Pathogenic missense variants of CSNK2B associated with Poirier-Bienvenu neurodevelopmental disorder impact differently on CK2 holoenzyme formation
-
Hanna Kavaliova



Abstract
Poirier-Bienvenu neurodevelopmental syndrome is a neurodevelopmental disorder associated with de novo variants of the CSNK2B gene, characterized by intellectual disability, developmental delay, frequent seizures and more. While the majority of variants are nonsense variants leading to abortion of protein translation and no or truncated CK2β, many pathogenic missense variants also exist. We investigated the effect of four variants on CK2 holoenzyme formation and activity. We show that variants in the Zinc-finger region leads to reduced protein stability and altered subcellular localization. The instability is partly mediated by proteasomal and lysosomal degradation. We further show that homodimerization of these CK2β variants (p.Arg111Pro, p.Cys137Phe), localized within the Zinc-finger domain, is significantly reduced, while CK2α binding appears not affected. Other variants, p.Asp32Asn and p.Arg86Cys, did not affect stability or CK2β/α binding. For these mutants, the key to understanding the pathological mechanism may depend on external factors, such as altered protein–protein interaction. We conclude that Zinc-finger domain variants appear to destabilize the protein and affect holoenzyme formation, effectively reducing the pool of competent holoCK2. In the context of POBINDS, our findings suggest that Zinc-finger domain variants are likely to affect cells similarly to truncating and splicing variants with reduced translation of full-length CK2β.
1 Introduction
Poirier-Bienvenu neurodevelopmental syndrome (OMIM #6187D32N, POBINDS) is a rare neurodevelopmental disorder associated with de novo variants of the CSNK2B gene (Poirier et al. 2017). The symptomatic profile of POBINDS is variable, but is characterized by intellectual disability, developmental delay, hypotonia and early-onset seizures, as well as growth deficits, dysmorphism and cranial size abnormalities (Ernst et al. 2021; Li et al. 2023).
The CSNK2B gene encodes for the regulatory subunit β of the serine/threonine kinase CK2. CK2 is a ubiquitous and constitutively active tetrameric enzyme composed of two catalytic α or α′ subunits and a dimer of two β regulatory subunits (Niefind et al. 2001).
It is known to phosphorylate more than 300 substrates with both ATP and GTP (Meggio and Pinna 2003) and has been implicated in many physiological and pathological processes, ranging from brain and heart development, cell proliferation and survival, and modulation of neurotransmitter signaling to cancer (Castello et al. 2018; Götz and Montenarh 2017; Rebholz et al. 2009; Ruzzene and Pinna 2010). CK2β(−/−) mouse embryos die early in gestation at E7.5, while heterozygous mice are viable and do not differ significantly from wild type mice in CK2 expression levels, however, a proportion, approximately 20 % of the heterozygous embryos do not survive to birth and, in the non-surviving group, a western blot gel shift of CK2β has been observed, suggesting alterations in post-translational modifications (Blond et al. 2005).
The β subunit has a two-fold function: it is required for CK2 holoenzyme (ααββ) formation (Niefind et al. 2001) and to stabilize the holoenzyme, and it regulates catalytic activity or substrate specificity (Meggio et al. 1992). Major structural hallmarks of the CK2β sequence are shown in Figure 1A (adapted from Unni et al. 2022). The first step in the holoenzyme complex formation is the dimerization of two β subunits via their Zinc-finger regions (amino acids (AA) 105–146), which contain four conserved cysteine residues (Cys109, Cys114, Cys137 and Cys140) that can coordinate Zn2+ ions (Chantalat 1999). Once the dimer is formed, both β subunits together constitute an interface for the recruitment of two CK2α chains to form the CK2 tetramer (Niefind et al. 2001). CK2β possesses an acidic loop (Asp55-Asp70), which can interact with the basic cluster of a neighbouring CK2α molecule, as part of a secondary interaction between different holoenzymes (Niefind and Issinger 2005). This basic cluster within the kinase domain is crucial for substrate binding and recognition, affecting both substrate specificity and activity in general. Through this interaction, the acidic CK2β loop acts as a pseudo-substrate that competitively hinders the access different substrates to the active site, thereby down-regulating CK2 activity (Niefind et al. 2001).
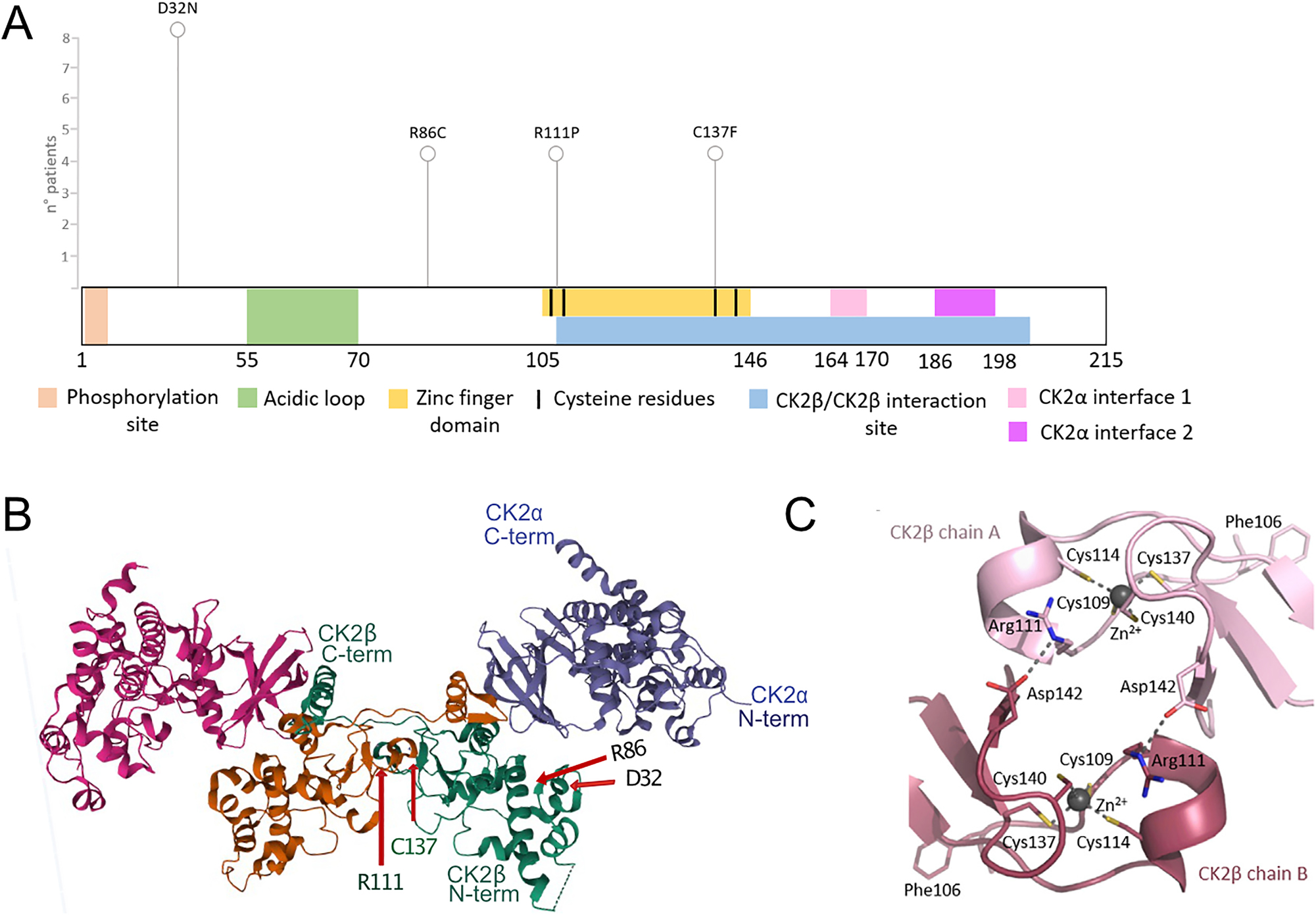
Localization of POBINDS variants in the CK2β protein. Linear scheme of CK2β protein, with functional domains and variants annotated (A). 3-D structure of CK2 holoenzyme, (https://www.rcsb.org/structure/4DGL, Lolli et al. 2012), with the four amino acids (D32, R86, R111, C137) labelled (B), reproduction of crystal structure of the contact area of two CK2β chains, with the set of four Zn2+-coordinating cysteines marked for each chain (C), (Unni et al. 2022) (Figure 7F), with permission of the authors.
However, CK2β has also been reported to exist as a dimer and to be involved in CK2α-independent cellular functions. It has also been shown that CK2β can interact with several partners without the involvement of CK2α, notably c-Mos, Chk1, PAK and A-Raf (Chen et al. 1997; Guerra et al. 2003; Hagemann et al. 1997; Mentzel et al. 2009).
To date, 57 CK2β pathological POBINDS variants have been reported (ClinVar), found all along the amino acid sequence, without defined disease hotspots; however, the majority of variants seems to be in non-coding regions, and are majorly splice variants which often lead to truncated proteins (Ballardin et al. 2022). Additionally, a number of deletion mutations have been described (Zhang et al. 2024), supporting the notion of a haploinsufficiency mechanism of disease aetiology.
With regards to the missense variants leading to single amino acid exchange, very little is known about how disease phenotypes might be mediated: two variants, p.Asn32Asn and p.Asn32His, located in the KEN box-like motif, have recently been linked to the “intellectual disability-craniodigital syndrome”, or IDCS, a grouping of disorders with overlapping clinical features. The authors argue that IDCS caused by these two CK2β variants (p.Asp32Asn; p.Asp32His) is different from POBINDS, as the mutant proteins have been suggested to act in a dominant negative manner, altering the substrate profile of the CK2 holoenzyme via changes in the activation of the Wnt signalling pathway (Asif et al. 2022). Another KEN box-variant, p.Leu39Arg, was, in contrast, linked to POBINDS (Di Stazio et al. 2023).
To date, the consequences of the different missense CK2β variants, and how they contribute to the pathophysiology of the disease, have not been well studied. Our aim was to assess four POBINDS-linked missense variants – p.Asp32Asn, p.Arg86Cys, p.Arg111Pro, p.Cys137Phe, as representatives of sites that are multiply recorded in databases, in multiple patients, and that are distributed over the primary amino acid sequence. We wanted to study the effect of overexpression of these point mutants on protein expression, localization, homodimerization and on the interaction of the mutant protein with CK2α.
2 Results
2.1 Expression and subcellular localization
POBINDS is mainly caused by nonsense and frameshift variants, but a large number of missense variants spanning the entire protein sequence have also been associated with the disease. We selected four disease-associated variants based on the frequency of the affected amino acid in the literature and the ClinVar database but also based on their localization along the CK2β protein sequence (Figure 1A). The p.Asp32Asn variant is localized to the N-terminal domain (AA 1–54) containing the autophosphorylation site; the p.Arg86Cys mutant is C-terminal to the acidic loop and is one of the highly conserved residues exposed on the surface, facing away from the rest of the protein. p.Arg111Pro is within the Zinc-finger domain, and finally, p.Cys137Phe variant itself provides the sulfhydryl group for zinc coordination that stabilizes the β–β homodimer (Figure 1B and C). In all figures and legends, for simplicity reasons, the one-letter amino acid code is used (D32N, R86C, R111P, C137F).
We transfected the mutant CK2β proteins into Cos7 and HEK293T cells and observed that while WT (wild type) and p.Asp32Asn expressed at similar levels, the p.Arg111Pro and p.Cys137Phe variants are much less expressed, while p.Arg86Cys variant expression is slightly but not significantly reduced (Figure 2A–C). Furthermore, WT, p.Asp32Asn and p.Arg86Cys mutants were predominantly localized to the nucleus, while for p.Arg111Pro and p.Cys137Phe we observed a retention in the cytosol and a more punctate staining (Figure 2D and E). In cells expressing WT, p.Asp32Asn and p.Arg86Cys, the percentage of cells with predominantly nuclear HA signal was 78.5 %, 77.5 %, and 81.9 %, respectively, while for p.Arg111Pro and p.Cys137Phe it was 44.8 % and 24.4 %, respectively (Figure 2D and E). Consistently, for the p.Arg111Pro and p.Cys137Phe variants, we counted 44.2 % and 62.0 % of cells with equal HA signal in cytosol and nucleus, respectively, and also a small number of cells with predominantly cytosolic staining.
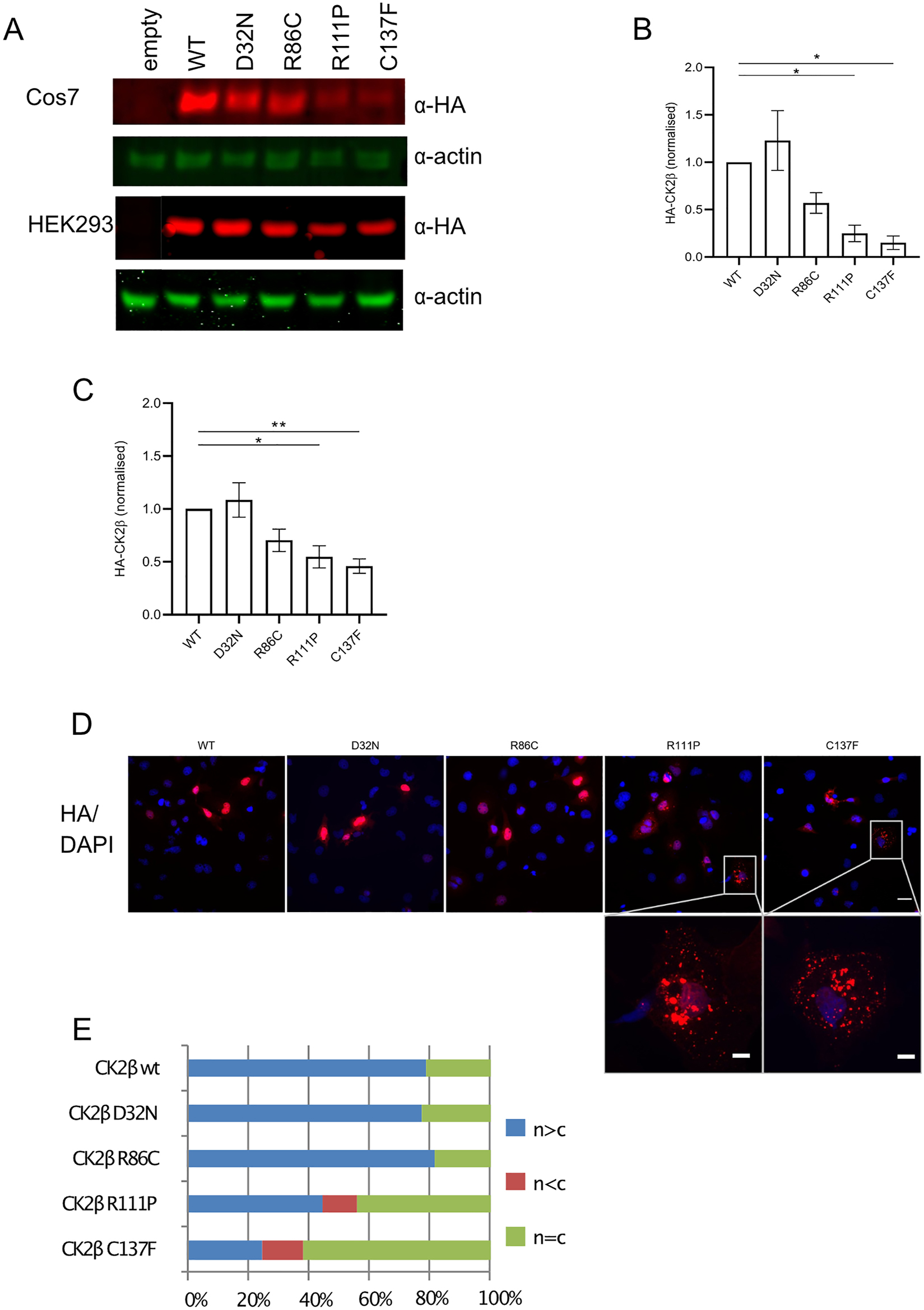
Expression and subcellular localization of POBINDS linked mutant proteins. Immunoblotting of Cos7 and HEK293T lysates overexpressing WT and mutant HA-tagged CK2β mutants and β-actin (A) and quantification thereof (B) (Cos7), (C) (Hek293T). Cos7 cells, stained with anti-HA antibody and DAPI (D). White bar represents 30 and 10 μm in the zoomed image. Quantification of CK2α subcellular localization. For all conditions, more than 200 cells were counted, and three independent experiments performed (E).
Using the translational inhibitor cycloheximide (CHX), we directly addressed the question of reduced stability of the p.Arg111Pro and p.Cys137Phe variants. We analysed lysates after 3 and 6 h of CHX treatment by Western blotting, using anti-HA and anti-CK2β antibodies, in order to compare the stabilities of overexpressed and endogenous proteins (Figure 3A). All overexpressed CK2β proteins were more labile than endogenous CK2β (Figure 3A and B). For example, within 3 h WT HA-tagged CK2β was reduced to 50 % of the level at time = 0. The slopes of the degradation curves were significantly steeper for the p.Arg111Pro and p.Cys137Phe variants, especially after 3 h of incubation, and were not significantly reduced for the p.Arg86Cys variant (Figure 3B), reflecting the results of the steady-state readout without CHX treatment (Figure 2B and C).
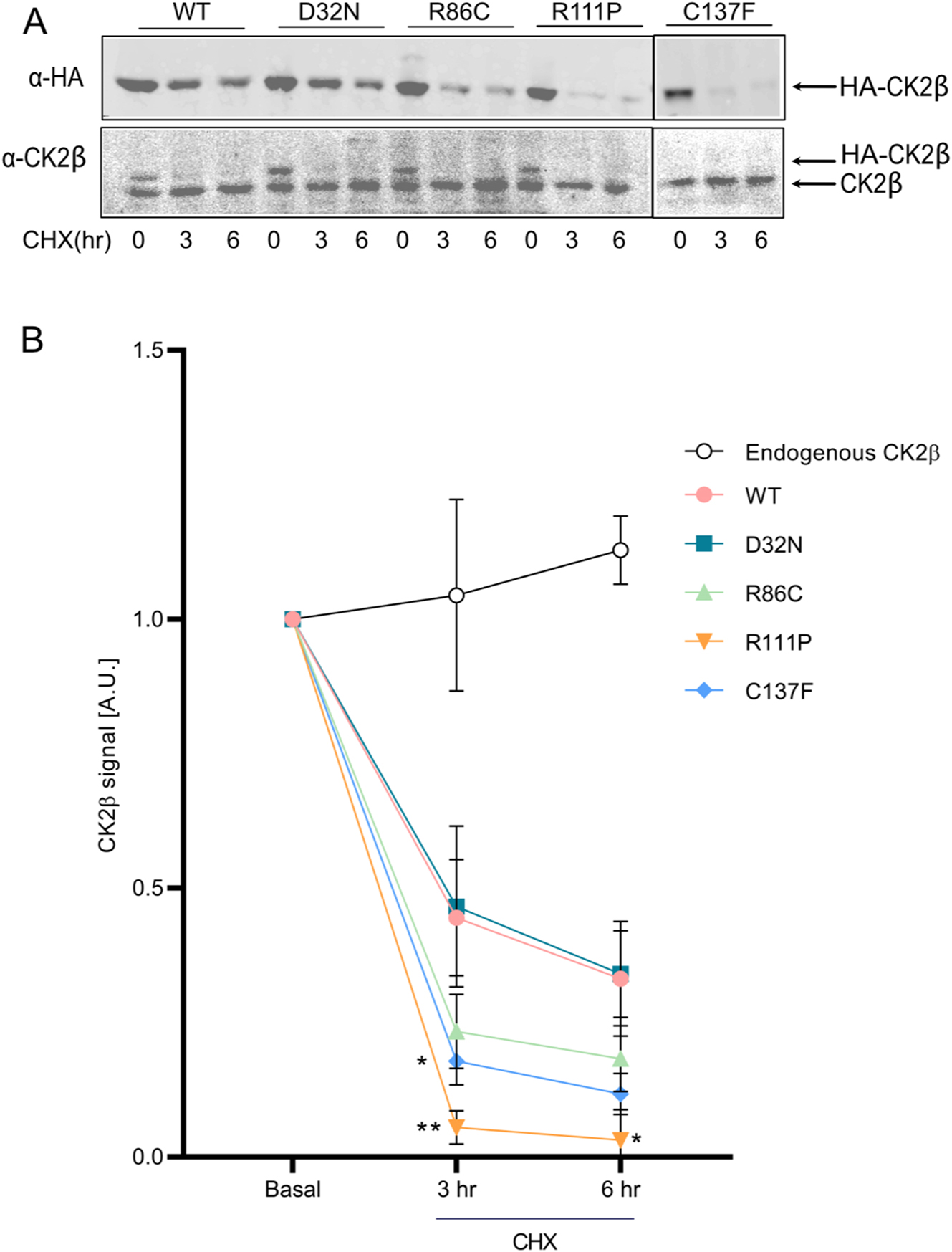
Stability of mutant proteins. Cycloheximide (100 μg/ml) was added to HEK293T cells expressing WT and mutant CK2β proteins in a chase experiment over 6 h. 40 µg of protein lysate was analysed by Western blotting using anti-HA antibody. Results from three independent experiments were quantified, with basal expression levels normalized to 1. Graphs show mean value ± SEM (B). Statistical analysis was performed using two-way ANOVA test with Dunnett post-test.
We wanted to determine whether the proteasomal activity was involved in the degradation of the p.Arg111Pro and p.Cys137Phe variants, since the role of the proteasome in the degradation of misfolded proteins in the unfolded protein response (UPR) pathway is well established (Hetz et al. 2020). To this end, the proteasome inhibitor MG132 was applied to HEK293T cells expressing HA-tagged WT and mutant CK2β. MG132 stabilized all over-expressed proteins while, again, similar to the CHX experiment, it had little effect on endogenous CK2β, as previously shown (Canton et al. 2001) (Figure 4A and B). The fold changes of HA expression, i.e. the ratios of HA-signal in the presence versus in the absence of MG132, clearly indicated that overexpressed proteins were susceptible to proteasomal degradation, with highest ratios of 4.1 and 4.6 observed for the p.Arg111Pro and p.Cys137Phe variants, although the results were not statistically different. Furthermore, the p.Asp32Asn variant showed a trend towards a reduced ratio, corresponding to a reduced susceptibility to proteasomal degradation (Figure 4A and B).
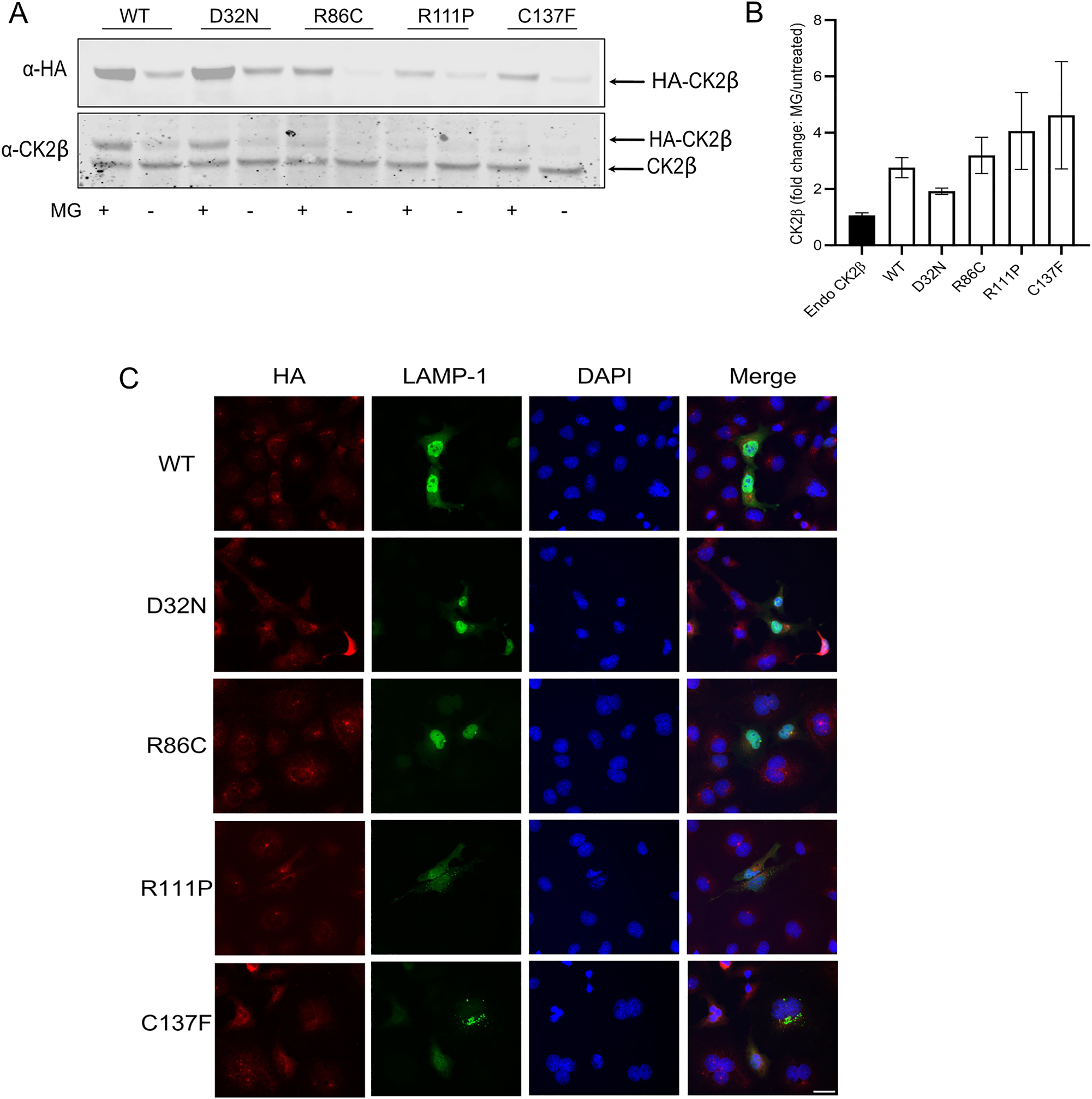
Degradation of mutant proteins. MG132 (10 µM) was added to HEK293T cells expressing WT and mutant CK2β proteins overnight (16 h). 60 µg of protein lysate was analysed by Western blotting using anti-HA antibody (A). Results from three independent experiments were quantified, fold changes calculated as the ratio of HA signal with/without MG132 and plotted. Graphs show mean value ± SEM (B). Statistical analysis was performed using two-way ANOVA test with Dunnett post-test. Immunofluorescence imaging of Cos7 cells, expressing WT and mutant CK2β, stained with anti-HA, anti-Lamp1 antibodies and DAPI (C). White bar represents 30 μm.
Proteasomal degradation canonically involves fusion of the proteosome with lysosomes, but lysosomal degradation can occur independently of the proteasome (Kim et al. 2024). Thus, we wanted to probe co-localization of the unstable variants p.Arg111Pro and p.Cys137Phe with the lysosomal marker Lamp1, based on the punctate staining observed in the cytosol. We observed partial co-labelling; however, the overlap was not complete (Figure 4C). We therefore suggest that the degradation of the mutant proteins must involve other pathways.
2.2 CK2β homodimerization is affected in POBINDS-associated variants
The CK2 holoenzyme formation comprises a first step of CK2β-β dimerization, followed by CK2α recruitment (Chantalat 1999; Raaf et al. 2008). Based on the location of the CK2β p.Arg111 and p.Cys137 amino acids within the β–β interaction region, where four Zinc-finger-forming cysteines are either present in close proximity, or where p.Cys137 itself provides the sulfhydryl group for bridging, we hypothesized that β–β homodimerization might be impaired. We performed co-immunoprecipitation assays to address this question. We expressed GFP-WT CK2β and HA-tagged wildtype and CK2β mutants in HEK293T cells, we precipitated GFP-WT CK2β and blotted for the presence of HA-tagged versions of CK2β (Figure 5A). As expected, both the p.Cys137Phe and p.Arg111Pro variants co-precipitated less with WT-CK2β mutants than HA-tagged CK2β wildtype, whereas no difference was observed for the p.Asp32Asn and p.Arg86Cys variants (Figure 5A and B).
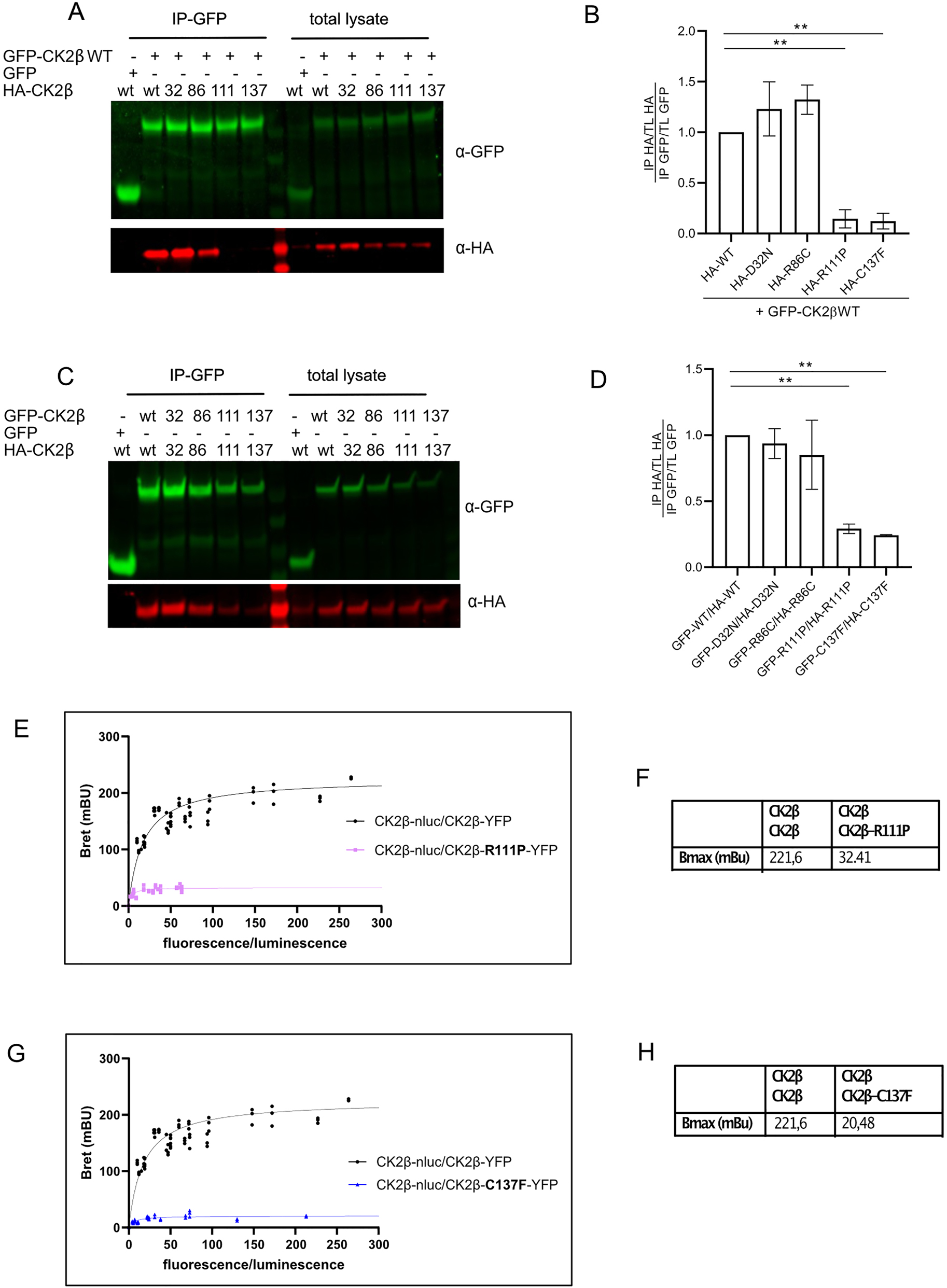
CK2β homodimer formation of mutant proteins. GFP-tagged CK2β (WT) and the series of HA-tagged CK2β mutants were expressed in HEK293T cells for 24 h. After lysis, 400 µg of lysate were used for immunoprecipitation using anti-GFP magnetic beads and washed precipitates probed with anti-HA and anti-GFP antibodies (A). Quantification of three independent experiments (B). GFP-tagged CK2β (WT and mutants) and the series of HA-tagged CK2β WT and mutants were expressed in Hek293 cells for 24 h. After lysis, 400 µg were used for immunoprecipitation using anti-GFP magnetic beads and washed precipitates probed with anti-HA and anti-GFP antibodies (C). Quantification of three independent experiments (D). BRET saturation curves of homodimerization of CK2β-β in HEK293T cells transiently transfected with a constant DNA amount of CK2β-Nluc and increasing quantities of CK2β R111P-YFP mutant. The curves were fitted using a non-linear regression equation assuming a single binding site (GrapPadPrism). Results were expressed in milliBRET units (mBU, with 1 mBU corresponding to the BRET values multiplied by 1,000) (E), and for the combination of CK2β-Nluc CK2β R137F-YFP (G). BretMax values for the interaction CK2β WT with R111P (F) and C137F (H). Data were obtained with at least three separate experiments with triplicate determinations. Statistical analysis was performed using two-way ANOVA test with Dunnett post-test (B, D) or with Newman Keuls post-test (F, H).
In POBINDS, about 50 % of the CK2β expressed is expected to be mutant, so in cells two mutant CK2β molecules will also try to form dimers. We wondered whether the dimer formation of two mutant proteins would be even more impaired.
To this end, we expressed both GFP- and HA-tagged mutant pairs, precipitated the GFP-tagged CK2β and blotted for the presence of HA-CK2β (Figure 5C and D). As a result, dimerization of only the p.Arg111Pro and p.Cys137Phe proteins was again significantly impaired, although the reduction compared to WT-WT interaction was less than when testing WT pairing with a mutant molecule. However, these two experiments cannot be directly compared due to differences in the expression of the various constructs.
To validate our findings by another approach, bioluminescence resonance energy transfer (BRET) saturation assays were developed to monitor, quantify, and compare the formation of CK2β-CK2β dimers. BRET saturation experiments have previously been successfully conducted to investigate the relative affinities of various interacting protein pairs (El Khamlichi et al. 2019; Guillemain et al. 2020; Mercier et al. 2002; Wang et al. 2005) and to estimate the proportions of dimeric and monomeric fractions of the two BRET partners (Mercier et al. 2002; Couturier and Jockers 2003).
The BRET signal is expressed as a function of the fluorescence/luminescence measured, which corresponds to the expression of the acceptor (YFP) and the donor (NLuc). Two measures are extracted from saturation curves: BRETmax which represents the maximal BRET signal reached at donor saturation and informs about the relative amounts of dimers, and BRET50, which corresponds to the [YFP]/[luc] ratio at 50 % of BRETmax and has been suggested as an indicator of the relative affinities between donor and acceptor fusion proteins.
We transiently transfected HEK293T with expression vectors for CK2β WT-NanoLuciferase and YFP-tagged CK2β p.Arg111Pro or p.Cys137Phe mutants, by applying escalating doses of YFP-CK2β plasmids and keeping the amount of the donor Luciferase plasmid constant. As a result, BRET assays confirmed the loss of β–β dimerization for both mutants. Compared to the CK2β/CK2β dimerization (BRETmax 221.6 mBu), BRETmax was reduced by factors of 7.4 to 32.4 mBu and by 11.9x to 20.5 mBu, for p.Arg111Pro and p.Cys137Phe, respectively (Figure 5E–H). The strongly reduced BRET is indicative of a lack of dimerization of mutants. We were unable to determine BRET50 values for the mutants due to the very low BRET signal at all Fluo/Luc ratios.
We wanted to determine if CK2α recruitment into the holoenzyme was affected by POBINDS variants. To do this, we again used the BRET technology (CK2α-Luc, CK2β mutants-YFP). We found that none of the variants showed a difference compared to WT, in BRETmax and BRET50 (Figure 6A–E). This was unexpected since it is known that CK2β-CK2β dimerization is a prerequisite for holoenzyme formation, and therefore we expected to see a clear reduction in CK2α recruitment to p.Arg111Pro and p.Cys137Phe mutants.
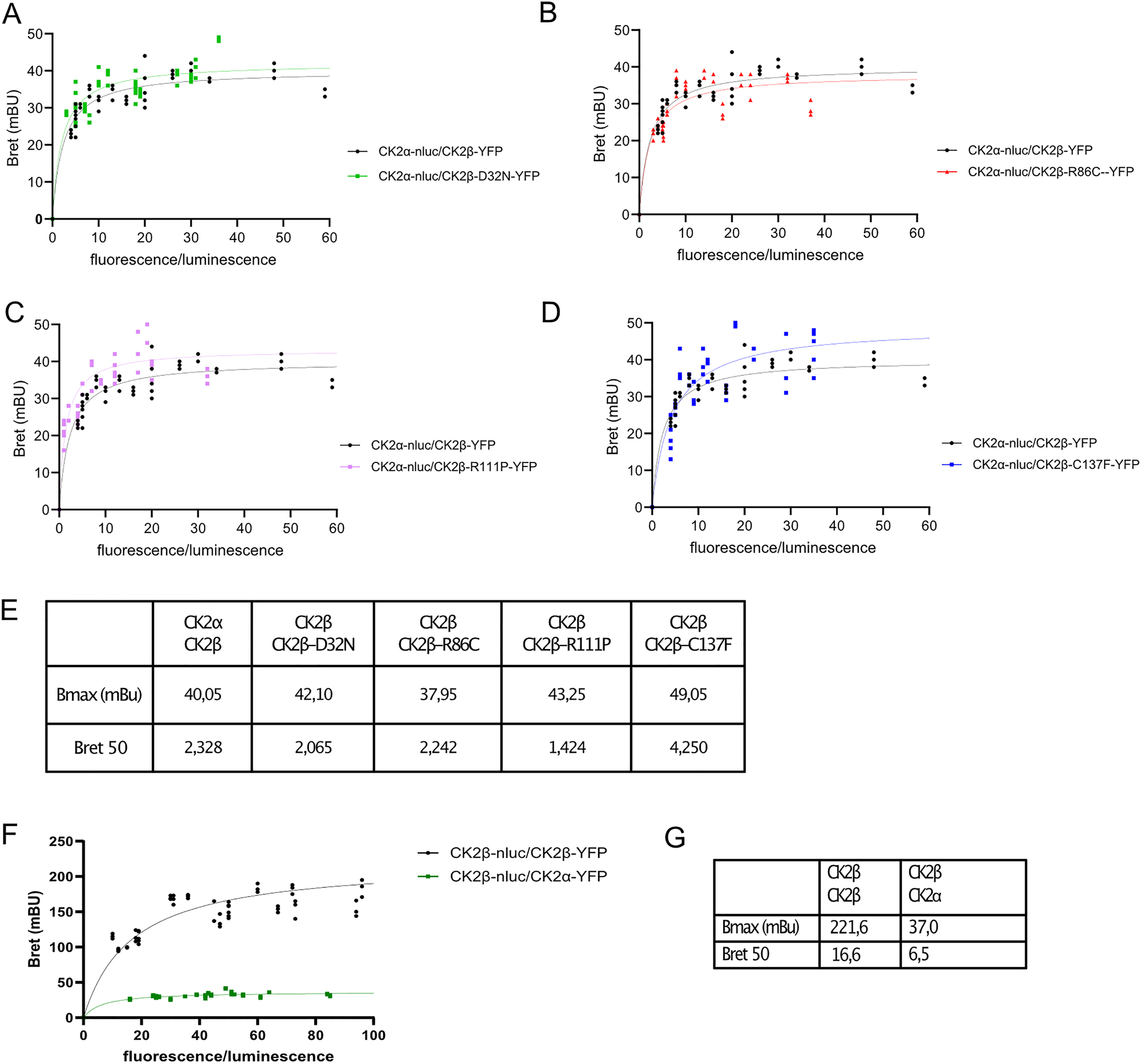
CK2α-β formation is not affected by CK2β mutants. BRET saturation curves of heterodimerization of CK2α and CK2β in HEK293T cells. BRET signals were performed on HEK293T cells transiently transfected with a constant DNA amount of CK2α-Nluc and increasing quantities of CK2β-YFP. The curves were fitted using a non-linear regression equation assuming a single binding site (GraphPadPrism). Results were expressed in milliBRET units (mBU, with 1 mBU corresponding to the BRET values multiplied by 1,000). Comparison CK2α-CK2β WT with CK2α-CK2βD32N (A), with R86C (B), with R111P (C) and with C137F (D). Data were obtained with at least three separate experiments with triplicate determinations. Parameters derived from BRET saturation curves of the heterodimerization of CK2. Table listing Bmax and Bret50 values for individual combinations (E). BRET saturation curves comparing CK2α-β and CK2β-β interaction between wildtype CK2 subunits (F), table listing Bmax and Bret50 values (G).
Comparing the affinity of the CK2α-CK2β versus CK2β-CK2β interaction in our cellular BRET system, the saturation curves are qualitatively highly distinct: the CK2β/CK2β curve shows a clear response to expression levels with high maximal BRET (222 mBu) and a high BRET50, whereas CK2α/CK2β curve has reduced BRETmax of 37 mBU, suggesting that there are more CK2β/CK2β dimers compared to CK2α/CK2β dimers (Figure 6F and G). The BRET50 values were slightly lower for CK2α/CK2β (BRET50 = 6.5) compared to CK2β/CK2β (BRET50 = 16.6). The variation in BRET50 values suggests that CK2β may in vivo have slightly higher affinity for binding to CK2α compared to CK2β.
2.3 CK2α activity when complexed with POBINDS-variant proteins
Finally, we wanted to determine the activity of CK2 holoenzyme, composed of at least one mutant CK2β molecule, to phosphorylate a substrate peptide in vitro. We expressed HA-tagged wildtype and CK2β mutants, precipitated them using anti-HA-agarose and performed an in vitro kinase assay on the washed immunocomplexes. For the p.Arg111Pro and p.Cys137Phe mutants, we observed a significant reduction in ADP generation, correlating with ATP consumption, a surrogate for CK2 activity in the ADP-Glo system (Figure 7A). This is not surprising given the low expression of these two mutants, as shown in Figure 1A. Quantification of the presence of CK2α in the precipitates, shows no significant changes in co-precipitated CK2α when normalized to CK2β (Figure 7B), mirroring the findings of the BRET assay in Figure 6. The normalization of ADP generation by the amount of co-precipitated CK2α, restored kinase activity for all mutants, indicating that the activity towards the specific substrate peptide is unaffected by the presence of one or more mutant CK2β molecules in the holoenzyme (Figure 7C). As control, CK2α is not co-precipitated in the absence of HA-CK2β, testifying to the specificity of the precipitation and indicative that the activity measured is due to CK2α-β interaction (Figure 7D).
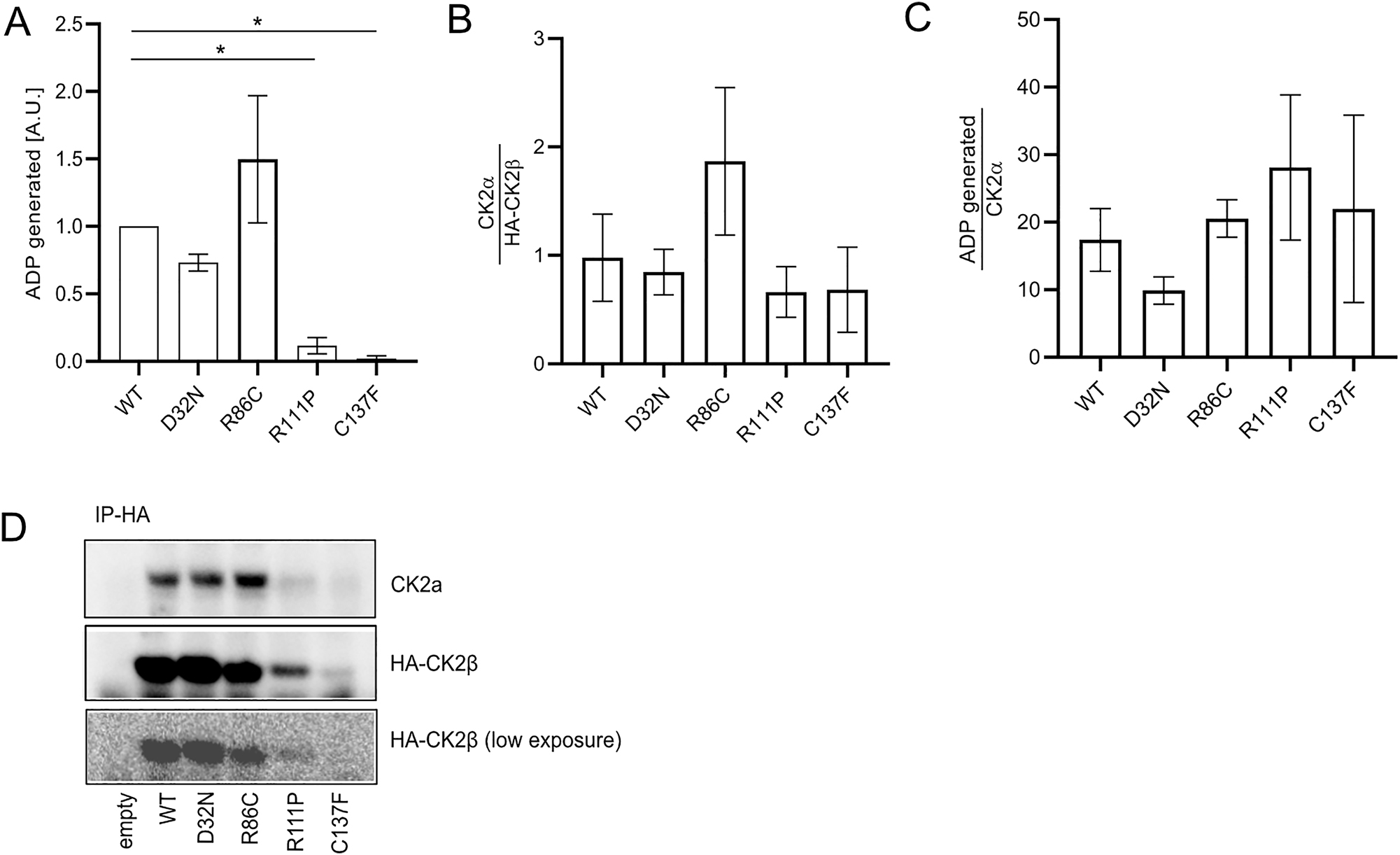
In vitro kinase activity of mutant immunoprecipitates. HA-CK2β WT and mutants were expressed in Cos7 cells. 400 μg of cell lysates were used for immunoprecipitation using anti-HA magnetic beads, followed by a kinase assay performed with washed immunoprecipitates. ADP generation was plotted (A). The immunoprecipitates were then separated on a SDS-PAGE gel and amounts of HA-CK2β and CK2α in precipitates were quantified (B). ADP generation was normalized by amount of co-precipitated CK2α (C). Results from three independent experiments were used for quantification. Representative western blot showing CK2α and HA-CK2β after α-HA immunoprecipitation (D). Graphs show mean value ± SEM. Statistical analysis was performed using one-way ANOVA test with Dunnett post-test.
3 Discussion
3.1 Stability of variants
We observed a generally reduced stability of exogenously expressed compared to endogenuous CK2β, at steady state and when chased with translational inhibitor cycloheximide. One difference between the two types of protein that may explain this, is the HA-tag on the exogenous CK2β. Importantly, we detected increased instability specifically for p.Arg111Pro and p.Cys137Phe variants, a finding which is corroborated by previous results showing that a Zinc-finger-deficient mutant (Myc-CK2β Cys109Ser/Cys114Ser) is less stable than its WT counterpart (Canton et al. 2001). In this publication, the authors additionally observed reduced phosphorylation of this mutant, rendering it more labile. We further show that in our cell system, CK2β is majorly localized to the nucleus, confirming prior work (Lorenz et al. 1993), but the unstable mutants p.Arg111Pro and p.Cys137Phe are retained in the cytosol.
The mechanism of degradation involves the proteasome which has a stronger effect in the p.Arg111Pro and p.Cys137Phe variants, as well as lysosomal degradation, where the lysosomal marker Lamp1 partially co-localizes with these two mutants. However, we believe that other degradation mechanisms may also be involved. Interestingly, the most stable mutant is the p.Asp32Asn variant, which has been associated to a new intellectual disability-craniodigital syndrome (IDCS) distinct from POBINDS (Asif et al. 2022). In patient cells, the authors observed increased CK2β levels in the nucleus and cytoplasm, together with altered protein-protein interaction and dysregulation of canonical Wnt signalling, leading the authors to postulate a dominant negative effect (Asif et al. 2022). In our cellular system, the p.Asp32Asn mutant does not express to a higher extent than WT protein, but it appears to be the most stable compared to the other mutants tested. Another variant located adjacently in the KEN box, p.Leu39Arg was shown to be less stable, leading to reduced net CK2 activity (Di Stazio et al. 2023). These findings indicate that the precise location of a variant, even within a small motif, is clearly important for exerting biological effects.
3.2 CK2β dimerization and CK2α-β dimerization
Using co-immunoprecipitation and BRET assays, we found that the p.Arg111Pro and p.Cys137Phe variants have a significantly reduced dimerization capacity. This is not surprising, given that p.Cys137 is itself involved in Zn2+ binding, while the p.Arg111 is closely adjacent to p.Cys109, another of the four Zinc-finger competent cysteines in the β–β interaction domain. The β–β dimerization is mediated by many hydrophobic interactions between zinc ribbon motifs (composed of three-stranded β sheets, coordinating one Zn2+) for each of the two β monomers (Chantalat 1999; Filhol et al. 2005). It is therefore noteworthy to state that mutation of only one residue within this domain almost completely abolishes the dimerization. This finding indeed goes beyond Canton et al. (2001), who observed the impairment of CK2β homodimerization only for a double mutant myc-CK2β p.Cys109Ser/p.Cys114Ser (directly concerning two zinc-binding cysteine side chains) and not an interconnecting residue like Arg111.
The finding that none of the CK2β variants had a direct effect on the recruitment of CK2α into the holoenzyme, was initially unexpected since it has been established that β–β dimerization is needed to form a shared interface where two β subunits interact with two distinct CK2α subunits. This interface is provided by two different C-terminal segments (AA 164–170/interface region 1 and AA 186–198/interface region 2, as shown in Figure 1A) (Niefind et al. 2001).
We can speculate that endogenous CK2β may interact, albeit weakly, with the CK2β mutant, which could be sufficient to recruit CK2α and induce a BRET signal. However, the affinity of wildtype CK2β to itself outweighs binding affinity to zinc-binding motif mutant CK2β, and therefore such dimers are unlikely to occur.
Alternatively, we suggest that the interaction with CK2α may directly occur via monomeric CK2β, which would indicate that α-β dimers exist in our cellular system.
Besides the BRET results, we first observed a reduced co-immunoprecipitation of CK2α protein and activity with CK2β p.Arg111Pro and p.Cys137Phe variants, but this difference was rescued upon normalization, suggesting that at the molecular level, mutant CK2β p.Arg111Pro and p.Cys137Phe are able to interact with CK2α in our cellular overexpression system.
Previous studies had shown that the C-terminal region of CK2β (AA 171–215) stabilizes the CK2α-CK2β interaction (Bidwai et al. 1995; Reed et al. 1994; Sarno et al. 1999). In particular, the region encompassing AA186-193 in interface region 2, bound to CK2α, and a peptide containing this segment was efficient to antagonize holo-complex formation (Laudet et al. 2007; Raaf et al. 2009). Our variants are N-terminal to the crucial interface region 1 and 2, yet the lack of dimerization suggests these regions will fold differently. Structural analysis of CK2β monomer has, to our knowledge, only been performed on C-terminally truncated CK2β (Chantalat 1999).
The question of whether monomeric full-length CK2β can interact with CK2α was addressed previously: In vitro, in the presence of reducing sulfhydryl-modifying reagents, α-β complexes were detected, which, however, did not display activity (Meggio et al. 2000). The difference in catalytic activity could be explained by the in vitro system and the presence of reducing agent, which is not the case in our precipitation experiments.
In contrast, in a Cos7 cell overexpression system, a doubly mutated p.Cys109Ser/Cys114Ser CK2β which does not dimerize, did not co-precipitate with overexpressed HA-CK2α′ (Canton et al. 2001). Further, pulldowns between GST-CK2 α′ and this in vitro transcribed/translated CK2β mutant failed to detect an interaction. In these experiments, the precipitates were not normalized to input, and the input was not shown. Thus, the detection limit may not have been reached due to the low expression/high instability of the CK2β p.Cys109Ser/p.Cys114Ser mutant. Further, as binding partner, CK2α′ was used, which is known to have a one magnitude fold lower affinity for CK2β, compared to CK2α (Bischoff et al. 2011). However, in a yeast-two hybrid system, affinity of the same p.Cys109Ser/p.Cys114Ser non-dimerizing human CK2β mutant to human CK2α was demonstrated (Canton et al. 2001). Thus, we hypothesize that besides instability of the mutant protein, the difference in expression systems (yeast and in vitro translation) could underlie the contradictory results: the yeast proteome is highly similar compared to the mammalian proteome and may alter protein folding through chaperones and post-translational modifications. From this perspective, our results align with the results in yeast.
Regarding the BRET curves of CK2β/CK2β and CK2α/CKβ, the highest BRET signal for CK2β/CK2β (BRETmax = 222 mBu) suggests that there are more CK2β/CK2β dimers compared to CK2α/CKβ (BRETmax = 37 mBu). But these differences could also reflect variations in the orientation of the YFP/RLuc moieties within the proteins. BRET50 values were slightly lower for CK2α/CKβ (BRET50 = 6.5) compared to CK2β/CK2β (BRET50 = 16.6), suggesting that CK2β may have a slightly higher affinity for binding to CK2α compared to CK2β. This contrasts with the knowledge about CK2 subunit interactions showing that CK2β has a stronger affinity to itself, compared to CK2α, which already presents very high affinity to CK2β, Kd being in the nanomolar range (Bischoff et al. 2011). However, affinity experiments have thus far been performed in in vitro systems. Here, we evaluated the physical association of CK2 subunits in intact living cells which differ from in vitro methods that rely on purified proteins. In order to define the affinities in a more quantitative manner, one would have to perform future BRET experiments that allow better curve fitting at even very low fluorescence/luminescence ratios. Finally, the differences observed between BRET and in vitro experiments could be due to the tags, Nanoluciferase and YFP, which may alter the affinity between subunits. Thus, this question needs to be further studied in the future.
3.3 Activity
We did not detect any changes in CK2 activity as a consequence of the variants studied. We used a well-characterized acidic substrate peptide in the in vitro kinase assay. However, one peptide may not be sufficient to detect subtle changes in activity. For example, disease-associated variants of the CK2α subunit, which cause a neurodevelopmental disorder known as OCNDS, have highlighted the importance of expanding the peptide array to capture changes in substrate binding (Werner et al. 2022).
In addition, CK2β is able to recruit substrates (Appel et al. 1995; Bojanowski et al. 1993; Leroy et al. 1999) and bind, as a homodimer, to other proteins and kinases independently of CK2α (Chen et al. 1997; Guerra et al. 2003; Hagemann et al. 1997). Therefore, it is of great importance to study different missense variants in protein-protein interaction assays, such as proximity-labelling, Bio-ID and others, which could also provide insights into the effect of POBINDS missense variants on the interactome and phosphorylation fingerprint of holoCK2 in POBINDS.
One consideration is that our experiments were performed in immortalized cells that already produce a steady state amount of CK2β. It is possible that overexpression per se disrupts the balance between dimerization and holoenzyme formation. CK2β is a highly conserved regulatory subunit and excessive synthesis of CK2β will form dimers that disrupt the overall structure and function of CK2 (Graham and Litchfield 2000; Niefind et al. 2001). However, when comparing the amount of overexpressed HA-CK2β to endogenous CK2β in our experimental system, the exogenous protein levels are always lower and therefore overexpression is not expected to significantly alter homeostatic processes.
We have shown that the precipitation of CK2α is specific and dependent on the presence of HA-CK2β, therefore the ATP hydrolysis activity does not stem from monomeric CK2α, devoid of CK2β. For mutant proteins p.Arg111Pro and p.Cys137Phe this finding argues again that CK2α-β heterodimers can form and be catalytically active. One factor to be taken into consideration is potential co-precipitation of CK2α′, however, we fail to detect this isoform with the pan-CK2α specific antibody, suggesting that if it were precipitated, it would be below our detection limit.
What are the implications of this finding for cells from POBINDS patients carrying these missense variants? If the Zinc-finger mutant proteins are less stable, then even with residual α-β binding, one would expect a reduced net activity of CK2 holoenzyme per amount of tissue. This would suggest that missense variants of the Zinc-finger domain may, at least in part, have a similar effect on CK2 activity as nonsense or splice variants, namely a reduction in the pool of functional holoCK2. Thus, these mutants could be considered to act in a haplo-insufficient manner. Further work is needed to confirm this hypothesis.
Our findings indicate that disruption of β–β dimerization does not necessarily prevent CK2α binding or catalytic activity. While it has previously been shown that preformation of the homodimer is necessary for holoenzyme formation, in the case of CK2β p.Arg111Pro and p.Cys137Phe variants, we still observed α–β interaction by Co-IP and BRET assays.
Based on our results, we further hypothesize that the p.Asp32Asn and p.Arg86Cys, both exposed on the surface and suggested to be important for ligand/substrate binding (Chantalat 1999) may act in a qualitatively different manner, since these variants are as stable as the WT protein, do not affect β–β dimerization, holoenzyme formation or activity towards a peptide, but may alter the profile of protein-protein interaction and in substrate binding.
Our work highlights the need to individually investigate missense variants in order to allow for genotype-phenotype correlations to better understand and treat this neurodevelopmental disorder.
4 Materials and methods
4.1 Plasmids and site-directed mutagenesis
Rat CK2β coding sequence (BC078807) was cloned into PIRESneo3 vector in frame with a N-terminal HA-tag (Rebholz et al. 2009). For BRET assays, coding sequences for rat CK2α (BC091130) was cloned into pEYFP-N1, with EYFP at the C-terminus, and CK2β into pRLuc-N1 and pEYFP-N1 vectors, with RLuc or EYFP at the N-terminus.
Site directed mutagenesis was performed by the Biochemistry and Biophysics (B&B) core facility of the Institute of Psychiatry and Neuroscience of Paris (IPNP). Mutagenesis targeting CK2β was carried out using the Q5® Site-Directed Mutagenesis Kit (NEB, France) in conjunction with NEBaseChanger V1 software (NEB, France). To construct the pEYFP-Ck2β WT and mutant plasmids, the Ck2β genes and the pEYFP plasmid were amplified with Q5® polymerase (NEB, France). The resulting amplicons were assembled into the linearized plasmid using the NEBuilder® HiFi DNA Assembly Kit (NEB, France).
4.2 Cell culture and protein extraction
Human Embryonic Kidney (HEK293T) cells were obtained from Evelyne Bloch-Gallego, Institut Cochin, Paris, France. Cos-7 cells (ATCC #CRL-1651) were obtained from Thierry Galli, IPNP, Paris, France. Cells were grown in DMEM 10 % FBS and transfected with Lipofectamine 2000 (Invitrogen, 52887) according to manufacturer’s instructions. 24 h post transfection, cells were washed with phosphate buffered saline (PBS 0.1 M) and lysis buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1 % NP-40, 5 mM EDTA, 1 mM EGTA) containing protease and phosphatase inhibitor cocktails (Roche, 66373700 and 04906837001) was added in each plate/well. Cells were scraped, pooled, and collected. Lysates were centrifuged at 13,000 rpm for 20 min at 4 °C and the supernatant was collected for western blot or immunoprecipitation experiments.
For CHX experiments, cycloheximide (100 μg/ml) was added for 3 or 6 h, cells were immediately lysed thereafter. Untreated cells were lysed at the start of drug incubation.
For MG132 experiments, MG312 (10 µM) was added to cells for 16 h before lysis.
4.3 Western blot analysis
Proteins were separated on a 4–12 % Bis–Tris protein gels by SDS-PAGE and blotted onto either nitrocellulose or PVDF membranes. Non-specific binding sites were blocked either with 5 % non-fat dry milk in PBS/0.1 % Tween20 or with 3 % BSA in PBS/0.1 % Tween20. Membranes were incubated with primary antibodies for 2 h or overnight. The following antibodies were used: anti-HA (Cell Signalling, 3724S), anti-GFP (Abcam, ab1218), anti-β-actin (Santa Cruz, sc-81178), anti-CK2α (Cell Signalling, 2656S) and anti-CK2β (Bethyl, A301-983A). For BRET test, anti-GFP antibody (Cell Signalling, 2956) was used to recognize YFP-tagged proteins. Bands were visualized either by incubation in ECL (Amersham) for chemiluminescence detection by ChemiDoc system (BioRad) or with the Odyssey CLx imaging system (LI-COR) for fluorescence detection. Relative protein amounts were quantified with ImageJ software.
4.4 Immunocytofluorescence
Cells were fixed in 4 % paraformaldehyde for 10 min, rinsed with PBS and then permeabilized with 0.5 % TritonX-100 for 5 min. Cells were blocked with 3 % BSA in PBS for 1 h at room temperature, then incubated for 2 h at room temperature or overnight at 4 °C with the primary antibody diluted in 3 % BSA/PBS. The following antibodies were used: anti-HA (Cell Signalling, 3724S) and anti-Lamp1 (DB Biosciences, 555798). After three washes with PBS, cells were incubated with anti-mouse Alexa Fluor 568, anti-rabbit Alexa Fluor 568 and anti-mouse Alexa Fluor 488 antibody (Invitrogen), diluted in 3 % BSA/PBS during 2 h at room temperature in the dark. After three washes with PBS, cells were mounted using DAPI mounting medium (Prolong, Invitrogen). Immunofluorescent staining was imaged with a Zeiss Axioplan fluorescence microscope and analysed with ImageJ software.
4.5 Immunoprecipitation and immunoblot analysis
HEK293T and Cos7 cells were lysed in lysis buffer (50 mM Tris-HCl pH 7.6, 150 mM NaCl, 5 mM EDTA, 1 mM EGTA, 1 % Triton X-100, protease and phosphatase inhibitor cocktails (Roche, 66373700 and 04906837001). Whole cell extracts were cleared by centrifugation. Protein content was determined using the bicinchoninic acid (BCA) protein assay kit (Thermo Fisher Scientific, 23225), with a bovine serum albumin (BSA) standard series. 400 µg of lysate were used for precipitation: GFP-CK2β was immunoprecipitated using anti-GFP-magnetic antibody for 16 h (ABclonal, AE079). Immune complexes were washed on a magnetic rack and subjected to analysis by SDS/PAGE and immunoblotting. The antigen-antibody complexes were incubated with anti-HA (Cell Signalling, 3724S), anti-GFP (Abcam, ab1218) or pan-CK2α antibody (Cell Signaling #2656), followed by incubation with goat anti-Rabbit IgG (licorbio IRDye 680RD, 926-68071) or goat anti-Mouse IgG (Licorbio IRDye 800CW, 926-32210). Fluorescence was detected by the Odyssey infrared imaging system (LI-COR), and quantified using ImageJ software.
4.6 In vitro kinase assay (IVK)
Wildtype and mutant proteins were expressed in Cos7 cells and lysed in lysis buffer, LB (50 mM Tris-HCl pH 7.6, 150 mM NaCl, 5 mM EDTA, 1 mM EGTA, 1 % Triton X-100, protease and phosphatase inhibitor cocktails (Roche, 66373700 and 04906837001). Immunoprecipitation using anti-HA antibody (0.5 μg per assay point, Cell Signalling, 3724S) was performed at 4 °C for 3 h, followed by two washes in LB and two washes in kinase 1x buffer (MOPS pH 7.4 (50 mM), β-glycerophosphate (10 mM), EGTA (2 mM) and MgCl2 (12 mM)). The immunoprecipitates were divided into two equal parts. Incubation took place at 30 °C for 20 min in kinase buffer containing ATP (150 μM), and either no peptide (control) or CK2 peptide RRRADDSDDDDD (150 uM, Abcam, 204855). Then the ADP-Glo™ (Promega) and detection reagents were added, according to manufacturer’s instructions (Promega ADP glow V9101) and luminescence was measured using the SpectraMax plate reader (Molecular Devices). The value obtained from the control (no peptide) was considered background phosphorylation (autophosphorylation plus eventual phosphorylation of protein(s) co-precipitated with CK2) and was subtracted from the value obtained in the sample incubated with peptide. Precipitates were then analysed by western blotting as described above.
4.7 BRET
HEK293T cells were transfected with the calcium phosphate precipitation method. 48 h after transfection, cells were washed twice with ice-cold PBS and resuspended in HBSS buffer. Intact cells were distributed in 96-well microplates (Optiplate, Perkin Elmer). Furimazine substrate (Interchim, B2W800) was added at a final concentration of 5 μM, and readings were performed with a Mithras LB 943 Multireader (Berthold), which allows the sequential integration of luminescence signals detected with two filter settings (NLuc filter, 480 ± 10 nm; YFP filter, 540 ± 20 nm). Emission signals at 540 nm were divided by emission signals at 480 nm. The BRET ratio was defined as the difference between the emission ratio obtained with co-transfected NLuc and YFP fusion proteins and that obtained with the NLuc fusion protein alone. The results were expressed in milliBRET units (mBU, with 1 mBU corresponding to the BRET ratio values multiplied by 1,000). BRETmax is the maximal BRET signal obtained in milliBRET units and BRET50 represents the ratio of acceptor and donor receptors (acceptor/donor) yielding 50 % of maximum BRET signal. All BRET, luminescence, and fluorescence measurements were performed at 21 °C using a Mithras LB 943 microplate analyser (Berthold). All data analyses were carried out with the GraphPad Prism 9.5.1 software for Windows (GraphPad Software Inc, San Diego, CA, USA). Concentration response curves were fitted by nonlinear regression and saturation curves by a hyperbolic one-binding site equation. The method provided estimates for BRETmax and BRET50 values and corresponding SEM. Each experiment was repeated at least three times.
4.8 Statistics
Data are presented as mean ± standard error of mean (SEM). Statistical differences were assessed using a parametric unpaired Student’s t-tests or one-way or two-away ANOVA with multiple comparisons in Dunnett post-tests or one-way ANOVA followed by the Newman Keuls multiple comparisons test for BRET analysis. A p-value below 0.05 was considered to be statistically significant, with *p < 0.05, **p < 0.01 and ***p < 0.001.
Funding source: H2020 Marie Skłodowska-Curie Actions
Award Identifier / Grant number: 894207
Funding source: Agence Nationale de la Recherche
Award Identifier / Grant number: ANR-22-CPJ2-0086-01
Acknowledgments
The authors would like to thank Jose Cruz Gamero for technical assistance and critical comments to the manuscript, D. Tareste, E. Boedec at the Biochemistry and Biophysics (B&B) core facility of the Institute of Psychiatry and Neuroscience of Paris (IPNP) for use of plate reader (SpectraMax, Molecular Devices), for plasmid generation and site-directed mutagenesis. Imaging was carried out at the NeurImag facility (Inserm 1266 and Université Paris Cité).
-
Research ethics: Not applicable.
-
Informed consent: Not applicable.
-
Author contributions: HK, BL and LC performed the studies, analyzed data. DB and HR co-wrote the manuscript. SML, TB, HK contributed to the manuscript. HR conceptualized the study and supervised all experiments.
-
Use of Large Language Models, AI and Machine Learning Tools: Not applicable.
-
Conflict of interest: The authors state no conflict of interest.
-
Research funding: The work was supported by the Horizon 2020 MSCA-fellowship 894207 (HR) and the ANR-22-CPJ2-0086-01 (HR).
-
Data availability: Original data that is not shown will be shared upon request.
References
Appel, K., Wagner, P., Boldyreff, B., Issinger, O.G., and Montenarh, M. (1995). Mapping of the interaction sites of the growth suppressor protein p53 with the regulatory β-subunit of protein kinase CK2. Oncogene 11: 1971–1978.Search in Google Scholar
Asif, M., Kaygusuz, E., Shinawi, M., Nickelsen, A., Hsieh, T.-C., Wagle, P., Budde, B.S., Hochscherf, J., Abdullah, U., Höning, S., et al.. (2022). De novo variants of CSNK2B cause a new intellectual disability-craniodigital syndrome by disrupting the canonical Wnt signaling pathway. HGG Adv. 3: 100111, https://doi.org/10.1016/j.xhgg.2022.100111.Search in Google Scholar PubMed PubMed Central
Ballardin, D., Cruz-Gamero, J.M., Bienvenu, T., and Rebholz, H. (2022). Comparing two neurodevelopmental disorders linked to CK2: Okur-Chung neurodevelopmental syndrome and Poirier-Bienvenu neurodevelopmental syndrome-two sides of the same coin? Front. Mol. Biosci. 9: 850559, https://doi.org/10.3389/fmolb.2022.850559.Search in Google Scholar PubMed PubMed Central
Bidwai, A.P., Reed, J.C., and Glover, C.V.C. (1995). Cloning and disruption of CKB1, the gene encoding the 38-kDa β subunit of Saccharomyces cerevisiae casein kinase II (CKII). J. Biol. Chem. 270: 10395–10404, https://doi.org/10.1074/jbc.270.18.10395.Search in Google Scholar PubMed
Bischoff, N., Olsen, B., Raaf, J., Bretner, M., Issinger, O.-G., and Niefind, K. (2011). Structure of the human protein kinase CK2 catalytic subunit CK2α′ and interaction thermodynamics with the regulatory subunit CK2β. J. Biol. Chem. 407: 1–12, https://doi.org/10.1016/j.jmb.2011.01.020.Search in Google Scholar PubMed
Blond, O., Jensen, H.H., Buchou, T., Cochet, C., Issinger, O.-G., and Boldyreff, B. (2005). Knocking out the regulatory beta subunit of protein kinase CK2 in mice: gene dosage effects in ES cells and embryos. Mol. Cell. Biochem. 274: 31–37, https://doi.org/10.1007/s11010-005-3117-x.Search in Google Scholar PubMed
Bojanowski, K., Filhol, O., Cochet, C., Chambaz, E.M., and Larsen, A.K. (1993). DNA topoisomerase II and casein kinase II associate in a molecular complex that is catalytically active. J. Biol. Chem. 268: 22920–22926, https://doi.org/10.1016/s0021-9258(18)41614-6.Search in Google Scholar
Canton, D.A., Zhang, C., and Litchfield, D.W. (2001). Assembly of protein kinase CK2: investigation of complex formation between catalytic and regulatory subunits using a zinc-finger-deficient mutant of CK2beta. Biochem. J. 358: 87–94, https://doi.org/10.1042/0264-6021:3580087.10.1042/bj3580087Search in Google Scholar
Castello, J., LeFrancois, B., Flajolet, M., Greengard, P., Friedman, E., and Rebholz, H. (2018). CK2 regulates 5-HT4 receptor signaling and modulates depressive-like behavior. Mol. Psychiatr. 23: 872–882, https://doi.org/10.1038/mp.2017.240.Search in Google Scholar PubMed
Chantalat, L. (1999). Crystal structure of the human protein kinase CK2 regulatory subunit reveals its zinc finger-mediated dimerization. EMBO J. 18: 2930–2940, https://doi.org/10.1093/emboj/18.11.2930.Search in Google Scholar PubMed PubMed Central
Chen, M., Li, D., Krebs, E.G., and Cooper, J.A. (1997). The casein kinase II β subunit binds to Mos and inhibits Mos activity. Mol. Cell. Biol. 17: 1904–1912, https://doi.org/10.1128/MCB.17.4.1904.Search in Google Scholar PubMed PubMed Central
Couturier, C. and Jockers, R. (2003). Activation of the leptin receptor by a ligand-induced conformational change of constitutive receptor dimers. J. Biol. Chem. 278: 26604–26611, https://doi.org/10.1074/jbc.M302002200.Search in Google Scholar PubMed
Di Stazio, M., Zanus, C., Faletra, F., Pesaresi, A., Ziccardi, I., Morgan, A., Girotto, G., Costa, P., Carrozzi, M., d’Adamo, A.P., et al.. (2023). Haploinsufficiency as a foreground pathomechanism of Poirer-Bienvenu syndrome and novel insights underlying the phenotypic continuum of CSNK2B-associated disorders. Genes 14: 250, https://doi.org/10.3390/genes14020250.Search in Google Scholar PubMed PubMed Central
El Khamlichi, C., Reverchon-Assadi, F., Hervouet-Coste, N., Blot, L., Reiter, E., and Morisset-Lopez, S. (2019). Bioluminescence resonance energy transfer as a method to study protein-protein interactions: application to G protein coupled receptor biology. Molecules 24: 537, https://doi.org/10.3390/molecules24030537.Search in Google Scholar PubMed PubMed Central
Ernst, M.E., Baugh, E.H., Thomas, A., Bier, L., Lippa, N., Stong, N., Mulhern, M.S., Kushary, S., Akman, C.I., Heinzen, E.L., et al.. (2021). CSNK2B: a broad spectrum of neurodevelopmental disability and epilepsy severity. Epilepsia 62, https://doi.org/10.1111/epi.16931.Search in Google Scholar PubMed PubMed Central
Filhol, O., Benitez, M.J., and Cochet, C. (2005) A zinc ribbon motif is essential for the formation of functional tetrameric protein kinase CK2. In: Iuchi, S., and Kuldell, N. (Eds.). Zinc finger proteins: from atomic contact to cellular function. Springer, USA, pp. 121–127.10.1007/0-387-27421-9_18Search in Google Scholar
Götz, C. and Montenarh, M. (2017). Protein kinase CK2 in development and differentiation. Biomed Rep. 6: 127–133, https://doi.org/10.3892/br.2016.829.Search in Google Scholar PubMed PubMed Central
Graham, K.C. and Litchfield, D.W. (2000). The regulatory beta subunit of protein kinase CK2 mediates formation of tetrameric CK2 complexes. J. Biol. Chem. 275: 5003–5010, https://doi.org/10.1074/jbc.275.7.5003.Search in Google Scholar PubMed
Guerra, B., Issinger, O.-G., and Wang, J.Y. (2003). Modulation of human checkpoint kinase Chk1 by the regulatory β-subunit of protein kinase CK2. Oncogene 22: 4933–4942, https://doi.org/10.1038/sj.onc.1206721.Search in Google Scholar PubMed
Guillemain, A., Laouarem, Y., Cobret, L., Štefok, D., Chen, W., Bloch, S., Zahaf, A., Blot, L., Reverchon, F., Normand, T., et al.. (2020). LINGO family receptors are differentially expressed in the mouse brain and form native multimeric complexes. FASEB J. 34: 13641–13653, https://doi.org/10.1096/fj.202000826R.Search in Google Scholar PubMed
Hagemann, C., Kalmes, A., Wixler, V., Wixler, L., Schuster, T., and Rapp, U.R. (1997). The regulatory subunit of protein kinase CK2 is a specific A-Raf activator. FEBS Lett. 403: 200–202, https://doi.org/10.1016/S0014-5793(97)00011-2.Search in Google Scholar PubMed
Hetz, C., Zhang, K., and Kaufman, R.J. (2020). Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 21: 421–438, https://doi.org/10.1038/s41580-020-0250-z.Search in Google Scholar PubMed PubMed Central
Kim, J., Byun, I., Kim, D.Y., Joh, H., Kim, H.J., and Lee, M.J. (2024). Targeted protein degradation directly engaging lysosomes or proteasomes. Chem. Soc. Rev. 53: 3253–3272, https://doi.org/10.1039/D3CS00344B.Search in Google Scholar
Laudet, B., Barette, C., Dulery, V., Renaudet, O., Dumy, P., Metz, A., Prudent, R., Deshiere, A., Dideberg, O., Filhol, O., et al.. (2007). Structure-based design of small peptide inhibitors of protein kinase CK2 subunit interaction. Biochem. J. 408: 363–373, https://doi.org/10.1042/BJ20070825.Search in Google Scholar PubMed PubMed Central
Leroy, D., Alghisi, G.C., Roberts, E., Filhol-Cochet, O., and Gasser, S.M. (1999). Mutations in the C-terminal domain of topoisomerase II affect meiotic function and interaction with the casein kinase 2 beta subunit. Mol. Cell. Biochem. 191: 85–95, https://doi.org/10.1007/978-1-4419-8624-5_11.Search in Google Scholar
Li, D., Zhou, B., Tian, X., Chen, X., Wang, Y., Hao, S., Zhang, C., and Hui, L. (2023). Genetic analysis and literature review of a Poirier-Bienvenu neurodevelopmental syndrome family line caused by a de novo frameshift variant in CSNK2B. Mol. Genet. Genomic Med. 12: e2327, https://doi.org/10.1002/mgg3.2327.Search in Google Scholar PubMed PubMed Central
Lolli, G., Pinna, L.A., and Battistutta, R. (2012). Structural determinants of protein kinase CK2 regulation by autoinhibitory polymerization. ACS Chem. Biol. 7: 1158–1163, https://doi.org/10.1021/cb300054n.Search in Google Scholar PubMed
Lorenz, P., Pepperkok, R., Ansorge, W., and Pyerin, W. (1993). Cell biological studies with monoclonal and polyclonal antibodies against human casein kinase II subunit beta demonstrate participation of the kinase in mitogenic signaling. J. Biol. Chem. 268: 2733–2739, https://doi.org/10.1016/s0021-9258(18)53835-7.Search in Google Scholar
Meggio, F. and Pinna, L.A. (2003). One-thousand-and-one substrates of protein kinase CK2? FASEB J. 17: 349–368, https://doi.org/10.1096/fj.02-0473rev.Search in Google Scholar PubMed
Meggio, F., Boldyreff, B., Marin, O., Pinna, L.A., and Issinger, O.G. (1992). Role of the beta subunit of casein kinase-2 on the stability and specificity of the recombinant reconstituted holoenzyme. Eur. J. Biochem. 204: 293–297, https://doi.org/10.1111/j.1432-1033.1992.tb16636.x.Search in Google Scholar PubMed
Meggio, F., Ruzzene, M., Sarno, S., Pagano, M.A., and Pinna, L.A. (2000). pCMB treatment reveals the essential role of cysteinyl residues in conferring functional competence to the regulatory subunit of protein kinase CK2. Biochem. Biophys. Res. Commun. 267: 427–432, https://doi.org/10.1006/bbrc.1999.1924.Search in Google Scholar PubMed
Mentzel, B., Jauch, E., and Raabe, T. (2009). CK2β interacts with and regulates p21-activated kinases in Drosophila. Biochem. Biophys. Res. Commun. 379: 637–642, https://doi.org/10.1016/j.bbrc.2008.12.136.Search in Google Scholar PubMed
Mercier, J.-F., Salahpour, A., Angers, S., Breit, A., and Bouvier, M. (2002). Quantitative assessment of β1- and β2-adrenergic receptor Homo- and heterodimerization by bioluminescence resonance energy transfer. J. Biol. Chem. 277: 44925–44931, https://doi.org/10.1074/jbc.M205767200.Search in Google Scholar PubMed
Niefind, K. and Issinger, O.-G. (2005). Primary and secondary interactions between CK2alpha and CK2beta lead to ring-like structures in the crystals of the CK2 holoenzyme. Mol. Cell. Biochem. 274: 3–14, https://doi.org/10.1007/s11010-005-3114-0.Search in Google Scholar PubMed
Niefind, K., Guerra, B., Ermakowa, I., and Issinger, O.-G. (2001). Crystal structure of human protein kinase CK2: insights into basic properties of the CK2 holoenzyme. EMBO J. 20: 5320–5331, https://doi.org/10.1093/emboj/20.19.5320.Search in Google Scholar PubMed PubMed Central
Poirier, K., Hubert, L., Viot, G., Rio, M., Billuart, P., Besmond, C., and Bienvenu, T. (2017). CSNK2B splice site mutations in patients cause intellectual disability with or without myoclonic epilepsy. Hum. Mutat. 38: 932–941, https://doi.org/10.1002/humu.23270.Search in Google Scholar PubMed
Raaf, J., Brunstein, E., Issinger, O.-G., and Niefind, K. (2008). The interaction of CK2alpha and CK2beta, the subunits of protein kinase CK2, requires CK2beta in a preformed conformation and is enthalpically driven. Protein Sci. 17: 2180–2186, https://doi.org/10.1110/ps.037770.108.Search in Google Scholar PubMed PubMed Central
Raaf, J., Issinger, O.-G., and Niefind, K. (2009). First inactive conformation of CK2α, the catalytic subunit of protein kinase CK2. J. Mol. Biol. 386: 1212–1221, https://doi.org/10.1016/j.jmb.2009.01.033.Search in Google Scholar PubMed
Rebholz, H., Nishi, A., Liebscher, S., Nairn, A.C., Flajolet, M., and Greengard, P. (2009). CK2 negatively regulates Galphas signaling. Proc. Natl. Acad. Sci. U.S.A. 106: 14096–14101, https://doi.org/10.1073/pnas.0906857106.Search in Google Scholar PubMed PubMed Central
Reed, J.C., Bidwai, A.P., and Glover, C.V. (1994). Cloning and disruption of CKB2, the gene encoding the 32-kDa regulatory beta′-subunit of Saccharomyces cerevisiae casein kinase II. J. Biol. Chem. 269: 18192–18200, https://doi.org/10.1016/S0021-9258(17)32434-1.Search in Google Scholar
Ruzzene, M. and Pinna, L.A. (2010). Addiction to protein kinase CK2: a common denominator of diverse cancer cells? Biochim. Biophys. Acta 1804: 499–504, https://doi.org/10.1016/j.bbapap.2009.07.018.Search in Google Scholar PubMed
Sarno, S., Vaglio, P., Cesaro, L., Marin, O., and Pinna, L.A. (1999). A multifunctional network of basic residues confers unique properties to protein kinase CK2. Mol. Cell. Biochem. 191: 13–19, https://doi.org/10.1007/978-1-4419-8624-5_2.Search in Google Scholar
Unni, P., Friend, J., Weinberg, J., Okur, V., Hochscherf, J., and Dominguez, I. (2022). Predictive functional, statistical and structural analysis of CSNK2A1 and CSNK2B variants linked to neurodevelopmental diseases. Front. Mol. Biosci. 9: 851547, https://doi.org/10.3389/fmolb.2022.851547.Search in Google Scholar PubMed PubMed Central
Wang, D., Sun, X., Bohn, L.M., and Sadée, W. (2005). Opioid receptor homo- and heterodimerization in living cells by quantitative bioluminescence resonance energy transfer. Mol. Pharmacol. 67: 2173–2184, https://doi.org/10.1124/mol.104.010272.Search in Google Scholar PubMed
Werner, C., Gast, A., Lindenblatt, D., Nickelsen, A., Niefind, K., Jose, J., and Hochscherf, J. (2022). Structural and enzymological evidence for an altered substrate specificity in Okur-Chung neurodevelopmental syndrome mutant CK2αLys198Arg. Front. Mol. Biosci. 9, https://doi.org/10.3389/fmolb.2022.831693.Search in Google Scholar PubMed PubMed Central
Zhang, X., Lu, H., Ji, Y., and Sun, W. (2024). Case report: novel deletions in the 6p21.33 involving the CSNK2B gene in patients with Poirier-Bienvenu neurodevelopmental syndrome and literature review. Front. Med. 11: 1441573, https://doi.org/10.3389/fmed.2024.1441573.Search in Google Scholar PubMed PubMed Central
© 2025 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Frontmatter
- 70 years of CK2: still exciting, essential – and enigmatic!
- Protein kinase CK2 contributes to glucose homeostasis
- CK2 control of human papillomavirus life cycles
- Time- and dose-dependent effects of CIGB-300 on the proteome of lung squamous cell carcinoma
- A CK2α′ mutant indicating why CK2α and CK2α′, the isoforms of the catalytic subunit of human protein kinase CK2, deviate in affinity to CK2β
- Rapid method for evaluation of CK2 enzymatic activity and CK2α/CK2β-interaction in Escherichia coli cell lysates
- Exploring the biological potential of the brominated indenoindole MC11 and its interaction with protein kinase CK2
- Pathogenic missense variants of CSNK2B associated with Poirier-Bienvenu neurodevelopmental disorder impact differently on CK2 holoenzyme formation
Articles in the same Issue
- Frontmatter
- 70 years of CK2: still exciting, essential – and enigmatic!
- Protein kinase CK2 contributes to glucose homeostasis
- CK2 control of human papillomavirus life cycles
- Time- and dose-dependent effects of CIGB-300 on the proteome of lung squamous cell carcinoma
- A CK2α′ mutant indicating why CK2α and CK2α′, the isoforms of the catalytic subunit of human protein kinase CK2, deviate in affinity to CK2β
- Rapid method for evaluation of CK2 enzymatic activity and CK2α/CK2β-interaction in Escherichia coli cell lysates
- Exploring the biological potential of the brominated indenoindole MC11 and its interaction with protein kinase CK2
- Pathogenic missense variants of CSNK2B associated with Poirier-Bienvenu neurodevelopmental disorder impact differently on CK2 holoenzyme formation