcinoma in a child: unusual presentation with seizures. J Pediatr Surg 1984;19:194–6.10.1016/S0022-3468(84)80448-0Search in Google Scholar
12. Meier DE, Snyder WH, Dickson BA, Margraf LR, Guzzetta PC. Parathyroid carcinoma in a child. J Pediatr Surg 1999;34: 606–8.10.1016/S0022-3468(99)90084-2Search in Google Scholar
13. Fiedler AG, Rossi C, Gingalewski CA. Parathyroid carcinoma in a child: an unusual case of an ectopically located malignant parathyroid gland with tumor invading the thymus. J Pediatr Surg 2009;44:1649–52.10.1016/j.jpedsurg.2009.04.024Search in Google Scholar PubMed
14. Vinodh M, Rajeshwari A. Parathyroid carcinoma presenting as genu valgum. Indian Pediatr 2012;49:156.10.1007/s13312-012-0014-8Search in Google Scholar PubMed
15. Kim YS. Parathyroid carcinoma with lung metastasis in a thirteen-year-old girl. J Korean Surg Soc 2012;82:385.10.4174/jkss.2012.82.6.385Search in Google Scholar PubMed PubMed Central
16. Cetani F, Ambrogini E, Viacava P, Pardi E, Fanelli G, et al. Should parafibromin staining replace HRTP2 gene analysis as an additional tool for histologic diagnosis of parathyroid carcinoma? Eur J Endocrinol 2007;156:547–54.10.1530/EJE-06-0720Search in Google Scholar PubMed
17. Shattuck TM, Välimäki S, Obara T, Gaz RD, Clark OH, et al. Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N Engl J Med 2003;349:1722–9.10.1056/NEJMoa031237Search in Google Scholar PubMed
18. Shane E. Clinical review 122: parathyroid carcinoma. J Clin Endocrinol Metab 2001;86:485–93.10.1210/jcem.86.2.7207Search in Google Scholar PubMed
19. Givi B, Shah JP. Parathyroid carcinoma. Clin Oncol 2010;22: 498–507.10.1016/j.clon.2010.04.007Search in Google Scholar PubMed PubMed Central
20. Silverberg SJ, Shane E, de la Cruz L, Dempster DW, Feldman F, et al. Skeletal disease in primary hyperparathyroidism. J Bone Miner Res 1989;4:283–91.10.1002/jbmr.5650040302Search in Google Scholar PubMed
21. El Scheich T, Marquard J, Westhoff B, Schneider A, Cupisti K, et al. Approach to the management of slipped capital femoral epiphysis and primary hyperparathyroidism. J Pediatr Endocrinol Metab 2012;25:239–44.10.1515/jpem-2012-0003Search in Google Scholar
22. Bone LB, Roach JW, Ward WT, Worthen HG. Slipped capital femoral epiphysis associated with hyperparathyroidism. J Pediatr Orthop 1985;5:589–92.10.1097/01241398-198509000-00017Search in Google Scholar PubMed
23. Kinoshita J, Kaneda K, Matsuno T, Hosokawa Y, Nagashio R. Slipped capital femoral epiphysis associated with hyperparathyroidism. A case report. Int Orthop 1995;19:245–7.10.1007/BF00185232Search in Google Scholar PubMed
24. Chiroff RT, Sears KA, Slaughter WH. Slipped capital femoral epiphyses and parathyroid adenoma. J Bone Joint Surg 1974;56:1063–7.10.2106/00004623-197456050-00019Search in Google Scholar
25. Yang WE, Shih CH, Wang KC, Jeng LB. Slipped capital femoral epiphyses in a patient with primary hyperparathyroidism. J Formos Med Assoc 1997;96:549–52.Search in Google Scholar
26. Madeira IR, Machado M, Maya MC, Sztajnbok FR, Bordallo MA. [Primary hyperparathyroidism associated to slipped capital femoral epiphysis in a teenager]. Arq Bras Endocrinol Metabol 2005;49:314–8.10.1590/S0004-27302005000200021Search in Google Scholar
27. Khiari K, Cherif L, Ben Abdallah N, Maazoun I, Hadj Ali I, et al. [Slipped capital femoral epiphysis associated with hyperparathyroidism. A case report]. Ann Med Interne 2003;154:544–6.Search in Google Scholar
28. Kim TS, Choi WS. Bilateral slipped capital femoral epiphysis due to primary hyperparathyroidism – a case report. J Korean Orthop Assoc 2009;44:565.10.4055/jkoa.2009.44.5.565Search in Google Scholar
29. Bhadada SK, Menon AS, Bhansali A, Das S, Gill SS, et al. An unusual cause of limping in primary hyperparathyroidism. Endocrinologist 2009;19:122–3.10.1097/TEN.0b013e3181a58878Search in Google Scholar
30. Bennett JT, Alexander HH, Morrissy RT. Parathyroid adenoma presenting as a pathologic fracture of the femoral neck in an adolescent. J Pediatr Orthop 1986;6:473–6.10.1097/01241398-198607000-00017Search in Google Scholar PubMed
31. Peck D. Slipped capital femoral epiphysis: diagnosis and management. Am Fam Physician 2010;82:258–62.Search in Google Scholar
32. Lehmann CL, Arons RR, Loder RT, Vitale MG. The epidemiology of slipped capital femoral epiphysis: an update. J Pediatr Orthop 2006;26:286–90.10.1097/01.bpo.0000217718.10728.70Search in Google Scholar PubMed
33. Loder RT, Aronsson DD, Weinstein SL, Breur GJ, Ganz R, et al. Slipped capital femoral epiphysis. Instr Course Lect 2008;57:473–98.Search in Google Scholar
34. Gholve PA, Cameron DB, Millis MB. Slipped capital femoral epiphysis update. Curr Opin Pediatr 2009;21:39–45.10.1097/MOP.0b013e328320aceaSearch in Google Scholar PubMed
35. Sainio A, Järvseläinen H. Extracellular matrix macromolecules in tumour micvoenvironment with special reference to desmoplastic reaction and the role of matrix proteoglycans and hyaluronan. J Carcinog Mutagen 2013;S13:002.10.4172/2157-2518.S13-002Search in Google Scholar
36. Marcocci C, Cetani F, Rubin MR, Silverberg SJ, Pinchera A, et al. Parathyroid carcinoma. J Bone Miner Res 2008;23:1869–80.10.1007/978-1-84628-881-4_23Search in Google Scholar
37. Koea JB, Shaw JH. Parathyroid cancer: biology and management. Surg Oncol 1999;8:155–65.10.1016/S0960-7404(99)00037-7Search in Google Scholar
38. Mittendorf EA, McHenry CR. Parathyroid carcinoma. J Surg Oncol 2005;89:136–42.10.1002/jso.20182Search in Google Scholar PubMed
39. Wu J, Nwariaku F, Snyder WH, Remaley AT, Sokoll LJ. National academy of clinical biochemistry guideline for the use of tumor markers in parathyroid gland adenomas and carcinomas. Clin Biochem 2008;41:210–21.Search in Google Scholar
40. Carpten JD, Robbins CM, Villablanca A, Forsberg L, Presciuttini S, et al. HRPT2, encoding parafibromin, is mutated in hyperparathyroidism–jaw tumor syndrome. Nat Genet 2002;32:676–80.10.1038/ng1048Search in Google Scholar PubMed
41. Howell VM, Gill A, Clarkson A, Nelson AE, Dunne R, et al. Accuracy of combined protein gene product 9.5 and parafibromin markers for immunohistochemical diagnosis of parathyroid carcinoma. J Clin Endocrinol Metab 2009;94:434–41.10.1210/jc.2008-1740Search in Google Scholar PubMed
42. Howell VM, Haven CJ, Kahnoski K, Khoo SK, Petillo D, et al. HRPT2 mutations are associated with malignancy in sporadic parathyroid tumours. J Med Genet 2003;40:657–63.10.1136/jmg.40.9.657Search in Google Scholar PubMed PubMed Central
43. Cetani F, Pardi E, Borsari S, Viacava P, Dipollina G, et al. Genetic analyses of the HRPT2 Gene in primary hyperparathyroidism: germline and somatic mutations in familial and sporadic parathyroid tumors. J Clin Endocrinol Metab 2004;89: 5583–91.10.1210/jc.2004-0294Search in Google Scholar PubMed
44. Krebs LJ, Shattuck TM, Arnold A. HRPT2 mutational analysis of typical sporadic parathyroid adenomas. J Clin Endocrinol Metab 2005;90:5015–7.10.1210/jc.2005-0717Search in Google Scholar PubMed
45. Rozenblatt-rosen O, Hughes CM, Nannepaga SJ, Shanmugam KS, Copeland TD, et al. The parafibromin tumor suppressor protein is part of a human Paf1 complex the parafibromin tumor suppressor protein is part of a human paf1 complex. Mol Cell Biol 2005;25:612–20.10.1128/MCB.25.2.612-620.2005Search in Google Scholar PubMed PubMed Central
46. Woodard GE, Lin L, Zhang JH, Agarwal SK, Marx SJ, et al. Parafibromin, product of the hyperparathyroidism-jaw tumor syndrome gene HRPT2, regulates cyclin D1/PRAD1 expression. Oncogene 2004;24:1272–6.10.1038/sj.onc.1208274Search in Google Scholar PubMed
47. Newey PJ, Bowl MR, Cranston T, Thakker RV. Cell division cycle protein 73 homolog (CDC73) mutations in the hyperparathyroidism-jaw tumor syndrome (HPT-JT) and parathyroid tumors. Hum Mutat 2010;31:295–307.10.1002/humu.21188Search in Google Scholar PubMed
48. Korpi-Hyövälti E, Cranston T, Ryhänen E, Arola J, Aittomälo L, et al. CDC73 intragenic deletion in familial primary hyperparathyroidism associated with parathyroid carcinoma. J Clin Endocrinol Metab 2014;99:3044–8.10.1210/jc.2014-1481Search in Google Scholar PubMed PubMed Central
49. Bricaire L, Odou MF, Cardot-Bauters C, Delemer B, North MO, et al. Frequent large germline HRPT2 deletions in a French national cohort of patients with primary hyperparathyroidism. J Clin Endocrinol Metab 2013;98:E403–8.10.1210/jc.2012-2789Search in Google Scholar PubMed
50. Cetani F, Pardi E, Ambrogini E, Viacava P, Borsari S, et al. Different somatic alterations of the HRPT2 gene in a patient with recurrent sporadic primary hyperparathyroidism carrying an HRPT2 germline mutation. Endocr Relat Cancer 2007;14:493–9.10.1677/ERC-06-0092Search in Google Scholar PubMed
51. Wang O, Wang C, Nie M, Cui Q, Guan H, et al. Novel HRPT2/CDC73 gene mutations and loss of expression of parafibromin in Chinese patients with clinically sporadic parathyroid carcinomas. PLoS One 2012;7:e45567.10.1371/journal.pone.0045567Search in Google Scholar PubMed PubMed Central
52. Gill A. Understanding the genetic basis of parathyroid carcinoma. Endocr Pathol 2014;25:30–4.10.1007/s12022-013-9294-3Search in Google Scholar PubMed
53. Witteveen JE, Hamdy NA, Dekkers OM, Kievit J, van Wezel T, et al. Downregulation of CASR expression and global loss of parafibromin staining are strong negative determinants of prognosis in parathyroid carcinoma. Mod Pathol 2011;24:688–97.10.1038/modpathol.2010.236Search in Google Scholar PubMed
©2016 Walter de Gruyter GmbH, Berlin/Boston
Articles in the same Issue
- Frontmatter
- Review
- A germline mutation of HRPT2/CDC73 (70 G>T) in an adolescent female with parathyroid carcinoma: first case report and a review of the literature
- Original Articles
- Seroprotection status of hepatitis B and measles vaccines in children with type 1 diabetes mellitus
- Prevalence and awareness of functional and structural foot abnormalities in children and adolescents with type 1 diabetes
- WHO European Childhood Obesity Surveillance Initiative in Serbia: a prevalence of overweight and obesity among 6–9-year-old school children
- Pilot study of newborn screening of inborn error of metabolism using tandem mass spectrometry in Malaysia: outcome and challenges
- In children with autoimmune thyroid diseases the association with Down syndrome can modify the clustering of extra-thyroidal autoimmune disorders
- Identification of monogenic gene mutations in Japanese subjects diagnosed with type 1B diabetes between >5 and 15.1 years of age
- Referral pattern of children with short stature to a pediatric endocrine clinic in Kuwait
- Factors associated with post-menarcheal growth: results of a longitudinal study in Chilean girls from different socioeconomic statuses
- Percentiles for anthropometric measures in Iranian children and adolescents: the CASPIAN-IV study
- Hormone disorder and vitamin deficiency in attention deficit hyperactivity disorder (ADHD) and autism spectrum disorders (ASDs)
- Case Reports
- Hyperinsulinism-hyperammonemia syndrome: a de novo mutation of the GLUD1 gene in twins and a review of the literature
- Novel homozygous likely-pathogenic intronic variant in INS causing permanent neonatal diabetes in siblings
- A girl with permanent neonatal diabetes due to KCNJ11 mutation presented with Mauriac syndrome after improper adjustment in sulfonylurea dosage over 6 years
- Treatment experience and long-term follow-up data in two severe neonatal hyperparathyroidism cases
- A novel splice site mutation of FGD1 gene in an Aarskog-Scott syndrome patient with a large anterior fontanel
Articles in the same Issue
- Frontmatter
- Review
- A germline mutation of HRPT2/CDC73 (70 G>T) in an adolescent female with parathyroid carcinoma: first case report and a review of the literature
- Original Articles
- Seroprotection status of hepatitis B and measles vaccines in children with type 1 diabetes mellitus
- Prevalence and awareness of functional and structural foot abnormalities in children and adolescents with type 1 diabetes
- WHO European Childhood Obesity Surveillance Initiative in Serbia: a prevalence of overweight and obesity among 6–9-year-old school children
- Pilot study of newborn screening of inborn error of metabolism using tandem mass spectrometry in Malaysia: outcome and challenges
- In children with autoimmune thyroid diseases the association with Down syndrome can modify the clustering of extra-thyroidal autoimmune disorders
- Identification of monogenic gene mutations in Japanese subjects diagnosed with type 1B diabetes between >5 and 15.1 years of age
- Referral pattern of children with short stature to a pediatric endocrine clinic in Kuwait
- Factors associated with post-menarcheal growth: results of a longitudinal study in Chilean girls from different socioeconomic statuses
- Percentiles for anthropometric measures in Iranian children and adolescents: the CASPIAN-IV study
- Hormone disorder and vitamin deficiency in attention deficit hyperactivity disorder (ADHD) and autism spectrum disorders (ASDs)
- Case Reports
- Hyperinsulinism-hyperammonemia syndrome: a de novo mutation of the GLUD1 gene in twins and a review of the literature
- Novel homozygous likely-pathogenic intronic variant in INS causing permanent neonatal diabetes in siblings
- A girl with permanent neonatal diabetes due to KCNJ11 mutation presented with Mauriac syndrome after improper adjustment in sulfonylurea dosage over 6 years
- Treatment experience and long-term follow-up data in two severe neonatal hyperparathyroidism cases
- A novel splice site mutation of FGD1 gene in an Aarskog-Scott syndrome patient with a large anterior fontanel