Acyclic analogs of nucleosides based on tris(hydroxymethyl)phosphine oxide: synthesis and incorporation into short DNA oligomers
-
Barbara Nawrot
 ,
Olga Michalak
,
Olga Michalak

Abstract
Tris-(hydroxymethyl)phosphine oxide (THPO) to a certain extent resembles a part of 2′-deoxyribofuranose, although it exists in an acyclic form only and the oxygen atom at the THPO phosphorus center provides additional hydration site or acceptor of hydrogen bonds. After proper protection of hydroxyl groups, THPO was functionalized with nucleobases and converted into phosphoramidite monomers suitable for incorporation into growing oligonucleotide chains within the solid phase synthesis protocol. The resultant THPO-DNA analogs show reduced affinity to complementary DNA strands, and are resistant towards snake venom and calf spleen exonucleases.
Introduction
In principle, oligonucleotide analogs are modified within the internucleotide bonds (e.g. phosphorothioates [1, 2], and phosphorodithioates [3], methylphosphonates [4], boranophosphates [5], methylboranophosphonates [6], phosphoramidates/phosphorothioamidates [7], and methylphosphoramidates [8]), heterocyclic bases (e.g. 2-thiouracil [9], 6-thioguanine [10]) or within the sugar moiety (e.g. 2′-O-alkyl [11], LNA [12], 2′-F [13–15]). They are widely applied in cell and molecular biology as gene expression inhibitors in antisense or RNAi strategies [16–20], and are useful tools in enzymatic studies of nucleases, topoisomerases or transferases [2, 21, 22]. Beneficially, certain analogs of oligonucleotides intended to be used in antisense or RNAi strategies exert enhanced affinity towards target messenger RNA, or increased nucleolytic stability. However, there are persisting problems in therapeutic applications of oligonucleotides, such as poor cellular uptake or off-target effects, so there is still a need for novel nucleic acids analogs free of those shortcomings. In this frame, we previously developed a new DNA analog containing an acyclic unit (originated from bis(hydroxymethyl)phosphinic acids, BHPA, 2) replaced for the deoxyribose ring [23, 24]. The nucleobase in 2 could occupy the position identical to that in a natural nucleoside or a more distant one, depending on the number of methylene units n (Figure 1). BHPA related analogs were also obtained in the form of a double-anionic derivative 3 deprived of the nucleobases [23, 24]. The BHPA-DNA analogs are slightly cytotoxic towards HUVEC and HeLa cells, and are resistant to nucleolytic degradation. The nucleoside analogs, where the 3′-, 4′- and 5′-carbon atoms of the original sugar moiety are represented by either the BHPA or tris(hydroxymethyl)phosphine oxide (THPO) moiety, have been already synthesized and their antiviral properties investigated [25]. Hypothetically, introduction of a nucleobase to THPO would also create a DNA-like unit able to form the Watson-Crick hydrogen bonds with a nucleoside in a complementary DNA or RNA strand. Moreover, the DNA oligomers modified with the THPO units would contain more flexible sugar-phosphate backbone and additional hydration sites (the acceptor oxygen atoms at the THPO phosphorus centers) compared to the natural DNA. In this paper, we present an approach for the synthesis of suitably protected phosphoramidite derivatives of THPO-related acyclic nucleosides and their use as monomers for synthesis of short DNA oligomers of the structure 4. Structural characteristics of the acyclic THPO-DNA analogs, as well as their hybridization properties and stability against selected exonucleases are described.

A DNA unit (1), bis(hydroxymethyl)phosphinic acid (BHPA) based acyclic analog of natural DNA (2, X = O or N), its abasic double-anionic form (3), and a tris-(hydroxymethyl)phosphine oxide (THPO) based analog (4).
Results and discussion
Synthesis of protected THPO-acyclic nucleoside phosphoramidites 11
Protection of two hydroxyl groups in THPO
For selective protection of only one hydroxyl group, tris(hydroxymethyl)phosphine oxide 5 was treated with one molar equivalent of 4,4′-dimethoxytrityl chloride (DMT-Cl) in pyridine at room temperature (Scheme 1). The O-DMT derivative 6 was obtained in 51% yield after silica gel chromatography. The second hydroxyl group in 6 was protected with tert-butyldimethylsilyl (TBDMS) moiety upon treatment with one molar equivalent of TBDMS-Cl (in the presence of a four-fold molar excess of imidazole) furnishing compound 7A (R = TBDMS). Compound 6 was also treated with benzoyl chloride (Bz-Cl) in pyridine to yield the O-DMT-O′-Bz derivative 7B(R = Bz). When 5 was allowed to react with a two-fold molar excess of DMT-Cl, O,O′-di-DMT derivative 7C (R = DMT) was obtained in good yield.

Transformation of tris(hydroxymethyl)phosphine oxide into acyclic nucleoside phosphoramidites.
Conditions: (i) 1 eq. DMT-Cl/pyridine, 20 h, room temperature (RT); (ii) for 7A: TBDMS-Cl, imidazole, CH3CN, 48 h, RT; for 7B: BzCl, pyridine, 24 h, RT; for 7C: 2 eq. DMT-Cl/pyridine, 20 h, RT; (iii) DMAP, p-TsCl/CH2Cl2, 2 h, 0–5°C; (iv) cytosine, NaH, DMF, 60°C, 3 h; (v) BzCl, pyridine, 24 h, RT; (vi): N3-benzoylthymine, N6-benzoyladenine or N2-isobutyryl-O6-diphenylcarbamoylguanine, PhP3, DIAD, THF, 48 h; (vii): for 8a,c,d: 1 m (t-Bu)4NF in THF, 24 h, RT; for 8a′: 28% aq. NH4OH, 24 h; for 8b: 1 M p-TsOH/MeOH, TLC control, pyridine; (viii) 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite, CH2Cl2, RT.
Coupling of the protected THPO derivatives with nucleobases
Compound 7A was used as a starting material for the PPh3/DIAD promoted coupling (the Mitsunobu reaction conditions) [26] with N3-benzoyl-thymidine [27], N6-benzoyl-adenine [28, 29], or N2-isobutyryl-O6-diphenylcarbamoyl-guanine [30, 31]. The first reaction gave the derivative 8a (BPG= N3-Bz-Thy) in 95% yield (Table 1). Similarly effective was the condensation of 7B with the same nucleobase, which produced 8a′.
Spectral characteristics and yields of THPO derivatives 8, 10 and 11.
| Compound | R | 31P NMR δ (ppm) | FAB MS (m/z) | Yield (%) | |
|---|---|---|---|---|---|
| [M+H]+ | [M-H]- | ||||
| 8a | TBDMS | 38.52 | 912.8d | 758.5 | 92a |
| 8a′ | Bz | 40.87 | 921.5d | 767.5 | 95a |
| 8b | DMT | 41.15 | 942.2 | 940.3 | 44b |
| 8c | TBDMS | 42.02 | 778.5 | 776.4 | 51 |
| 8d | TBDMS | 40.89 | 955.2 | 953.2 | 90 |
| 10a | 38.40 | – | 653.5 | 38 | |
| 10b | 45.15 | 640.3 | 638.2 | 60 | |
| 10c | 42.39 | 664.3 | 662.0 | 84 | |
| 10d | 36.92 | 841.5 | 839.2 | 51 | |
| 10e | 45.46 | 551.6 | 549.0 | 60c, 41c | |
| 10f | 43.64 | 646.4 | 644.3 | 40 | |
| 11b | 152.54; 152.34; 152.03f 39.76; 39.64; 39.46; 39.33g | NDe | ND | 45 | |
| 11c | 151.88; 151.72; 151.57f 151.41; 38.10; 37.77g | ND | ND | 77 | |
| 11e | 150.95; 150.71; 150.64;f 37.20; 37.02; 36.90; 36.72g | ND | ND | 70 | |
| 11f | 151.91; 151.59; 151.27;f 39.54; 39.22; 38.99; 38.68g | ND | ND | 41 | |
aThe product was in ca. 30% contaminated with Ph3PO.bThe combined yield of transformations 9→8g→8b. cThe yields of two transformations: 8a→10e – 60%, and 8a′→10e – 41%. dThe [M+153]+ ion was registered for 8a and 8a′ in a complex with m-nitrobenzyl alcohol (NBA) matrix molecule (molecular mass 153). eND – Not determined. fResonances at δ>150 ppm correspond to the phosphorus atom in the phosphoramidite part of the molecule. gResonances at δ<40 ppm correspond to the phosphorus atom in the THPO part of the molecule.
The coupling of 7A with N6-benzoyl-adenine was more challenging, since two products (most likely regioisomers) at ca. 1:1 ratio were obtained. Chromatographic separation delivered the required N9-adenine derivative 8c (fast-eluting, BPG= N6-Bz-Ade) in 51% yield. Its structure was confirmed by a 2D NMR analysis in a heteronuclear multiple bonds coherence (HMBC) experiment, since the spin-spin coupling between the hydrogen atom at C1′ in the THPO moiety and C4 and C8 atoms of adenine confirmed the presence of the P-C-N9 linkage (Figure 2).
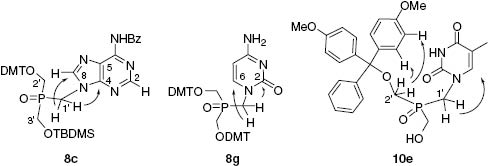
The detected spin-spin coupling (by a HMBC 2D NMR experiment) between hydrogen atoms at C1′ in the THPO moiety and selected neighboring carbon atoms in 8c, 8g, and 10e.
The reaction of 7A with N2-isobutyryl-O6-diphenylcarbamoylguanine furnished two products at a 95:5 ratio (as determined by 31P NMR), and the major product (efficiently isolated in 90% yield by silica gel chromatography) was the required N9-derivative 8d (BPG= N2-iBu-O6-Dpc-Gua) the structure of which was confirmed as previously described [25].
Unfortunately, under the Mitsunobu reaction conditions, N4-benzoylcytosine did not react with any compound 7 and the required compound 8b was not obtained, probably because of insufficient acidity of the N1-H function. Therefore, cytosine was treated with NaH in DMF to generate anion, which was allowed to react with O,O′-bis-DMT-O″-tosyl-derivative 9 (obtained from 7C by routine tosylation with tosyl chloride) furnishing a 95:5 mixture of regioisomers. The required N1-substituted regioisomer 8g (BPG= Cyt) was isolated by silica gel column chromatography and its structure was confirmed by 2D HMQC and HMBC analysis, as shown previously [25] (Figure 2). It was further benzoylated at the exoamine function by treatment with benzoyl chloride in pyridine to yield 8b (BPG= N4-Bz-Cyt).
Deprotection of one hydroxyl group in derivatives 8
In the next step, a set of derivatives 8, suitably protected at the base moieties, were deprotected under various conditions, depending on a protecting group to be removed to yield compounds possessing one OH function free and another one a DMT-protected (10). The TBDMS group of 8a,c,d was removed by treatment with 1.2 molar excess of TBAF reagent in THF, giving rise to 10a,c,d, respectively (Table 1). However, the treatment with TBAF partially removed the N3-Bz group in 10a, so a minor compound 10e was also obtained and chromatographically isolated. The structure of 10e (Figure 2) was confirmed by 2D HMBC analysis and possible correlation contacts between the hydrogens at C1′ and neighboring atoms were visible. The NOESY spectrum showed the NOE contacts between the hydrogens at C2′ and hydrogen atoms of the DMT group. Deprotection of 8d with TBAF produced also a minor compound 10f, resulting from the removal of the N6-Dpc group. The O′-Bz group of the THPO moiety in 8a′ was removed by treatment with 28% aqueous NH4OH, but this treatment also quantitatively removed the N3-Bz group, thus 10e was obtained as a sole product. One DMT protecting group from bis-DMT derivative 8b was removed by TLC-controlled treatment with 1 mp-TsOH/MeOH, terminated by addition of pyridine. After solvents removal and silica gel chromatography the resulting 10b was obtained in 60% yield.
Phosphitylation of derivatives 10
Phosphitylation of 10b,c,e,f was carried out in anhydrous dichloromethane using 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite (1.2 eq.) in the presence of diisopropylethylamine (3.0 eq.) at room temperature, followed by silica gel column chromatography. The corresponding acyclic nucleoside amidites 11b,c,e,f (Scheme 1) were obtained in 41-77% yields (Table 1). Their 31P NMR spectra contain two groups of signals with the chemical shifts δ in a range of 37–39 ppm (phosphine oxide moieties) and >150 ppm (phosphoramidite groups). From the stereochemical point of view, each compound 11 comprises of four diastereomers (RPSP, RPRP, SPRP, SPSP) and 3–4 resonance lines are observed in each group of signals, depending on differences in chemical shifts.
Synthesis of chimeric oligomers containing the THPO motif
Phosphoramidite derivatives of THPO-based acyclic nucleosides 11b, 11c, 11e and 11f, together with commercial phosphoramidites of suitably protected thymidine, 2′-deoxycytidine, 2′-deoxyadenosine and 2′-deoxyguanosine were used for the automated solid phase synthesis of chimeric DNA oligomers 12-21 (Table 2, the lower-case italicized letters a, c, g and t represent the modified acyclic units bearing Ade, Cyt, Gua or Thy nucleobases, respectively) [32]. The syntheses were performed on a succinyl-linked LCAA CPG solid support using an ABI 394 synthesizer (Applied Biosystems Inc., Foster City, CA, USA). The only modification made in the manufacturer’s protocol was a prolonged coupling time (up to 600 s) applied for the monomers 11. For monomers 11c and 11e the coupling efficiency (determined by the DMT-cation assay) was in the range of 98–99%, while for 11b and 11f the efficiency dropped below 90%.
Mass spectrometry and chromatographic characteristics of chimeric DNA oligomers possessing tris(hydroxymethyl)phosphine oxide units (THPO), marked by lower-case italicized letters.
| No. | Oligomer sequence 5′→3′ | Molecular mass | MALDI-TOF m/z | RP-HPLC, Rt(min)a | |
|---|---|---|---|---|---|
| DMT-on | DMT-off | ||||
| 12 | T9tT9 | 5720 | 5721 | 23.74 | 23.16 |
| 13 | T8tttT8 | 5730 | 5732 | 23.60 | 23.11 |
| 14 | T9aT9 | 5730 | 5729 | 21.40 | 22.91 |
| 15 | T8aaaT8 | 5760 | 5758 | 21.26 | 22.81 |
| 16 | T9gT9 | 5746 | 5751 | 23.04 | 19.56 |
| 17 | T8gggT8 | 5808 | 5809 | 22.57 | 19.05 |
| 18 | T9cT9 | 5706 | 5713 | 22.14 | 19.12 |
| 19 | T9ccT8 | 5697 | 5696 | 21.96 | 20.61 |
| 20 | atttaattat | 3077 | 3076 | 20.00/20.62 | 18.69/18.90 |
| 21 | ataattaaat | 3097 | 3095 | 18.71/19.47 | 17.19/17.01 |
aRP-HPLC purification was performed on a C18 column (4.6×250 mm, ThermoQuest) with a linear gradient of buffer A (0.1 m triethylammonium bicarbonate, pH 7.5) and buffer B (40% acetonitrile in 0.1 m triethylammonium bicarbonate, pH 7.5), flow rate 1 mL/min; a gradient of B for DMT-on 15–100%, DMT-off 0–100%, over 30 min.
After synthesis of 12-19, standard cleavage from the solid support and deprotection of phosphate groups and nucleobases was done (28% aqueous NH4OH, 55°C, 16 h). Notably, in the case of fully modified decamers 20 and 21, containing only the THPO-based units a, c, g, or t), an LCAA-CPG support with a universal linker was used [33], for which the synthesis started with a phosphoramidite monomer 11e. The assembled oligomers were then cleaved from the support with a 20% ethanolic solution of gaseous ammonia (55°C, 4 h), followed by the treatment with 28% aq. NH4OH/40% aq. MeNH2 (room temperature, 16 h) [34, 35].
To purify the oligomers, a two-step RP-HPLC (DMT-on/DMT-off) was applied [36]. The DMT-tagged oligomers (isolated during the first step) were detritylated with 50% aq. acetic acid and purified in the DMT-off form. The respective retention times (Rt) are given in Table 2. Illustrative RP-HPLC profiles for 5′-T9gT9-3′ (16) in its DMT-on and DMT-off forms are shown in Figure 3. The structures of all oligomers listed in Table 2 were confirmed by MALDI-TOF mass spectrometry.

Reverse-phase HPLC profiles for purification of 16 in its DMT-on (A) and DMT-off form (B), and for DMT-21 oligomer (C).
Interestingly, the RP-HPLC profiles recorded for DMT-20 and DMT-21 showed two closely eluted main peaks of similar intensity (Figure 3C). These four products (two pairs: DMT-20-fast and DMT-20-slow; DMT-21-fast and DMT-21-slow) were collected separately and detritylated. Interestingly, within each pair of fully deprotected products the RP-HPLC retention times were almost identical and mass spectrometry analysis showed identical m/z values (Table 2). Moreover, all four detritylated samples exhibited identical electrophoretic mobility in 20% polyacrylamide/7 m urea gel (data not shown). Therefore, we concluded that each pair consists of molecules of the same sequence, which differ in absolute configuration of the P-stereogenic centers present in the THPO acyclic nucleotide first from the 5′-end. Similar observations were noted in earlier works on P-chiral, 5′-DMT protected oligo(nucleoside phosphorothioate)s [37]. Perhaps for the same reason, a relatively small difference in the chromatographic mobility was also seen after removal of the DMT group (Table 2). This assumption is supported by reports that short oligo(nucleoside phosphorothioate)s (trimers, tetramers and pentamers) were chromatographically separated into diastereomeric species but the efficiency of this process strongly depended upon the sequence of nucleobases and composition of the buffered eluent [38].
Physicochemical characterization of oligomers 12-21
Hybridization properties of THPO-chimeric DNA oligomers
Thermal stability of complexes formed by oligomers 12-19 with complementary single stranded DNA and RNA oligomers (THPO/DNA and THPO/RNA, respectively) and that of the duplex 20/21was determined by UV-monitored thermal melting measurements and the results (the Tm values) are given in Table 3. DNA/DNA and DNA/RNA duplexes isosequential with the investigated duplexes were used as the reference complexes.
Tm and ΔTm* values for complexes of chimeric oligomers 12-19 with complementary DNA and RNA strands and for the duplex 20/21, measured in 10 mm Tris-HCl, pH 7.4, 10 mm MgCl2, 100 mm NaCl.
| No. | Oligomer sequence 5′→3′ | Tma (°C) | ΔTm* (°C) | Tma (°C) | ΔTm* (°C) | ||
|---|---|---|---|---|---|---|---|
| THPO/DNA | DNA/DNA | THPO/RNA | DNA/RNA | ||||
| 12 | T9tT9 | 45.0 | 51.0 | -6.0 | 34.4 | 41.4 | -7.0 |
| 13 | T8tttT8 | 35.5 | 51.0 | -5.2 | 28.0 | 41.4 | -4.5 |
| 14 | T9aT9 | 45.9 | 50.4 | -4.5 | |||
| 15 | T8aaaT8 | 38.3 | 50.8 | -4.2 | |||
| 16 | T9gT9 | 45.6 | 52.8 | -7.2 | |||
| 17 | T8gggT8 | 44.3 | 56.8 | -4.2 | |||
| 18 | T9cT9 | 45.4 | 52.3 | -6.9 | |||
| 19 | T9ccT8 | 38.5 | 53.5 | -7.5 | |||
| 20/21 | 9.9b | 17.4 | -0.4c | ||||
The ΔTm* is a difference between the Tm found for THPO/DNA or THPO/RNA duplex and a corresponding reference DNA/DNA or DNA/RNA duplex, respectively, calculated per one THPO-modified unit. aThe measurement error ±1.0°C. bNo DNA oligomer added. cCalculated for the total of 20 (2×10) modified units.
The values of the Tm parameter for duplexes formed by chimeric oligomers 12–19 with DNA and RNA templates are lower than those of the reference non-modified duplexes (ΔTm*= -4.2 ÷ -7.2°C). This distinctly lower affinity of the investigated DNA analogs towards the complementary strands probably results from their higher flexibility in comparison to natural DNA molecules, which brings the loss of entropy [39, 40]. It has been shown that a rigid structure of DNA analog (like e.g. LNA) provides better affinity to a complementary RNA strand [41], although one has to keep in mind that the conformationally flexible peptide nucleic acid analogs (PNA) exhibit extremely high affinity toward their DNA and RNA complements [42].
Fully THPO-modified 10-mers 20 and 21were designed as complementary to each other. Interestingly, the thermal stability of a 20/21 duplex is by ca. 8°C lower than that of the reference non-modified DNA/DNA complex (Tm = 9.9°C vs. 17.4°C), although in terms of the decrease per a modified unit the loss of stability is, formally, rather small (ΔTm*= -7.5°C/20 = -0.4°C). This low stability of 20/21 may be attributed to the fact, that the THPO based acyclic nucleosides 10 are obtained on a non-stereoselective way as mixtures of two P-enantiomeric forms. This feature is transferred into the phosphoramidite monomers 11, thus the decamers 20 and 21 are synthesized as the mixtures of 210 diastereomers, while each diastereomer may exert different hybridization properties. Of course, the presence of 210 stereoisomers of 20 and of the same number of stereoisomers of 21 makes the whole hybridizing system extremely complicated. However, the assumption that P-diastereomeric inhomogeneity is a major factor decreasing the thermal stability of 20/21 is supported by the fact that similarly flexible GNA (glycol nucleic acids) obtained form enantiomerically pure acyclic glycol nucleosides forms quite stable double stranded structures, compared to the respective ds DNA duplexes [43].
Circular dichroism analysis of duplexes formed by oligomers 20 and 21
The helical structures of duplexes formed by 20 and 21 with complementary DNA templates, as well as the structure of the homo-THPO duplex 20/21, were examined by circular dichroism spectroscopy (CD) (Figure 4). The CD spectra for 20 and 21 alone indicate that these single-stranded oligomers do not adopt any ordered helical structure. To analyze the duplexes, the 1:1 equimolar mixtures of complementary oligomers (in 10 mm Tris-HCl, pH 7.4, 10 mm MgCl2, 100 mm NaCl buffer) were annealed by slow cooling from 95°C, down to the room temperature. The CD spectra recorded for 20/5′-d(ATAATTAAAT)-3′ and 21/5′-d(ATTTAATTAT)-3′ duplexes as well as for the 20/21 duplex exhibit positive Cotton effects at λ = 276 nm, and negative Cotton effects at λ = 253 nm. All three spectra are characteristic for the B-type DNA duplex helical structures, with the most ordered structure of the 20/21 duplex. Interestingly, in CD spectra of 20/21 and 21/5′-d(ATTTAATTAT)-3′ the two isoelliptic points are present at similar wavelengths (at 235 and 265 nm), while the CD spectrum of 20/5′-d(ATAATTAAAT)-3′ very much differs from those previously mentioned, especially at the short-wave region (220–250 nm, no isoelliptic point below 260 nm). This difference is quite surprising because at first glance 20 and 21 differ by only two nucleobases (4×Ade, 6×Thy vs. 6×Ade, 4×Thy).

The CD spectra of the duplexes: 20/21(circles), 20/5′-d(ATAATTAAAT)-3′ (triangles) and 21/5′-d(ATTTAATTAT)-3′ (crosses), as well as single stranded THPO-oligomers 20 and 21(solid line) (2 μm duplex in a 10 mm Tris-HCl, pH 7.4, 10 mm MgCl2, 100 mm NaCl buffer).
Stability of the oligomer 13 (5′-T8tttT8-3′) towards selected 5′- and 3′-exonucleases
Chimeric oligomer 13 (5′-T8tttT8-3′), possessing three THPO-thymine units (t) located in the central part of the homothymidine nonadecamer, was tested as a substrate for 3′- and 5′-exonucleases, i.e. for snake venom phosphodiesterase (svPDE or PDE I) and for calf spleen phosphodiesterase (csPDE, PDE II), respectively. The digestion products were identified by MALDI-TOF MS analysis (Figure 5). From the samples of 13 treated with PDE I (reaction A) or PDE II (reaction B) small aliquots were taken after 15, 45 and 60 min, or after 10, 30 and 90 min, respectively.

MALDI-TOF mass spectrometry analysis of the products of the enzymatic hydrolysis of oligomer 13 with snake venom phosphodiesterase (PDE I, panel A) and with calf spleen phosphodiesterase (PDE II, panel B).
The time of incubation is indicated above the consecutive plots.
Both sets of spectra show the ladders of products, which differ by m/z 304, corresponding to the products of consecutive removal of PT (with PDE I) or TP (with PDE II) from the parent oligonucleotide 13 (m/z 5732). After 45 min of the reaction A, accumulation of the oligonucleotide 5′-T8ttt-3′ (a band at m/z 3298) was observed. No further degradation of this product was observed over next 6 h (data not shown). After 60 min of the reaction B, accumulation of the oligonucleotide 5′-TtttT8-3′ (a band at m/z 3602) was observed, so the reaction stopped at the last natural nucleotide upstream of the modification site. Thus, the THPO modified unit inserted within the DNA chain is recognized by both 3′- and 5′-exonucleases tested. Consequently, the use of the THPO units at the 3′- and 5′-ends of therapeutic nucleic acids will make them resistant towards cellular exonucleases and will improve their pharmacokinetics. Similar abasic phosphinic acid ‘clamps’(BHPA, 3) were already successfully applied in in vitro experiments for protection of deoxyribozymes (directed towards the HIV-1 viral RNA sequence) in HIV-1 infected cells (unpublished results), and of deoxyribozymes designed to cleave bcr-abl mRNA fragments [44].
Conclusions
A series of novel DNA analogs was obtained by incorporation of the tris(hydroxymethyl) phosphine oxide (THPO) derived residues (acylic nucleoside analogs) into an oligonucleotide chain. Structural flexibility and loss of entropy make these chimeric oligomers unable to form thermally stable duplexes with their complementary RNA and DNA strands. However, the duplex formed by two fully modified complementary THPO-oligomers adopts a helical structure (by CD analysis) and its thermal stability is only slightly lower than that of its isosequential DNA duplex. Oligomers with the THPO units are stable toward two 3′- and 5′-exonucleases tested (hydrolysis stops at the modification site), and thus can be used to increase the stability of therapeutic oligonucleotides in body fluids.
Experimental
Synthesis of an O-DMT derivative of THPO (6)
To a solution of 10 g (0.07 mol) of tris(hydroxymethyl)phosphine oxide (5) in pyridine (100 mL) 4,4′-dimethoxytrityl chloride (DMT-Cl) (24 g, 0.07 mol) was added and the mixture was stirred for 20 h at room temperature. Pyridine was evaporated, the residue was (three times) dissolved in ethanol and evaporated to dryness. This step was repeated with toluene. The desired product 6 was obtained in 51% yield (16 g, 0.036 mol) by chromatographic separation on a silica gel column (CHCl3/MeOH, 100:0→97:3, v/v, and 0.1% vol. of Et3N). 1H NMR (200 MHz, CDCl3): δ 7.41–6.74 (m, 13H, aromatic protons), 4.19 (m, JP-H= 3 Hz, 4H, CH2OH), 3.74 (s, 3H, OCH3), 3.73 (s, 3H, OCH3), 3.64 (d, JP-H= 6 Hz, 2H, CH2ODMT); 31P NMR (CDCl3): δ 46.51; MS FAB (m/z): 544.3 [M+102 (Et3NH)]+.
Synthesis of an O-DMT-O′-TBDMS derivative of THPO (7A)
To a solution of 2 g (4.5 mmol) of 6 in acetonitrile (60 mL), imidazole (1.2 g, 18 mmol) was added, followed by TBDMS-Cl (0.68 g, 4.5 mmol), and the mixture was stirred for 20 h at room temperature. Then, triethylamine (5 mL) was added and the solvent was evaporated. The desired (DMT-oxymethyl)(tert-butyldimethylsilyloxymethyl)(hydroxymethyl)phosphine oxide 7A was obtained in 45% yield (1.1 g, 2.0 mmol) by chromatographic separation on a silica gel column (CHCl3/MeOH, 100:0→95:5, v/v). 1H NMR (200 MHz, CDCl3): δ 7.46–6.80 (m, 13H, aromatic protons), 4.14–4.07 (m, 4H, CH2OTBDMS), 3.78 (s, 3H, OCH3), 3.77 (s, 3H, OCH3), 3.62–3.53 (m, 2H, CH2ODMT), 0.81 (s, 9H, 3×CH3), 0.06 (s, 3H, Si-CH3), 0.02 (s, 3H, Si-CH3); 31P NMR (CDCl3): δ 44.55; MS FAB (m/z): 579.4 [M+Na]+, 709.3 [M+153]+.
Synthesis of an O-DMT-O′-Bz derivative of THPO (7B)
To a solution of 3.5 g (7.8 mmol) of 6 in pyridine (50 mL) benzoyl chloride (1.1 g, 7.8 mmol) was added and the mixture was stirred for 16 h at room temperature. Pyridine was evaporated, the residue was (three times) dissolved in ethanol and evaporated to dryness. This step was repeated with toluene. The desired (DMT-oxymethyl)(benzoyl-oxymethyl)(hydroxymethyl)phosphine oxide 7B was obtained in 49% yield (2.1 g, 3.9 mmol) by chromatographic separation on a silica gel column (CHCl3:MeOH, 100:0→95:5, v/v). 1H NMR (CDCl3): δ 8.12–6.73 (m, 18H, aromatic protons), 4.90–4.77 (m, 2H), 4.24 (s, 2H), 3.79–3.73 (m, J = 7 Hz, J = 3 Hz, 2H), 3.75 (s, 3H, OCH3), 3.74 (s, 3H, OCH3); 31P NMR (CDCl3): δ 42.40; FAB MS (m/z): 699.2 [M+153]+.
Synthesis of a O,O′-di-DMT derivative of THPO (7C)
To a solution of 1.4 g (10 mmol) of tris(hydroxymethyl)phosphine oxide (5) in pyridine (150 mL) 4,4′-dimethoxytrityl chloride (DMT-Cl) (6.8 g, 20 mmol) was added and the mixture was stirred for 20 h at room temperature. Pyridine was evaporated, the residue was (three times) dissolved in ethanol and evaporated to dryness. This step was repeated with toluene. The desired (bis-(DMT-oxymethyl))(hydroxymethyl)phosphine oxide 7C was obtained in 50% yield (3.11 g, 5 mmol) by chromatographic separation on a silica gel column (CHCl3/MeOH, 100:0→98:2, v/v). 1H NMR (200 MHz, CDCl3): δ 7.72–6.51 (m, 26H, aromatic protons), 4.02 (d, J = 4.7 Hz, 2H), 3.90–3.80 (m, 2H), 3.77 (s, 6H, 2×OCH3), 3.76 (s, 6H, 2×OCH3), 3.54 (dd, J = 5.5 Hz, J = 12.4 Hz, 2H); 31P NMR (200 MHz, CDCl3): δ 43.87; FAB MS (m/z): 767.3 [M+Na]+, 897.6 [M+153]+.
Synthesis of an (N2-isobutyryl-O4-diphenylocarbamoyl-guanine) derivative of 7A (8d)
A suspension of N2-isobutyryl-O6-diphenylocarbamoyl-guanine (163 mg, 0.39 mmol) in anhydrous THF was heated under reflux for 20 min and cooled to room temperature. Then 7A (166 mg, 0.3 mmol), triphenylphosphine (165 mg, 0.63 mmol) and DIAD (diisopropyl azodicarboxylate) (124 μL, 0.63 mmol) were added (argon atmosphere, daylight protected). After 48 h the solvent was evaporated. The desired ((N2-isobutyryl-O4-diphenylocarbamoyl-guanine)-9-methyl)(DMT-oxymethyl)(tert-butyldimethylsilyloxymethyl)phosphine oxide (8d) was obtained in 90% yield (257 mg, 0.26 mmol) by chromatographic separation on a silica gel column (CHCl3/MeOH, 100:0→95:5, v/v). 1H NMR (200 MHz, CDCl3): δ 8.21 (s, 1H, H-8), 7.81–6.78 (m, 23H, aromatic protons), 4.75 ppm (d, 2H, JP-H= 6.7 Hz, CH2N), 4.15 (dd, 2H, J = 2.5 Hz, J = 5.5 Hz, CH2Si), 3.75 (s, 3H, OCH3), 3.74 (s, 3H, OCH3), 3.57 (d, 2H, JP-H= 6.2 Hz, CH2ODMT), 2.95–2.81 (m, 1H, CH(CH3)2), 1.22 (d, 6H, J = 6.8 Hz, 2×CH3), 0.76 (s, 9H, 3×CH3), -0.03 (s, 3H, Si-CH3), -0.05 (s, 3H, Si-CH3); 31P NMR (CDCl3): δ 40.89; FAB MS (m/z): [M+H]+ 955.2, [M-H]- 953.2.
Synthesis of an N3-benzoyl-thymine derivative of 7A (8a)
To a solution of N3-benzoyl-thymine (110 mg, 0.48 mmol) in anhydrous THF, 7A (200 mg, 0.37 mmol), triphenylphosphine (204 mg, 0.77 mmol) and DIAD (152 μL, 0.77 mmol) were added (argon atmosphere, daylight protected). After 48 h the solvent was evaporated. The desired product [(N3-benzoyl-thymine)-1-methyl](DMT-oxymethyl)(tert-butyldimethylsilyl-oxymethyl)phosphine oxide (8a) was obtained in 92% yield (260 mg, 0.34 mmol) by chromatographic separation on a silica gel column (CHCl3/MeOH, 100:0→95:5, v/v).
1H NMR (200 MHz, CDCl3): δ 7.91–6.33 (m, 19H, aromatic protons, H6), 5.08–4.91 (m, 2H, CH2N), 4.05 (d, 2H, J = 5.4 Hz, CH2Si), 3.78 (s, 3H, OCH3), 3.77 (s, 3H, OCH3), 3.61 (d, 2H, J = 6.3 Hz, CH2ODMT), 1.93 (d, J = 1 Hz, CH3), 0.77 (s, 9H, 3×CH3), 0.01 (s, 3H, CH3), -0.02 (s, 3H, CH3); 31P NMR (CDCl3): δ 40.87; FAB MS (m/z): [M+154]+921.5, [M-H]‑ 767.5.
Synthesis of N6-benzoyl-adenine derivative of 7A (8c)
To a solution of N6-benzoyl-adenine (115 mg, 0.48 mmol) in anhydrous THF, 7A (200 mg, 0.37 mmol), triphenylphosphine (204 mg, 0.77 mmol) and DIAD (152 μL, 0.77 mmol) were added (argon atmosphere, daylight protected). After 48 h the solvent was evaporated. The desired product [(N6-benzoyl-adenine)-9-methyl](DMT-oxymethyl)(tert-butyldimethylsilyl-oxymethyl)phosphine oxide (8c) was obtained in 51% yield (148 mg, 0.19 mmol) by chromatographic separation on a silica gel column (CHCl3/MeOH, 100:0→95:5, v/v).
1H NMR (200 MHz, CDCl3): δ 8.93 (s, 1H, NH), 8.76 (s, 1H, H-2), 8.32 (s, 1H, H-8), 7.96–6.68 (m, 18H, aromatic protons), 4.82 (d, 2H, J = 6.5 Hz, CH2N), 4.08 (dd, J = 5 Hz, J = 14 Hz, 1H, CH2Si), 4.02 (dd, J = 6.5 Hz, J = 14 Hz, 1H, CH2Si), 3.78 (s, 3H, OCH3), 3.77 (s, 3H, OCH3), 3.60–3.59 (m, 2H, CH2ODMT), 0.80 ( s, 9H, 3×CH3), 0.01 (s, 3H, CH3), -0.02 (s, 3H, CH3); 31P NMR (CDCl3): δ 42.02; FAB MS (m/z): [M+H]+ 778.5, [M-H]- 776.4.
Synthesis of an N3-benzoylthymine derivative of 7B (8a′)
To a solution of N3-benzoyl-thymine (110 mg, 0.48 mmol) in anhydrous THF, 7B (202 mg, 0.37 mmol), triphenylphosphine (204 mg, 0.77 mmol) and DIAD (152 μL, 0.77 mmol) were added (argon atmosphere, daylight protected). After 48 h the solvent was evaporated. The desired product ((N3-benzoylthymine)-1-methyl)(DMT-oxymethyl)(tert-butyldimethylsilyl-oxymethyl)phosphine oxide (8a′) was obtained in 90% yield (253 mg, 0.33 mmol) by chromatographic separation on a silica gel column (CHCl3/MeOH, 100:0→95:5, v/v).
1H NMR (200 MHz, CDCl3): δ 7.93–6.73 (m, 24H, aromatic protons + H6); 4.88 (dd, J = 5 Hz, J = 14 Hz, 1H, CH′N), 4.75 (dd, J = 4 Hz, J = 14 Hz, 1H, CH″N), 4.58 (dd, J = 4 Hz, J = 16 Hz, 1H, CH″OBz), 4.13 (dd, J = 6 Hz, J = 16 Hz, 1H, CH′OBz), 3.82–3.71 (m, CH2ODMT), 3.75 (s, 3H, OCH3), 3.74 (s, 3H, OCH3), 1.90 (d, J = 1 Hz, CH3); 31P NMR (CDCl3): δ 38.52; FAB MS (m/z): [M-H]- 758.5, [M+153]+912.8.
Synthesis of an O,O′-di-DMT-O″-tosyl derivative of THPO (9): Conversion of 7C to 9
To a solution of 7C (400 mg, 0.53 mmol) and DMAP (410 mg, 3.36 mmol), in anhydrous methylene chloride (50 mL, cooled to 0°C) p-toluenesulfonic chloride (303 mg, 1.59 mmol) was added. The mixture was kept at 0–5°C for 2 h and then was diluted with dichloromethane (100 mL) and washed with saturated aqueous NaHCO3. The organic layer was dried with MgSO4 and evaporated to dryness. The desired [bis (DMT-oxymethyl)](p-toluenesulfonyloxymethyl)phosphine oxide 9 was obtained in 40% yield (193 mg, 0.21 mmol) by chromatographic separation on a silica gel column using CHCl3 as an eluent.
1H NMR (200 MHz, CDCl3): δ 7.69–6.66 (m, 20H, aromatic protons), 4.35 (d, J = 6 Hz, 2H, CH2OTs), 3.78 (s, 6H, 2×OCH3), 3.77 (s, 6H, 2×OCH3), 3.70–3.52 (m, 4H, CH2ODMT), 2.43 (s, 3H, CH3); 31P NMR (CDCl3): δ 39.87; FAB MS (m/z): [M-H]-898.1, [M+Na]+ 922.5.
Synthesis of an O,O′-di-DMT-cytosine derivative of THPO (8g)
To a solution of cytosine (101 mg, 0.91 mmol) in anhydrous DMF (30 mL) sodium hydride (22 mg, 0.91 mmol, 60% suspension in mineral oil) was added. The mixture was kept at room temperature for 20 min and then a solution of 9 (672 mg, 0.75 mmol) in DMF (10 mL) was added dropwise. The mixture was kept at 60°C for 3 h and the solvent was evaporated under reduced pressure. The desired (cytosine-1-methyl)[bis(DMT-oxymethyl)]phosphine oxide (8g) was obtained in 60% yield (376 mg, 0.45 mmol) by chromatographic separation on a silica gel column (CHCl3/MeOH, 100:0→90:10, v/v). 1H NMR (500 MHz, CDCl3): δ 7.32–6.66 (m, 27H, aromatic protons, H6), 5.59 (d, J = 7.0 Hz, 1H, H-5), 4.12 (d, J = 5.6 Hz, 2H, CH2N), 3.81 (dd, J = 8.5 Hz, J = 12.5 Hz, CH2ODMT), 3.73 (s, 6H, 3×OCH3), 3.72 (s, 6H, 3×OCH3), 3.52 (dd, J = 5.0 Hz, J = 12.5 Hz, 2H, CH2ODMT); 31P NMR (CDCl3): δ 41.92; 13C NMR (125 MHz, CDCl3): δ 165.8 (C4), 156.2 (C-2), 145.2 (C6), 95.1 (C5), 88.4 (d, JCP= 12 Hz, C-Ph), 58.6 (d, JCP= 83 Hz, CH2O), 44.3 (d, JCP= 67 Hz, CH2N), 55.1 (CH3O), 143.6–113.3 (36C, 6×Ph); FAB MS (m/z): [M+H]+ 838.2, [M-H]- 836.5.
Synthesis of an O,O′-bis-DMT-N4-benzoylcytosine derivative of THPO (8b)
To a solution of (cytosine-1-methyl)[bis(DMT-oxymethyl)]phosphine oxide (8g) (350 mg, 0.42 mmol) in anhydrous pyridine (20 mL, cooled to 0°C) benzoyl chloride (240 μL, 2.09 mmol) was added dropwise. The mixture was kept at room temperature for 16 h and then water (12 μL) and 28% aqueous NH4OH (12 μL) were added. After 0.5 h the solvent was evaporated under reduced pressure. The residue was diluted with dichloromethane (100 mL) and washed with saturated aqueous NaHCO3. The organic layer was dried with MgSO4 and evaporated to dryness. The desired [(N4-benzoylcytosine)-1-methyl][bis(DMT-oxymethyl)]phosphine oxide (8b) was obtained in 74% yield (289 mg, 0.31 mmol) by chromatographic separation on a silica gel column (CHCl3:MeOH, 100:0→90:10, v/v).
1H NMR (200 MHz, CDCl3): δ 8.62–6.73 ppm (m, 33H, aromatic protons, H6, H5); 4.40 (d, J = 7 Hz, 2H, CH2N), 3.8–3.7 (m, 2H, CH2ODMT), 3.74 (s, 6H, 2×OCH3), 3.73 (s, 6H, 2×OCH3), 3.60 (dd, J = 6.4 Hz, J = 12.8 Hz, 2H, CH2ODMT); 31P NMR (CDCl3): δ 41.15; FAB MS (m/z): [M+H]+942.2, [M-H]-940.3.
General procedure for removal of TBDMS protecting group in 8a,c,d,f: synthesis of 10a,c-f
To a solution of 8 (8a,c,d,f, 200 mg, ≈ 0.25 mmol) in anhydrous THF (3 mL), 1 m THF solution of tetra-n-butylammonium fluoride (311 μL, 0.31 mmol) was added. After 24 h the solvent was evaporated and the residue was dissolved in water and extracted with chloroform (3 times). The organic layer was dried with MgSO4 and evaporated to dryness. The products 10a,c-f were isolated by chromatography on a silica gel column (CHCl3/MeOH, 100:0→90:10, v/v).
[(N3-Benzoylthymine)-1-methyl](DMT-oxymethyl)(hydroxymethyl)phosphine oxide (10a)
Yield 39%; 1H NMR (200 MHz, CDCl3): δ 7.97–6.73 (m, 19H, aromatic protons, H6), 4.56 (dd, J = 15.5 Hz, J = 5.0 Hz, 1H), 4.11 (dd, J = 15.5 Hz, J = 6.0 Hz, 1H), 4.02–3.88 (m, 2H), 3.79 (s, 3H, OCH3), 3.78 (s, 3H, OCH3), 3.71–3.67 (m, 2H), 1.92 (d, 3H, J = 1.1 Hz, CH3); 31P NMR (CDCl3): δ 38.40; FAB MS (m/z): [M-H]- 653.5.
[(N6-Benzoyladenine)-9-methyl](DMT-oxymethyl)(hydroxymethyl)phosphine oxide (10c)
Yield 84%; 1H NMR (200 MHz, CDCl3): δ 8.75 (s, 1H, H-2), 8.16 (s, 1H, H-8), 8.04–6.80 (m, 18H, aromatic protons), 4.91 (dd, J = 3 Hz, J = 16 Hz, 1H, CH2N), 4.77 (dd, J = 8.5 Hz, J = 16 Hz, 1H, CH2N), 3.88 (d, J = 4.0 Hz, 2H, CH2OH), 3.78 (s, 3H, OCH3), 3.77 (s, 3H, OCH3), 3.75–3.65 (m, J = 5.0 Hz, J = 7.3 Hz, 2H, CH2ODMT); 31P NMR (CDCl3): δ 42.39; FAB MS (m/z): [M+H]+664.3, [M-H]- 662.0.
[(N2-Isobutyryl-O6-diphenylcarbamoylguanine]-9-methyl)(DMT-oxymethyl)(hydroxymethyl)phosphine oxide (10d)
Yield 51%; 1H NMR (200 MHz, CDCl3): δ 7.42 (s, 1H, H-8), 7.39-6.73 (m, 23H, aromatic protons), 4.83 (d, JP-H= 5 Hz, 2H, CH2N), 4.45 ppm (dd, J = 9 Hz, J = 16 Hz, 1H, CH′OH), 4.22 (dd, J = 5 Hz, J = 16 Hz, 1H,CH″ OH ), 3.79 (s, 3H, OCH3), 3.78 (s, 3H, OCH3), 3.40 (dd, JP-H= 7.2 Hz, J = 13 Hz, 1H, CH′ODMT), 3.25 (dd, JP-H= 4.1 Hz, J = 13 Hz, 1H, CH″ODMT), 2.90–2.28 (m, 1H, CH(CH3)2), 0.98 (d, J = 4.6 Hz, 6H, 2×CH3), 31P NMR (CDCl3): δ 36.91; FAB MS (m/z): [M+H]+ 841.5, [M-H]- 839.2.
(Thymine-1-methyl)(DMT-oxymethyl)(hydroxymethyl)phosphine oxide (10e)
Yield 60% (combined for two steps); 1HNMR (500 MHz, CDCl3): δ 7.42-6.14 (m, 14H, aromatic protons, H6), 4.34 (dd, 2H, J = 5 Hz, J = 15.0 Hz, CH2N), 4.10 (dd, J = 3.6 Hz, J = 9.5 Hz, 2H, CH2OH), 3.79 (s, 3H, OCH3), 3.78 (s, 3H, OCH3), 3.61 (dd, 2H, J = 6.0 Hz, J = 12.8 Hz, CH2ODMT), 1.79 (d, 3H, J = 1.1 Hz, CH3); 31P NMR (CDCl3): δ 45.46; 13C NMR (125 MHz, CDCl3): δ 166.6 (C2), 160.4, 154.3 (18, 3×Ph), 152.8 (C4), 143.0 (C6), 111.6 (C5), 89.9 (d, JCP = 11 Hz, C-Ph), 59.2 (d, JCP = 81 Hz, CH2O), 58.3 (d, JCP = 80 Hz, CH2O ), 55.8 (CH3O), 44.4 (d, JC-P = 66 Hz, CH2N), 12.3 (CH3); FAB MS (m/z): [M+H]+551.6, [M-H]- 549.0.
[(N2-Isobutyrylguanine)-9-methyl](DMT-oxymethyl)(hydroxymethyl)phosphine oxide (10f)
Yield 40%; 1H NMR (200 MHz, CDCl3): δ 7.76 (s, 1H, H-8); 7.37–6.08 (m, 13H, aromatic protons), 4.75–4.64 (d, 2H, J = 7.0 Hz, J = 16 Hz, CH2N), 4.08 (d, 2H, CH2OH), 3.74 (s, 3H, OCH3), 3.73 (s, 3H, OCH3), 3.59 (s, 2H, CH2ODMT), 2.94–2.81 (m, 1H, CH(CH3)2), 1.14 (d, 6H, J = 6.7 Hz, 2×CH3); 31P NMR (CDCl3): δ 43.64; FAB MS (m/z): [M+H]+ 646.4, [M-H]-644.3.
Removal of the benzoyl protecting group in 8a′: synthesis of 10e
To a solution of 8a′ (200 mg, 0.26 mmol) in methanol (10 mL), 28% aqueous NH4OH (10 mL) was added. After 24 h the solvent was evaporated and the residue was evaporated with ethanol and then with toluene (three times). The desired (thymine-1-methyl)(DMT-oxymethyl)(hydroxymethyl)phosphine oxide 10e was obtained in 41% yield (82 mg, 0.15 mmol) by chromatographic separation on a silica gel column (CHCl3/MeOH, 100:0→95:5, v/v).
Removal of the DMT protecting group in 8b: synthesis of 10b
To a solution of 8b (170 mg, 0.18 mmol) in dichloromethane (5 mL), 1 m methanolic solution of p-tolueneosulfonic acid (180 μL, 0.18 mmol) was added. The reaction progress was monitored by TLC. The mixture was quenched with pyridine (1 mL) and the volatile components were evaporated. The desired ((N4-benzoyl-cytosine)-1-methyl)(DMT-oxymethyl)(hydroxymethyl)phosphine oxide 10b was obtained in 60% yield (69 mg, 0.11 mmol) by chromatographic separation on a silica gel column (CHCl3/MeOH, 100:0→95:5, v/v). 1HNMR (200 MHz, CDCl3): δ 8.09–6.69 (m, 20H, aromatic protons, H6, H5); 4.85 (d, J = 15.2, 2H); 4.29 (dd, J = 9.8 Hz, J = 16 Hz, 2H); 3.74 (s, 3H, OCH3); 3.73 (s, 3H, OCH3); 3.23–3.09 (m, 2H); 31P NMR (CDCl3): δ 45.15; FAB MS (m/z): [M+H]+640.3, [M-H]- 638.2.
General procedure for phosphitylation of compounds 10: synthesis of 11b,c,e,f
To a solution of 10 (0.15 mmol) in a mixture of CH3CN (1 mL) and CH2Cl2 (1 mL), N,N-diisopropylamine (78 μL, 0.45 mmol) was added, followed by 2-cyanoethyl-N,N-diisopropyloaminochlorophosphite (35 μL, 0.18 mmol). The reaction progress was monitored by TLC. After ca. 1 h the volatile components were evaporated. The desired products were isolated by chromatographic separation on a silica gel column (CHCl3/MeOH, 100:0→98:2, v/v).
(Thymine-1-methyl)](DMT-oxymethyl)(2-cyanoethoxy-N,N-diisopropylaminophosphineoxymethyl)phosphine oxide (11e)
Yield 70%; 31P NMR (CDCl3): δ 150.95, 150.71, 150.64, 37.20, 37.02, 36.90, 36.72.
[(N4-Benzoylcytosine)-1-methyl](DMT-oxymethyl)(2-cyanoethoxy-N,N-diisopropylaminophosphineoxymethyl)phosphine oxide (11b)
Yield 45%; 31P NMR (CDCl3): δ 152.54; 152.34, 152.03; 39.76; 39.64; 39.46; 39.33.
[(N6-Benzoyladenine)-9-methyl](DMT-oxymethyl)(2-cyanoethoxy-N,N-diisopropylaminophosphine-oxymethyl)phosphine oxide (11c)
Yield 77%; 31P NMR (CDCl3): δ 151.88, 151.72, 151.57, 151.41, 38.10, 37.77.
[(N2-Isobutyrylguanine)-9-methyl](DMT-oxymethyl) (2-cyanoethoxy-N,N-diisopropylaminophosphineoxymethyl)phosphine oxide (11f)
Yield 41%; 31P NMR (CDCl3): δ 151.91, 151.59, 151.27, 39.54, 39.22, 38.99, 38.68.
Synthesis of oligomers 12-21
Chimeric THP-DNA oligomers 12-19 and homo-THPO oligomers 20 and 21 were prepared on an ABI 394 synthesizer (Applied Biosystems Inc., Foster City, CA, USA) at a 1 μmol scale. Acetonitrile solutions of phosphoramidites 11b, 11c, 11e and 11f, and of commercial thymidine, 2′-deoxycytidine, 2′-deoxyadenosine and 2′-deoxyguanosine phosphoramidites, at concentrations 0.08–0.12 m were used. For the monomers 11, a coupling time of 600 s was applied. The syntheses of oligomers 12-19 were performed on a thymidine succinyl-linked LCAA CPG solid support, and for oligomers 20 and 21, the LCAA-CPG support with a universal linker was used. The cleavage of oligomers 12-19 was done in 28% aqueous NH4OH over 16 h at 55°C. Fully modified oligomers 20 and 21were cleaved from the support with a 20% ethanolic solution of ammonia (55°C, 4 h), followed by the treatment with 28% aq. NH4OH/40% aq. MeNH2 (room temperature, 16 h). All oligomers were purified by a two-step RP-HPLC (DMT-on/DMT-off). The DMT-tagged oligomers (isolated during the first step) were detritylated with 50% aq. acetic acid and purified in the DMT-off form. RP-HPLC purification was performed on a C18 column (4.6×250 mm, ThermoQuest) with a linear gradient of a buffer A (0.1 m triethylammonium bicarbonate, pH 7.5) and a buffer B (40% acetonitrile in 0.1 m triethylammonium bicarbonate, pH 7.5); a gradient for DMT-on analysis: 15–100% B, for DMT-off: 0–100% B over 30 min, flow rate 1 mL/min.
Assays for enzymatic digestion of THPO-DNA oligomers 13
Digestion of the chimeric oligomer 13with snake venom and calf spleen phosphodiesterases (PDE I and PDE II, respectively) was performed as described earlier [8, 24]. Briefly, to a 1 μL aliquot of a 5 μm solution of oligonucleotide 13 diluted with water (3 μL), a solution of PDE I from Crotalus durissus (1 μL, EC 3.1.15.1, Boehringer Mannheim GmbH, Germany, 0.1 mU/μL) or PDE II from calf spleen (1 μL, EC 3.1.16.1, Sigma, St. Louis, MO, USA, 0.1 μg/μL) was added. The mixtures were incubated at 37°C. The 1 μL samples were withdrawn from the reaction mixture after 0, 5, 15 and 45 min (for PDE I) or after 0, 10, 30 and 60 min (for PDE II), and analyzed by MALDI-TOF MS.
MALDI-TOF mass spectrometry measurements
Samples (1 μL) withdrawn from the digestion reaction mixtures were loaded on a sample plate, mixed with the matrix solution [1 μL of an 8/1 (v/v) mixture of 2,4,6-trihydroxyacetophenone (10 μg/mL in ethanol) and diammonium citrate (50 μg/mL in water)] and left for crystallization. MALDI-TOF spectra were recorded on a Voyager-Elite instrument (PerSeptive Biosystems, CT, USA) in a reflector mode, at a resolution of 2000. The m/z negative ion peaks are shown in the spectra.
Melting temperature (Tm) measurements
The Tm measurements were performed on a Cintra 40 instrument (GBC Australia). The samples (2 μm concentration of duplexes) were prepared by hybridization of modified oligomers (in a 10 mm Tris-HCl, pH 7.4, 10 mm MgCl2, 100 mm NaCl buffer) with the complementary single stranded DNA or RNA as listed in Table 3. Before melting, the duplexes were annealed from 95°C to 5°C at a temperature gradient of 0.5°C/min. and kept at 5°C for 5 min. Melting was performed up to 86°C at a temperature gradient of 0.2°C/min. The melting temperatures Tm were calculated using the first order derivative method.
Circular dichroism measurements
Samples of the duplexes 20/21, 20/5′-d(ATAATTAAAT)-3′ and 21/5′-d(ATTTAATTAT)-3′, as well as single stranded THPO-oligomers 20 and 21were prepared at 2 μm concentration in a 10 mm Tris-HCl, pH 7.4, 10 mm MgCl2, 100 mm NaCl buffer. The spectra were recorded on a Jobin Yvon CD6 dichrograf. Measurements were made at room temperature using 0.5 cm path-length quartz cuvettes of 1 mL capacity, 2 nm bandwidth and 1-2 s integration time. Each spectrum was smoothed with a 9- or 15-point algorithm (included in the manufacturer’s software, version 2.2.1) after averaging of at least three scans.
Acknowledgments
Financial support from the Statutory Funds of Centre of Molecular and Macromolecular Studies of the Polish Academy of Sciences is gratefully acknowledged. The authors thank Dr. Piotr Guga for critical reading of the manuscript and fruitful discussion.
Dedication: We dedicate this paper to the memory of Dr. Kyo Watanabe, whom we sorely miss. We will keep him in our hearts as a respected scientist, a unique person, a great colleague, and a very special friend of Poland and the Polish people.
References
[1] Eckstein, F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 2014, 24, 374–387.10.1089/nat.2014.0506Suche in Google Scholar
[2] Nagarajan, R.; Kwon, K.; Nawrot, B.; Stec, W. J.; Stivers, J. T. Catalytic phosphoryl interactions of topoisomerase IB. Biochemistry. 2005, 44, 11476–11485.10.1021/bi050796kSuche in Google Scholar
[3] Yang, X.; Sierant, M.; Janicka, M.; Peczek, L.; Martinez, C.; Hassell, T.; Li, N.; Li, X.;Wang, T.; Nawrot, B. Gene silencing activity of siRNA molecules containing phosphorodithioate substitutions. ACS Chem. Biol. 2012, 7, 1214–1220.10.1021/cb300078eSuche in Google Scholar
[4] Miller, P. S.; Cassidy, R. A.; Hamma, T.; Kondo, N. S. Studies on anti-human immunodeficiency virus oligonucleotides that have alternating methylphosphonate/phosphodiester linkages. Pharmacol. Ther.2000, 85, 159–163.10.1016/S0163-7258(99)00054-6Suche in Google Scholar
[5] Hall, A. H.; Wan, J.; Spesock, A.; Sergueeva, Z.; Shaw, B. R.; Alexander, K. A. High potency silencing by single-stranded boranophosphate siRNA. Nucleic Acids Res. 2006, 34, 2773–2781.10.1093/nar/gkl339Suche in Google Scholar
[6] Krishna, H.; Caruthers, M. H. Solid-phase synthesis, thermal denaturation studies, nuclease resistance, and cellular uptake of oligodeoxyribonucleoside)methylborane phosphine-DNA chimeras. J. Am. Chem. Soc. 2011, 133, 9844–9854.10.1021/ja201314qSuche in Google Scholar
[7] Gryaznov, S. M. Oligonucleotide n3′→p5′ phosphoramidates and thio-phoshoramidates as potential therapeutic agents. Chem. Biodivers. 2010, 7, 477–493.10.1002/cbdv.200900187Suche in Google Scholar
[8] Nawrot, B.; Boczkowska, M.; Wójcik, M.; Sochacki, M.; Kazmierski, S.; Stec, W. J. Novel internucleotide 3′-NH-P(CH3)(O)-O-5′ linkage. Oligo(deoxyribonucleoside methanephosphonamidates); synthesis, structure and hybridization properties. Nucleic Acids Res. 1998, 26, 2650–2658.10.1093/nar/26.11.2650Suche in Google Scholar
[9] Sipa, K.; Sochacka, E.; Kazmierczak-Baranska, J.; Maszewska, M.; Janicka, M.; Nowak G.; Nawrot, B. Effect of base modifications on structure, thermodynamic stability, and gene silencing activity of short interfering RNA. RNA2007, 13, 1301–1316.10.1261/rna.538907Suche in Google Scholar
[10] Nawrot, B.; Widera, K.; Wojcik, M.; Rebowska, B.; Nowak, G.; Stec, W. J. Mapping of the functional phosphate groups in the catalytic core of deoxyribozyme 10-23. FEBS J. 2007, 274, 1062–1072.10.1111/j.1742-4658.2007.05655.xSuche in Google Scholar
[11] Kraynack, B. A.; Baker, B. F. Small interfering RNAs containing full 2′-O-methylribonucleotide-modified sense strands display Argonaute2/eIF2C2-dependent activity. RNA. 2006, 12, 163–176.10.1261/rna.2150806Suche in Google Scholar
[12] Jepsen, J. S.; Wengel, J. LNA-antisense rivals siRNA for gene silencing. Curr. Opin. Drug Discov. Dev. 2004, 7, 188–194.Suche in Google Scholar
[13] Ferrari, N.; Bergeron, D.; Tedeschi, A. L.; Mangos, M. M.; Paquet, L.; Renzi, P. M.; Damha, M. J. Characterization of antisense oligonucleotides comprising 2′-deoxy-2′-fluoro-beta-D-arabinonucleic acid (FANA): specificity, potency, and duration of activity. Ann. N.Y. Acad. Sci.2006, 1082, 91–102.10.1196/annals.1348.032Suche in Google Scholar
[14] Pallan, P. S.; Greene, E. M.; Jicman, P. A.; Pandey, R. K.; Manoharan, M.; Rozners, E.; Egli, M. Unexpected origins of the enhanced pairing affinity of 2′-fluoro-modified RNA. Nucleic Acids Res. 2011, 39, 3482–3495.10.1093/nar/gkq1270Suche in Google Scholar
[15] Sierant, M.; Sobczak, M.; Janicka, M.; Paduszynska, A.; Piotrzkowska, D. Biological and physicochemical characterization of siRNAs modified with 2′,2′-difluoro-2′-deoxycytidine (gemcitabine). New J. Chem. 2010, 34, 918–924.10.1039/b9nj00746fSuche in Google Scholar
[16] Dirin, M.; Winkler, J. Influence of diverse chemical modifications on the ADME characteristics and toxicology of antisense oligonucleotides. Expert Opin. Biol. Ther. 2013, 13, 875–888.10.1517/14712598.2013.774366Suche in Google Scholar
[17] Engels, J. W. Gene silencing by chemically modified siRNAs. Nat. Biotechnol. 2013, 30, 302–307.10.1016/j.nbt.2012.07.002Suche in Google Scholar
[18] Nawrot, B.; Sipa, K. Chemical and structural diversity of siRNA molecules. Curr. Top. Med. Chem. 2006, 6, 913–925.10.2174/156802606777303658Suche in Google Scholar
[19] Valenzuela, R. A.; Suter, S. R.; Ball-Jones, A. A.; Ibarra-Soza, J. M.; Zheng, Y.; Beal, P. A. Base modification strategies to modulate immune stimulation by an siRNA. Chembiochem. 2015, 16, 262–267.10.1002/cbic.201402551Suche in Google Scholar
[20] Wu, S. Y.; Yang, X.; Gharpure, K. M.; Hatakeyama, H.; Egli, M.; McGuire, M. H.; Nagaraja, A. S.; Miyake, T. M.; Rupaimoole, R.; Pecot, C. V.; et al. 2′-OMe-phosphorodithioate-modified siRNAs show increased loading into the RISC complex and enhanced anti-tumour activity. Nat. Commun. 2014, 5, 3459–3488.10.1038/ncomms4459Suche in Google Scholar
[21] Rowley, P. A.; Kachroo, A. H.; Ma, C. H.; Maciaszek, A. D.; Guga, P.; Jayaram, M. Stereospecific suppression of active site mutants by methylphosphonate substituted substrates reveals the stereochemical course of site-specific DNA recombination. Nucleic Acids Res. 2015,43, 6023–6037.10.1093/nar/gkv513Suche in Google Scholar
[22] Guga, P.; Koziołkiewicz, M. Phosphorothioate nucleotides and oligonucleotides – recent progress in synthesis and application. Chem. Biodivers. 2011, 8, 1642–1681.10.1002/cbdv.201100130Suche in Google Scholar
[23] Nawrot, B.; Michalak, O.; Nowak, M.; Okruszek, A.; Dera, M.; Stec, W. J. Bis(hydroxymethyl)phosphinic acid analogues of acyclic nucleosides; synthesis and incorporation into short DNA oligomers. Tetrahedron Lett.2002, 43, 5397–5400.10.1016/S0040-4039(02)01067-5Suche in Google Scholar
[24] Nawrot, B.; Michalak, O.; Janicka, M.; Maszewska, M.; Wojcik, M.; Nowak, G.; Mikolajczyk, B.; Stec, W. J. Novel nucleic acid analogs with a chimeric phosphinate /phosphate backbone; synthesis and biophysical properties. Arkivoc2004, (iii) 151–175.10.3998/ark.5550190.0005.314Suche in Google Scholar
[25] Nawrot, B.; Michalak, O.; De Clercq, E.; Stec, W. J. Analogues of acyclic nucleosides derived from tris-(hydroxymethyl)phosphine oxide or bis-(hydroxymethyl)phosphinic acid coupled to DNA nucleobases. Antivir. Chem. Chemother.2004, 15, 319–328.10.1177/095632020401500604Suche in Google Scholar
[26] Dembinski, R. Recent advances in the Mitsunobu reaction: modified reagents and the quest for chromatography-free separation. Eur. J. Org. Chem. 2004, 13, 2763–2772.10.1002/ejoc.200400003Suche in Google Scholar
[27] Cruickshank, K. A.; Jiricny, J.; Reese, C. B. The benzoylation of uracil and thymine. Tetrahedron Lett. 1984, 25, 681–684.10.1016/S0040-4039(00)99971-4Suche in Google Scholar
[28] Thomson, S. A.; Josey, J. A.; Cadilla, R.; Gaul, M. D.; Hassman, C. F.; Luzzio, M. J.; Pipe, A. J.; Reed, K.L.; Wiethe, R. W.; Noble, S. A. Fmoc mediated synthesis of peptide nucleic acids. Tetrahedron1995, 51,6179–6194.10.1016/0040-4020(95)00286-HSuche in Google Scholar
[29] Will, D. W.; Breipohl, G.; Langner,D.; Knolle, J.; Uhlmann, E. The synthesis of polyamide nucleic acids using a novel monomethoxytrityl protecting-group strategy. Tetrahedron1995, 51, 12069–12082.10.1016/0040-4020(95)00766-2Suche in Google Scholar
[30] Jenny, T. F.; Schneider, K. C.; Benner, S. A. N2-Isobutyryl-O6- [2-(p-Nitrophenyl)Ethyl]Guanine: A New Building Block for the Efficient Synthesis of Carbocyclic Guanosine Analogs. Nucleosides Nucleotides1992, 11, 1257–1261.10.1080/07328319208018340Suche in Google Scholar
[31] Robins, M. J.; Zou, R.; Guo, Z.; Wnuk, S. F. Nucleic acid related compounds. 93. A solution for the historic problem of regioselective sugar−base coupling to produce 9-glycosylguanines or 7-glycosylguanines . J. Org. Chem. 1996, 61, 9207–9212.10.1021/jo9617023Suche in Google Scholar
[32] Caruthers, M. H. Gene synthesis machines: DNA chemistry and its uses. Science1985, 230, 281–285.10.1126/science.3863253Suche in Google Scholar PubMed
[33] Scott, S.; Hardy, P.; Sheppard, R. C.; McLean, M. J. A universal support for oligonucleotide synthesis. In Innovations and Perspectives in Solid Phase Synthesis, 3rd International Symposium, 1994; Epton R., Ed. Mayflower Worldwide, 1994; pp 115–124.Suche in Google Scholar
[34] Azhayev, A. V.; Antopolsky, M. L. Amide group assisted 3′-dephosphorylation of oligonucleotides synthesized on universal A-supports. Tetrahedron2001, 57, 4977–4986.10.1016/S0040-4020(01)00409-4Suche in Google Scholar
[35] Glen Research universal support description and usage datasheet http://www.glenresearch.com/GlenReports/GR20-110.htmlSuche in Google Scholar
[36] Zon, G.; Stec, W. J. In Oligonucleotides and Analogues. A Practical Approach. Eckstein, F., Ed. IRL Press Oxford University Press: Oxford, 1991; pp 87–100.10.1093/oso/9780199632800.003.0004Suche in Google Scholar
[37] Koziołkiewicz, M.; Uznański, B.; Stec, W. J.; Zon, G. P-chiral analogues of oligodeoxyribonucleotides: synthesis, stereochemistry and enzyme studies. Chemica Scripta1986, 26, 251–260.Suche in Google Scholar
[38] Murakami, A.; Tamura, Y.; Wada, H.; Makino, K. Separation and characterization of diastereoisomers of antisense oligodeoxyribonucleoside phosphorothioates. Analytical Biochem. 1994, 233, 285–290.10.1006/abio.1994.1586Suche in Google Scholar
[39] Habus, I.; Agrawal, S. Oligonucleotides containing acyclic nucleoside analogues with carbamate internucleoside linkages. Nucleosides Nucleotides1995, 14, 1853–1859.10.1080/15257779508010708Suche in Google Scholar
[40] Schneider, K. Ch.; Benner, S.A. Oligonucleotides containing flexible nucleoside analogs. J. Am. Chem. Soc. 1990, 112, 453–455.10.1021/ja00157a073Suche in Google Scholar
[41] Koshkin, A. A.; Singh, S. K.; Nielsen, P.; Rajwanshi, V. K.; Kumar, R.; Meldgaard, M.; Olsen, C. E.; Wengel, J. LNA (Locked Nucleic Acid): synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition. Tetrahedron1998, 54, 3607–3630.10.1016/S0040-4020(98)00094-5Suche in Google Scholar
[42] Demidov, V. V.; Yavnilovich, M. V.; Belotserkovskii, B. P.; Frank-Kamenetskii, M. D.; Nielsen, P. E. Kinetics and mechanism of polyamide (“peptide”) nucleic acid binding to duplex DNA. Proc. Natl. Acad. Sci. USA1995, 92, 2637–2641.10.1073/pnas.92.7.2637Suche in Google Scholar
[43] Zhang, L.; Peritz, A.; Meggers, E. A simple glycol nucleic acid. J. Am. Chem. Soc. 2005, 127, 4174–4175.10.1021/ja042564zSuche in Google Scholar
[44] Warashina, M.; Nawrot, B.; Obika, S.; Wozniak, L. A.; Kuwabara, T.; Imanishi, T.; Stec, W. J.; Taira, K. Effect of Modifications on the Intracellular Activity of a DNA Enzyme. In Synthetic Nucleic Acids as Inhibitors of Gene Expression: Mechanisms, Applications and Therapeutic Implications, ed. Khachigian, L., CRC Press, Hauppauge, NY, 2004; pp. 95–113.10.1201/9781420037890.ch6Suche in Google Scholar
©2015 by De Gruyter
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Artikel in diesem Heft
- Frontmatter
- Guest Editorial
- Dedication to Kyoichi A. Watanabe
- Review
- From ribavirin to NAD analogues and back to ribavirin in search for anticancer agents
- Preliminary Communications
- 5′-Norcarbocyclic analogues of furano[2,3-d]pyrimidine nucleosides
- Fluorescent 1,2,3-triazole derivative of 3′-deoxy-3-azidothymidine: synthesis and absorption/emission spectra
- Research Articles
- Synthesis and characterization of N-glucosylated dithiadiazepine derivatives through carbon-sulfur bond formation
- Synthesis of 8-alkoxy-1,3-dimethyl-2, 6-dioxopurin-7-yl-substituted acetohydrazides and butanehydrazides as analgesic and anti-inflammatory agents
- 13C NMR spectroscopy of heterocycles: 3,5-diaryl-4-bromoisoxazoles
- Synthesis and anti-proliferative activity of pyridine O-galactosides and 4-fluorobenzoyl analogues
- Optimized synthesis of 3′-O-aminothymidine and evaluation of its oxime derivative as an anti-HIV agent
- Synthesis and antimicrobial properties of 5,5′-modified 2′,5′-dideoxyuridines
- Acyclic analogs of nucleosides based on tris(hydroxymethyl)phosphine oxide: synthesis and incorporation into short DNA oligomers
- Synthesis and antiviral evaluation of 2′,3′-dideoxy-2′,3′-difluoro-D-arabinofuranosyl 2,6-disubstituted purine nucleosides
Artikel in diesem Heft
- Frontmatter
- Guest Editorial
- Dedication to Kyoichi A. Watanabe
- Review
- From ribavirin to NAD analogues and back to ribavirin in search for anticancer agents
- Preliminary Communications
- 5′-Norcarbocyclic analogues of furano[2,3-d]pyrimidine nucleosides
- Fluorescent 1,2,3-triazole derivative of 3′-deoxy-3-azidothymidine: synthesis and absorption/emission spectra
- Research Articles
- Synthesis and characterization of N-glucosylated dithiadiazepine derivatives through carbon-sulfur bond formation
- Synthesis of 8-alkoxy-1,3-dimethyl-2, 6-dioxopurin-7-yl-substituted acetohydrazides and butanehydrazides as analgesic and anti-inflammatory agents
- 13C NMR spectroscopy of heterocycles: 3,5-diaryl-4-bromoisoxazoles
- Synthesis and anti-proliferative activity of pyridine O-galactosides and 4-fluorobenzoyl analogues
- Optimized synthesis of 3′-O-aminothymidine and evaluation of its oxime derivative as an anti-HIV agent
- Synthesis and antimicrobial properties of 5,5′-modified 2′,5′-dideoxyuridines
- Acyclic analogs of nucleosides based on tris(hydroxymethyl)phosphine oxide: synthesis and incorporation into short DNA oligomers
- Synthesis and antiviral evaluation of 2′,3′-dideoxy-2′,3′-difluoro-D-arabinofuranosyl 2,6-disubstituted purine nucleosides