C1-Substituted N-tert-butoxycarbonyl-5-syn-tert-butyldimethylsilyloxymethyl-2-azabicyclo[2.1.1]hexanes as conformationally constrained β-amino acid precursors
-
Grant R. Krow
 ,
Kevin C. Cannon
,
Kevin C. Cannon

Abstract
Regioselective introduction and transformation of substituents at the C1 carbon of N-tert-butoxycarbonyl-5-syn-tert-butyldimethylsilyloxymethyl-2-azabicyclo[2.1.1]hexane (7) is described. These azabicycles are precursors to conformationally constrained β-amino acids with potential to form oligomers with definite secondary structures. Selected examples of these precursors are converted into their corresponding amino acid derivatives.
Introduction
Oligomers of 2,2-disubstituted pyrrolidine-4-carboxylic acids (1) have shown circular dichorism-based evidence of secondary structure despite the absence of hydrogen bonding [1], [2], [3], [4] (Figure 1).
![Figure 1 Disubstituted pyrrolidine carboxylic acids 1, 2 and C1-substituted 2-azabicyclo[2.1.1]hexane 3.](/document/doi/10.1515/hc-2016-0046/asset/graphic/j_hc-2016-0046_fig_001.jpg)
Disubstituted pyrrolidine carboxylic acids 1, 2 and C1-substituted 2-azabicyclo[2.1.1]hexane 3.
The combination of the ring structure and the presence of two substituents at C2 of 1 has been attributed as the source of the oligomers’ secondary structure. The two C2 substituents sterically force the amide carbonyl to be in an s-trans conformation (Figure 2).

s-Trans and s-cis conformations of the amide carbonyl of 2,2-disubstituted pyrrolidine-4-carboxylic acids incorporated into oligomers.
In this context, 2,2-disubstituted pyrrolidine-3-carboxylic acids 2 have been proposed and predicted to show a stronger rotamer bias for resulting β-peptides, but the difficulty in introducing substituents at the hindered C2 position of 2 made the syntheses of these β-amino acids and their corresponding β-peptides unsuccessful [2]. In response to the difficulty in synthesizing 2, we have proposed the synthesis and application of C1-substituted 2-azabicyclo[2.1.1]hexanes 3 which can be considered as more rigid analogs of 2. Oligomers of four to eight β-amino acids synthesized from the unsubstituted 2-azabicyclo[2.1.1]hexane (3, R1=H) and the C6-substituted 2-azabicyclo[2.1.1]hexanes both show increasingly ordered secondary folding structure with increasing oligomer length [5], [6]. The C1 substituent and the 1,3-methano bridge may mimic the expected s-trans conformational bias expected for the oligomers of disubstituted carboxylic acid 2 (Figure 3). In this work, we prepared precursors of 3, which vary at the C1 substituent (R1) and converted selected examples into their respective β-amino acids.
![Figure 3 s-Trans and s-cis conformations of the amide carbonyl of C1-substituted 2-azabicyclo[2.1.1]hexanes 3 incorporated into oligomers.](/document/doi/10.1515/hc-2016-0046/asset/graphic/j_hc-2016-0046_fig_003.jpg)
s-Trans and s-cis conformations of the amide carbonyl of C1-substituted 2-azabicyclo[2.1.1]hexanes 3 incorporated into oligomers.
Results and discussion
The synthesis of 3 started with the key compound 4 followed by reduction of ester 5 with lithium aluminum hydride (LAH). Protection of the alcohol functionality in the resultant product 6 with tert-butyldimethylsilyl chloride (TBDMSCl) yielded compound 7 in 63% yield (Scheme 1).

Synthesis of protected alcohol 7.
Ester 5 was prepared in four steps starting with the photochemical cross-addition of N-BOC-N-allyl-N-vinyl amide 4 followed by oxidation of the resulting syn ketone by a known procedure [7]. Utilizing Krow’s already established protocol of regioselective introduction of electrophilic substituents selectively at the C1 bridgehead of N-BOC-2-azabicyclo[2.1.1]hexanes [8], [9], vs. the C3 carbon, a variety of functional groups were incorporated in 7 (Scheme 2).

Introduction of electrophilic substituents selectively at the C1 bridgehead of 7.
Regioselective deprotonation of 7 at C1 with sec-butylithium (s-BuLi) in the presence of TMEDA at 0°C followed by iodomethane quench of the anion resulted in the preparation of methyl-substituted compound 8a in 70% yield. The use of other electrophiles such as bromoethane and 1-bromobutane did not yield any of the expected products; only unreacted 7 was isolated. To exclude steric factors as the source of the failure of the above two alkylations, bulkier trimethylsilyl chloride was employed as an electrophile, which resulted in the formation of silylated product 8b. Compounds 8c (R=CH2OCH3) and 8d (R=CH2-CH=CH2) were also prepared by quenching the C1-anion of 7 with methoxymethyl chloride and allyl bromide, respectively.
Surprisingly, when benzyl bromide, a more reactive electrophile, was used, 1-bromo-substituted azabicycle 8e was isolated as the major product (63%). An ion-radical mechanism is probably operative in the incorporation of bromine in the azabicycle [10]. However, a benzyl-substituted product 8f was isolated in low yields (14%) employing 4-methoxybenzyl bromide as the electrophile.
Freshly distilled N, N-dimethylformamide (DMF) was used to synthesize the desired 1-formyl-substituted azabicycle 8g; the use of reagent grade DMF produced no product. Incorporation of benzoyl and 4-fluorobenzoyl (8h and 8i) substituents was also accomplished although the latter product was isolated in a lower yield (76% and 33%, respectively).
Reactions of the lithium anion of 7 with benzyl and methyl chloroformates gave different results. The former gave only the anticipated benzyl ester 8j (46%), while the latter resulted in the preparation of methyl ester 8k along with the dimeric ketone by-product 9k. The formation of 9k may be explained by the subsequent reaction of the methyl ester 8k with the lithium anion of 7. To circumvent the formation of 9k, carbon dioxide was used as electrophile resulting exclusively in the synthesis of carboxylic acid 8l, which was then directly converted into the desired ester 8k by treatment with TMSCHN2 [11] in an overall yield of 83%.
The intermediate carboxylic acid 8l was also coupled with benzylamine in the presence of 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDCI*HCl) and 1-hydroxybenzotriazole hydrate (HOBt) in acetonitrile to yield benzyl amide 8m (Scheme 3) [12].

Conversion of acid 8l into amide 8m.
Our next goal was to modify the functional groups already introduced at C1 in the above-generated β-amino acid precursors into biologically relevant side chains. The formyl group in compound 8g was used as a key intermediate for these alterations. Reduction of 8g with NaBH4 gave alcohol 10 (Scheme 4). Treatment of 10 with methanesulfonyl chloride (MsCl) in the presence of triethylamine gave mesylate 11 [13]. Attempts to purify mesylate 11 by chromatography were unsuccessful; crude mesylate was therefore treated with sodium azide to give 12–14 although in low yields. The formation of compound 12 is a consequence of the neighboring carbamate participation in the displacement of the mesylate. The formation of a similar carbamate has been previously reported by Malpass [13]. The exclusive formation of 12 is also shown in Scheme 4 when KCN and 18-crown-6 ether [4] were used for an anticipated nucleophilic displacement of the mesylate with cyanide in 11.

Synthesis of mesylate 11 and attempted nucleophilic substitutions with KCN and NaN3.
Reaction of 8g with triphenylmethyl bromide [14] and n-BuLi yielded alkene 15 in 52% yield (Scheme 5). Alkene 15 was also obtained in a slightly better yield (59%) employing the Tebbe reagent [15]. Hydroboration-oxidation of the alkene side chain in 15 gave the corresponding alcohol 16 [16]. Chain extension in 8g was carried out via a Wittig reaction with methyl(triphenylphosphoranylidene) acetate leading to the isolation of vinyl ester 17 (Scheme 6).

Synthesis of alcohol 16.

Conversion of aldehyde 8g into alcohol 19.
Palladium-catalyzed hydrogenation of the double bond in 17 both in protic and in aprotic solvents [17] yielded cyclobutane 20 which is a result of N-C1 bond cleavage of the bicycle (Scheme 6). Fortunately, reduction of the same double bond with diimide (generated in situ from tosylhydrazine in the presence of TMEDA) [18] gave the desired product 18 with an intact [2.1.1] azabicycle core. Further, the ester group in 18 was reduced to the corresponding alcohol 19 by treatment with LAH.
The final step in the synthesis of constrained β-amino acid derivatives was oxidation of the TBDMS-protected hydroxylmethyl group at C5 to the corresponding carboxylic acid. In two selected cases (substituent at C5=COPh, CO2Me), successful oxidation with Jones reagent [19] demonstrated that there was no necessity to remove the TBDMS protecting group on the oxygen prior to oxidation (Scheme 7).

Direct oxidation of protected alcohols 8h and 8k.
As shown in Scheme 7, both compounds 8h and 8k were oxidized to the corresponding acids 21h and 21k with CrO3 in H2SO4 in reasonable yields. Acid 21h was crystallized from hexane/dichloromethane and isolated as a white crystalline solid. Figure 4 shows the ORTEP of 21h with 30% probability thermal ellipsoids. The crystal structure confirmed the endo-configuration of the carboxylic acid on C5 (designated as C3 on the ORTEP).
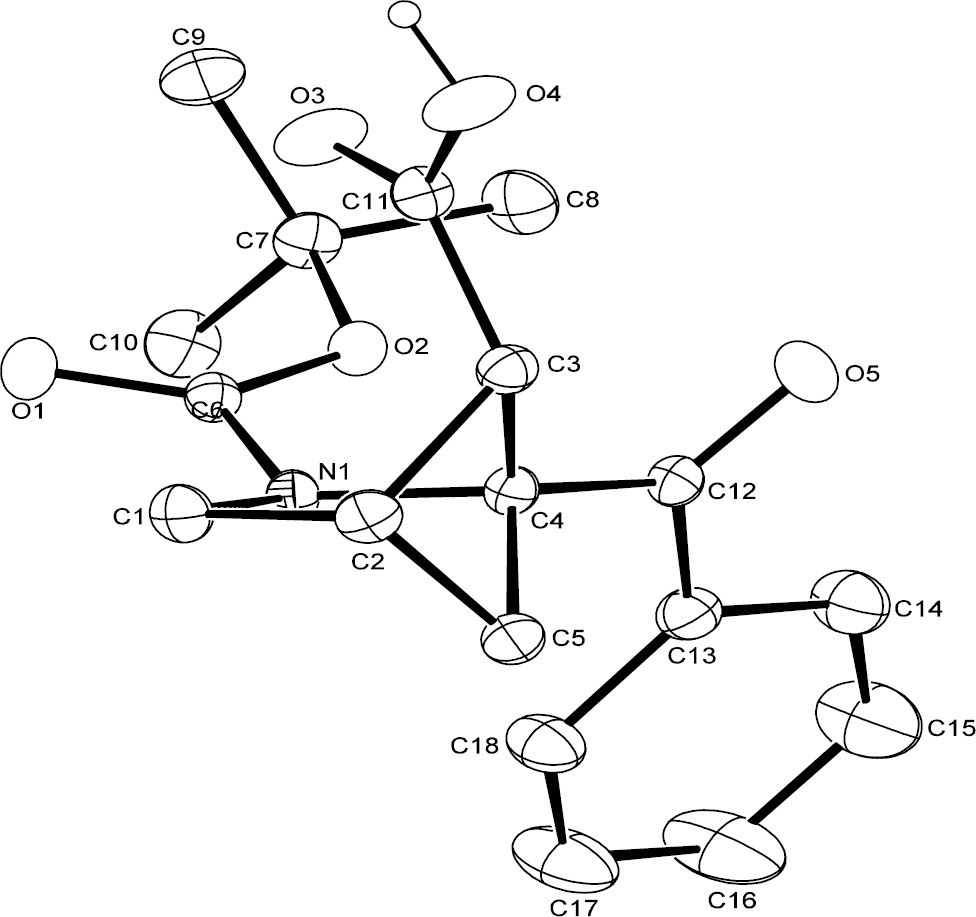
ORTEP of 21h with 30% probability thermal ellipsoids.
The β-carboxylic acid derivative 21k can be visualized as a constrained analog of aspartic acid (Figure 5).

Structural similarity of 21k with aspartic acid.
Despite the successful oxidation of 8h and 8k to the corresponding acids, attempts to oxidize 8a with Jones reagent to produce a constrained analog of homoalanine (21a) resulted in decomposition of the starting material. Likewise, oxidation under basic conditions with NaOCl and catalytic 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) [20] was also futile.
Synthesis of 21a was eventually accomplished by deprotection of the TBDMS group of 8a to yield 22 [21] followed by oxidation with tetrapropylammonium perruthenate (TPAP) and N-methylmorpholine-N-oxide (NMO) [22]; product 21a was obtained in a moderate yield of 33% (Scheme 8). It can be hypothesized that the presence of the methyl group at C1 may stabilize a carbocation intermediate involved in the formation of ring cleavage products, but further investigation of the compatibility of C1-substituted 2-azabicyclo[2.1.1]hexanes toward oxidative reaction conditions is warranted.

Synthesis of acid 21a.
Conclusions
A variety of non-polar (alkyl, allyl and vinyl), polar (hydroxymethyl), potentially acidic (carboxymethyl and benzyloxycarbonyl), potentially basic (azido) and other substituents (bromo, benzoyl, formyl and MOM) were introduced regioselectively at C1 of 2-azabicyclo[2.1.1]hexanes 3. Chain homologation was also carried out with C1-substituted CHO (8g). In some cases, standard functional group transformations failed or were realized in low yields due to steric congestion and neighboring group effects. Three precursors 8a, 8h and 8k were converted into their corresponding acids. These compounds can be visualized as β-amino acids with restricted degrees of conformational freedom; they have the potential to form oligomers with definite secondary structures.
Experimental
Thin-layer chromatography was performed on precoated plates of silica gel GF 250 μm. Column chromatography was performed on silica gel, Merck grade 60 (230–400 mesh). Reagent-grade chemicals were obtained from commercial suppliers, and reagent-grade solvents were used without further purification. Tetrahydrofuran, dichloromethane and DMF were distilled from the solvent dispensing system designed by Meyer under an argon atmosphere. DMF was additionally distilled from Linde type 4A molecular sieves. All reactions were performed under an argon atmosphere. Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker 300 at 298 K observing 1H and 13C resonances at 300 MHz and 75 MHz, respectively. The spectrometer was locked to either the deuterium or carbon resonance of CDCl3 and all chemical shifts were referenced to residual CHCl3. High resolution mass spectral data were acquired with a Bruker Daltonics 7 tesla Fourier transform ion cyclotron resonance (FTICR) mass spectrometer by the use of electrospray ionization. External calibration was accomplished with oligomers of polypropylene glycol, PPG 425. Melting points are uncorrected; a Thomas Hoover capillary melting point apparatus was used.
N-(tert-butoxycarbonyl)-5-syn-hydroxymethyl-2-azabicyclo[2.1.1]hexane (6)
To a solution of 5 (260 mg, 1.08 mmol) in dry THF (13 mL) at −78°C was added 1 M solution of LAH (701 μL, 0.7 mmol). The mixture was stirred at −78°C for 1 h, then brought to room temperature and stirred for an additional 2 h. The reaction was quenched by adding water (25 μL), followed by 15% NaOH (25 μL) and H2O (100 μL). The resulting solution was dried and concentrated to give 187 mg (81%) of alcohol 6; Rf=0.30 (ethyl acetate/hexane, 2:1); 1H NMR: δ 4.40 (d, J=7.2 Hz, 1H, H1), 3.41 (m, 4H, OCH2 and 2H3), 2.82 (m, 1H, H4), 2.25 (m, 1H, H5), 2.00 (br, 1H, OH), 1.80 (d, J=7.4 Hz, 1H, H6anti), 1.45 (s, 9H), 1.40 (d, J=7.4 Hz, H6syn); 13C NMR: δ 157.9, 79.7, 61.2, 60.8, 57.8, 49.6, 46.5, 45.3, 38.8, 37.7, 28.3. HR-MS. Calcd for C11H20NO3 (M+H): m/z 214.1443. Found: m/z 214.1445.
N-(tert-butoxycarbonyl)-5-syn-(tert-butyldimethylsilyloxymethyl)-2-azabicyclo[2.1.1]hexane (7)
To a solution of 6 (482 mg, 2.3 mmol) in dry CH2Cl2 (15 mL) under argon was added imidazole (770 mg, 11.3 mmol) followed by TBDMSCl (211 mg, 2.7 mmol) in small portions. The mixture was stirred at room temperature for 16 h, then transferred into a separating funnel and washed with water (10 mL), brine (10 mL) and dried with Na2SO4. Solvent was removed in vacuo to give 514 mg of the crude product. Flash chromatography on silica gel gave 475 mg (63%) of 7; Rf=0.60 (hexane/ether, 2:1); 1H NMR: δ 4.24 (br d, J=6.0 Hz, H1), 3.30 (m, 2H, OCH2), 3.20 (d, J=9.2 Hz, H3), 3.12 (d, J=9.2 Hz, H3), 2.70 (m, H4), 2.13 (m, H5), 1.67 (m, 1H, H6anti), 1.44 (s, 9H, BOC), 1.26 (d, J=7.2 Hz, H6syn), 0.86 (s, 9H, TBDMS), 0.00 (s, 6H, TBDMS); 13C NMR: δ 154.6, 77.4, 58.6, 55.4, 49.6, 39.0, 36.2, 30.7, 28.6, 26.2, 18.6,7.5. HR-MS. Calcd for C17H34NO3Si (M+H): m/z 328.2313. Found: m/z 328.2308. Calcd for C17H33NO3SiNa (M+Na): m/z 350.2136. Found: m/z 350.2127.
Synthesis of compounds 8a–l
To a solution of 7 (1 equiv) in dry ether (15 mL) at 0°C was added TMEDA (1.1 equiv) and the mixture was stirred for 15 min. To the resulting solution was added s-BuLi (1.4 M in cyclohexane, 1.2 equiv) dropwise. The mixture was stirred for 2 h at 0°C, treated dropwise with the electrophile (5 equiv) indicated below, slowly allowed to warm to room temperature and quenched with a saturated solution of NH4Cl. The aqueous phase was extracted with ether three times, and the combined organic phases were washed with water, brine, dried (Na2SO4) and concentrated. Product 8a–m was purified by silica gel flash chromatography eluting with ethyl acetate/heptanes (1:5).
N-(tert-butoxycarbonyl)-1-methyl-5-syn-(tert-butyldimethylsilyloxymethyl)-2-azabicyclo[2.1.1]hexane (8a)
This compound was obtained with methyl iodide as the electrophile; yield 70%; Rf=0.38 (hexane/ether, 4:1); 1H NMR: δ 3.43 (dd, J=10.8, 6.8 Hz, 1H, OCH2), 3.32 (dd, J=10.8, 6.8 Hz, 1H, OCH2), 3.29 (d, J=9.4 Hz, 1H, H3), 3.23 (d, J=9.4 Hz, 1H, H3), 2.58 (t, J=3 Hz, H4), 1.94 (ddd, J=6.8, 6.8, 2.8 Hz, H5), 1.64 (s, 3H, Me), 1.53 (ddd, J=7.2, 3.2, 1.2 Hz, H6anti), 1.46 (s, 10H, BOC), 1.43 (d, J=7.2 Hz, 1H, H6syn), 0.89 (s, 9H, TBDMS), 0.03 (s, 6H, TBDMS); 13C NMR: δ 156.6 (C=O), 79.2 (O-tert-Bu), 71.6 (OCH2), 59.7 and 59.5 (C3), 54.2 (C1), 49.5 (C5), 43.8 (C4), 35.6 (C6), 28.9 (BOC), 26.7 (CH3), 26.3 (BOC), 18.6 (TBDMS), 7.5 (TBDMS). HR-MS. Calcd for C18H35NO3SiNa (M+Na): m/z 364.2292. Found: m/z 364.2284.
N-(tert-butoxycarbonyl)-1-trimethylsilyl-5-syn-(tert-butyldimethylsilyloxymethyl)-2-azabicyclo[2.1.1]hexane (8b)
This compound was obtained with trimethylsilyl chloride as the electrophile; yield 89%; Rf=0.78 (hexane/ether, 3:1); 1H NMR: δ 3.36 (dd, J=10.8, 6.4 Hz, 1H, OCH2), 3.25 (d, J=8.8 Hz, 1H, H3), 3.19 (d, J=8.8 Hz, 1H, H3), 3.16 (dd, J=10.8, 6.4 Hz, 1H, OCH2), 2.79 (brt, J=2.8 Hz, H4), 2.17 (ddd, J=6.4, 6.4, 2.8 Hz, 1H, H5), 1.66 (ddd, J=6.8, 2.8, 1.2 Hz, H6anti), 1.45 (s, 9H, BOC), 1.20 (d, J=6.8 Hz, 1H, H6syn), 0.86 (s, 9H, TBDMS), 0.12 (s, 9H, TBDMS), 0.01 (s, 6H, TMS); 13C NMR: δ 155.7, 65.8, 59.5, 51.9, 48.2, 40.0, 39.9, 28.6, 26.0,18.3, –0.87, –5.40, -5.50. HR-MS. Calcd for C20H42NO3Si2 (M+H): m/z 400.2698. Found: m/z 400.2688.
N-(tert-butoxycarbonyl)-2-azabicyclo-1-methoxymethyl-5-syn-(tert-butyldimethylsilyloxymethyl)[2.1.1]hexane (8c)
This compound was obtained with chloromethylmethyl ether as the electrophile; yield 30%; Rf=0.53 (ethylacetate/hexane, 2:1); 1H NMR: δ 3.94 (d, J=11.3 Hz, 1H, OCH2), 3.87 (m, 1H, OCH2), 3.36 (s, 3H, OCH3), 3.26 (m, 4H, OCH2 and 2H3), 2.58 (dd, J=3.0 Hz, 1H, H4), 2.24 (ddd, J=6.9, 6.3, 3.0 Hz, 1H, H5), 1.81 (ddd, J=7.0, 3.0, 1.5 Hz, 1H, H6anti), 1.43 (s, 9H, BOC), 1.32 (d, J=7.0 Hz, 1H, H6syn), 0.86 (s, 9H, TBDMS), 0.00 (s, 6H, TBDMS); 13C NMR: δ 155.8, 79.2, 70.8, 59.2, 58.8, 49.8, 49.2, 39.5, 39.0, 35.1, 28.6, 25.9. HR-MS. Calcd for C19H37NO4Si (M+H): m/z 372.3565. Found: m/z 372.3568. Calcd for C19H37NO4SiNa (M+Na): m/z 394.2384. Found: m/z 394.2388.
N-(tert-butoxycarbonyl)-1-allyl-5-syn-(tert-butyldimethylsilyloxymethyl)-2-azabicyclo[2.1.1]hexane (8d)
This compound was obtained with allyl bromide as the electrophile; yield 11%; Rf=0.72 (hexane/ether, 4:1); 1H NMR: δ 5.85 (m, 1H), 5.10 (d, J=16.4 Hz, 1H), 5.00 (dd, J=12.5 Hz, 1H), 3.41 (m, 1H, OCH2), 3.25 (m, OCH2, 2H3, 3H), 2.95 (dd, J=7.9, 12.0 Hz, 1H, CH2), 2.86 (dd, J=7.9, 12.0 Hz, 1H, CH2), 2.58 (br, H4), 2.00 (m, 1H, H5), 1.42 (m, 1H, H6anti), 1.40 (s, 9H, BOC), 1.25 (d, J=6.9 Hz, H6syn), 0.95 (s, 9H, TBDMS), 0.00 (s, 6H, TBDMS); 13C NMR: δ 158.0, 135.9, 117.3, 74.7, 61.0, 59.3, 57.2, 51.5, 49.8, 40.8, 36.0, 35.5, 28.9, 26.3, 18.6, 0.00. HR-MS. Calcd for C20H37NO3SiNa (M+Na): m/z 390.2434. Found: m/z 390.2434.
N-(tert-butoxycarbonyl)-1-bromo-5-syn-tert-butyldimethylsilyloxymethyl-2-azabicyclo[2.1.1]hexane (8e)
This compound was obtained with benzyl bromide as the electrophile; yield 63%; Rf=0.62 (hexane/ethyl acetate, 4:1); 1H NMR: δ 3.63 (dd, J=11.1, 5.1 Hz, 1H, OCH2), 3.40 (m, 3H, OCH2 and 2H3), 2.89 (br, 1H, H4), 2.41 (m, 1H, H5), 2.05 (m, H6anti), 1.89 (d, J=7.2 Hz, H6syn), 1.46 (s, 9H, BOC), 0.87 (s, 9H, TBDMS), 0.04 (s, 6H, TBDMS); 13C NMR: δ 156.9, 80.8, 67.3, 58.5, 58.4, 47.9, 46.7, 36.2, 28.9, 26.5, 18.6, –5.1. HR-MS. Calcd for C17H32NO3SiNa79Br (M+Na): m/z 428.1232. Found: m/z 428.1224. Calcd for C17H32NO3SiNa81Br (M+Na): m/z 430.1212. Found: m/z 430.1220.
N-(tert-butoxycarbonyl)-1-(p-methoxybenzyl)-5-syn-(tert-butyldimethylsilyloxymethyl)-2-azabicyclo[2.1.1]hexane (8f)
This compound was obtained in a mixture with 8e using p-methoxybenzyl bromide as the electrophile; yield 30%; Rf=0.47 (hexane/ethyl acetate, 4:1); 1H NMR: δ 6.84 (m, 2H, Ph), 6.77 (m, 2H, Ph), 3.74 (s, 3H, OMe), 3.42 (dd, J=10.0, 7.5 Hz, 1H, OCH2), 3.33 (m, 5H, OCH2, CH2PMB, 2H3), 3.23 (m, 1H, CH2PMB), 2.41 (br t, J=3.1 Hz, H4), 1.85 (m, H5), 1.43 (s, 9H, BOC), 1.26 (dd, J=7.7, 2.6 Hz, 1H, H6anti), 1.20 (d, J=7.7 Hz, 1H, H6syn), 0.86 (s, 9H, TBDMS), 0.01 (s, 6H, TBDMS); 13C NMR: δ 158.4, 131.9, 131.5, 114.5, 113.8, 79.5, 76.4, 59.6, 55.6, 50.3, 50.1, 47.8, 40.1, 36.5, 35.6, 29.0, 26.2, 18.7, –5.0. HR-MS. Calcd for C25H42NO4Si (M+H): m/z 448.2878. Found: m/z 448.2896.
N-(tert-butoxycarbonyl)-1-formyl-5-syn-(tert-butyldimethylsilyloxymethyl)-2-azabicyclo[2.1.1]hexane (8g)
This compound was obtained using DMF as the electrophile; yield 52%; Rf=0.40 (hexane/ether, 2:1); 1H NMR: δ 9.80 (s, 1H, CHO), 3.58 (dd, J=10.4, 6.8 Hz, 1H, OCH2), 3.43 (brd, J=9.2 Hz, H3), 3.39 (brd, J=9.2 Hz, H3), 3.38 (dd, J=10.4, 6.8 Hz, 1H, OCH2), 2.67 (m, H4), 2.40 (ddd, J=7.2, 7.0, 3.1 Hz, H5), 1.95 (ddd, J=7.4, 3.0, 1.2 Hz, H6anti), 1.45 (s and m, 10H, BOC and H6syn), 0.88 (s, 9H, TBDMS), 0.03 (s, 6H, TBDMS); 13C NMR: δ 194.4, 156.6 (C=O), 79.7, 58.6, 55.4, 49.6, 39.0, 36.2, 30.7, 28.6, 26.2,18.6, 7.5. HR-MS. Calcd for C18H33NO4SiNa (M+Na): m/z 378.2086. Found: m/z 378.2076.
N-(tert-butoxycarbonyl)-1-benzoyl-5-syn-(tert-butyl-dimethylsilyloxymethyl)-2-azabicyclo[2.1.1]hexane (8h)
This compound was obtained using benzoyl chloride as the electrophile; yield 76%; Rf=0.57 (hexane/ethyl acetate, 2:1); 1H NMR: δ 8.09 (m, 2H, Ph), 7.51 (m, 1H, Ph), 7.40 (m, 2H, Ph), 3.81 (br, 1H, OCH2,), 3.62 (s, 2H, H3), 3.23 (br, 1H, OCH2,), 2.83 (br, 1H, H4), 2.57 (br, 1H, H5), 2.00 (br, 1H, H6anti), 1.36 (br, 1H, H6syn), 1.09 (s, 9H, BOC), 0.86 (s, 9H, TBDMS), 0.03 (s, 6H, TBDMS); 13C NMR: δ 193.0, 156.1, 141.2, 127.8, 127.6, 127.2, 81.5, 59.0, 57.9, 51.2, 49.9, 45.0, 35.6, 28.5, 28.0/ 27.5, 25.9/ 25.7, 18.3, –5.3, –5.5. HR-MS. Calcd for C24H37NO4SiNa (M+Na): m/z 454.2384. Found: m/z 454.2398.
N-(tert-butoxycarbonyl)-1-(4-fluorobenzoyl)-5-syn-(tert-butyldimethylsilyloxymethyl)-2-azabicyclo[2.1.1]hexane (8i)
This compound was obtained using p-fluorobenzoyl fluoride as the electrophile; yield 33%; Rf=0.67 (hexane/ether, 4:1); 1H NMR: δ 8.15 (m, 2H, Ph), 7.09 (m, 2H, Ph), 3.83 (br, 1H, OCH2), 3.63 (s, 2H, 2H3), 3.22 (br, 1H, OCH2), 2.85 (br, 1H, H4), 2.57 (br, 1H, H5), 2.01 (br, 1H, H6anti), 1.36 (br, 1H, H6syn), 1.06 (s, 9H, BOC), 0.85 (s, 9H, TBDMS), 0.05 (s, 6H, TBDMS); 13C NMR: δ 191.8, 157.2, 131.3, 132.0,115.4, 115.2, 81.7, 57.9, 51.1, 50.0, 45.5, 35.6, 28.5, 27.6, 25.9, 18.3, –5.5, –5.3. HR-MS. Calcd for C25H36O4SiF (M+H): m/z 450.2471. Found: m/z 450.2466. Calcd for C24H36O4SiFNa (M+Na): m/z 472.2292. Found: m/z 472.2292.
N-(tert-butoxycarbonyl)-1-benzyloxycarbonyl-5-syn-(tert-butyldimethylsilyloxymethyl)-2-azabicyclo[2.1.1]hexane (8j)
This compound was obtained with benzyl chloroformate as the electrophile; yield 46%; Rf=0.48 (hexane/ether, 3:1); 1H NMR: δ 7.35 (m, 5H, Ph), 5.20 and 5.18 (2d, J=7.5 Hz, 2H, CH2Ph), 3.70 (dd, J=10.8, 4.2 Hz, 1H, OCH2), 3.42 (s, 2H, 2H3), 3.32 (m, OCH2), 2.73 (br, 1H, H4), 2.40 (m, H5), 1.93 (dd, J=7.2, 2.4 Hz, 1H, H6anti), 1.66 (d, J=7.2 Hz, H6syn), 1.42 (s, 9H, BOC), 0.88 (s, 9H, TBDMS), 0.06 (s, 6H, TBDMS); 13C NMR: δ 168.0, 157.3, 135.8, 128.5, 128.1, 127.9, 80.7, 66.4, 59.0, 58.1, 52.5, 48.9, 41.0, 35.8, 28.5, 28.2, 25.9, 18.3, –5.4. HR-MS. Calcd for C26H40NO5Si (M+H): m/z 462.2620. Found: m/z 462.2686. Calcd for C26H39NO5SiNa (M+Na): m/z 484.2495. Found: m/z 484.2505.
N-(tert-butoxycarbonyl)-1-methoxycarbonyl-5-syn-(tert-butyldimethylsilyloxymethyl)-2-azabicyclo[2.1.1]hexane (8k)
This compound was obtained as a mixture with 9k using methyl chloroformate as the electrophile.
Compound 8k Yield 35%; Rf=0.67 (hexane/ethylacetate, 2:1); 1H NMR: δ 3.76 (s, 3H, OMe), 3.70 (dd, J=9.5, 4.5 Hz, OCH2), 3.37 (m, 3H, 2H3, OCH2), 2.73 (dd, J=3.0, 2.5 Hz, H4) 2.38 (m, H5), 1.92 (ddd, J=8.0, 3.0, 1.0 Hz, H6anti), 1.64 (d, J=8.0 Hz, H6syn), 1.43 (s, 9H, BOC), 0.88 (s, 9H, TBDMS), 0.04 (s, 6H, TBDMS); 13C NMR: δ 169.0, 157.6, 78.7, 59.4, 58.5, 52.1, 49.3, 41.4, 39.4, 36.1, 30.7, 28.7, 18.6, –5.0. HR-MS. Calcd for C19H35NO5SiNa (M+Na): m/z 204.2182. Found: m/z 204.2180.
Compound 9k (Scheme 2) Yield 9%; Rf=0.42 (hexane/ethylacetate, 4:1); 1H NMR: δ 3.90 (m, 2H), 3.40 (m, 2H), 2.70 (m, 2H), 2.17 (m, 8H), 1.69 (s, 18H), 1.43 (s, 18H), 0.02 (s, 12H); 13C NMR: δ 198.0, 155.0/154.4, 77.5, 71.7, 56.5/56.2, 49.6, 47.5/47.2, 39.4, 33.6/33.1, 26.3, 22.9, 15.3, –5.4. HR-MS. Calcd for C35H65N2O7Si2 (M+H): m/z 681.4309. Found: 681.4330. Calcd for C35H64N2O7Si2Na (M+Na): m/z 703.2150. Found: m/z 703.2131.
N-(tert-butoxycarbonyl)-1-carboxy-5-syn-tert-butyldimethylsilyloxymethyl-2-azabicyclo[2.1.1]hexane (8l)
This compound was obtained using carbon dioxide as the electrophile (bubbling for 15 min); yield 48% of crude acid 8l. Without further purification, the crude acid was dissolved in a mixture of isopropanol and hexane (1:1, 20 mL) and charged with TMSCHN2. The solution was stirred at room temperature for 1 h. Concentration followed by silica gel chromatography gave the methyl ester 8k in a yield of 97%.
N-(tert-butoxycarbonyl)-2-azabicyclo-1-(benzocarbamoyl)-5-syn-(hydroxymethyl)[2.1.1]hexane (8m)
To a solution of the acid 8l (55 mg, 0.15 mmol) in CH3CN (9 mL) at 0°C was added EDC*HCl (40 mg, 0.21 mmol), HOBt (28 mg, 0.21 mmol) followed by benzylamine (32 mg, 0.30 mmol). The resulting solution was slowly warmed to room temperature and stirred for 12 h. Workup was done by diluting the mixture with ether (15 mL), washing with 1N HCl (3×5 mL) and drying with Na2SO4. Removal of the solvent gave 38 mg (75%) of 8m; Rf=0.33 (ethyl acetate/hexane, 4:1); 1H NMR: δ 7.33 (m, 5H, Ph), 6.45 (br, 1H, NH), 4.51 (br d, J=5.1 Hz, CH2Bn), 4.47 (br, 1H, CH2Bn), 3.48 (dd, J=11.6, 3.9 Hz, 1H, OCH2), 3.36 (m, 3H, OCH2, 2H3), 2.66 (br, 1H4), 2.49 (m, H5), 1.94 (dd, J=7.2, 2.8 Hz, 1H, H6anti), 1.74 (d, J=7.2 Hz, H6syn), 1.47 (s, 9H, BOC); 13C NMR: δ 168.9, 157.4, 132.0, 128.8, 127.9, 127.6, 78.6, 67.2, 58.2, 51.3, 49.4, 43.8, 36.0, 35.2, 28.2, 25.7. HR-MS. Calcd for C19H26N2O4Na (M+Na): m/z 369.1790. Found: m/z 369.1786.
N-(tert-butoxycarbonyl)-1-hydroxymethyl- 5-syn-(tert-butyldimethylsilyloxymethyl)-2-azabicyclo [2.1.1] hexane (10)
To a solution of the crude aldehyde 8g (56 mg, 0.16 mmol) in dry MeOH (8 mL) at 0°C was added NaBH4 (30 mg, 0.08 mmol) in small portions. The solution was stirred at 0°C for 15 min, concentrated in vacuo and the residue was dissolved in ether (15 mL). The ether solution was washed with water (2×5 mL) and dried with Na2SO4. Solvent was then removed in vacuo to give 38 mg (68%) of 10; Rf=0.21 (hexane/ether, 2:1); 1H NMR: δ 4.62 (br, 1H, OH), 4.00 (d, J=13.2, 6.6 Hz, 1H, CH2OH), 3.97 (br dd, J=12.6, 7.5 Hz, 1H, CH2OH), 3.50 (m, 1H, H3), 3.24 (m and s, 3H, 2CH2 OTBDMS and H3), 2.60 (m, 1H, H4), 2.15 (ddd, J=7.2, 6.9, 3.0 Hz, H5), 1.85 (d, J=7.4 Hz, H6anti), 1.79 (d, J=7.4 Hz, H6syn), 1.45 (s, 9H, BOC), 0.88 (s, 9H, TBDMS), 0.03 (s, 6H, TBDMS); 13C NMR: δ 161.0, 79.7, 61.4, 58.8, 51.7, 49.2, 39.9, 35.1, 31.0, 28.5, 25.9, 18.3, 7.5. HR-MS. Calcd for C18H36NO4SiNa (M+H): m/z 358.2410. Found: m/z 358.2414. Calcd for C18H35NO4SiNa (M+Na): m/z 380.2235. Found: m/z 380.2233.
3-Oxa-5-aza-tricyclo-5-syn-(tert-butyldimethylsilyloxymethyl)[5.1.1.01,5]nonan-4-one (12)
To a solution of 10 (98 mg, 0.3 mmol) in dry CH2Cl2 (15 mL) was added Et3N dropwise (153 mg, 1.5 mmol) followed by MsCl (68 mg, 0.6 mmol). The resulting solution was stirred for 3 h at room temperature. Washing with water (2×5 mL) and drying with sodium sulfate followed by concentration gave 108 mg (97%) of mesylate 11. To a solution of mesylate 11 (46 mg, 0.1 mmol) in acetonitrile (6 mL) was added KCN (37 mg, 0.5 mmol) and 18-crown-6 ether (6 mg). The resulting solution was heated at 70°C for 12 h. Solvent was removed in vacuo to give 68 mg of crude product which was purified by preparative TLC to give 21 mg (66%) of 12; Rf=0.35 (hexane/ethyl acetate, 2:1); 1H NMR: δ 4.36 (d, J=9.4 Hz, 1H, OCH2), 4.29 (d, J=9.4 Hz, 1H, OCH2), 3.52 (m, 2H, OCH2), 3.30 (d, J=9 Hz, H3), 3.18 (d, J=9 Hz, H3), 2.86 (dd, J=2.1, 2.4 Hz, 1H, H4), 2.41 (m, H5), 1.79 (dd, J=7.3, 3.3 Hz, H6anti), 1.64 (d, J=7.3 Hz, H6syn), 0.87 (s, 9H, TBDMS), 0.05 (s, 6H, TBDMS); 13C NMR: δ 156.7, 74.8, 65.8, 58.3, 52.3, 44.0, 41.3, 41.1, 25.8, 18.2, –5.5. HR-MS. Calcd for C14H26NO3Si (M+H): m/z 284.1681. Found: m/z 284.1675.
N-(tert-butoxycarbonyl)-2-azabicyclo-5-syn-(tert-butyldimethylsilyloxymethyl)-1-azidomethyl[2.1.1]hexane (13)
A solution of mesylate 11 (117 mg, 0.3 mmol) and sodium azide (186 mg, 3.0 mmol) in DMF (5 mL) was stirred at 70°C for 12 h. Workup was done by diluting the mixture with ether (20 mL) and washing with water (4×5 mL). Ether was removed in vacuo to give 315 mg of a crude mixture which was purified on silica gel chromatography to give 6 mg (6%) of 13, 4 mg (5%) of 14 and 7 mg (10%) of 12.
Compound 13 Rf=0.27 (hexane/ethyl acetate, 2:1); 1H NMR: δ 4.06 (br, 1H, CH2OH), 3.90 (d, J=14.1 Hz, 1H, CH2OH), 3.39 (m, 1H, CH2ON3), 3.35 (m, 1H, CH2ON3), 3.32 (m, 1H, H3), 3.26 (d, J=8.7 Hz, 1H, H3), 2.63 (br, H4), 2.22 (m, H5), 1.70 (m, 1H, H6anti), 1.47 (s and m, 9H, H6syn and BOC), 0.87 (s, 9H, TBDMS), 0.03 (s, 6H, TBDMS); 13C NMR: δ 156.7, 80.0, 72.8, 58.4, 51.7, 51.4, 49.7, 40.4, 35.8, 28.5, 18.0, –0.05. HR-MS. Calcd for C18H35N4O3Si (M+H): m/z 383.2473. Found: m/z 383.2473.
Compound 14 Rf=0.80 (hexane/ethyl acetate, 2:1); 1H NMR: δ 4.20 (br d, J=13.2 Hz, 1H, CH2OH), 3.84 (br d, J=13.2 Hz, 1H, CH2OH), 3.42 (m, 1H, CH2ON3), 3.35 (m, CH2ON3), 3.30 (d, J=9.3 Hz, 1H, H3), 3.23 (d, J=9.3 Hz, 1H, H3), 2.62 (br, H4), 2.12 (m, H5), 1.69 (brd, J=7.5 Hz, 1H, H6anti), 1.52 (br, 1H, OH), 1.40 (s and m, H6syn and BOC); 13C NMR: δ 155.7, 75.5, 73.0, 58.8, 58.5, 51.0, 49.6, 49.1, 39.9, 35.1, 28.7, 28.3. HR-MS. Calcd for C12H21N4O3 (M+H): m/z 269.1608. Found: m/z 269.1604.
N-(tert-Butoxycarbonyl)-1-vinyl-5-syn-(tert-butyldimethylsilyloxymethyl)-2-azabicyclo[2.1.1]hexane (15)
Procedure 1 To a solution of aldehyde 8g (57 mg, 0.16 mmol) in CH2Cl2 (10 mL) at 0°C was added Tebbe reagent (319 μL, 0.16 mmol). The resultant solution was slowly warmed to room temperature and stirred for 12 h. The mixture was then diluted with ether and charged with 10% NaOH (1 mL). The organic solution was dried with sodium sulfate and filtered on celite. Concentration gave 67 mg of crude product 15; silica gel flash chromatography furnished 33 mg (59%) of pure compound 15.
Procedure 2 To a solution of methyl(triphenylphosphonium) bromide (292 mg, 0.82 mmol) in dry THF (15 mL) at 0°C under argon was added n-BuLi (457 μL, 0.73 mmol, 1.6 M in hexane). The resulting bright yellow mixture was stirred for 0.5 h at 0°C. Compound 8g (100 mg, 0.28 mmol) in dry THF (15 mL) was cannulated to the mixture and the resulting solution was slowly brought to room temperature and stirred for 20 h under argon. Ether (10 mL) was added and the mixture was stirred for 10 min followed by addition of water (10 mL) and stirring for an additional 10 min. The organic layer was separated and the aqueous layer was extracted with ether (2×10 mL). The combined organic layers were washed with brine (5 mL), dried with Na2SO4 and concentrated to yield 158 mg of crude product. Silica gel flash chromatography gave 51 mg (52%) of pure compound 15; Rf=0.69 (hexane/ether, 4:1); 1H NMR: δ 6.45 (br m, 1H), 5.16 (dd, J=18, 2 Hz, 1H), 5.10 (brd, J=10.5 Hz, 1H), 3.53 (dd, J=11, 7 Hz, OCH2), 3.34 (m, H3 and OCH2, 2H), 3.29 (d, J=8.5 Hz, 1H, H3), 2.62 (t, J=3.0, 3.0 Hz, H4), 2.20 (ddd, J=7.5, 7.5, 3.5 Hz, H5), 1.73 (d, J=7.5, 3.0, 2.0, 1H, H6anti), 1.55 (d, J=7.0 Hz, H6syn), 1.45 (s, 9H, BOC), 0.89 (s, 9H, TBDMS), 0.04 (s, 6H, TBDMS); 13C NMR: δ 158.0, 135.9, 117.3, 74.7, 73.0, 57.2, 51.5, 49.8, 40.8, 35.5, 28.9, 26.3, 18.6, –5.00. HR-MS. Calcd for C19H35NO3SiNa (M+H): m/z 354.2459. Found: m/z 354.2454. Calcd for C19H34NO2SiNa (M+H-H2O): m/z 336.2364. Found: m/z 336.2348.
N-(tert-butoxycarbonyl)-1-(2-hydroxyethyl)-5-syn- (tert-butyldimethylsilyloxymethyl)-2-azabicyclo [2.1.1]hexane (16)
To a solution of 15 (40 mg, 0.11 mmol) in THF (8 mL) at 0°C was added borane in THF (340 μL, 0.34 mmol). The mixture was stirred for 7 h at room temperature and charged with 2N NaOH (220 μL, 0.44 mmol) followed by 30% H2O2 (0.44 mmol) at −78°C. Potassium carbonate was added to saturate the aqueous phase. The aqueous layer was extracted with ether (10×5 mL). The ether layers were combined and dried with Na2SO4. Concentration and chromatography gave 9.1 mg (22%) of alcohol 16; Rf=0.29 (hexane/ether, 3:1); 1H NMR: δ 3.75 (m, 2H, CH2OH), 3.44 (dd, J=10.4, 7.6 Hz, 1H, CH2OTBDMS), 3.37 (dd, J=10.8, 5.2 Hz, 1H, CH2OTBDMS), 3.28 (d, J=9.2 Hz, H3), 3.19 (d, J=9.2 Hz, H3), 2.53 (brt, J=3.2, 2.8 Hz, H4), 2.37 (m, 2H, CH2), 2.10 (m, H5), 1.63 (m, H6anti), 1.39 (s and m, 10 H, BOC and H6syn), –0.83 (s, 9H, TBDMS), 0.00 (s, 6H, TBDMS); 13C NMR: δ 156.2, 79.4, 73.7, 59.9, 59.5, 52.5, 49.2, 41.3, 35.1, 33.8, 28.6, 25.9, 18.3, –5.5. HR-MS. Calcd for C19H38NSiO4 (M+H): m/z 372.2565. Found: m/z 372.2577. Calcd for C19H37NSiO4Na (M+Na): m/z 394.2384. Found: m/z 394.2406.
N-(tert-butoxycarbonyl)-1-[(2-methoxycarbonyl)-1-vinyl]-5-syn-(tert-butyldimethylsilyloxymethyl)- 2-azabicyclo[2.1.1]hexane (17)
To a solution of 8g (130 mg, 0.37 mmol) in dry CH2Cl2 (15 mL) was added methyl (triphenylphosphoranylidene)acetate (183 mg, 0.55 mmol) under argon. The resulting solution was stirred at 40°C for 2 days. Solvent was removed in vacuo and the crude product was purified by silica gel flash chromatography to give 29 mg (22%) of unreacted aldehyde 8g and 61 mg (41%) of 17; Rf=0.42 (hexane/ethyl acetate, 4:1); 1H NMR: δ 7.40 (br d, J=15.3 Hz, 1H), 5.90 (dd, J=15.9, 3 Hz, 1H), 3.69 (s, 3H, OCH3), 3.36 (m, 4H, 2H3, CH2OH), 2.64 (br, H4), 2.21 (br, H5), 1.74 (br, H6anti), 1.57 (d, J=7.2 Hz, H6syn), 1.41 (s, 9H, BOC), 0.89 (s, 9H, OTBDMS), 0.00 (6H, OTBDMS); 13C NMR: δ 167.1, 156.7, 145.0, 120.0, 80.4, 73.5, 59.0, 54.8, 51.8, 49.2, 42.8, 36.0, 28.7, 20.2, 18.5, –5.0. HR-MS. Calcd for C21H38NO5Si (M+H): m/z 412.2520. Found: m/z 412.2514.
N-(tert-butoxycarbonyl)-1-[(2-methoxycarbonyl)-1-ethyl]-5-syn-(tert-butyldimethylsilyloxy-methyl)-2-azabicyclo [2.1.1]hexane (18)
To a solution of compound 17 (45 mg, 0.11 mmol) in toluene (8 mL) was added TsNHNH2 (163 mg, 0.88 mmol) and TMEDA (197 μL, 1.3 mmol). The mixture was heated under reflux for 12 h, then cooled to room temperature, washed with H2O (5 mL), brine (5 mL) and dried with MgSO4. After concentration, the residue was purified by chromatography on silica gel to give 39 mg (87%) of product 18; Rf=0.64 (hexane/ether, 4:1); 1H NMR: δ 3.65 (s, 3H, OMe), 3.39 (dd, J=10.2, 6.8 Hz, 1H, OCH2), 3.27 (m, 3H, 2H3, OCH2), 2.58 (br, 1H, H4), 2.46 (m, 2H, CH2), 2.28 (m, 2H, CH2), 2.02 (ddd, J=6.8, 6.8, 2.2 Hz, 1H, H5), 1.53 (m, 1H, H6anti), 1.45 (s, 9H, BOC), 1.38 (d, J=7.2 Hz, 1H, H6syn), 0.86 (s, 9H, TBDMS), 0.04 (s, 6H, TBDMS); 13C NMR: δ 174.0, 156.0, 79.3, 74.3, 58.9, 52.3, 51.4, 49.4, 40.9, 34.9, 30.9, 28.5, 26.5, 25.8, 18.2, –5.4, –5.5. HR-MS. Calcd for C21H39NO5SiNa (M+Na): m/z 436.2495. Found: m/z 436.2503.
N-(tert-butoxycarbonyl)-1-(3-hydroxypropyl)-5-syn- (tert-butyldimethylsilyloxymethyl)-2-azabicyclo[2.1.1]hexane (19)
A solution of 18 (17 mg, 0.04 mmol) in THF (2 mL) was stirred and treated with LAH (13 μL, 0.03 mmol) at –78°C. The mixture was slowly warmed to room temperature, stirred for an additional 1 h, diluted with ether (10 mL) and washed with water (2×5 mL). Filtration and drying with Na2SO4 followed by concentration gave 14 mg (88%) of 19; Rf=0.38 (hexane/ether, 4:1); 1H NMR: δ 3.64 (m, 2H, OCH2), 3.43 (dd, J=10.8, 6.8 Hz, 1H), 3.28 (m, 3H), 2.58 (br t, J=3.2, 2.8 Hz, 1H, H4), 2.38 (br, 1H, OH), 2.04 (m, 3H, H5 and CH2), 1.67 (m, 2H, CH2), 1.57 (ddd, J=7.0, 3.2, 1.6 Hz, 1H, H6anti), 1.39 (d, J=7.0 Hz, 1H, H6syn), 1.45 (s, 9H, BOC), 0.87 (s, 9H, TBDMS), 0.02 (s, 6H, TBDMS); 13C NMR: δ 156.1, 79.1, 75.1, 62.7, 59.0, 52.2, 49.5, 40.8, 34.8, 29.4, 28.5 and 28.2, 26.8, 25.9 and 25.7, 18.2, –5.4, –5.5; HR-MS. Calcd for C20H39NO4SiNa (M+Na): m/z 408.2546. Found: m/z 408.2548.
Methyl 3-{3-[(tert-butoxycarbonylamino)methyl]-2-(2-hydroxymethyl)-1-cyclobutyl}propanoate (20)
To a solution of 17 (17 mg, 0.04 mmol) in ethyl acetate (8 mL) was added Pd/C (3 mg) and the mixture was stirred at room temperature for 1 h. The catalyst was filtered off and the solution was concentrated to give 9 mg (75%) of 20; Rf=0.24 (hexane/ethyl acetate, 2:1); 1H NMR: δ 3.95 (t, J=10.6 Hz, 1H, CH2OH), 3.81 (dd, J=10.4, 4.8 Hz, 1H, CH2OH), 3.65 (s, 3H, OMe), 3.29 (dd, J=14.0, 4.2 Hz, 1H, CH2NHBOC), 3.10 (dd, J=14.0, 9.6 Hz, 1H, CH2NHBOC), 2.58 (m, 1H), 2.45 (m ,1H), 2.25 (m, 1H), 2.20 (m, 2H), 2.11 (m, 1H), 1.80 (br, 1H, OH), 1.70 (m, 2H), 1.52 (m, 1H), 1.40 (s, 9H, BOC), 1.37 (m, 1H); 13C NMR: δ 173.9, 156.1, 79.5, 59.6, 51.9 and 51.8, 41.8, 41.2, 34.6, 33.3, 32.4, 30.0, 28.8, 26.4, 14.5. HR-MS. Calcd for C15H27NO5Na (M+Na): 324.1787. Found: m/z 324.1772.
N-(tert-butoxycarbonyl)-1-benzoyl-5-syn-carboxy-2-azabicyclo[2.1.1]hexane (21h)
A solution of 8h (33 mg, 0.08 mmol) in acetone (5.0 mL) was treated dropwise at 0°C with Jones reagent (60 μL, 0.16 mmol, 2.7 M CrO3 in 4N H2SO4). The mixture was maintained at 0°C for 1 h, followed by slow addition of isopropanol (3.0 mL) to quench excess oxidant. The resultant green biphasic mixture was stirred vigorously for 1 h at 0°C, then filtered over a pad of celite and the filter cake was washed with ethyl acetate (3×5 mL). The organic layer was washed with water (3×15 mL), dried over Na2SO4 and concentrated in vacuo to give a yellow oil. The oil was diluted with ethyl acetate (10 mL) and stirred vigorously with 10% NaOH for 5 min. The mixture was transferred to a separatory funnel, the aqueous layer was removed and the organic layer was washed with 10% NaOH (2×5 mL). The combined basic aqueous layers were cooled to 0°C, acidified to pH 1.0 with 1.0 N HCl and extracted with ethyl acetate (5×10 mL). The extract was dried over Na2SO4 and concentrated in vacuo to yield compound 21h as an off-white solid (19 mg, 76%); mp 133°C –134°C; 1H NMR: δ 8.08 (m, 1H, Ph), 7.59 (m, 1H, Ph), 7.47 (dd, 2H, J=7.8, 7.5 Hz, 2H), 3.95 (m, H5), 3.65 (m, 1H), 3.13 (br, 2H), 2.09 (m, 2H); 13C NMR: δ 195.8/ 195.7, 156.8, 133.6, 129.0, 128.4, 82.1, 78.5, 58.1, 51.4, 49.8, 45.7, 36.0, 28.3/ 28.1, 27.4. HR-MS. Calcd for C18H21NO5 (M+Na): m/z 354.1317. Found: m/z 354.1307.
N-(tert-butoxycarbonyl)-1-methoxycarbonyl-5-syn-carboxy-2-azabicyclo[2.1.1]hexane (21k)
To a solution of silyl ether 8k (80.4 mg, 0.209 mmol) in acetone (3.0 mL) at 0°C was added slowly Jones reagent (0.20 mL, 0.54 mmol, 2.7 M CrO3 in 4N H2SO4). The mixture was maintained at 0°C for 1 h and then slowly treated with isopropanol (5.0 mL) to quench excess oxidant. The resultant green biphasic mixture was stirred vigorously for 1 h at 0°C, then filtered over a pad of celite and rinsed with ethyl acetate (3×15 mL). The organic layer was washed with water (3×15 mL), dried over Na2SO4 and concentrated in vacuo to give a yellow oil. The oil was diluted with ethyl acetate (10 mL) and stirred vigorously with 10% NaOH for 5 min. The mixture was transferred to a separatory funnel, then the aqueous layer was removed and the organic layer was washed with 10% NaOH (2×10 mL). The combined basic aqueous layers were cooled to 0°C, acidified to pH 1.0 with 1.0 N HCl and extracted with ethyl acetate (5×10 mL). Concentration in vacuo furnished compound 21k as an off-white solid (51.0 mg, 85%); 1H NMR: δ 3.88 (s, 3H), 3.70 (d, J=8.9 Hz, 1H3), 3.43 (d, J=8.9 Hz, 1H3), 3.04 (brs, 1H4), 3.00 (br, 1H5), 1.98 (ddd, J=7.4, 3.0, 1.5 Hz, H6anti), 1.75 (d, J=7.4 Hz, H6syn), 1.42 (s, 9H); 13C NMR: δ 170.3, 169.7, 155.9, 81.2, 70.6, 53.1, 51.7, 49.3, 41.2, 37.6, 28.9. HR-MS. m/z 286.1306, Calcd for C13H20NO6 (M+H) 286.1285; m/z 308.1131, Calcd for C13H19NO6Na (M+Na) 308.1104.
N-(tert-butoxycarbonyl)-1-methyl-5-syn-(hydroxymethyl)-2-azabicyclo[2.1.1]hexane (22)
To a solution of 8a (179 mg, 0.53 mmol) in dry THF (16 mL) at 0°C was added TBAF (1.6 mL, 1.6 mmol) dropwise over a period of 5 min. The resulting solution was warmed to room temperature and stirred for 16 h. The mixture was diluted with ether (10 mL) and washed with water (5 mL), brine (5 mL) and dried with Na2SO4. Solvent was removed in vacuo to give 107 mg (89%) of crude compound 22 which was taken to the next step without further purification; Rf=0.26 (hexane/ether, 1:1); 1H NMR: δ 3.43 (dd, J=10.8, 6.8 Hz, 1H, CH2OH), 3.32 (dd, J=10.8, 6.8 Hz, 1H, CH2OH), 3.28 (d, J=9.2 Hz, 1H, H3), 3.22 (d, J=9.2 Hz, 1H, H3), 2.57 (brt, J=3.0 Hz, 1H, H4), 1.94 (ddd, J=6.8, 6.8, 2.8 Hz, 1H, H5), 1.64 (s, 3H, Me), 1.51 (ddd, J=6.8, 2.8, 1.6 Hz, 1H6anti), 1.45 (s, 9H, BOC), 1.42 (d, J=6.8 Hz, 1H, H6syn), 0.87 (s, 9H, TBDMS), 0.03 (s, 6H, TBDMS); 13C NMR: δ 156.9, 79.2, 71.1, 58.8, 53.2/53.0, 49.5, 43.5, 35.3/35.1, 28.5, 18.2. HR-MS. Calcd for C12H21O3NNa (M+Na): m/z 250.1419. Found: m/z 250.1425.
N-(tert-butoxycarbonyl)-1-methyl-5-syn-carboxy-2-azabicyclo[2.1.1]hexane (21a)
To a solution of alcohol 22 (23 mg, 0.10 mmol) and NMO (23.7 mg, 0.20 mmol) in anhydrous acetonitrile (4 mL) was added TPAP (3.6 mg, 0.01 mmol) in one portion. The resulting dark mixture was stirred at room temperature for 1 h before being treated with additional portions of TPAP (3 mg) and NMO (23.7 mg). The mixture was then immediately quenched with water (5 μL) and concentrated. The residue was treated with dichloromethane (10 mL) and the mixture was washed with 1 N NaOH (10 mL). The layers were separated and the aqueous layer was acidified to pH 3 and extracted with ethyl acetate (3×5 mL). Solvent was removed to give 8 mg (33%) of acid 21a; 1H NMR: δ 8.64 (br, 1H, CO2H), 3.65 (d, J=9.0 Hz, H3), 3.33 (d, J=9.0 Hz, H3), 2.92 (br, H4), 2.56 (br, H5), 1.77 (s, 3H, Me), 1.59 (m, 1H, H6anti), 1.48 (d, J=8.1 Hz, 1H, H6syn), 1.44 (s, 9H, BOC); 13C NMR: δ 169.7, 155.9, 81.2, 70.6, 53.1, 51.7, 49.3, 41.2, 37.6, 28.2. HR-MS. Calcd for C12H20NO4 (M+H): m/z 242.2915. Found: m/z 242.2874.
References
[1] Huck, B. R.; Fisk, J. D.; Guzei, I. A.; Carlson, H. A.; Gellman, S. H. Secondary structural preferences of 2,2-disubstituted pyrrolidine-4-carboxylic acid oligomers: β-peptide foldamers that cannot form internal hydrogen bonds. J. Am. Chem. Soc. 2003, 125, 9035–9037.10.1021/ja034561cSearch in Google Scholar
[2] Huck, B. R. The development of novel β-peptide foldamers: 1. Tertiary amine β-peptide oligomers. 2. Chimeric α/β-peptide hairpins. Ph.D. thesis, University of Wisconsin-Madison, 2002.Search in Google Scholar
[3] Huck, B. R.; Langenhan, J. M.; Gellman, S. H. Non-hydrogen-bonded secondary structure in β-peptides: evidence from circular dichorism of (S)-pyrrolidine-3-carboxylic acid oligomers and (S)-nipecotic acid oligomers. Org. Lett. 1999, 1, 1717–1720.10.1021/ol9909482Search in Google Scholar
[4] Huck, B. R.; Gellman, S. H. Synthesis of 2,2-disubstituted pyrrolidine-4-carboxylic acid derivatives and their incorporation into β-peptide oligomers. J. Org. Chem.2005, 70, 3353–3362.10.1021/jo048639zSearch in Google Scholar
[5] Liu, N. Novel non-hydrogen-bonded 5-syn-carboxymethanopyrrolidine β-peptide oligomers. Ph.D. thesis, Temple University, 2008.Search in Google Scholar
[6] Hu, Z. Constrained β-prolines: 1. Methanopyrrolidine β-amino acids: synthesis and characterization of novel C6-substituted analogues and peptide oligomers. 2. Synthesis of 2,2-disubstituted pyrrolidine-3-carboxylic acids. Ph.D. thesis, Temple University, 2015.Search in Google Scholar
[7] Krow, G. R.; Lin, G.; Herzon, S. B.; Thomas, A. M.; Moore, K. P.; Huang, Q.; Carroll, P. J. Convenient preparation of 2,4-methanopyrrolidine and 5-carboxy-2,4-methanopyrrolidines. J. Org. Chem.2003, 68, 7562–7564.10.1021/jo0348672Search in Google Scholar
[8] Krow, G. R.; Herzon, S. B.; Lin, G.; Qui, F.; Sonnet, P. E. Complex-induced proximity effects. Temperature-dependent regiochemical diversity in lithiation-electrophilic substitution reactions of N-BOC-2-azabicyclo[2.1.1]hexane. Org. Lett. 2002, 4, 3151–3154.10.1021/ol026509bSearch in Google Scholar
[9] Kwak, Y. S.; Winkler, J. D. Synthesis of 6-azabicyclo[3.2.1]octan-3-ones via vinylogous imide photochemistry: an approach to the synthesis of the hetisine alkaloids. J. Am. Chem. Soc.2001, 123, 7429–7430.10.1021/ja010542wSearch in Google Scholar
[10] Bailey, W. F.; Patricia, J. J. The mechanism of the lithium - halogen interchange reaction: a review of the literature. J. Organomet. Chem.1988, 352, 1–46.10.1016/0022-328X(88)83017-1Search in Google Scholar
[11] Hashimoto, N.; Aoyama, T.; Shioiri, T. New methods and reagents in organic synthesis. A simple efficient preparation of methyl esters with trimethylsilyldiazomethane (TMSCHN2) and its application to gas chromatographic analysis of fatty acids. Chem. Pharm. Bull.1981, 29, 1475–1478.10.1248/cpb.29.1475Search in Google Scholar
[12] Sadowsky, J. D.; Fairlie, W. D.; Hadley, E. B.; Lee, H. S.; Umezawa, N.; Nikolovska, C. Z.; Wang, S.; Huang, D. C. S.; Tomita, Y.; Gellman, S. H. (α/β+α)-Peptide antagonists of BH3 domain/Bcl-xLrecognition: toward general strategies for foldamer-based inhibition of protein-protein interactions. J. Am. Chem. Soc.2007, 129, 139–154.10.1021/ja0662523Search in Google Scholar PubMed
[13] Malpass, J. R.; Patel, A. B.; Davies, J. W.; Fulford, S. Y. Modification of 1-substituents in the 2-azabicyclo[2.1.1]hexane ring system; approaches to potential nicotinic acetylcholine receptor ligands from 2,4-methanoproline derivatives. J. Org. Chem.2003, 68, 9348–9355.10.1021/jo035199nSearch in Google Scholar PubMed
[14] Evans, G. B.; Furneaux, R. H.; Lenz, D. H.; Painter, G. F.; Schramm, V. L.; Singh, V.; Tyler, P. C. Second generation transition state analogue inhibitors of human 5‘-methylthioadenosine phosphorylase. J. Med. Chem.2005, 48, 4679–4689.10.1021/jm050269zSearch in Google Scholar
[15] Cuellar, M. A.; Salas, C.; Cortes, M. J.; Morello, A.; Diego, M. J.; Preite, M. D. Synthesis and in vitro trypanocide activity of several polycyclic drimane-quinone derivatives. Bioorg. Med. Chem.2003, 11, 2489–2497.10.1016/S0968-0896(03)00193-7Search in Google Scholar
[16] Brown, H. C.; Unni, M. K. Hydroboration. XXV. The hydroboration of 3-butenyl derivatives containing representative substituents. J. Am. Chem. Soc.1968, 90, 2902–2905.10.1021/ja01013a030Search in Google Scholar
[17] Poon, C.; Chiu, P. A synthesis of the tetracyclic carboskeleton of isaindigotidione. Tet. Lett.2004, 45, 2985–2988.10.1016/j.tetlet.2004.02.052Search in Google Scholar
[18] Yin, B. L.; Fan, J. F.; Gao, Y.; Wu, Y. L. Molecular diversity from tonghaosu analogues, selective reduction of theendo-cyclic double bond of tonghaosu analogues and the synthesis of cyclopentenone derivatives. Synlett2003, 3, 399–401.10.1055/s-2003-37131Search in Google Scholar
[19] Lescop, C.; Mevellec, L.; Huet, F. A new synthesis of 2-azabicyclo[2.1.1]hexanes. J. Org. Chem.2001, 66, 4187–4193.10.1021/jo001790ySearch in Google Scholar PubMed
[20] Anelli, P. F.; Biffi, C.; Montanari, F.; Quici, S. Fast and selective oxidation of primary alcohols to aldehydes or to carboxylic acids and of secondary alcohols to ketones mediated by oxoammonium salts under two-phase conditions. J. Org. Chem.1987, 52, 2559–2562.10.1002/chin.198749121Search in Google Scholar
[21] Corey, E. J.; Venkateswarlu, A. Protection of hydroxyl groups as tert-butyldimethylsilyl derivative. J. Am. Chem. Soc.1972, 94, 6190–6191.10.1021/ja00772a043Search in Google Scholar
[22] Hu, T.; Panek, J. S. Total synthesis of (−)-motuporin. J. Org. Chem.1999, 64, 3000–3001.10.1021/jo9904617Search in Google Scholar PubMed
©2016 Walter de Gruyter GmbH, Berlin/Boston
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Articles in the same Issue
- Frontmatter
- Research Articles
- Pyrimidinethione as a building block in heterocyclic synthesis: synthesis of pyrano[2,3-d]pyrimidine, chromeno[2,3-d]pyrimidine, pyrido[3′,2′:5,6]pyrano[2,3-b]pyridine, and pyrimido[5′,4′:5,6]pyrano[2,3-d]pyrimidine derivatives
- C1-Substituted N-tert-butoxycarbonyl-5-syn-tert-butyldimethylsilyloxymethyl-2-azabicyclo[2.1.1]hexanes as conformationally constrained β-amino acid precursors
- Synthesis and characterization of 1,3,4-thiadiazole-2,5-dithio crown ethers
- Oxidative reaction of 2-aminopyridine-3-sulfonyl chlorides with tertiary amines
- New 8-substituted BODIPY-based chromophores: synthesis, optical and electrochemical properties
- Heterocyclization of 5,6-disubstituted 3-alkenyl-2-thioxothieno[2,3-d]pyrimidin-4-one with p-alkoxyphenyltellurium trichloride
- Synthesis and biological evaluation of 4-(2′,4′-difluorobiphenyl-4-yl)-6-arylpyrimidin- 2-amine derivatives
- Synthesis and antimicrobial properties of cycloheptyl substituted benzimidazolium salts and their silver(I) carbene complexes
- Solvent-free microwave-assisted synthesis and biological evaluation of 2,2-dimethylchroman-4-one based benzofurans
Articles in the same Issue
- Frontmatter
- Research Articles
- Pyrimidinethione as a building block in heterocyclic synthesis: synthesis of pyrano[2,3-d]pyrimidine, chromeno[2,3-d]pyrimidine, pyrido[3′,2′:5,6]pyrano[2,3-b]pyridine, and pyrimido[5′,4′:5,6]pyrano[2,3-d]pyrimidine derivatives
- C1-Substituted N-tert-butoxycarbonyl-5-syn-tert-butyldimethylsilyloxymethyl-2-azabicyclo[2.1.1]hexanes as conformationally constrained β-amino acid precursors
- Synthesis and characterization of 1,3,4-thiadiazole-2,5-dithio crown ethers
- Oxidative reaction of 2-aminopyridine-3-sulfonyl chlorides with tertiary amines
- New 8-substituted BODIPY-based chromophores: synthesis, optical and electrochemical properties
- Heterocyclization of 5,6-disubstituted 3-alkenyl-2-thioxothieno[2,3-d]pyrimidin-4-one with p-alkoxyphenyltellurium trichloride
- Synthesis and biological evaluation of 4-(2′,4′-difluorobiphenyl-4-yl)-6-arylpyrimidin- 2-amine derivatives
- Synthesis and antimicrobial properties of cycloheptyl substituted benzimidazolium salts and their silver(I) carbene complexes
- Solvent-free microwave-assisted synthesis and biological evaluation of 2,2-dimethylchroman-4-one based benzofurans