Synthesis and characterization of (4-arm-star-PMMA)/PMMA-g-SiO2 hybrid nanocomposites
-
Do Quang Tham


Abstract
This study provides a route to prepare 4-arm star poly(methyl methacrylate) (4sPMMA)/PMMA grafted SiO2 (PMMA-g-SiO2) hybrid nanocomposites that can be used as 3D printing material and filler for dental materials. First, 4sPMMA was synthesized via atom transfer radical polymerization with low metal catalyst concentration. Modified colloidal silica nanoparticles (MCSPs) were synthesized by grafting 3-methoxypropyl trimethoxysilane (MPS) onto the surface of colloidal silica nanoparticles (CSPs) and then dispersed in the solution of methyl methacrylate monomer in dioxane. The mixture of 4sPMMA and MCSPs solutions was degassed and replaced in an oil bath at 70–75°C; the reaction was continued with α,α′-azobis(isobutyronitrile) as an initiator for 24 h to form 4sPMMA/PMMA-g-SiO2 hybrid nanocomposites. Viscosity measurement showed that viscosity of the hybrid was increased with increasing MPS loading used in modification of CSPs, which verified that PMMA had been grafted onto MCSPs. Fourier transform infrared spectra of the hybrid nanocomposites demonstrated the strong molecular interaction between MCSPs and polymer matrix, and 1H NMR spectra confirmed the formation of PMMA-g-SiO2. Field emission scanning electron microscopy and transmission electron microscopy images revealed that MCSPs were well dispersed in polymer matrix with the size of about 20–30 nm. Thermal stability of the hybrid nanocomposites was improved compared with PMMA made from free radical polymerization.
1. Introduction
Poly(methyl methacrylate) (PMMA) is widely used in various applications such as optical, biomedical, denture and prosthetic materials due to its many advantageous properties [1], [2], [3]. However, for every certain application, PMMA has some drawbacks such as poor toughness, low thermal stability, lack of bioactivity and high polymerization shrinkage [3], [4]. There are now numerous studies that focused on the use of nanofillers to overcome these drawbacks of PMMA [5], [6], [7], [8], [9], [10], [11], [12], [13], [14]. Some studies have shown that high nanofiller content can lead to intensive agglomeration and subsequently decrease some mechanical properties of PMMA [13], [14]. In order to get enhanced interaction between PMMA matrix and the nanofillers, much attention has been paid to the chemical modification of nanofillers either with trialkoxysilane coupling agents or by grafting polymer chains to the surface of nanofillers [8], [9], [10], [11]. Among the various nanofillers, silica nanoparticles (SNPs) have been used more often to enhance the physical, mechanical and thermal stabilities of PMMA because of their attractive features such as low toxicity, large specific surface area, high mechanical properties and easy functionalization [15], [16]. SNPs are also known to improve bioactivity and biocompatibility of PMMA applied in denture and prosthetic materials [17], [18], [19], [20]. In the studies mentioned above, SNPs in powder forms are the most frequently used; this might be because of easy functionalization and simple purification process. It is well known that SNPs are prone to agglomeration that is irreversible; redispersion is difficult to achieve the primary nanoscale of SNPs [21]. However, conventional nanoscale dispersion (below 100 nm) of SNPs in polymer matrix with lower agglomeration can be also obtained [14], [18].
Recently, colloidal silica nanoparticles (CSPs) have attracted great attention because of their wide range of applications [22]. The most facile method to produce CSPs is Stöber method, which allows controlled synthesis of spherical, monodispersed nanosilica [15], [22], [23]. When modified by silane coupling agents, CSPs can be easily stabilized in organic solvents or monomers to prevent agglomeration. For this reason, many synthetic approaches have used unmodified or modified CSPs for preparing polymer/silica hybrid nanocomposites, as well as PMMA hybrid composites in particular [5], [6], [7], [24], [25], [26], [27], [28], [29], [30], [31]. Among these approaches, free-radical in situ polymerization exhibits some advantages, including easy handling, relatively quick process and ability to obtain high performance products [6].
Therefore, in this study, CSPs were synthesized via Stöber method, and the modification process was carried out in the same solution by adding 3-(methacryloxy)propyltrimethoxysilane (MPS). In order to maintain nano size distribution of CSPs, solvent exchange and dialysis processes were applied in the purification. After methyl methacrylate (MMA) was added to unmodified or modified colloidal silica solutions, the in situ reaction of PMMA hybrid was performed in the presence of 4-arm-star PMMA prepolymer(4sPMMA) which had been prepared by atom transfer radical polymerization (ATRP). ATRP was performed with low concentration of catalysts, the removal of which can be done by using a silica gel chromatography column. Experiments were carried out in a green manner in the Advanced Nano Material Laboratory, Department of Polymer Science and Engineering at the Pusan National University, Korea. The Lab was equipped with standard instruments for minimum waste to air and water source. All organic and inorganic wastes after experiments were collected, classified, reused as much as possible and finally treated in standard routes according to Korean waste management law.
Fourier transform infrared (FTIR), proton nuclear magnetic resonance (1H NMR), thermal gravimetric analysis (TGA), scanning/transmission electron microscopies (SEM/TEM) and viscosity measurement were employed to characterize the hybrid nanocomposites, and the results are discussed.
2. Materials and methods
2.1. Materials
MMA (99%, Sigma-Aldrich, Milwaukee, USA) and 3-methacryloxy-propyltrimethoxy-silane (MPS, 98%, Tokyo Kasei, Kogyo Co., Tokyo, Japan) were passed through a column filled with basic alumina to remove the inhibitor. Dioxane (1,4-dioxane, 99%, Junsei Chemical, Tokyo, Japan) was distilled with calcium hydride and then stored with drying agent in a bottle. Methanol (MeOH, 99.9%, Carlo Erba Reactifs, Val de Reuil, France), tetraethyl orthosilicate (TEOS, 95%, Kasei), ammonia solution (28%, Junsei), diethyl ether (99%, Dae-Jung, Shiheung, South Korea) and α,α′-azobis(isobutyronitrile) (AIBN, 98%, Junsei) were used as received without further purification. Pentaerythritol (99%), 2-bromoisobutyryl bromide (99%), triethylamine (99%, Junsei), chloroform, CuBr2, Tris[2-(dimethylamino)-ethyl] amine (Me6TREN, 99%), tin(II) 2-ethylhexanoate (Sn(EH)2, 95%), anhydrous tetrahydrofuran (>99.8%) and other compounds were purchased from Sigma-Aldrich (USA) or Dae-Jung company (Korea) and used without further purification.
2.2. General synthesis procedures
2.2.1 Synthesis of colloidal silica nanoparticles
TEOS (40 g, 0.182 mol) and methanol (75 g) were placed in a three-neck flask equipped with magnetic bar. The flask was placed in an oil bath over a magnetic stirrer with controlled temperature of 50±2°C. Water (12.6 g) and 0.83 g of ammonia solution (28%) were then added to the mixture, and the reaction was allowed to continue for 24 h. Colloidal silica solution was obtained and stored in a bottle at room temperature. Dynamic light scattering analysis showed average particle size of 30 nm with narrow distribution.
2.2.2 Modification of colloidal silica
Two grams of CSPs (in about 20 g of colloidal solution) was dispersed in 20 ml dioxane in a two-necked flask equipped with magnetic bar. Some amount of MPS was dissolved in 2 ml MeOH in a vial, and the mixture was injected slowly into the two-necked flask. The reaction was allowed for 24 h at room temperature. Ammonia, methanol and ethanol were removed by using rotary evaporator; dioxane was added to obtain a total of 20 g of final solution. Unreacted MPS or hydrolyzed MPS was removed by using dialysis membrane with 14,000 Da cut-off molecular weight against dioxane solvent for 48 h with periodic bath changes. When colloidal silica was modified with different MPS contents of 0.3, 0.45, 0.6, 1 and 1.5 g per 1 g of SiO2, the modified colloidal silica samples were coded as MCSP1, MCSP2, MCSP3, MCSP4 and MCSP5, respectively. Dynamic light scattering analysis showed average particle size of approximately 30 nm with narrow distribution. Unmodified colloidal silica sample was coded as CSP0.
2.2.3 Synthesis of pentaerythritoltetrakis[2-bromoisobutyrate] (4PT- Br)
To a 500 ml round-bottom flask, pentaerythritol (15 g, 11.0 mmol), triethylamine (100 ml, 82.0 mmol) and chloroform (100 ml) were added and stirred for 15 min. The flask was continuously purged with nitrogen gas and cooled down to 0°C. Then, 2-bromoisobutyryl bromide (10 ml, 0.12 mmol) was slowly injected via a purged syringe for 60 min. The solution was stirred and allowed to react for 24 h at room temperature. After the reaction was completed, the solution of the product was washed with aqueous 5% NaOH solution, 1% HCl solution and deionized water, followed by extraction with ethyl acetate. The organic layer was collected and dried with MgSO4. After filtration and evaporation of the solvent, the product of pentaerythritoltetrakis (2-bromoisobutylrate) was obtained from recrystallization in ethyl ether as white powder, which was then dried in a vacuum oven. 1H NMR (in CDCl3) δ: 4.316 ppm (s, 8H, C-CH2O), 1.93 ppm (s, 24H, Br-C-CH3).
2.2.4 Synthesis of 4sPMMA by ATRP
1,4-Dioxane (25.6 ml), MMA (25.6 ml, 237.3 mmol), CuBr2 (15.2 μmol), Me6TREN (152 μmol), Sn(EH)2 (152 μmol) and 4PT-Br (434.3 mg, 593 μmol) were placed in a 250 ml Schlenk flask with magnetic stir bar. The flask was degassed by three freeze-pump-thaw cycles, stirred to get a uniform dispersion, backfilled with N2 and sealed. The flask was immerged in oil bath with magnetic stirring at 70°C. ATRP was terminated after 24 h by opening the flask and exposing the catalyst to air. The mixture was diluted with dioxane and passed through a column filled with a neutral silica gel layer to remove the copper complex. The polymer was recovered by precipitation from methanol. The polymer product was dried under vacuum to a constant weight and its molecular weight was analyzed by gel permeation chromatography.
2.2.5 Synthesis of 4sPMMA/PMMA-g-SiO2 hybrid nanocomposites
In a 100 ml flask, 3.9 g of modified colloidal silica solutions containing 0.39 g modified silica (MCSP1, 2, 3, 4, 5) were mixed with MMA (0.91 g) and AIBN (0.15 wt.% compared with MMA weight). In another 50 ml flask with magnetic bar, 3.03 g of 4sPMMA (Mn~30k) was dissolved completely in 20 ml dioxane for 1 h at 50°C and cooled to room temperature for 2 h. These two solutions were mixed together, degassed 3 times, purged with N2 gas and placed in a preheated oil bath at 70°C for 24 h. The hybrid nanocomposites solutions were precipitated in methanol, collected and washed three times with methanol and dried at 50°C in a vacuum oven for 48 h. By using CSP0 and different MCSPs, the different hybrid nanocomposites were obtained and coded as HybM0, HybM1, HybM2, HybM3, HybM4 and HybM5.
2.3 Characterizations
FTIR spectra of samples were recorded on an Agilent Technologies Cary 600 series FTIR spectrometer at room temperature, in the range of 4000–400 cm−1 with a resolution of 4 cm−1 and average of 32 scans, using KBr pellet method. Before FTIR sampling, the colloidal silica or modified colloidal silica solutions were dried to obtain powder samples; these samples were washed two times with methanol and diethyl ether and then dried again in order to ensure the complete removal of unreacted MPS. TGA was performed at heating rate of 10°C/min, from 30 to 800°C with nitrogen gas flow of 40 ml/min by using TGA Q50 V20.13 Build-39 instrument. Viscosity of hybrid nanocompositess solutions before and after reaction was measured by using a Brookfield instrument with rotor speed of 100 rpm at controlled temperature of 23°C. The 1H NMR spectra of samples were recorded on an FT-500 MHz Unity-Inova 500 spectrophotometer. The morphology of the hybrid nanocomposites films were investigated by field emission scanning electron microscopy (FESEM, Zeiss Supra 25) and TEM (Hitachi H7600). All characterizations mentioned above were carried out at Pusan National University, Korea.
3. Results and discussion
3.1 FTIR spectra of 4sPMMA/PMMA-g-SiO2 hybrid nanocomposites
FTIR spectra of CSP0, MCSP3, 4sPMMA, HybM0 and HybM3 samples are presented in Figure 1. There are absorption peaks at 1098 and 465 cm−1 in the spectra of MCSP3 (Figure 1B) which are assigned to vibration of Si-O groups of silica. The stretching vibration peaks of C=C groups at 1637 cm−1, C=O at 1712 cm−1, C–H groups at 2962 cm−1 and 2856 cm−1 are also observed, respectively [32], [33]. Compared to FTIR spectrum of CSP0, these results revealed that MPS silane has been incorporated on the surface of CSPs [as depicted in Figure 2, Eq. (2)]. In Figure 1C, all peaks are assigned to specific groups of PMMA, such as C-H stretching vibrations at 2844, 2852 and 2896 cm−1, C=O stretch at 1731 cm−1, deformation mode of C-H at 1484–1389 cm−1 range and stretching mode of C-O-C at 1243–1272 and 1149–1194 cm−1 ranges [25], [34].
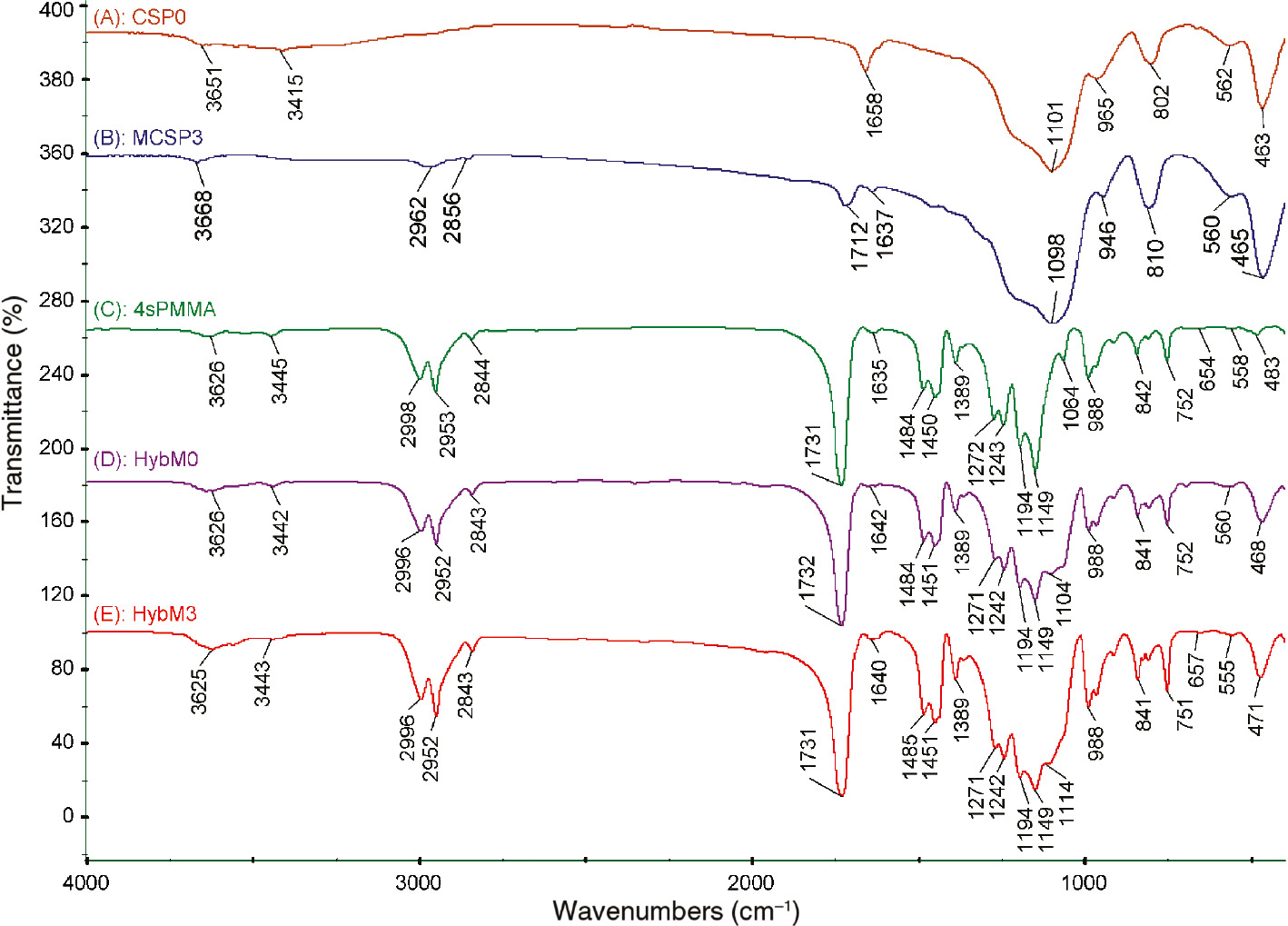
FTIR spectra of (A) unmodified CSPs (CSP0), (B) modified CSPs (MCSP3), (C) neat 4sPMMA and (D, E) hybrid nanocomposites (D: HybM0 (4sPMMA/(PMMA/CSP0)); E: HybM3 (4sPMMA/PMMA grafted MCSP3)).
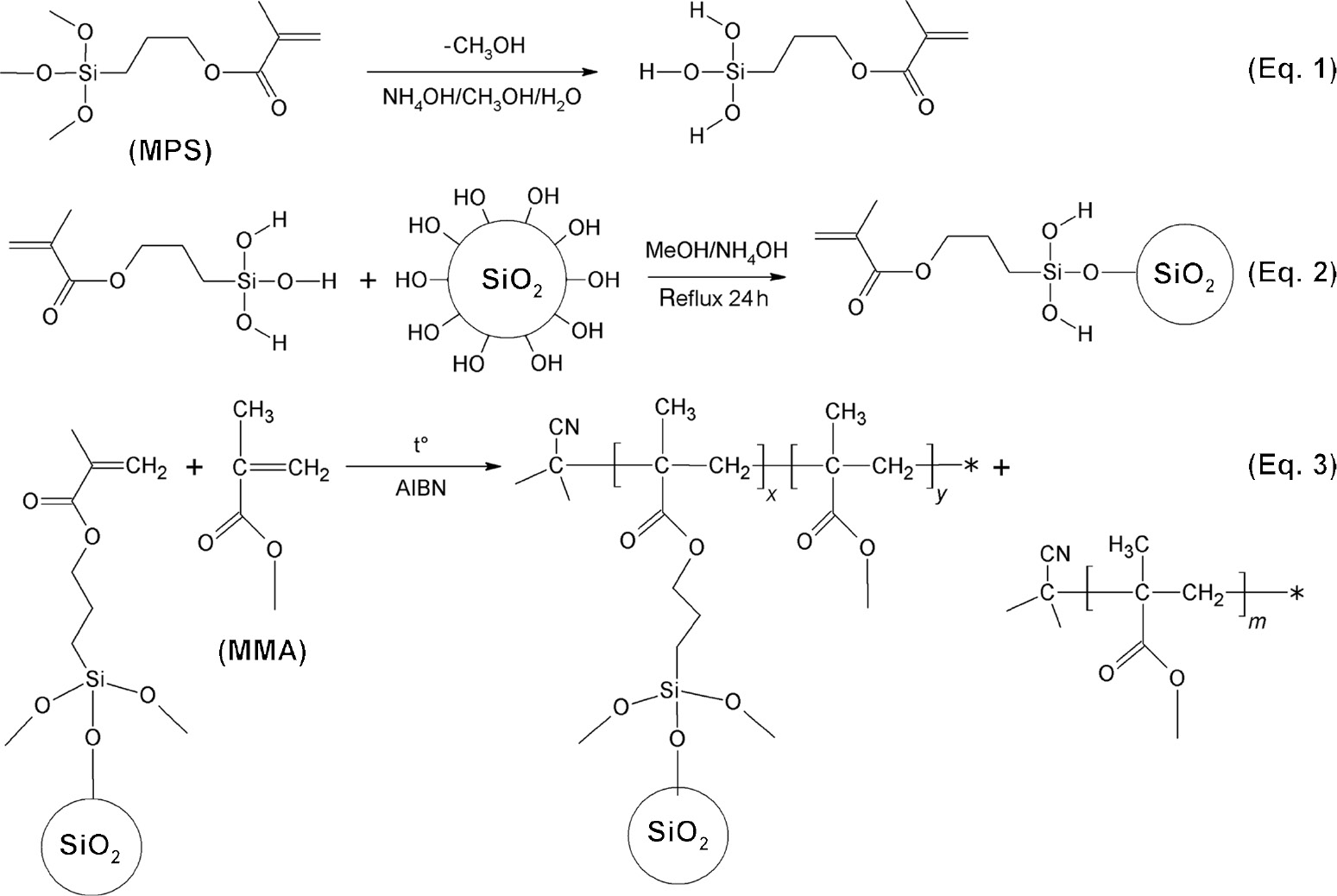
Reactions scheme for the formation of PMMA-g-SiO2 hybrid particles.
Compared to FTIR spectrum of 4sPMMA, there are pronounced peaks appearing at around 470 and 1100 cm−1 which are attributed to stretching (ν) and bending (δ) modes of Si-O (Si-O-C, Si-O-Si) in spectra of HybM0 and HybM3 hybrid nanocomposites. Table 1 shows that there are peak shifts (Δ) of ν and δ modes of the hybrid nanocomposites compared with those of CSP0. In the case of HybM0, smaller Δδ and Δν shifts of 5 and 6 cm−1 may be related to the formation of hydrogen bonds between C=O or C-O-C groups of PMMA and surface SiOH groups of CSP0. For the HybM3 sample, stronger Δδ and Δν shifts of 8 and 16 cm−1 are observed, which can be explained by the stronger interaction due to the formation of covalent bonding between PMMA and MCSPs as depicted in Figure 2. The MMA monomers can react with the vinyl groups of MCSPs to form PMMA-g-SiO2 using AIBN as an initiator.
Specific assignment in FTIR spectra of modified CSPs and hybrid nanocomposites.
| Sample code | SiOSi/SiOC stretching | SiO bending | Sample code | SiOSi/SiOC stretching | SiO bending |
|---|---|---|---|---|---|
| HybM0 | 1103.7 | 467.5 | CSP0 | 1100.8 | 463.1 |
| HybM1 | 1111.9 | 472.0 | MCS1 | 1099.5 | 465.6 |
| HybM2 | 1112.8 | 471.6 | MCS2 | 1099.2 | 465.5 |
| HybM3 | 1114.3 | 471.1 | MCS3 | 1097.6 | 465.9 |
| HybM4 | 1115.1 | 471.3 | MCS4 | 1097.1 | 466.5 |
| HybM5 | 1115.7 | 470.7 | MCS5 | 1097.6 | 467.4 |
3.2 Proton NMR study
Figure 3 represents 1H NMR spectra of pure MPS and 4sPMMA in solvent of DMSO-d6. The spectrum of pure MPS shows two peaks at 6.01 and 5.65 ppm attributed to hydrogen atoms in vinyl groups and the peaks at 4.02 ppm, 3.45 ppm, 1.86 ppm, 1.65 ppm and 0.61 ppm which are assigned to O-CH2, O-CH3, -C-CH3, C-CH2-C and Si-CH2 groups, respectively [35].
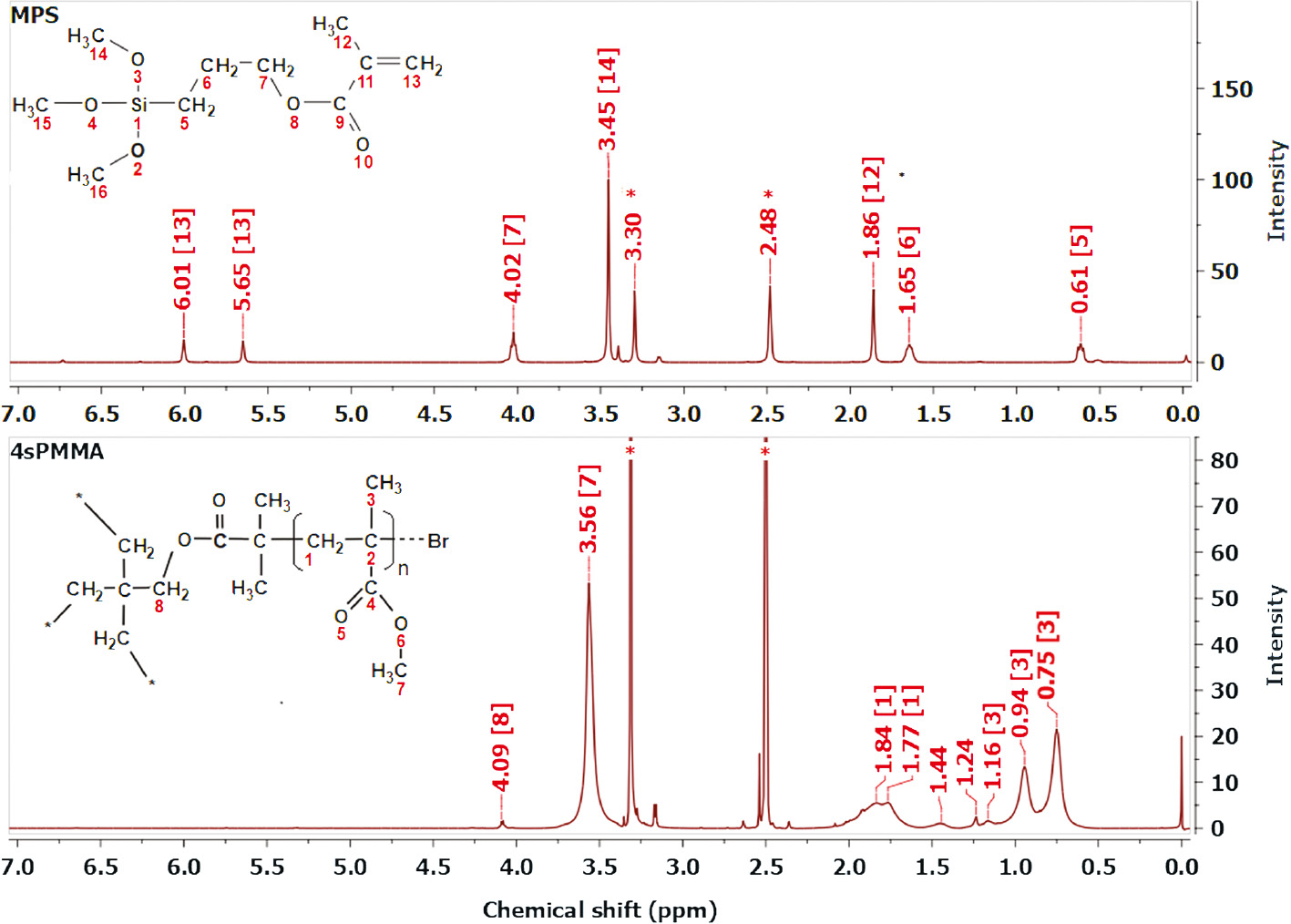
1H NMR spectra of MPS and 4sPMMA in DMSO-d6 (*, residual solvent).
In the spectrum of 4sPMMA, the peak at 4.09 is attributed to O-CH2 groups of the 4-arm star initiator (4Br-PT), the three groups of peaks at 3.65 ppm, (1.87; 1.77 ppm) and (1.16; 0.94; 0.75 ppm) are attributed to O-CH3, -CH2- and C-CH3 groups of PMMA, respectively [36]. In these spectra, the peaks at 3.30 ppm and 2.48 ppm are of residual solvents.
In a spectrum of extracted HybM3 sample called PMMA-g-SiO2 nanocomposites (homopolymers were extracted out of the sample by using acetone as solvent), there are apparent peaks at 3.56 ppm (O-CH3), 1.84; 1.77 ppm (-CH2-), 1.16; 0.94; 0.75 ppm (-C-CH3) and 4.02 ppm (-CH2-O-), which is supporting evidence of the formation of PMMA-g-SiO2 via copolymerization of MMA and vinyl groups of hydrolyzed MPS on the surface of silica particles (Figure 4).
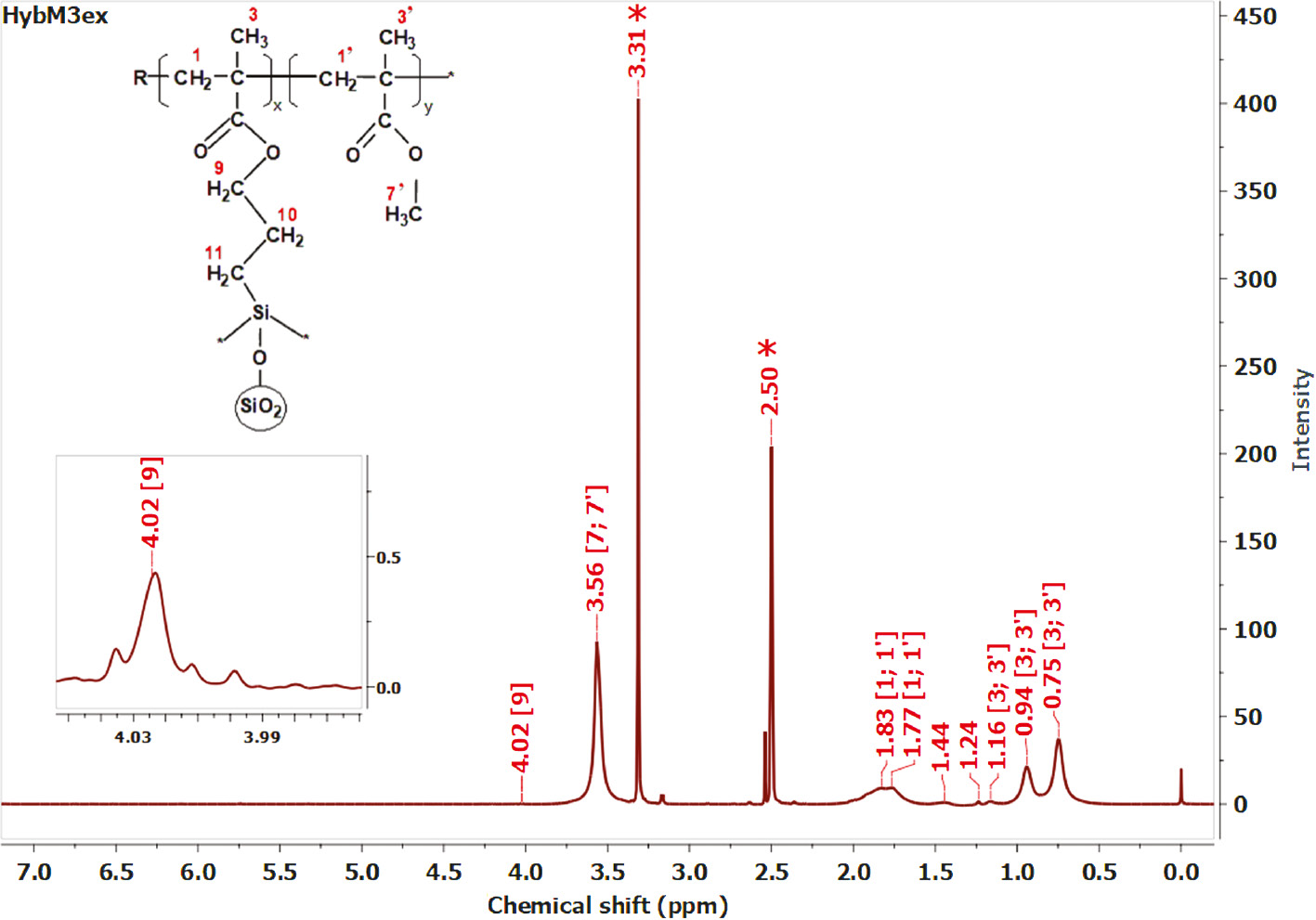
1H NMR spectrum of PMMA-g-SiO2 nanocomposite in DMSO-d6 (*, residual solvent).
3.3 Viscosity study
Table 2 displays the viscosities of the hybrid nanocomposites before and after reaction. The results showed that the viscosities of hybrid solutions before reaction were approximately equal to 32.6–32.9 cP. After reaction was completed, the viscosity of the hybrid solutions increases. For HybM0 hybrid solution, the increase of viscosity was only due to the formation of PMMA, leading to the very weak hydrogen bond interaction between CSPs and polymers. Meanwhile, the viscosity of other hybrid solutions increases with increasing MPS loading used in modification of CSPs, meaning that there is a formation of PMMA-g-SiO2 particles, apart from the formation of PMMA homopolymer as shown in Figure 2. Because the hydrolyzed MPS had been grafted at many sites on the surface of CSPs, when MMA monomers react with these sites of MCSPs to form polymer, the particles can be linked to each other. In other words, the more vinyl groups of MCSPs, the higher cross-linked structure of PMMA-g-SiO2 can be formed.
Viscosity of hybrid nanocomposites before and after reaction.
| Sample code | Viscosity (cP) | |
|---|---|---|
| Before reaction | After reaction | |
| HybM0 | 32.6±0.3 | 45.7±0.3 |
| HybM1 | 32.7±0.5 | 46.8±0.3 |
| HybM2 | 32.7±0.3 | 47.9±0.5 |
| HybM3 | 32.8±0.3 | 50.9±0.4 |
| HybM4 | 32.8±0.4 | 52.2±0.5 |
| HybM5 | 32.9±0.6 | 53.2±0.6 |
3.4 Morphological study
The morphology of silica particles in the HybM0 and HybM3 hybrid nanocomposites are investigated by FESEM images in Figure 5A and B. It can be found that CSPs and MCSPs disperse regularly in PMMA matrices, with nanoscale of about 20–30 nm, and maintain their spherical shapes. For the HybM0 sample, it is easy to distinguish the bare silica particles with polymer matrix by the contrasting gray color. Meanwhile, the MCSPs disperse in the HybM3 hybrid nanocomposite with less contrast color. This could be explained by the formation of PMMA layers around silica particles which reduced the difference of electron interaction between inorganic and organic phases in FESEM observation.

FESEM images of (A) 4sPMMA/(PMMA/CSP0) (HybM0) and (B) 4sPMMA/PMMA grafted MCSP3 (HybM3) hybrid nanocomposites.
The dispersion morphology of MCSPs in the hybrid nanocomposites was observed with TEM image displayed in Figure 6. The TEM image of HybM3 hybrid nanocomposite confirms that PMMA-g-SiO2 particles are well embedded in PMMA matrices; the size of silica particles is about 20–30 nm. It can be also seen that some MCSPs are linked to each other to form clusters. However, the presence of 4sPMMA reduced the formation of big clusters because 4sPMMA chains could penetrate into the cluster during reaction.
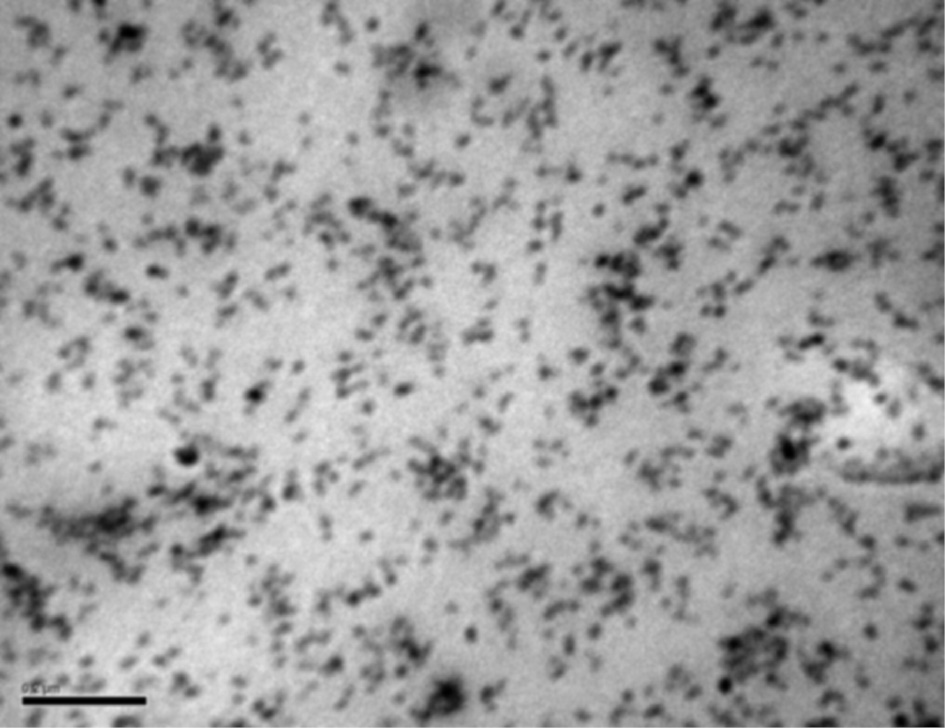
TEM images of HybM3 hybrid nanocomposite (black scale bar 200 nm).
3.5 Thermogravimetric analysis
Figure 7 shows TGA diagrams of free radical polymerization PMMA (fr-PMMA), 4sPMMA and HybM3 hybrid nanocomposites, and the thermal characteristics are summarized in Table 3. The results show that fr-PMMA degrades at considerably low temperature with onset point (Tonset) of 180°C. This is attributed to the presence of double bonds at the ends of PMMA chains synthesized by free radical polymerization [37]. The TGA curve of 4sPMMA shifts to higher temperature with higher Tonset of 254°C, which may be due to 4sPMMA with mainly bromide end groups (showed in Figure 3). The Tonset of the hybrid nanocomposites is slightly lower than that of 4sPMMA. The results indicate that thermal stability of hybrid nanocomposite is much higher than pure fr-PMMA, although it also contains fr-PMMA with theoretical amount of 21 wt.%.
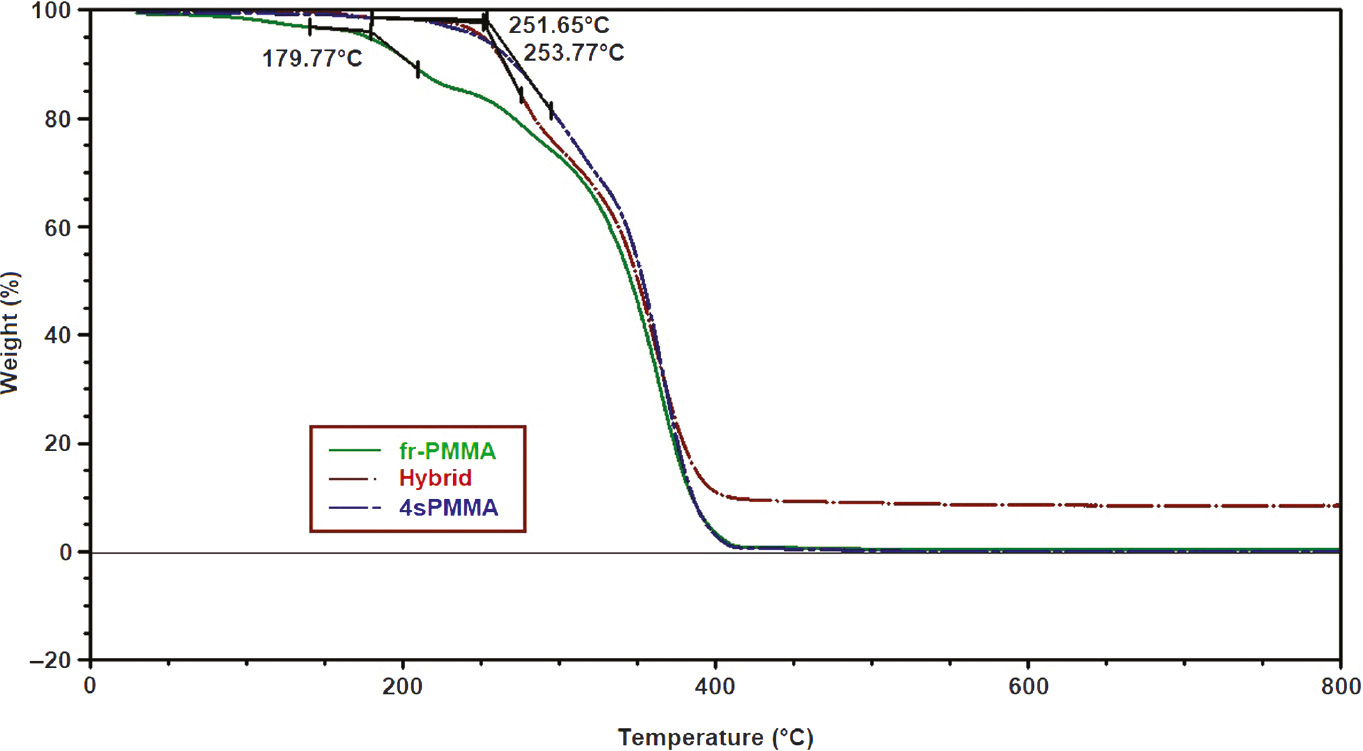
TGA diagrams of fr-PMMA, 4sPMMA and HybM3 hybrid nanocomposites.
Thermal characteristic of fr-PMMA, 4sPMMA and hybrid HybM3 samples under nitrogen at heating rate of 10°C min−1.
| Sample code | Tonset (°C) | TG weight (%) at temperature | |||
|---|---|---|---|---|---|
| 300°C | 350°C | 400°C | 800°C | ||
| fr-PMMA | 179.5 | 72.89 | 46.27 | 3.06 | 0 |
| 4sPMMA | 253.8 | 79.61 | 54.15 | 3.16 | 0 |
| HybM3 | 245.0 | 74.78 | 50.99 | 11.62 | 9.11 |
4. Conclusions
The hybrid nanocomposites of 4sPMMA/PMMA-g-SiO2 were synthesized by in situ solution blending. FTIR, 1H NMR spectroscopies and viscosity measurements indicated that PMMA has been grafted on modified colloidal silica particles to form PMMA-g-SiO2 particles. FESEM and TEM images revealed that SNPs were well dispersed in polymer matrices with size of about 20–30 nm. Thermal stability of the hybrid nanocomposites was improved compared with fr-PMMA.
Acknowledgments
This research was supported by the Human Resource Training Program for Regional Innovation and Creativity (NRF-2015H1C1A1035709) and the Basic Science Research Program through National Research Foundation of Korea (NRF) funded by the Ministry of Education (2017R1D1A1B04032465). The authors also thank the BK21 PLUS Program for partial financial support.
References
[1] He H, Chen S, Bai J, Zheng H, Wu B, Ma M, Shi Y, Wang X. RSC Adv. 2016, 6, 34685–34691.Search in Google Scholar
[2] Wang W, Liao S, Zhu Y, Liu M, Zhao Q, Fu Y. J. Nanomater. 2015, 2015, doi: 10.1155/2015/408643.10.1155/2015/408643Search in Google Scholar
[3] Yu YY, Chen CY, Chen WC. Polymer 2003, 44, 593–601.10.1016/S0032-3861(02)00824-8Search in Google Scholar
[4] Szanka A, Szarka G, Iván B. Polymer 2013, 54, 6073–6077.10.1016/j.polymer.2013.09.025Search in Google Scholar
[5] Hu Y-H, Chen C-Y, Wang C-C. Polym. Degrad. Stab. 2004, 84, 545–553.10.1016/j.polymdegradstab.2004.02.001Search in Google Scholar
[6] Yang F, Nelson GL. J. Appl. Polym. Sci. 2004, 91, 3844–3850.10.1002/app.13573Search in Google Scholar
[7] Alvarado-Rivera J, Muñoz-Saldaña J, Ramírez-Bon R. J. Sol-Gel Sci. Technol. 2010, 54, 312–318.10.1007/s10971-010-2196-7Search in Google Scholar
[8] Hammer P, dos Santos FC, Cerrutti BM, Pulcinelli SH, Santilli CV. Prog. Org. Coat. 2013, 76, 601–608.10.1016/j.porgcoat.2012.11.015Search in Google Scholar
[9] Mammeri F, Teyssandier J, Connan C, Le Bourhis E, Chehimi MM. RSC Adv. 2012, 2, 2462–2468.10.1039/c2ra00937dSearch in Google Scholar
[10] Otsuka T, Chujo Y. Polym. J. 2010, 42, 58–65.10.1038/pj.2009.309Search in Google Scholar
[11] Khaled SM, Sui R, Charpentier PA, Rizkalla AS. Langmuir 2007, 23, 3988–3995.10.1021/la062879nSearch in Google Scholar PubMed
[12] Nafisi S, Schäfer-Korting M, Maibach HI. In Agache’s Measuring the Skin: Non-invasive Investigations, Physiology, Normal Constants, Humbert, P, Fanian, F, Maibach, HI, Agache, P, Eds., Springer International Publishing: Cham, 2017, pp. 1141–1164.10.1007/978-3-319-32383-1_44Search in Google Scholar
[13] Balos S, Pilic B, Markovic D, Pavlicevic J, Luzanin O. J. Prosthet. Dent. 2014, 111, 327–334.10.1016/j.prosdent.2013.06.021Search in Google Scholar PubMed
[14] Salman AD, Jani GH, Fatalla AA. Biomed. Pharmacol. J. 2017, 10, 1525–1535.10.13005/bpj/1262Search in Google Scholar
[15] Zou H, Wu S, Shen J. Chem. Rev. 2008, 108, 3893–3957.10.1021/cr068035qSearch in Google Scholar PubMed
[16] Rahman IA, Padavettan V. J. Nanomater. 2012, 2012, doi: 10.1155/2012/132424.10.1155/2012/132424Search in Google Scholar
[17] Topouzi M, Kontonasaki E, Bikiaris D, Papadopoulou L, Paraskevopoulos KM, Koidis P. J. Mech. Behav. Biomed. Mater. 2017, 69, 213–222.10.1016/j.jmbbm.2017.01.013Search in Google Scholar PubMed
[18] Canché-Escamilla G, Duarte-Aranda S, Toledano M. Mater. Sci. Eng. C 2014, 42, 161–167.10.1016/j.msec.2014.05.016Search in Google Scholar PubMed
[19] Ravarian R, Wei H, Rawal A, Hook J, Chrzanowski W, Dehghani F. J. Mater. Chem. B 2013, 1, 1835–1845.10.1039/c2tb00251eSearch in Google Scholar PubMed
[20] Kim JW, Kim LU, Kim CK. Biomacromolecules 2007, 8, 215–222.10.1021/bm060560bSearch in Google Scholar PubMed
[21] Tadano T, Zhu R, Muroga Y, Hoshi T, Sasaki D, Yano S, Sawaguchi T. Polym. J. (Tokyo, Jpn.) 2014, 46, 342–348.10.1038/pj.2014.6Search in Google Scholar
[22] Hyde EDER, Seyfaee A, Neville F, Moreno-Atanasio R. Ind. Eng. Chem. Res. 2016, 55, 8891–8913.10.1021/acs.iecr.6b01839Search in Google Scholar
[23] Yamada H, Urata C, Aoyama Y, Osada S, Yamauchi Y, Kuroda K. Chem. Mater. 2012, 24, 1462–1471.10.1021/cm3001688Search in Google Scholar
[24] Joseph R, Zhang S, Ford WT. Macromolecules 1996, 29, 1305–1312.10.1021/ma951111zSearch in Google Scholar
[25] Chau JLH, Hsieh C-C, Lin Y-M, Li A-K. Prog. Org. Coat. 2008, 62, 436–439.10.1016/j.porgcoat.2008.02.005Search in Google Scholar
[26] Sugimoto H, Daimatsu K, Nakanishi E, Ogasawara Y, Yasumura T, Inomata K. Polymer 2006, 47, 3754–3759.10.1016/j.polymer.2006.04.002Search in Google Scholar
[27] Liu Y-L, Hsu C-Y, Hsu K-Y. Polymer 2005, 46, 1851–1856.10.1016/j.polymer.2005.01.009Search in Google Scholar
[28] Kashiwagi T, Morgan AB, Antonucci JM, VanLandingham MR, Harris RH, Awad WH, Shields JR. J. Appl. Polym. Sci. 2003, 89, 2072–2078.10.1002/app.12307Search in Google Scholar
[29] Liu X, Zhao H, Li L, Yan J, Zha L. J. Macromol. Sci., Part A: Pure Appl. Chem. 2006, 43, 1757–1764.10.1080/10601320600939502Search in Google Scholar
[30] Gu S, Shi Y, Wang L, Liu W, Song Z. Colloid Polym. Sci. 2014, 292, 267–273.10.1007/s00396-013-3109-4Search in Google Scholar
[31] Schoth A, Wagner C, Hecht LL, Winzen S, Muñoz-Espí R, Schuchmann HP, Landfester K. Colloid Polym. Sci. 2014, 292, 2427–2437.10.1007/s00396-014-3316-7Search in Google Scholar
[32] Bensaid MO, Ghalouci L, Hiadsi S, Lakhdari F, Benharrats N, Vergoten G. Vib. Spectrosc. 2014, 74, 20–32.10.1016/j.vibspec.2014.07.001Search in Google Scholar
[33] Pantoja M, Díaz-Benito B, Velasco F, Abenojar J, del Real JC. Appl. Surf. Sci. 2009, 255, 6386–6390.10.1016/j.apsusc.2009.02.022Search in Google Scholar
[34] Haris M, Kathiresan S, Mohan S. Pharma Chem. 2010, 2, 316–323.Search in Google Scholar
[35] Brochier Salon M-C, Abdelmouleh M, Boufi S, Belgacem MN, Gandini A. J. Colloid Interface Sci. 2005, 289, 249–261.10.1016/j.jcis.2005.03.070Search in Google Scholar PubMed
[36] Ueda J, Kamigaito M, Sawamoto M. Macromolecules 1998, 31, 6762–6768.10.1021/ma980608gSearch in Google Scholar
[37] Manring LE. Macromolecules 1989, 22, 2673–2677.10.1021/ma00196a024Search in Google Scholar
©2018 Walter de Gruyter GmbH, Berlin/Boston
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Articles in the same Issue
- Frontmatter
- In this issue
- ASAM-6
- The 6th Asian Symposium on Advanced Materials: Chemistry, Physics and Biomedicine of Functional and Novel Materials (ASAM-6; Hanoi, Vietnam, September 27–30, 2017)
- Synthesis and characterization of (4-arm-star-PMMA)/PMMA-g-SiO2 hybrid nanocomposites
- Factors influencing green strength of commercial natural rubber
- Removal of arsenic from water using crumpled graphite oxide
- Adsorption behavior of Cd2+ ions using hydroxyapatite (HAp) powder
- Study on characteristics, properties, and morphology of poly(lactic acid)/chitosan/hydroquinine green nanoparticles
- Original articles
- Green synthesis and stabilization of earthworm-like gold nanostructure and quasi-spherical shape using Caesalpinia sappan Linn. extract
- Catalytic performance of Ag, Au and Ag-Au nanoparticles synthesized by lichen extract
- Comparative kinetics of the alkali-catalyzed sunflower oil methanolysis with co-solvent under conventional and microwave heating with controlled cooling
- Facile nitration of aromatic compounds using Bi(NO3)3·5H2O/MgSO4 under mechanochemical conditions
- Optimization of the treatment process of zinc leaching residue by using the response surface method
- A new green process to produce activated alumina by spray pyrolysis
Articles in the same Issue
- Frontmatter
- In this issue
- ASAM-6
- The 6th Asian Symposium on Advanced Materials: Chemistry, Physics and Biomedicine of Functional and Novel Materials (ASAM-6; Hanoi, Vietnam, September 27–30, 2017)
- Synthesis and characterization of (4-arm-star-PMMA)/PMMA-g-SiO2 hybrid nanocomposites
- Factors influencing green strength of commercial natural rubber
- Removal of arsenic from water using crumpled graphite oxide
- Adsorption behavior of Cd2+ ions using hydroxyapatite (HAp) powder
- Study on characteristics, properties, and morphology of poly(lactic acid)/chitosan/hydroquinine green nanoparticles
- Original articles
- Green synthesis and stabilization of earthworm-like gold nanostructure and quasi-spherical shape using Caesalpinia sappan Linn. extract
- Catalytic performance of Ag, Au and Ag-Au nanoparticles synthesized by lichen extract
- Comparative kinetics of the alkali-catalyzed sunflower oil methanolysis with co-solvent under conventional and microwave heating with controlled cooling
- Facile nitration of aromatic compounds using Bi(NO3)3·5H2O/MgSO4 under mechanochemical conditions
- Optimization of the treatment process of zinc leaching residue by using the response surface method
- A new green process to produce activated alumina by spray pyrolysis