Facile nitration of aromatic compounds using Bi(NO3)3·5H2O/MgSO4 under mechanochemical conditions
-
Feng-Chao Guo


Abstract
Bi(NO3)3·5H2O/MgSO4 was developed as an efficient and green reagent for the nitration of aromatic compounds under mechanochemistry (or ball milling) condition. While aromatics with weak activating groups such as phenyl could be nitrated by this reagent with 100% conversion, aromatics with weak deactivating groups such as chloro- or bromo- could also be nitrated but with moderate conversion and regioselectivity with big para/ortho ratios. The in situ generated N2O4 or NO2 due to the decomposition of Bi(NO3)3·5H2O promoted by MgSO4 should be responsible for this mechanochemical nitration.
1 Introduction
Aromatic nitration is of great importance for both industrial applications and academic studies [1], [2], [3], [4]. While large amounts of simple nitroaromatic compounds such as nitrobenzenes and nitrotoluenes are manufactured by this mature industrial method [1], many kinds of nitroaromatic fine chemicals in relatively small amounts are still needed to be manufactured in order to meet the requirement of dyes, plastics, and pharmaceutical industry. Currently, most of these fine chemicals are manufactured by the traditional nitration methods which usually employ an excess of nitric acid or a mixture of nitric acid and sulfuric acid (so called “mixed acids”) as nitration reagents. These reactions suffer from drawbacks such as unselectivity and imperfect functional group tolerance. Besides, these reactions may cause explosion and also generate nitrogen oxide (NOx) fumes and large quantities of waste acids; the disposal of these spent liquors presents a serious environmental problem. Therefore, there has been a continuous effort to look out for new nitrating agents in order to make the nitration reaction safe, ecofriendly, and less costly. Recently, various new nitrating reagents such as organic nitrating compounds [3], polymer-supported nitrating salts [3], and various inorganic nitrate salts have been utilized as “green” nitrating reagents to produce nitroaromatic compounds. Such nitrate salts include Fe(NO3)3·9H2O [5], AgNO3 [6], Mg(NO3)2·6H2O [7], Me4NNO3 [8], NaNO3 [9], [10], [11], Ni(NO3)2·6H2O [12], Bi(NO3)3 [13], and Bi(NO3)3·5H2O [14], [15], [16], [17], [18], [19], [20], [21], [22], [23], [24], [25], [26], [27], [28], [29]. Among these nitrate salts, Bi(NO3)3·5H2O is an effective, inexpensive, low toxic, and easy to handle reagent. Several Bi(NO3)3·5H2O based nitrating reagents such as Bi(NO3)3·5H2O [14], [22], Bi(NO3)3·5H2O/AC2O [15], and Bi(NO3)3·5H2O/montmorillonite [17], [23] have been developed in recent years. However, most of these nitrating reagents were only effective for the nitration of highly activated aromatics, such as phenols and anilines. Recently, Laali [16] reported that either chlorobenzene or bromobenzene could be nitrated with good yield with Bi(NO3)3·5H2O in imidazolium ionic liquid at relatively high temperature (e.g. 85°C) and long reaction time (e.g. 15 h). They also reported that if the ionic liquid was replaced by 1,2-dichloroethane, the nitration could be also performed smoothly under refluxing for a very long time (e.g. 40–70 h). To our knowledge, this is the only report which mentioned that deactivated aromatics can be nitrated by Bi(NO3)3·5H2O, although expensive ionic liquids are employed or tedious reaction time is needed for regular solvents.
Along with the rapid development of green chemistry, mechanochemistry has attracted increasing attention from chemists [24], [25]. Mechanochemistry is mainly promoted by mechanical milling at a frequency of 5–60 Hz. A solvent-free mechanochemical organic reaction is a green process, which exhibits many advantages over its liquid-phase counterpart in terms of higher product yield, better selectivity, shorter reaction time, simple work-up procedure, elimination of harmful organic solvent, etc. Nowadays, no difficulty exists in scaling-up mechanochemical syntheses up to 200 g batches, and final large-scale industrial productions [26]. Considering these kinds of advantages of mechanochemical organic reaction as well as our early experiences about the mechanochemistry related to [60]fullerene or carbon nanotubes [27], [28], [29], we investigated the mechanochemical nitrating reaction of aromatic compounds by employing Bi(NO3)3·5H2O as the nitrating reagent. We found that along with the addition of MgSO4 as auxiliary, the nitration reaction was greatly improved and deactivated aromatics such as chlorobenzene and bromobenzene could be regioselectively nitrated in good yield and within a short time. In addition, this method can be explored to gram scale by taking planetary ball mill as a reactor.
2 Materials and methods
All chemicals were purchased from Sigma-Aldrich (Billerica, MA, USA), Alfa Aesar (Ward Hill, MA, USA), or TCI (Tokyo, Japan), and were used without further purification. The nitrating reaction was carried out either in a MM400 mixer ball mill (Retsch GmbH, Haan, Germany) or in a QM-3SP04 planetary ball mill (Nanjing NanDa Instrument, Nanjing, China). The 1H NMR and 13C NMR spectra were recorded using either a Bruker AV 400 or a Bruker AV 600 spectrometer (Bruker Corporation, Billerica, MA, USA) in CDCl3 with tetramethylsilane as the internal standard. The GC-MS measurements were carried out using an Agilent Technologies instrument equipped with a 6890N network GC system and 5973 network mass selective detector (Agilent Technologies, Santa Clara, CA, USA).
2.1 General procedure for the mechanochemical nitrating reaction
For the MM400 mixer ball mill, the stainless milling beaker (2.5 ml) was filled with one stainless milling ball (6.35 mm) and then the aromatic compound (0.02 mmol), Bi(NO3)3·5H2O (0.09 mmol), and 100 mg of MgSO4 were placed into the beaker sequentially. The mechanical reaction was performed at specific oscillation frequency (ν=20 Hz) and time (t=2 h). The reaction mixture was extracted by dichloromethane (3×5 ml). The solvent was evaporated and the residue was dried in vacuum. Unless specified, the components of the residue were analyzed and determined by 1H NMR and literature.
For the QM-3SP04 planetary ball mill, the stainless milling beaker (100 ml) was filled with 30 stainless milling ball (10 mm) and then the aromatic compound (2 mmol), Bi(NO3)3·5H2O (9 mmol), and 10 g of MgSO4 were placed into the beaker sequentially. The mechanical reaction was performed at a specific rotating speed (400 rpm) and for a certain time according to the different substrates. The reaction mixture was extracted by dichloromethane (3×50 ml). The solvent was evaporated and the residue was dried in vacuum. The components of the residue were analyzed and determined by 1H NMR and literature.
Entry 1: 4-Nitrobiphenyl [30] 1H NMR (600 MHz): δ=8.32–8.28 (d, 2H, m-Ar-H), 7.77–7.72 (m, 2H, o-Ar-H), 7.65–7.60 (m, 2H, o-Ar′-H), 7.54–7.48 (m, 2H, m-Ar′-H), 7.48–7.38 (m, 1H, p-Ar′-H). 2-Nitrobiphenyl[1] 1H NMR (600 MHz): δ=7.86 (dd, J=8.1, 1.1 Hz, 1H), 7.77–7.72 (m, 1H), 7.65–7.60 (m, 1H), 7.54–7.48 (m, 1H), 7.48–7.38 (m, 3H), 7.32 (ddd, J=9.5, 5.1, 2.8 Hz, 1H).
Entry 2: The reaction was performed in the same manner as described in the general procedure. The mixture was purified by column chromatography on silica gel with n-hexane-dichloromethane (1:1) as the eluent. 3-Nitro-4-methoxylbiphenyl [30] 1H NMR (600 MHz): δ=8.09–8.06 (m, 1H), 7.76 (dd, J=8.7, 2.0 Hz, 1H), 7.58–7.53 (m, 2H), 7.45 (t, J=7.7 Hz, 2H), 7.37 (t, J=7.4 Hz, 1H), 7.16 (d, J=8.7 Hz, 1H), 4.00 (s, 3H). 3,4′-Dinitro-4-methoxylbiphenyl 1H NMR (600 MHz): δ=8.35–8.30 (m, 2H), 8.14 (d, J=2.4 Hz, 1H), 7.82 (dd, J=8.7, 2.4 Hz, 2H), 7.75–7.71 (m, 1H), 7.25–7.22 (m, 1H), 4.04 (s, 3H). 13C NMR (100 MHz, CDCl3): δ=153.4, 147.3, 144.6, 139.9, 132.7, 131.1, 127.4, 124.5, 124.4, 114.3, 56.8. GC-MS: tR=12.30 min, calcd. For C13H10N2O5, (m/z)=274.06; found 274.10 (M+).
Entry 3: 4-Nitro-4′-bromobiphenyl [31] 1H NMR (400 MHz): δ=8.33–8.28 (m, 2H), 7.74–7.68 (m, 2H), 7.66–7.60 (m, 2H), 7.52–7.47 (m, 2H). 2-Nitro-4′-bromobiphenyl [31] 1H NMR (400 MHz): δ=7.89 (dd, J=8.1, 1.2 Hz, 1H), 7.66–7.60 (m, 1H), 7.58–7.52 (m, 2H), 7.52–7.47 (m, 1H), 7.41 (dd, J=7.6, 1.4 Hz, 1H), 7.22–7.17 (m, 2H).
Entry 4: 4-Nitrobenzenebutanoic acid [30] 1H NMR (400 MHz): δ=8.23–8.13 (m, 2H), 7.38 (t, J=8.3 Hz, 2H), 2.85–2.76 (m, 2H), 2.45 (dt, J=21.6, 7.3 Hz, 2H), 2.10–1.95 (m, 2H). 2-Nitrobenzenebutanoic acid [32] 1H NMR (400 MHz): δ=7.98–7.89 (m, 1H), 7.55 (td, J=7.5, 1.2 Hz, 1H), 7.38 (t, J=8.3 Hz, 2H), 2.97 (dd, J=8.8, 6.8 Hz, 2H), 2.45 (dt, J=21.6, 7.3 Hz, 2H), 2.10–1.95 (m, 2H).
Entry 5: The reaction was performed in the same manner as described in the general procedure. The mixture was purified by column chromatography on silica gel with n-hexane-dichloromethane (1:1) as the eluent. The products were not purified further and directly analyzed by 1H NMR. 4-Nitrochlorobenzene [30] 1H NMR (600 MHz): δ=8.22–8.16 (m, 2H), 7.58–7.49 (m, 2H). 2-Nitrochlorobenzene [30] 1H NMR (600 MHz): δ=7.92–7.85 (m, 1H), 7.58–7.49 (m, 2H), 7.46–7.40 (m, 1H).
Entry 6: The reaction was performed in the same manner as described in the general procedure. The mixture was purified by column chromatography on silica gel with n-hexane-dichloromethane (1:1) as the eluent. The products were not purified further and directly analyzed by 1H NMR. 4-Nitrobromobenzene [30] 1H NMR (600 MHz): δ=8.10 (d, J=8.3 Hz, 2H), 7.69 (d, J=8.9 Hz, 2H). 2-Nitrobromobenzene [30] 1H NMR (600 MHz): δ=7.84 (dd, J=8.3, 1.6 Hz, 1H), 7.75 (dd, J=7.8, 1.3 Hz, 1H), 7.50–7.42 (m, 2H).
Entry 7: The reaction was performed in the same manner as described in the general procedure. The mixture was purified by column chromatography on silica gel with n-hexane-dichloromethane (1:1) as the eluent. The products were not purified further and directly analyzed by 1H NMR. 1,2-Dichloro-3-nitrobenzene [30] 1H NMR (600 MHz): δ=7.70 (td, J=6.7, 3.3 Hz, 2H), 7.37 (dd, J=13.6, 5.4 Hz, 2H). 1,2-Dichloro-4-nitrobenzene [30] 1H NMR (600 MHz): δ=8.35 (d, J=2.5 Hz, 2H), 8.09 (dd, J=8.8, 2.6 Hz, 2H), 7.65 (d, J=8.8 Hz, 2H).
Entry 8: 4-Methyl-3-nitrobenzoic acid [30] 1H NMR (400 MHz): δ=8.68 (s, 1H), 8.20 (d, J=8.0 Hz, 1H), 7.49 (d, J=8.0 Hz, 1H), 2.69 (s, 3H). 4-Methylbenzonic acid [30] 1H NMR (400 MHz): δ=8.00 (d, J=8.4, 2H), 7.27 (d, J=8.4 Hz, 2H), 2.44 (s, 3H).
Entry 13: The reaction was performed in the same manner as described in the general procedure. The mixture was purified by column chromatography on silica gel with n-hexane-dichloromethane (1:1) as the eluent. 1-Nitronaphthalene [30] 1H NMR (600 MHz, CDCl3): δ=8.53 (d, J=8.7 Hz, 1H), 8.19 (d, J=7.6 Hz, 1H), 8.07 (d, J=8.1 Hz, 1H), 7.91 (d, J=8.2 Hz, 1H), 7.68 (dd, J=8.5, 7.1 Hz, 1H), 7.59 (dd, J=8.1, 7.0 Hz, 1H), 7.49 (t, J=7.9 Hz, 1H).
Entry 14: 1-Nitropyrene [33] 1H NMR (600 MHz, CDCl3): δ=8.72 (d, J=9.4 Hz, 1H), 8.53 (d, J=8.4 Hz, 1H), 8.21 (d, J=7.6 Hz, 1H), 8.18 (d, J=7.5 Hz, 1H), 8.13 (d, J=9.4 Hz, 1H), 8.09 (d, J=8.8 Hz, 1H), 8.04 (t, J=7.6 Hz, 1H), 7.98 (d, J=8.4 Hz, 1H), 7.92 (d, J=8.8 Hz, 1H).
3 Results and discussion
In our experiments, we take biphenyl as the representative substrate, because biphenyl is solid at room temperature. Initially, we mix 0.5 mmol of biphenyl with 5 mmol of Bi(NO3)3·5H2O in the milling ball and the mixture was milled at 20 Hz up to 10 h However, only unreacted biphenyl was recovered. This implies that biphenyl cannot be nitrated by Bi(NO3)3·5H2O itself under our mechanochemical reaction condition.
It was reported that the addition of milling auxiliaries would improve the mechanochemical reaction on both conversion and selectivity [34], [35], [36]. Following this idea, a few of the auxiliaries were used for the Bi(NO3)3·5H2O based mechanochemical nitration. Considering that there are five crystal waters in the structure of Bi(NO3)3·5H2O which may hinder the reaction, several drying agents were selected as auxiliaries.
We mixed 0.02 mmol of biphenyl and 0.06 mmol of Bi(NO3)3·5H2O together. In order to keep the volume of the mixture at same level for each reaction, 100 mg of the selected auxiliary was added. The mixture was ball-milled at 20 Hz for 2 h. While the reactions with auxiliaries such as SiO2, CaCl2, γ-alumna, CaSO4, and Na2SO4 failed to give any products, the reaction with MgSO4 as auxiliary did work. The mixture was purified by column chromatography on silica gel with n-hexane-dichloromethane (1:1, v/v) as the eluent. After removing the unreacted biphenyl, the product was not purified further and directly analyzed by 1H NMR. Although the 1H NMR spectrum (Figure 1) looks complicated, carefully comparing with the standard 1H NMR spectra of the potential products of o-nitrobiphenyl, p-nitrobiphenyl, as well as m-nitrobiphenyl [30] one can draw a very clear conclusion. As no peak can be found around 8.40 ppm, which is the characteristic chemical shift of HA of m-nitrobiphenyl, it can be concluded that no m-nitrobiphenyl is produced in the reaction. The chemical shifts with relatively same higher intensity (integration is ~1) at ~8.30, ~7.74 and ~7.60 ppm correspond very well to the protons of HA, HB and HC of p-nitrobiphenyl, respectively. The chemical shifts with relatively same lower intensity (integration is ~ 0.35, double for HE) at ~7.85, ~7.60 and ~7.32 ppm are the characteristic chemical shifts of HA, HB and HE of o-nitrobiphenyl, respectively. The chemical shifts between ~7.41 and ~7.52 ppm correspond to the proton signals of HD, HE of p-nitrobiphenyl, as well as HC and HD of o-nitrobiphenyl, respectively. In this context, the mechanochemical nitration of biphenyl and Bi(NO3)3·5H2O with MgSO4 as auxiliary produces two isomers: o-nitrobiphenyl and p-nitrobiphenyl. The conversion of the reaction is 39.4%, and the para/ortho ratio can be determined through the integration of the proton NMR spectrum as 59:41. Thus MgSO4 is an effective auxiliary for the mechanochemical nitrating reaction.
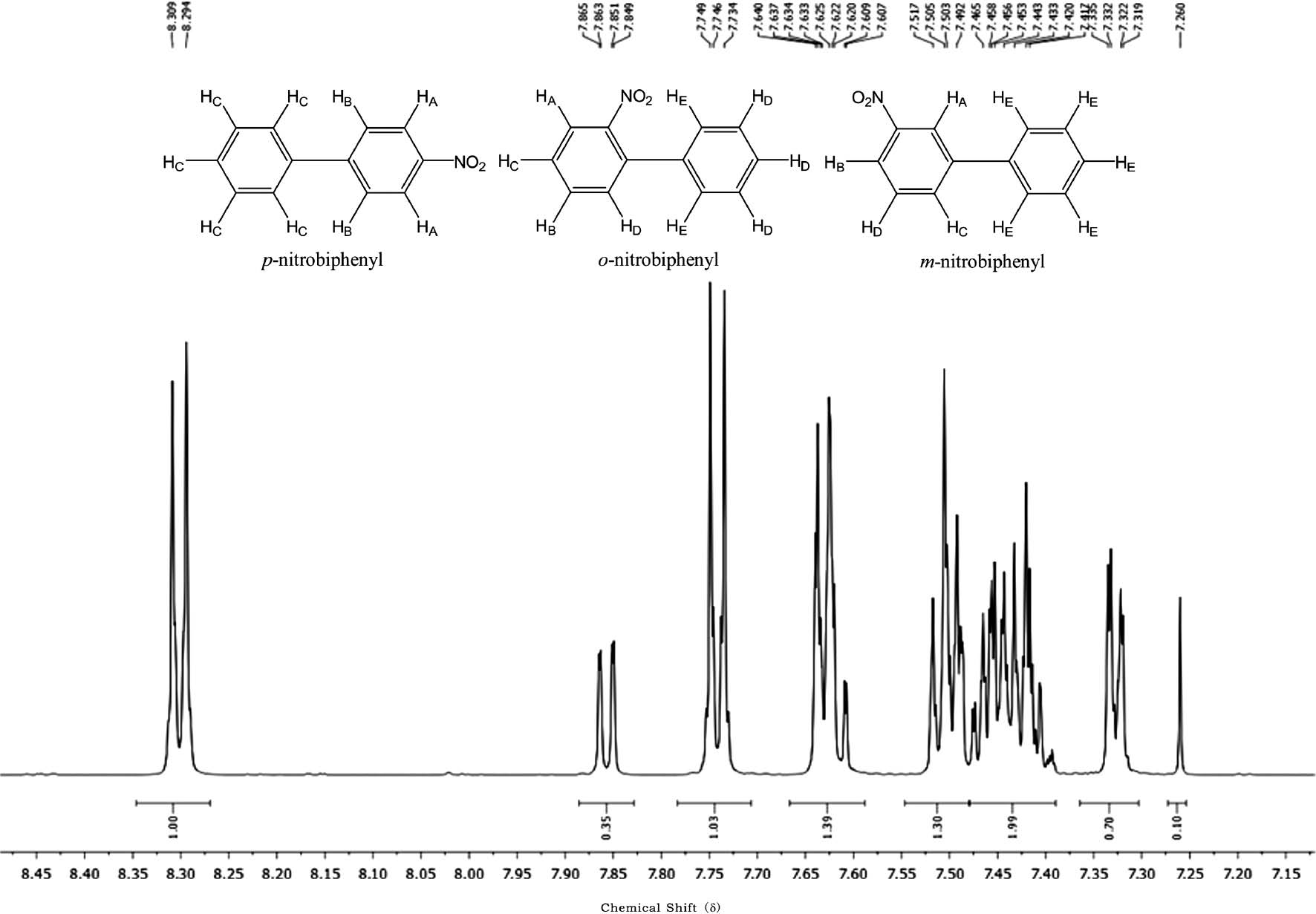
1H NMR spectrum of the reaction mixture of biphenyl and Bi(NO3)3·5H2O/MgSO4.
In order to get higher conversion of biphenyl, the effect of the MgSO4 loading was investigated (Figure 2). While keeping the molar ratio of biphenyl/Bi(NO3)3·5H2O as 1:3 (0.02 mmol vs. 0.06 mmol), the conversion of biphenyl is increased gradually with the increasing amount of MgSO4 (Figure 2A). When the amount of MgSO4 reached 100 mg, the conversion of biphenyl is about 65% and this conversion is kept almost unchanged when the amount of MgSO4 is increased to 120 mg. This implies that 100 mg of MgSO4 is the best loading for the reaction. In order to further achieve higher conversion of biphenyl, a larger molar ratio of 1:4.5 of biphenyl/Bi(NO3)3·5H2O (0.02 mmol vs. 0.09 mmol) was investigated under the same condition (Figure 2B). Clearly, 100% conversion of biphenyl is achieved when 100 mg of MgSO4 is employed. This conversion is kept unchanged when the amount of MgSO4 is increased to 120 mg. Thus under our mechanochemical nitrating condition, the best ratio is 0.02 mmol of biphenyl, 0.09 mmol of Bi(NO3)3·5H2O, and 100 mg of MgSO4.
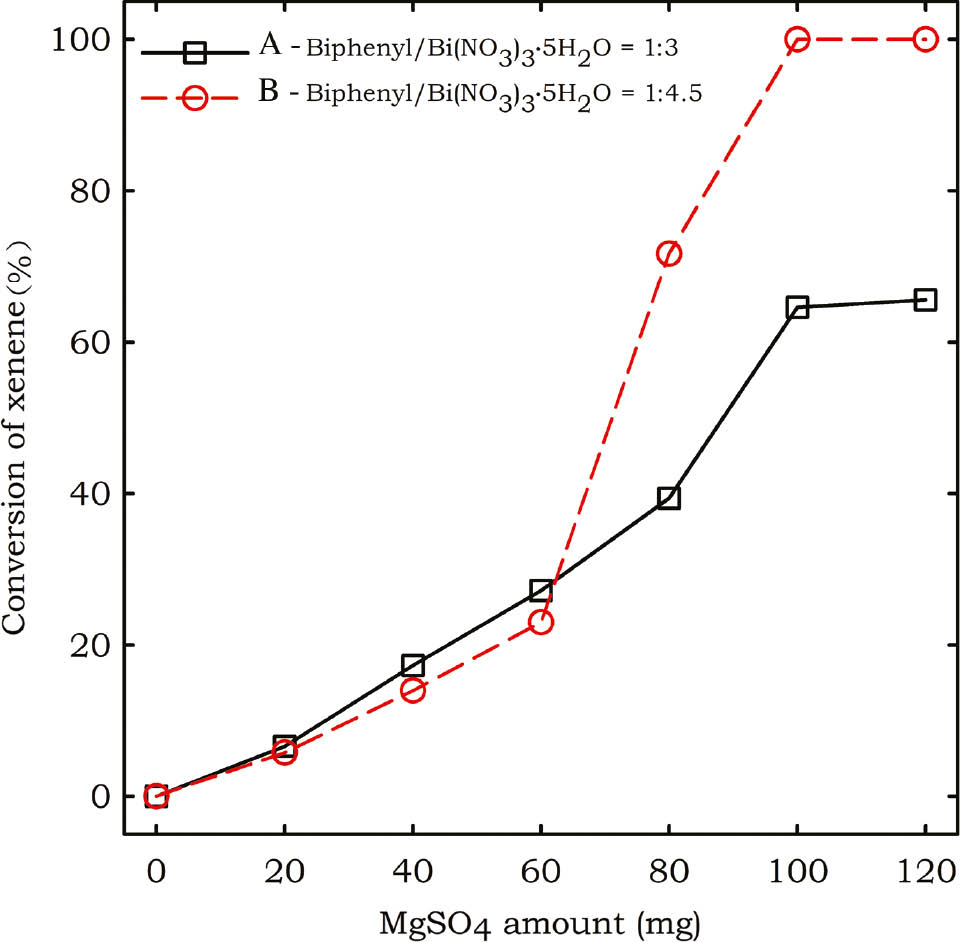
Influence of the MgSO4 amount on the conversion of biphenyl in the ball milling reaction.
In order to establish the generality of the methodology, the nitrating abilities of Bi(NO3)3·5H2O/MgSO4 system were investigated using a variety of aromatic compounds including activated and deactivated aromatics under the above optimized condition. Table 1 summarizes the results.
Bi(NO3)3·5H2O/MgSO4 nitration of aromatic compounds by ball milling (3′/13′ were performed by planetary ball mill, others were performed by mixer ball mill).
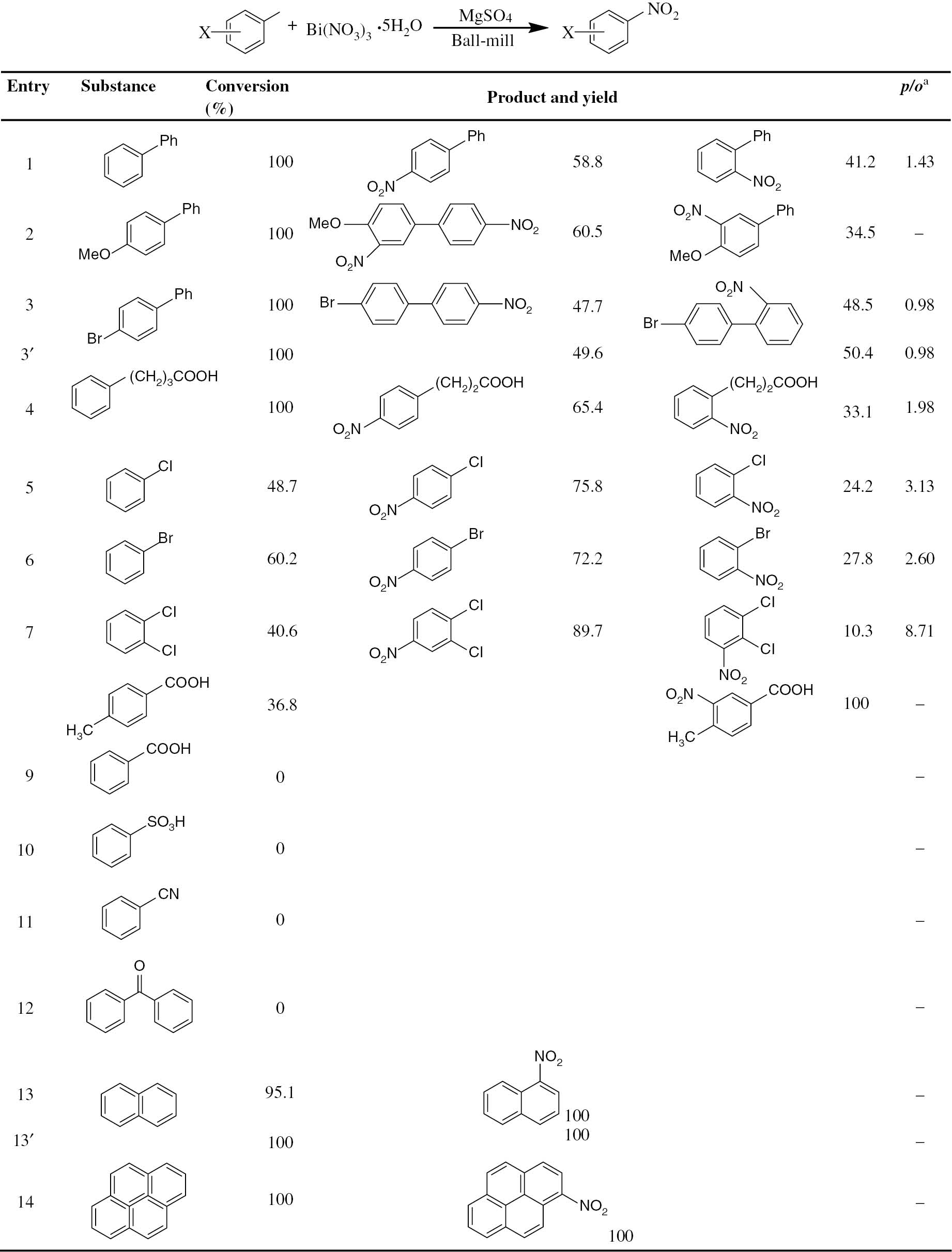
ap/o, para/ortho.
Apparently, under our mechanochemical nitrating condition, aromatics with weak activating electrophilic substituent such as phenyl (Entry 1) or alkyl (Entry 4) can perform nitrating very well. The conversion is 100%, giving only para- and ortho- products with para-compounds as the main products. For biphenyl with strong activating electrophilic substituent such as methoxyl (Entry 2), a bis-nitrated product is produced as the main product while the mono-nitrated product is also produced as expected. This suggests that aromatics with strong activation electrophilic substituents such as hydroxyl or animo group can be nitrated very easily under our mechanochemical nitrating condition. Meanwhile, polycyclic aromatics like naphthalene (Entry 13) and pyrene (Entry 14) can also perform nitrating reactions, resulting in mono-nitrating derivatives with quantitative yields. As for p-bromo modified biphenyl (Entry 3), the unsubstituted phenyl part reacts well and gives two nitrated products such as 4-nitro-4′-bromobiphenyl, and 2-nitro-4′-bromobiphenyl, respectively, which implies that it is not easy to nitrate aromatics with deactivating groups.
To our surprise, the nitration of either chlorobenzene (Entry 9) or bromobenzene (Entry 10) can be performed under the above mechanochemical nitrating condition, showing a moderate conversion around 50% with the para compounds as the main products. The ratio of para/ortho for nitrated chlorobenzene and bromobenzene is 3.13 and 2.60, respectively. These isomer ratios are very large compared with the counterparts of ordinary nitration reaction of chlorobenzene (e.g. 1.83) and bromobenzene (e.g. 1.30) by taking the mixed acids as the nitrating reagent [37]. As reported by Laali et al. [16], by taking Bi(NO3)3·5H2O as the nitrating reagent, the para/ortho ratio of nitrated chlorobenzene in ionic liquid (such as [bmim]PF6) or 1,2-dichloroethane is 1.08 and 1.56, respectively. For that of bromobenzene, the para/ortho ratio is 2.57 and 1.38, respectively. Thus by taking Bi(NO3)3·5H2O as the nitrating reagent and MgSO4 as the auxiliary, the mechanochemical nitrating reactions of chlorobenzene and bromobenzene are regioselective and the para derivatives are the major products. In the same condition, o-dichlorobenzene (Entry 7) can also be nitrated in 40.6% conversion to give the corresponding para- and ortho- substituted products with a para/ortho ratio of 8.71. Compared with Laali’s method in solution, our mechanochemical nitrating method provided another approach to realize the nitration of deactivated aromatics, which is solvent-free, time-saving, ecofriendly, easy to handle, and with better para regioselectivity.
While 4-methyl benzoic acid (Entry 8) can be nitrated to give 4-methyl-3-nitro benzoic acid in about 30% conversion and 100% yield, other aromatics modified with moderately deactivating substituents such as carboxylic group (Entry 9), sulfonic group (Entry 10), cyano group (Entry 11), and carbonyl group (Entry 12) cannot perform nitrating reaction in the above mechanochemical nitrating condition. This implies that the Bi(NO3)3·5H2O/MgSO4 system is not strong enough to nitrate deactivated aromatics.
In order to investigate whether the present method can be applied to a large-scale aromatic nitration, two substrates (Entry 3′ and 13′, Table 1) were tested using a QM-3SP04 planetary ball mill. Thus 100 molar times of each substrates was ball milled with Bi(NO3)3·5H2O/MgSO4 at the same molar ratio as described above. The reaction of 4-bromobiphenyl took 6 h to reach 100% conversion. The para/ortho ratio of the products is 0.98, exactly the same as the experimental results obtained above using the mixer ball mill. As for naphthalene, the reaction took only 40 min to get the final product of 1-nitronaphthalene quantitatively. The above results clearly show that the planetary ball mill works as well as the mixer ball mill, implying the possibility for applying this reaction in large scale.
It was reported by Hua et al. [22] that in their solid phase grinding reaction of phenols with Bi(NO3)3·5H2O at room temperature, the exothermic reaction resulted in higher temperature of 43°C. However, as for our experiments in planetary ball mill, the reaction was performed at an environmental temperature of 27.0°C, the temperatures of the mixtures for both two reactions were all between 30.4°C and 31.4°C, indicating no remarkable heat release under our reaction condition. Comparing with their reaction conditions, we believe that the addition of extra MgSO4 can distribute the reaction heat.
Currently, we do not know much about the mechanism of this reaction. During our experiments, we have observed the release of brown gas when the substances failed to give any nitrated product (for example, Entries 9–12 in Table 1). We believe this gas should be N2O4 or NO2. In this case, compared with other auxiliaries, we suggest that MgSO4 could somehow facilitate the thermal decomposition of Bi(NO3)3·5H2O, the in situ produced N2O4/NO2 should be responsible for the nitration, most probably, following the way reported by Kaupp and Schmegers [38]. More work still needs to be done in order to make this mechanism more clear.
In summary, we have developed a mechanochemical method for the nitration of aromatic compounds by taking Bi(NO3)3·5H2O as the nitrating agent and MgSO4 as the auxiliary. Aromatics with either weak activating groups or weak deactivating groups such as chloro- or bromo- could be nitrated with excellent or moderate yield. This method is green, effective, and could be performed on gram scale safely. Although the mechanism is not clear enough, we suggest that the in situ generated N2O4 or NO2 from the decomposition of Bi(NO3)3·5H2O should play a major role in this mechanochemical nitration.
Acknowledgments
The authors acknowledge the financial assistance from the Renmin University of China.
References
[1] Guggenheim,TL, Ed., Chemistry, Process Design, and Safety for the Nitration Industry, American Chemical Society: Washington DC, 2013.10.1021/bk-2013-1155Search in Google Scholar
[2] Yan G, Yang M. Org. Biomol. Chem. 2013, 11, 2554–2566.Search in Google Scholar
[3] Vekariya RH, Patel HD. Synth. Commun. 2014, 44, 2313–2335.10.1080/00397911.2014.896925Search in Google Scholar
[4] Kulkarni AA. Beilstein J. Org. Chem. 2014, 10, 405–425.10.3762/bjoc.10.38Search in Google Scholar PubMed PubMed Central
[5] Rajagopal R, Srinivasan KV. Synth. Commun. 2003, 33, 961–966.10.1081/SCC-120016360Search in Google Scholar
[6] Nowrouzi N, Mehranpour AM, Bashiri E, Shayan Z. Tetrahedron Lett. 2012, 53, 4841–4842.10.1016/j.tetlet.2012.06.126Search in Google Scholar
[7] Zolfigol MA, Ghaemi E, Madrakian E. Synth. Commun. 2000, 30, 1689–1694.10.1080/00397910008087210Search in Google Scholar
[8] Shackelford SA, Anderson MB, Christie LC, Goetzen T, Guzman MC, Hananel MA, Kornreich WD, Li H, Pathak VP, Rabinovich AK, Rajapakse RJ, Truesdale LK, Tsank SM, Vazir HN. J. Org. Chem. 2003, 68, 267–275.10.1021/jo026202qSearch in Google Scholar PubMed
[9] Zolfigol MA, Ghaemi E, Madrakian E. Molecules 2001, 6, 614–620.10.3390/60700614Search in Google Scholar
[10] Zolfigol MA, Madrakian E, Ghaemi E. Molecules 2002, 7, 734–741.10.3390/71000734Search in Google Scholar
[11] Zolfigol MA, Ghaemi E, Madrakian E. Synlett. 2003, 2003, 191–194.10.1055/s-2003-36791Search in Google Scholar
[12] Anuradha V, Srinivas PV, Aparna P, Rao JM. Tetrahedron Lett. 2006, 47, 4933–4935.10.1016/j.tetlet.2006.05.017Search in Google Scholar
[13] Hajipour AR, Zarei A, Ruoho AE. Russian J. Org. Chem. 2005, 41, 1493–1498.10.1007/s11178-005-0372-ySearch in Google Scholar
[14] Wasinska M, Korczewska A, Giurg M, Skarzewski J. Synth. Commun. 2015, 45, 143–150.10.1080/00397911.2014.954730Search in Google Scholar
[15] Lu Y, Li Y, Zhang R, Jin K, Duan C. Tetrahedron 2013, 69, 9422–9427.10.1016/j.tet.2013.08.076Search in Google Scholar
[16] Jacoway J, Kumar GGKSN, Laali KK. Tetrahedron Lett. 2012, 53, 6782–6785.10.1016/j.tetlet.2012.09.137Search in Google Scholar
[17] Ravi P, Tewari SP. Catal. Commun. 2012, 19, 37–41.10.1016/j.catcom.2011.12.016Search in Google Scholar
[18] Yadav RR, Vishwakarma RA, Bharate SB. Tetrahedron Lett. 2012, 53, 5958–5960.10.1016/j.tetlet.2012.08.121Search in Google Scholar
[19] Manna S, Maity S, Rana S, Agasti S, Maiti D. Org. Lett. 2012, 14, 1736–1739.10.1021/ol300325tSearch in Google Scholar PubMed
[20] Baghernejad B, Oskooie HA, Heravi MM, Beheshtiha YS. Chin. J. Chem. 2010, 28, 393–396.10.1002/cjoc.201090085Search in Google Scholar
[21] Badgujar DM, Talawar MB, Asthana SN, Venugopalan S, Rao AS, Mahulikar PPJ. J. Hazard. Mater. 2008, 152, 820–825.10.1016/j.jhazmat.2007.07.045Search in Google Scholar PubMed
[22] Sun HB, Hua R, Yin Y. J. Org. Chem. 2005, 70, 9071–9073.10.1021/jo0514669Search in Google Scholar
[23] Samajdar S, Becker FF, Banik BK. Tetrahedron Lett. 2000, 41, 8017–8020.10.1016/S0040-4039(00)01397-6Search in Google Scholar
[24] Do JL, Friscic T. ACS Cent. Sci. 2017, 3, 13–19.10.1021/acscentsci.6b00277Search in Google Scholar
[25] Stolle, A, Ed., Ball Milling Towards Green Synthesis: Applications, Projects, Challenges, The Royal Society of Chemistry: Cambridge, 2015.10.1039/9781782621980Search in Google Scholar
[26] Wang GW. Chem. Soc. Rev. 2013, 42, 7668–7700.10.1039/c3cs35526hSearch in Google Scholar
[27] Pan HL, Liu LQ, Guo ZX, Dai LM, Zhang FS, Zhu DB, Czerw R, Carroll DL. Nano Lett. 2003, 3, 29–32.10.1021/nl025856bSearch in Google Scholar
[28] Zhang P, Pan HL, Liu DF, Guo ZX, Zhang FS, Zhu DB. Synth. Commun. 2003, 33, 2469–2474.10.1081/SCC-120021836Search in Google Scholar
[29] Li XL, Liu LQ, Qin YJ, Wu W, Guo ZX, Dai LM, Zhu DB. Chem. Phys. Lett. 2003, 377, 32–36.10.1016/S0009-2614(03)01088-1Search in Google Scholar
[30] AIST: Integrated Spectral Database System of Organic Compounds. (Data were obtained from the National Institute of Advanced Industrial Scienceand Technology (Japan)).Search in Google Scholar
[31] Minard C, Palacio C, Cariou K, Dodd RH. Eur. J. Org. Chem. 2014, 14, 2942–2955.10.1002/ejoc.201400090Search in Google Scholar
[32] Strazzolini P, Giumanini AG, Runcio A, Scuccato M. J. Org. Chem. 1998, 63, 952–958.10.1021/jo9709763Search in Google Scholar
[33] Murudkar S, Mora AK, Singh PK, Nath S. J. Phys. Chem. A 2011, 115, 10762–10766.10.1021/jp205946cSearch in Google Scholar PubMed
[34] Schmidt R, Stolle A, Ondruschka B. Green Chem. 2012, 14, 1673–1679.10.1039/c2gc16508bSearch in Google Scholar
[35] Thorwirth R, Stolle A, Ondruschka B. Green Chem. 2010, 12, 985–991.10.1039/c000674bSearch in Google Scholar
[36] Thorwirth R, Bernhardt F, Stolle A, Ondruschka B, Asghari J. Chem. Eur. J. 2010, 16, 13236–13242.10.1002/chem.201001702Search in Google Scholar PubMed
[37] McMurry J. Organic Chemistry, 9th ed., Cengage Learning: Boston, 2016, p 494.Search in Google Scholar
[38] Kaupp G, Schmegers J. J. Org. Chem. 1995, 60, 5494–5503.10.1021/jo00122a031Search in Google Scholar
Supplementary Material
The online version of this article offers supplementary material (https://doi.org/10.1515/gps-2017-0069).
©2018 Walter de Gruyter GmbH, Berlin/Boston
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Articles in the same Issue
- Frontmatter
- In this issue
- ASAM-6
- The 6th Asian Symposium on Advanced Materials: Chemistry, Physics and Biomedicine of Functional and Novel Materials (ASAM-6; Hanoi, Vietnam, September 27–30, 2017)
- Synthesis and characterization of (4-arm-star-PMMA)/PMMA-g-SiO2 hybrid nanocomposites
- Factors influencing green strength of commercial natural rubber
- Removal of arsenic from water using crumpled graphite oxide
- Adsorption behavior of Cd2+ ions using hydroxyapatite (HAp) powder
- Study on characteristics, properties, and morphology of poly(lactic acid)/chitosan/hydroquinine green nanoparticles
- Original articles
- Green synthesis and stabilization of earthworm-like gold nanostructure and quasi-spherical shape using Caesalpinia sappan Linn. extract
- Catalytic performance of Ag, Au and Ag-Au nanoparticles synthesized by lichen extract
- Comparative kinetics of the alkali-catalyzed sunflower oil methanolysis with co-solvent under conventional and microwave heating with controlled cooling
- Facile nitration of aromatic compounds using Bi(NO3)3·5H2O/MgSO4 under mechanochemical conditions
- Optimization of the treatment process of zinc leaching residue by using the response surface method
- A new green process to produce activated alumina by spray pyrolysis
Articles in the same Issue
- Frontmatter
- In this issue
- ASAM-6
- The 6th Asian Symposium on Advanced Materials: Chemistry, Physics and Biomedicine of Functional and Novel Materials (ASAM-6; Hanoi, Vietnam, September 27–30, 2017)
- Synthesis and characterization of (4-arm-star-PMMA)/PMMA-g-SiO2 hybrid nanocomposites
- Factors influencing green strength of commercial natural rubber
- Removal of arsenic from water using crumpled graphite oxide
- Adsorption behavior of Cd2+ ions using hydroxyapatite (HAp) powder
- Study on characteristics, properties, and morphology of poly(lactic acid)/chitosan/hydroquinine green nanoparticles
- Original articles
- Green synthesis and stabilization of earthworm-like gold nanostructure and quasi-spherical shape using Caesalpinia sappan Linn. extract
- Catalytic performance of Ag, Au and Ag-Au nanoparticles synthesized by lichen extract
- Comparative kinetics of the alkali-catalyzed sunflower oil methanolysis with co-solvent under conventional and microwave heating with controlled cooling
- Facile nitration of aromatic compounds using Bi(NO3)3·5H2O/MgSO4 under mechanochemical conditions
- Optimization of the treatment process of zinc leaching residue by using the response surface method
- A new green process to produce activated alumina by spray pyrolysis