Endothelin systems in the brain: involvement in pathophysiological responses of damaged nerve tissues
-
Yutaka Koyama
Yutaka Koyama graduated from the Faculty of Pharmacy, Osaka University (Japan). He studied Neuropharmacology at the Graduate School of Pharmaceutical Sciences, Osaka University (1985–1989) and was awarded a PhD in Pharmaceutical Sciences. Thereafter, he worked at the Graduate School of Pharmaceutical Sciences, Osaka University as an Assistant Professor (1989–2001) and as an Associate Professor (2001–2006). During this career, he joined the Department of Pathology, at the College of Physicians and Surgeons, Columbia University as an invited researcher (1997–1998). In 2006, he became a Professor of Laboratory of Pharmacology, Faculty of Pharmacy, Osaka Ohtani University where he has been investigating pharmacological significance of glial cells as a target of neuroprotective drugs.


Abstract
In addition to their potent vasoconstriction effects, endothelins (ETs) show multiple actions in various tissues including the brain. The brain contains high levels of ETs, and their production is stimulated in many brain disorders. Accumulating evidence indicates that activation of brain ET receptors is involved in several pathophysiological responses in damaged brains. In this article, the roles of brain ET systems in relation to brain disorders are reviewed. In the acute phase of stroke, prolonged vasospasm of cerebral arteries and brain edema occur, both of which aggravate brain damage. Studies using ET antagonists show that activation of ETA receptors in the brain vascular smooth muscle induces vasospasm after stroke. Brain edema is induced by increased activity of vascular permeability factors, such as vascular endothelial growth factor and matrix metalloproteinases. Activation of ETB receptors stimulates astrocytic production of these permeability factors. Increases in reactive astrocytes are observed in neurodegenerative diseases and in the chronic phase of stroke, where they facilitate the repair of damaged nerve tissues by releasing neurotrophic factors. ETs promote the induction of reactive astrocytes through ETB receptors. ETs also stimulate the production of astrocytic neurotrophic factors. Recent studies have shown high expression of ETB receptors in neural progenitors. Activation of ETB receptors in neural progenitors promotes their proliferation and migration, suggesting roles for ETB receptors in neurogenesis. Much effort has been invested in the pursuit of novel drugs to induce protection or repair of damaged nerve tissues. From these studies, the pharmacological significance of brain ET systems as a possible target of neuroprotective drugs is anticipated.
Introduction
Since their discovery as a novel peptide family (1), the functions of endothelins (ETs) have been intensively investigated in the circulatory system, because of their potent vasoconstriction effects. However, soon after their discovery, it was shown that ET ligands and ET receptors are present in various tissues, and multiple functions for ET systems were postulated (2). Accumulating evidence indicates regulatory roles for ETs in functions other than vascular tone, such as hypertrophy, fibrosis, inflammation, and various other physiological and pathological functions. Several ET receptor agonists and antagonists have been developed (Table 1). At present, there are many selective ET agonists and antagonists, some of which are now used clinically (3, 4). Since the discovery of ETs, ET ligands and their receptors have been known to be highly expressed in the brain (5, 6). Therefore, specialized roles for ET systems in the nervous system have been postulated. In addition to roles in neurotransmission and embryonic development, investigations of brain ET systems have shown their significance in brain disorders including Alzheimer’s disease (AD) and stroke (7–9). Development of effective treatments for brain dysfunctions in neurodegenerative diseases and stroke is still greatly needed. Many neuroscientists are engaged in searches for novel targets of neuroprotective drugs. In this article, recent studies examining the possible roles of brain ET systems in the pathophysiological responses of damaged brains are reviewed.
Agonists and antagonists for ET receptors.
| Agonists | Antagonists | |
|---|---|---|
| ET non-selective | ET-1 | SB209670, bosentan, TAK-044, tezosentan |
| ETA selective | Sarafotoxin 6b | BQ123, darusentan, ambrisentan, sitaxsentan, clazosentan, S-0139, SB234551, Ro-61-1790 |
| ETB selective | Sarafotoxin 6c, IRL-1620, | BQ788, IRL-2500, |
| BQ3020, Ala1,3,11,15-ET-1 | A-192621, RES-701-1 |
ET ligands
The ET peptide family consists of three isopeptides: ET-1, ET-2, and ET-3 (Figure 1). These endogenous ET ligands have a structural similarity; they comprise 21 amino acids with two disulfide bonds. In humans, ET-1, ET-2, and ET-3 are encoded as large precursor proteins, prepro-ETs, by distinct genes. The biological actions of ETs are mediated by two types of receptors: ETA and ETB receptors. Because ET-1 is the most abundant isopeptide in many tissues, many investigations of ET ligands have focused on ET-1. ET-1 activates both ETA and ETB receptors, and induces biological effects including vasoconstriction and proliferation (2–4). ET-2 differs from ET-1 by two amino acids, but has similar receptor selectivity to ET-1. ET-2 is highly expressed in intestine, ovary, and pituitary glands (10–12). Owing to similar receptor selectivity, the biological actions of ET-2 were originally thought to overlap with ET-1. However, recent studies have shown that ET-2 has distinct actions from ET-1 (13, 14). ET-3, which differs from ET-1 by six amino acids, is abundant in the intestine, lung, and brain (2, 7). ET-3 has high affinity for ETB receptors, but not ETA receptors, indicating that it is an endogenous ETB ligand. Gene knockout of either ET-3 or ETB receptors in mice results in a similar impairment in enteric neuron development (15, 16). These mouse phenotypes resemble Hirschsprung’s disease, and in accordance with this, patients with Hirschsprung’s disease have a mutated ETB receptor gene (17–19).

Biosynthesis of human ETs from prepro-ETs.
ET-1 is translated as an inactive precursor protein called prepro-ET-1. Prepro-ET-1 is cleaved by dibasic pair-specific endopeptidases and converted to big-ET-1. Specific processing of big-ET-1 by endothelin-converting enzymes (ECEs) results in production of mature ET-1. There are three distinct ET family peptides, ET-1, ET-2, and ET-3, all of which consist of 16 amino acids and two intramolecular disulfide bonds and are produced by a similar process to ET-1.
As occurs for many peptide hormones, the active forms of ETs are produced by cleavage of their precursor peptides (Figure 1). For human ET-1, the 212 amino acid inactive precursor protein (prepro-ET-1) is translated from its mRNA. Prepro-ET-1 is cleaved by a dibasic pair-specific endopeptidase at Lys51-Arg52 and Arg92-Arg93 to produce a 38 amino acid precursor, big-ET-1. The active form of ET-1 is produced from big ET-1 after processing at Trp21-Val22 by specific proteases called endothelin converting enzymes (ECEs). Although the three ET isopeptides are encoded by distinct genes, production of ET-2 and ET-3 is mediated by a similar process to ET-1 (Figure 1) (20, 21).
Regulation of ET production
In vascular endothelial cells, mature ET-1 is continuously released through a constitutive pathway. The rate-limiting step of ET production is therefore thought to be at transcription of the prepro-ET-1 gene. Human prepro-ET-1 genes have five exons and a 5′-flanking region, spanning approximately 6.8 kb of DNA (Figure 2A) (22). Examination of regulation of ET-1 expression showed that transcription of prepro-ET-1 mRNA is regulated by various bioactive substances released by damaged tissues, including cytokines and hormones. Transforming growth factor β (TGFβ), thrombin, bradykinin, and tumor necrosis factor α (TNFα) stimulate transcription of the prepro-ET-1 gene in vascular endothelial cells (23–27). Some physiological and pathological conditions, such as hypoxia (28) and mechanical stress (29), also upregulate prepro-ET-1 mRNA levels. The ET-1 gene is under the transcriptional control of a TATA box at the position of 31 bases upstream from the transcription start site. Analysis of the 5′-flanking region revealed that the ET-1 gene has consensus sequences for binding of various transcription factors, including activator protein-1 (AP-1), GATA-2, Smad, hypoxia inducible factor-1α (HIF1α), and nuclear factor κB (NFκB) (30). These transcription factors coordinately couple intracellular signals triggered by extracellular stimuli to transcription of the prepro-ET-1 gene.
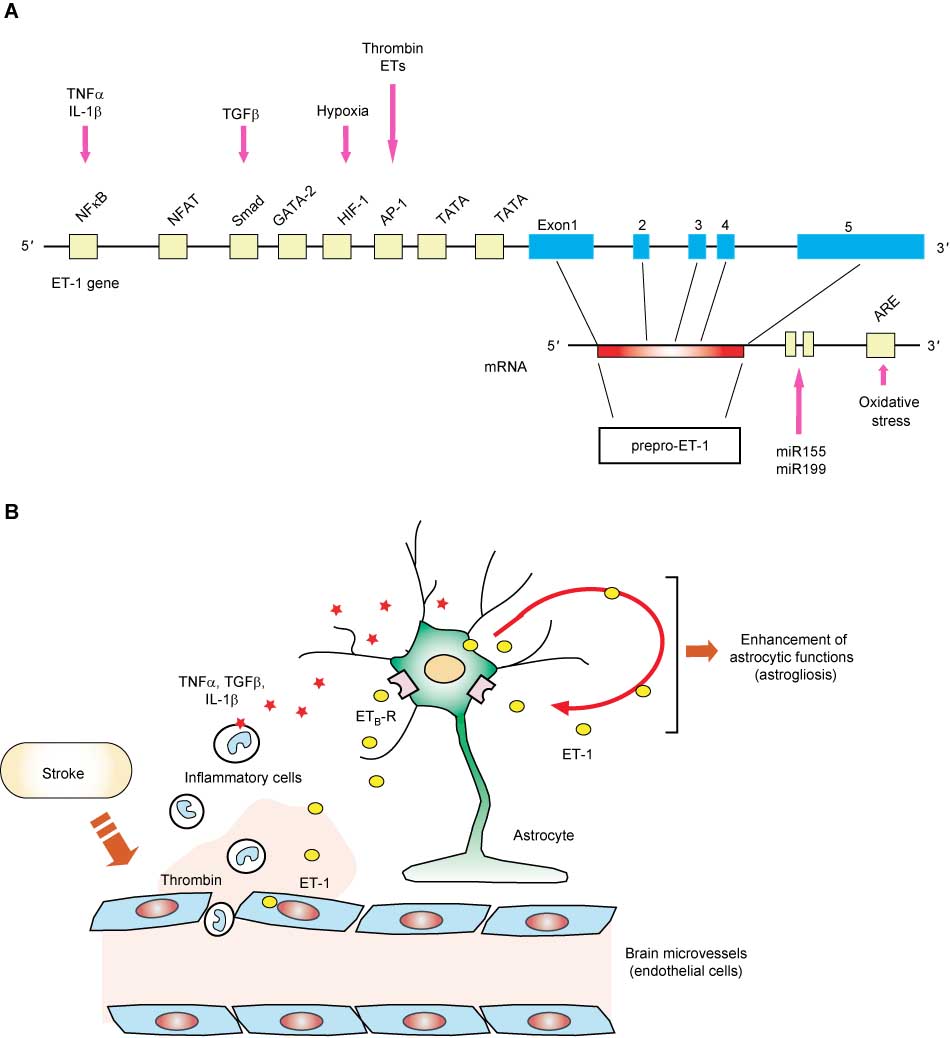
Signaling mechanisms stimulating ET-1 production.
(A) Transcriptional and post-transcriptional regulation of ET-1 gene expression. In the 5′-promoter region of the human ET-1 gene, recognition sites for several transcriptional factors, including NFκB, NFAT, GATA, HIFα, TATA, and AP-1, are present. Signaling molecules stimulating ET-1 production in vascular endothelial cells and astrocytes are indicated, along with transcription factors mediating their actions. In the 3′-untranslated region of prepro-ET-1 mRNA, an AU-rich element (ARE) and complementary sequences for microRNAs are present. Expression levels of prepro-ET-1 mRNA are also regulated by alteration of stability by ARE-binding proteins and microRNAs. (B) Expression of ET-1 in stroke. In the early stage of stroke, inflammatory cells enter nerve tissues across the disrupted blood–brain barrier. TNFα, IL-1β, and TGFβ are produced by blood-derived inflammatory cells. Increases in these cytokines, together with thrombin, stimulate ET-1 production in brain endothelial cells and astrocytes. Hypoxia following stroke induces brain ET-1 production. Increases in ET-1 stimulate several functions of astrocytes through ETB receptors, including astrocytic ET production via an autocrine mechanism.
Inoue et al. reported that prepro-ET-1 mRNA in vascular endothelial cells is rapidly degenerated with a half-life of approximately 15 min (22). Expression of prepro-ET-1 mRNA is also regulated by alterations of mRNA stability. Human prepro-ET-1 mRNA has an AU-rich element (ARE) in the 3′-untranslated region (22), which is required for ARE-binding proteins to degenerate transcripts. ARE-binding proteins, such as AU-binding factor 1 (AUF1) and glyceraldehyde-3-phosphate dehydrogenase (G3PDH), induce a rapid degradation of prepro-ET-1 mRNA (31, 32). Regulation by AUF1 and G3PDH are suggested to be involved in ET-1 production induced by heat shock and oxidative stress, respectively (31, 32). Recent studies proposed that expression of prepro-ET-1 mRNA is also regulated by microRNAs (miRNAs). Analysis of the 3′-untranslated region of prepro-ET-1 mRNA revealed the presence of complementary binding sequences for miRNAs. Yeligar et al. found that overexpression of miR199 and miR155 in endothelial cells reduced expression of prepro-ET-1 mRNA (33).
ET receptor subtypes
Receptors for ETs are classified into ETA and ETB types (Figure 3). These ET receptors differ in their selectivity for ET ligands. ETA receptors have ligand preferences of ET-1=ET-2>>ET-3, whereas ETB receptors show equal selectivity for the three ET ligands. High expression of ETA receptors is observed in the vascular smooth muscle and cardiocytes (4). Activation of vascular ETA receptors is responsible for the potent vasoconstriction caused by ETs. ETB receptors are abundantly expressed in the vascular endothelium, kidney, lung, and brain. Activation of ETB receptors in endothelial cells causes vasodilatation by releasing nitric oxide (NO) and prostacyclins (34). ETA and ETB receptors are therefore suggested to have opposing roles in the regulation of vascular tone (35). Both ET receptor subtypes are G-protein-coupled receptors. Through G-protein-mediated mechanisms, ETs induce activation of phospholipase C and increased cytosolic Ca2+ (36), which are the main signaling pathways for the regulation of vascular tone. ETs also activate mitogenic and survival signals in various cell types. These signal molecules include mitogen-activated protein kinases (MAPKs), Akt, and Src (36). Daub et al. first reported that stimulation of ET receptors activates epidermal growth factor (EGF) receptors by EGF-independent mechanisms (37). Such ‘transactivation’ of growth factor receptors by ETs has been observed in glomerular mesangial cells (38), vascular smooth muscle cells (39), and ovarian cells (40). The transactivation is thought to be involved in the action of ETs as mitogenic and survival factors.

Ligand selectivity of ET receptor subtypes.
ETA receptor subtype, which is predominantly expressed in smooth muscle cells and cardiocytes, has ligand selectivity for ET-1 and ET-2. ETB receptors are expressed in the vascular endothelium, kidney, lung, and brain. Ligand selectivity of ETB receptor is different from that of ETA type; ET-1, ET-2, and ET-3 equally activate it.
The ET system in the central nervous system
ET production in the brain
Nerve tissues abundantly express ET-1 and ET-3 (6, 7). In normal brain, immunohistochemical observations have shown the presence of ET-containing neurons in the spinal cord (41), perivascular neurons of the basilar artery (42), and in the hypothalamic-neurosecretory system (43). ET-1 induces excitation of neurons in the spinal cord and trigeminal system (44, 45). These findings suggest roles for ETs in neurotransmission, which Dashwood and Loesch recently presented a detailed review (46). Increases in brain ETs are observed in nerve injury animal models (47, 48). In human brain, increased levels of ET-1 in cerebrospinal fluid (CSF) are reported in stroke, head trauma, and neurodegenerative diseases (8, 49, 50). Because of their increased production in brain disorders, pathophysiological roles of ETs have also been examined. Immunohistochemical observations of damaged brains have shown that ETs are produced by brain microvessel endothelial cells and astrocytes (9). Factors such as TNFα, interleukin-1β (IL-1β), and thrombin, as well as hypoxia, which results in brain damage, induce ET-1 production in brain microvessel endothelial cells (51) and astrocytes (52–54). ETs are also known to stimulate astrocytic ET-1 production (55). ET-induced ET production indicates that astrocytic ET production is potentiated by an autocrine mechanism, via astrocyte-derived ET-1 (Figure 2B).
ECEs in the brain
Big-ETs are cleaved by ECEs to become active ETs. There are two subtypes of ECEs: ECE-1 and ECE-2, which share 59% amino acid homology (56). ECE-1 is widely expressed in various tissues including nerve tissues. In contrast to the ubiquitous expression of ECE-1, ECE-2 expression is restricted to nerve tissues. Rodriguez et al. found high levels of expression of ECE-2 mRNA in the midbrain, pituitary, hypothalamus, and cerebellum of mammalian brains (57). ECEs can also cleave other bioactive peptides, such as neuropeptides and amyloid β (Aβ) proteins. Based on this substrate selectivity, roles for brain ECEs other than ET production have been proposed. Accumulation of Aβ proteins, components of the amyloid plaques observed in brains of AD patients, is considered to be a pathogenesis of AD. Deletion mutations of either ECE-1 or ECE-2 increase Aβ1–40 and Aβ1–42 proteins in mouse brain (58). ECE-2 knockout mice show impaired learning and memory (57). In addition, upregulation of ECE-2 expression is reported in brains of AD patients (59). On the basis of these findings, brain ECEs are thought to regulate Aβ protein turnover and be involved in the pathogenesis of AD (60). Neuropeptides such as bradykinin, neurotensin, and substance P are also substrates of ECEs. Roosterman et al. proposed a novel hypothesis that degradation of neuropeptides by ECEs in endosomes promotes recycling and resensitization of internalized neuropeptide receptors (61). These neuropeptides cleaved by ECEs cause neurogenic inflammation. In fact, inhibition of ECEs is shown to decrease substance P-induced plasma extravasation in rats, suggesting that ECE inhibitors impair neurogenic inflammation by reducing resensitization of neuropeptide receptors (62).
ET receptors in the brain
Both ETA and ETB receptors are present in the brain, although with different cellular distributions. Brain ETA receptors are expressed in the vascular smooth muscle and mediate the potent vasoconstriction effects of ETs. Local application of ET-1 to the cerebral artery induced prolonged vasoconstriction and reduction of cerebral blood flow in rat and pig, through ETA receptors (63, 64). Owing to the likelihood of hypoxia, reduction of cerebral blood flow often leads to brain dysfunction. Taking advantage of its vasoconstriction effect, application of ET-1 to animal brains is used as a model for ischemic brain injury (63). In several brain disorders, production of ETs is increased (7, 8). ETA receptor-mediated reduction of cerebral blood flow is believed to play a role in the aggravation of brain dysfunction caused by ischemia in many brain disorders (see below). In some populations of neurons, ETA receptors have been shown to modulate neural transmission. For example, in isolated primary sensory neurons, ET-1 causes excitation and increases in cytosolic Ca2+ through ETA receptors (65). Consistent with ET-induced excitation of sensory neurons, administration of ET-1 caused pain-like behavior in rats, and the effect was antagonized by BQ123, an ETA antagonist (66). This action of ET-1 suggests a novel role of ETs, potentiating transduction of pain signals in somatosensory systems (67, 68).
ETB receptors are the prominent type in the brain. High expression of brain ETB receptors is observed in astrocytes (69–71). In addition, expression of astrocytic ETB receptors is upregulated after brain injury (70), which indicates that the roles of ETs in astrocytes are more significant in brain pathologies. ETs are known to be a potent mitogen of astrocytes; in cultured astrocytes, ETs induce cell cycle progression through ETB receptors (72, 73). Activation of ETB receptors also causes morphological alterations, accompanied by cytoskeletal reorganization (74, 75) and reduction of gap junctional communication (76, 77). Astrocytes produce and release various bioactive substances, for example, neurotrophic factors, cytokines, chemokines, NO, and vascular permeability factors, through which they interact with neurons and brain microvessels (78). Production of astrocyte-derived molecules is stimulated by activation of astrocytic ETB receptors (79). ETB receptors are also expressed in other brain cells. Recent studies have proposed roles for ETs in the development of oligodendrocytes and neurons (80, 81). Nishikawa et al. reported high expression of ETB in embryonic cortical neuronal progenitors (82).
Roles of ETs in brain pathology
Because expression of brain ET ligands and receptors increases in brain pathologies, many investigations of ETs have focused on brain disorders. There is accumulating experimental evidence that modulation of ET systems has beneficial effects on brain dysfunction in stroke and neurodegenerative diseases.
Vasospasm and brain edema
The concentration of ET-1 in CSF is increased after stroke (50, 83). Franceschini et al. reported a correlation between ET-1 concentration in CSF and brain infarct volume after subarachnoid hemorrhage (SAH) (84). Vasospasm of the cerebral artery often occurs after stroke. Because prolonged cerebral vasospasm aggravates ischemic brain damage, inhibition of vasospasm is a possible therapeutic strategy for neuroprotective drugs. In animal models of brain ischemia, ETA receptor antagonists prevent reduction of cerebral blood flow and improve ischemic brain damage (85–88). These findings indicate that activation of brain ETA receptors induces cerebral vasospasm in brain insults (Figure 4). After SAH and head trauma, vasogenic brain edema also occurs. Vasogenic brain edema is caused by influx of blood proteins across the disrupted blood–brain barrier (BBB). Accumulation of brain edematous fluid elevates intracranial pressure, which disrupts neuronal function and can result in death. In normal conditions, the BBB is maintained by low permeability of brain microvessel endothelial cells. The permeability of brain microvessels is not static, but is dynamically modulated by various permeability factors. In brain pathologies, such as SAH and head trauma, the actions of permeability factors are increased. By excess actions of permeability factors, the barrier functions of brain microvessels are disrupted, which results in vasogenic brain edema (89). Vascular endothelial growth factor (VEGF) and matrix metalloproteinases (MMPs) are the main factors which increase permeability of vascular endothelial cells. In brain pathologies, astrocytic production of MMPs and VEGF is increased (90). ETs stimulate production of MMPs and VEGF in cultured astrocytes and rat brain through ETB receptors (91–94). On the basis of these findings, ETs are proposed to be important factors in the development of vasogenic edema after stroke (Figure 4). Consistent with this idea, brain edema formation in rat models of brain ischemia and head trauma is reduced by ET antagonists (85, 95).

Involvement of ET receptors in vasospasm and brain edema formation after stroke.
In the early stage of stroke, increased ETs activate ETA receptors in the brain vascular smooth muscle and induce vasospasm. Prolonged vasospasm leads to brain ischemia. ETs also stimulate ETB receptors in astrocytes. Activation of ETB receptors increases production of vascular permeability factors (VEGF and MMPs) in astrocytes. ET-induced production of astrocytic vascular permeability factors induces disruption of the BBB, which results in brain edema formation. Thus, in the early stage of stroke, ETs are thought to aggravate brain damage by inducing vasospasm and brain edema.
As described above, studies using animal models suggest that administration of ET antagonists has beneficial effects on neuronal damage in the acute phase of stroke. On the basis of these results in animal models, clinical trials of ET antagonists in SAH patients have been conducted. Clazosentan, an ETA selective antagonist, was reported to reduce vasospasm and delayed ischemic neurological defects in SAH patients (96, 97). TAK-044, a non-selective ET antagonist, lowered the incidence of delayed neurological defects in SAH patients 3 months after the onset of symptoms (98).
Induction of reactive gliosis
In brain ischemia, head trauma, and neurodegenerative diseases, phenotypic conversion of astrocytes to the reactive type occurs, which is known as reactive gliosis (90). Reactive astrocytes are characterized by hypertrophy of the cell body and glial processes due to reorganization of cytoskeletal proteins. Conversion to reactive astrocytes is accompanied by functional alterations of astrocytes. Reactive astrocytes produce various bioactive substances, including neurotrophic factors, cytokines, chemokines, and proteases (78). Because these astrocyte-derived substances regulate neuronal viability, neuroinflammation, and repair of nerve tissues, preventing the induction of reactive astrocytes is a potential therapeutic target for many brain disorders. In addition to the enhanced production of bioactive substances, reactive astrocytes show a proliferative phenotype. Hyperplasia of reactive astrocytes leads to glial scar formation in damaged nerve tissues. Glial scars inhibit the repair of the damaged nervous system, by preventing axonal elongation and acting as a physical barrier to synaptogenesis. Administration of Ala1,3,11,15-ET-1, an ETB selective agonist, increased the number of reactive astrocytes in rat brain (99, 100). Induction of reactive astrocytes in stab wound brain injury was reduced by BQ788, an ETB antagonist (101). These findings indicate that, in brain disorders, activation of astrocytic ETB receptors is involved in phenotypic conversion to reactive astrocytes. Activation of ETB receptors reproduces functional alterations of reactive astrocytes in vitro. Proliferation of cultured astrocytes was stimulated by activation of ETB receptors (72, 73). In cultured astrocytes, ETs also induced reorganization of cytoskeletal proteins and altered morphology (74, 75), actions considered to be related to hyperplasia and hypertrophy of reactive astrocytes, respectively (79).
Expression of cyclin D proteins is increased in the late G1 phase and promotes G1/S phase cell cycle progression. Expression of astrocytic cyclin D1 and D3 was stimulated by activation of ETB receptors (77, 102). Examination of ETB receptor signals showed that different mechanisms play a role between astrocytic cyclin D1 and D3 expression. ET-induced astrocytic cyclin D1 expression was inhibited by disruption of cytoskeletal actin, suggesting an involvement of cell adhesion-independent mechanisms (102). In cell adhesion-independent mechanisms, extracellular signal-regulated kinases and protein kinase C mediated astrocytic cyclin D1 expression by ETs (102). By contrast, ET-induced expression of astrocytic cyclin D3 required integrity of cytoskeletal actin (77). ETs activated focal adhesion kinase (FAK), a key tyrosine kinase in cell adhesion-dependent proliferation, in cultured astrocytes (103). Expression of dominant-negative FAK mutants in cultured astrocytes prevented ET-induced G1/S cell cycle progression and cyclin D3 expression (102, 104). These findings indicate that ETs stimulate cyclin D3 expression and proliferation of cultured astrocytes through FAK-mediated adhesion-dependent mechanisms. Thus, ETs can activate both cell adhesion-dependent and -independent mechanisms in astrocytes. This cooperative action of two distinct signal pathways may underlie the potent mitogenic effects that ETs have on astrocytic proliferation.
Neuronal survival and neurogenesis
In addition to during development, the viability of neuronal cells in the adult brain is maintained by various trophic factors. A decline of these trophic signals increases the vulnerability of neuronal cells to other detrimental factors and cause neuronal death by apoptosis. Apoptotic neuronal death underlies the deficits in brain function in stroke and neurodegenerative diseases. In ETB-deficient rats, increases in apoptotic neurons were observed in the cerebellum (81) and dentate gyrus (105). Activation of ETB receptors showed antiapoptotic actions in cultured neurons of the olfactory bulb and cerebrum (106–108). As for mechanisms underlying the antiapoptotic actions of ETB receptor-triggered signals, inhibition of caspase-3 and voltage-dependent L-type Ca2+ channels was suggested (106–108). A histological observation on the dentate gyrus of rabbit and human meningitis by Ehrenreich et al. showed that increases in apoptotic death of dentate neurons were associated with reduction of neuronal ETB receptors (105). These findings indicate that activation of ETB receptors directly triggers trophic signals in some populations of neurons. In the hippocampus and subventricular zone of the lateral ventricle, new neurons are continuously generated from neural precursor cells. Recently, evidence has emerged that neurogenesis in the adult brain plays an important role in the repair of nerve tissues in brain disorders (109). Progenitors of cortical neurons show high levels of ETB receptors. Activation of ETB receptors in neural progenitors stimulated their proliferation and migration, indicating that ETs promote neurogenesis by directly acting on neural progenitors (82). These actions of ETs on neurons and neural progenitors suggest that activation of ETB receptors promotes repair of nerve tissues in the chronic phase of stroke and neurodegenerative diseases (Figure 5).
Viability of neurons and repair processes of damaged nerve tissues are supported by neurotrophins and glial cell-derived neurotrophic factor (GDNF). Neurotrophin family proteins, including nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF), which are produced by subpopulations of neurons in normal conditions, support the viability of cholinergic neurons in the basal forebrain, motor neurons, and hippocampal neurons (110). Recent studies showed that BDNF stimulates neurogenesis of the adult brain (111). GDNF supports survival of mid-brain dopaminergic neurons and has been suggested to have beneficial actions on patients with Parkinson’s disease (110). In neurodegenerative diseases and stroke, production of these neurotrophic factors is increased, where astrocytes are the main source of neurotrophic factors (78). Because of their potent ability to support neuronal viability, potentiation of neurotrophic factor production has been a promising therapeutic strategy for neuronal dysfunction in neurodegenerative diseases and stroke. Administration of Ala1,3,11,15-ET-1 into rat brain increased production of NGF, BDNF, neurotrophin-3, basic fibroblast growth factor, and GDNF in reactive astrocytes (100, 112). The effects of ETs on neurotrophic factor production were also observed in cultured astrocytes (100, 112–115). As well as having direct actions on neurons (106–108), ET-induced neurotrophic factor production suggests a possible role of astrocytic ETB receptors in supporting the viability and repair of damaged nerve tissues (Figure 5). A recent report by Leonard et al. showed that IRL1620, an ETB agonist, reduced neuronal damage and neurological defects in a rat brain ischemia model (116).

Possible roles of ETB receptors in survival and regeneration of damaged nerve tissues.
In some neuronal cells, direct activation of ETB receptors has been shown to induce antiapoptotic actions. Neuronal survival and regeneration is promoted by neurotrophic factors. Production of neurotrophic factors is stimulated by activation of astrocytic ETB receptors. Neural progenitors express ETB receptors. Proliferation and migration of neural progenitors are stimulated by ETs. These actions raise the possibility that activation of ETB receptors promotes repair of damaged nerve tissues in neurodegenerative diseases and in the chronic phase of stroke.
Outlook
For several decades, neuroscientists and pharmacologists have invested much effort into the invention of novel drugs to provide protection or repair to nerve tissues impaired by brain disorders. However, at present, it is not possible to say that effective drug treatments for many neurodegenerative diseases and brain insults have been established. Recent studies of brain ETs have clarified the involvement of ET systems in various pathophysiological responses of damaged brains. As a target of neuroprotective drugs, brain ET receptors could have two possible clinical applications in different states of brain pathologies. One possible application is to block brain ETA and ETB receptors in the acute phase of stroke. Blockage of brain ETA receptors ameliorates vasospasm of cerebral arteries and ischemic brain damage after stroke. In addition to ETA receptors, blockage of ETB receptors in astrocytes impairs brain edema formation by reduced production of VEGF and MMPs. The beneficial effects of ET antagonists have been shown in clinical trials for SAH patients. The other possible application of ET systems is to activate ETB receptors in the chronic phase of brain insults and in neurodegenerative diseases. In these states, promotion of nerve repair processes, that is, axonal elongation, synaptogenesis, and neurogenesis, is therapeutic, to allow recovery of brain functions. Beneficial actions of ETB agonists in chronic brain disorders can be supported by the findings that activation of brain ETB receptors caused production of neurotrophic factors, antiapoptotic effects in neuronal cells, and proliferation of neuronal progenitors. Already, many agonists and antagonists for ET receptors have been developed. Thus, it is expected that a novel drug for treatment of brain disorders may be discovered among these ET receptor agonists and antagonists.
Highlights
ET systems are involved in pathophysiological responses of damaged brain.
Brain microvessels and astrocytes produce ETs in response to brain injury.
ECEs cleave Aβ proteins and neuropeptides.
Activation of ETA receptors induces vasospasm of cerebral arteries after stroke.
ET antagonists have beneficial actions on neurological defects in SAH patients.
Activation of ETB receptors in astrocytes promotes conversion to reactive astrocytes.
Activation of ETB receptors stimulates production of astrocytic neurotrophic factors.
ETs stimulate proliferation and migration of neural progenitors.
Brain ET systems are expected to be a novel target of neuroprotective drugs.
About the author

Yutaka Koyama graduated from the Faculty of Pharmacy, Osaka University (Japan). He studied Neuropharmacology at the Graduate School of Pharmaceutical Sciences, Osaka University (1985–1989) and was awarded a PhD in Pharmaceutical Sciences. Thereafter, he worked at the Graduate School of Pharmaceutical Sciences, Osaka University as an Assistant Professor (1989–2001) and as an Associate Professor (2001–2006). During this career, he joined the Department of Pathology, at the College of Physicians and Surgeons, Columbia University as an invited researcher (1997–1998). In 2006, he became a Professor of Laboratory of Pharmacology, Faculty of Pharmacy, Osaka Ohtani University where he has been investigating pharmacological significance of glial cells as a target of neuroprotective drugs.
This work was supported by a Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science (21590108).
References
1. Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, Yazaki Y, Goto K, Masaki T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988; 332: 411–5.10.1038/332411a0Search in Google Scholar
2. Yanagisawa M, Masaki T. Molecular biology and biochemistry of the endothelins. Trends Pharmacol Sci 1989; 10: 374–8.10.1016/0165-6147(89)90011-4Search in Google Scholar
3. Barton M, Yanagisawa M. Endothelin: 20 years from discovery to therapy. Can J Physiol Pharmacol 2008; 86: 485–98.10.1139/Y08-059Search in Google Scholar
4. Goto K, Hama H, Kasuya Y. Molecular pharmacology and pathophysiological significance of endothelin. Jpn J Pharmacol 1996; 72: 261–90.10.1254/jjp.72.261Search in Google Scholar
5. Nunez DJ, Brown MJ, Davenport AP, Neylon CB, Schofield JP, Wyse RK. Endothelin-1 mRNA is widely expressed in porcine and human tissues. J Clin Invest 1990; 85: 1537–41.10.1172/JCI114601Search in Google Scholar
6. Matsumoto H, Suzuki N, Onda H, Fujino M. Abundance of endothelin-3 in rat intestine, pituitary gland and brain. Biochem Biophys Res Commun 1989; 164: 74–80.10.1016/0006-291X(89)91684-7Search in Google Scholar
7. Schinelli S. Pharmacology and physiopathology of the brain endothelin system: an overview. Curr Med Chem 2006; 13: 627–38.10.2174/092986706776055652Search in Google Scholar
8. Nie XJ, Olsson Y. Endothelin peptides in brain diseases. Rev Neurosci 1996; 7: 177–86.Search in Google Scholar
9. Ostrow LW, Sachs F. Mechanosensation and endothelin in astrocytes – hypothetical roles in CNS pathophysiology. Brain Res Brain Res Rev 2005; 48: 488–508.10.1016/j.brainresrev.2004.09.005Search in Google Scholar
10. Fang S, Ledlow A, Murray JA, Christensen J, Conklin JL. Vasoactive intestinal contractor: localization in the oppossum esophagus and effects on motor function. Gastroenterology 1994; 107: 1621–6.10.1016/0016-5085(94)90800-1Search in Google Scholar
11. Uchide T, Adur J, Fukamachi H, Saida K. Quantitative analysis of endothelin-1 and vasoactive intestinal contractor/endothelin-2 gene expression in rats by real-time reverse transcriptase polymerase chain reaction. J Cardiovasc Pharmacol 2000; 36 (5 Suppl 1): S5–8.10.1097/00005344-200036051-00004Search in Google Scholar PubMed
12. Masuo Y, Ishikawa Y, Kozakai T, Uchide T, Komatsu Y, Saida K. Vasoactive intestinal contractor/endothelin-2 gene expression in the murine central nervous system. Biochem Biophys Res Commun 2003; 300: 661–8.10.1016/S0006-291X(02)02872-3Search in Google Scholar
13. Grimshaw MJ, Naylor S, Balkwill FR. Endothelin-2 is a hypoxia-induced autocrine survival factor for breast cancer cells. Mol Cancer Ther 2002; 1: 1273–81.Search in Google Scholar
14. Takizawa S, Uchide T, Adur J, Kozakai T, Kotake-Nara E, Quan J, Saida K. Differential expression of endothelin-2 along the mouse intestinal tract. J Mol Endocrinol 2005; 35: 201–9.10.1677/jme.1.01787Search in Google Scholar
15. Baynash AG, Hosoda K, Giaid A, Richardson JA, Emoto N, Hammer RE, Yanagisawa M. Interaction of endothelin-3 with endothelin-B receptor is essential for development of epidermal melanocytes and enteric neurons. Cell 1994; 79: 1277–85.10.1016/0092-8674(94)90018-3Search in Google Scholar
16. Hosoda K, Hammer RE, Richardson JA, Baynash AG, Cheung JC, Giaid A, Yanagisawa M. Targeted and natural (piebald-lethal) mutations of endothelin-B receptor gene produce megacolon associated with spotted coat color in mice. Cell 1994; 79: 1267–76.10.1016/0092-8674(94)90017-5Search in Google Scholar
17. Amiel J, Attié T, Jan D, Pelet A, Edery P, Bidaud C, Lacombe D, Tam P, Simeoni J, Flori E, Nihoul-Fékété C, Munnich A, Lyonnet S. Heterozygous endothelin receptor B (EDNRB) mutations in isolated Hirschsprung disease. Hum Mol Genet 1996; 5: 355–7.10.1093/hmg/5.3.355Search in Google Scholar
18. Edery P, Attié T, Amiel J, Pelet A, Eng C, Hofstra RM, Martelli H, Bidaud C, Munnich A, Lyonnet S. Mutation of the endothelin-3 gene in the Waardenburg-Hirschsprung disease (Shah-Waardenburg syndrome). Nat Genet 1996; 12: 442–4.10.1038/ng0496-442Search in Google Scholar
19. Hofstra RM, Osinga J, Tan-Sindhunata G, Wu Y, Kamsteeg EJ, Stulp RP, van Ravenswaaij-Arts C, Majoor-Krakauer D, Angrist M, Chakravarti A, Meijers C, Buys CH. A homozygous mutation in the endothelin-3 gene associated with a combined Waardenburg type 2 and Hirschsprung phenotype (Shah-Waardenburg syndrome). Nat Genet 1996; 12: 445–7.10.1038/ng0496-445Search in Google Scholar
20. Bloch KD, Eddy RL, Shows TB, Quertermous T. cDNA cloning and chromosomal assignment of the gene encoding endothelin 3. J Biol Chem 1989; 264: 18156–61.10.1016/S0021-9258(19)84690-2Search in Google Scholar
21. Ohkubo S, Ogi K, Hosoya M, Matsumoto H, Suzuki N, Kimura C, Ondo H, Fujino M. Specific expression of human endothelin-2 (ET-2) gene in a renal adenocarcinoma cell line. Molecular cloning of cDNA encoding the precursor of ET-2 and its characterization. FEBS Lett 1990; 274: 136–40.10.1016/0014-5793(90)81348-RSearch in Google Scholar
22. Inoue A, Yanagisawa M, Takuwa Y, Mitsui Y, Kobayashi M, Masaki T. The human preproendothelin-1 gene. Complete nucleotide sequence and regulation of expression. J Biol Chem 1989; 264: 14954–9.10.1016/S0021-9258(18)63795-0Search in Google Scholar
23. Emori T, Hirata Y, Imai T, Ohta K, Kanno K, Eguchi S, Marumo F. Cellular mechanism of thrombin on endothelin-1 biosynthesis and release in bovine endothelial cell. Biochem Pharmacol 1992; 44: 2409–11.10.1016/0006-2952(92)90687-ESearch in Google Scholar
24. Kurihara H, Yoshizumi M, Sugiyama T, Takaku F, Yanagisawa M, Masaki T, Hamaoki M, Kato H, Yazaki Y. Transforming growth factor-β stimulates the expression of endothelin mRNA by vascular endothelial cells. Biochem Biophys Res Commun 1989; 159: 1435–40.10.1016/0006-291X(89)92270-5Search in Google Scholar
25. Marsden PA, Brenner BM. Transcriptional regulation of the endothelin-1 gene by TNF-α. Am J Physiol 1992; 262: C854–6110.1152/ajpcell.1992.262.4.C854Search in Google Scholar
26. Maemura K, Kurihara H, Morita T, Oh-hashi Y, Yazaki Y. Production of endothelin-1 in vascular endothelial cells is regulated by factors associated with vascular injury. Gerontology 1992; 38(Suppl 1): 29–35.10.1159/000213360Search in Google Scholar
27. Marsden PA, Dorfman DM, Collins T, Brenner BM, Orkin SH, Ballermann BJ. Regulated expression of endothelin 1 in glomerular capillary endothelial cells. Am J Physiol 1991; 261: F117–25.10.1152/ajprenal.1991.261.1.F117Search in Google Scholar
28. Hu J, Discher DJ, Bishopric NH, Webster KA. Hypoxia regulates expression of the endothelin-1 gene through a proximal hypoxia-inducible factor-1 binding site on the antisense strand. Biochem Biophys Res Commun 1998; 245: 894–9.10.1006/bbrc.1998.8543Search in Google Scholar
29. Yoshizumi M, Kurihara H, Sugiyama T, Takaku F, Yanagisawa M, Masaki T, Yazaki Y. Hemodynamic shear stress stimulates endothelin production by cultured endothelial cells. Biochem Biophys Res Commun 1989; 161: 859–64.10.1016/0006-291X(89)92679-XSearch in Google Scholar
30. Stow LR, Jacobs ME, Wingo CS, Cain BD. Endothelin-1 gene regulation. FASEB J 2011; 25: 16–28.10.1096/fj.10-161612Search in Google Scholar PubMed PubMed Central
31. Mawji IA, Robb GB, Tai SC, Marsden PA. Role of the 3′-untranslated region of human endothelin-1 in vascular endothelial cells. Contribution to transcript lability and the cellular heat shock response. J Biol Chem 2004; 79: 8655–67.10.1074/jbc.M312190200Search in Google Scholar PubMed
32. Rodríguez-Pascual F, Redondo-Horcajo M, Magán-Marchal N, Lagares D, Martínez-Ruiz A, Kleinert H, Lamas S. Glyceraldehyde-3-phosphate dehydrogenase regulates endothelin-1 expression by a novel, redox-sensitive mechanism involving mRNA stability. Mol Cell Biol 2008; 28: 7139–55.10.1128/MCB.01145-08Search in Google Scholar PubMed PubMed Central
33. Yeligar S, Tsukamoto H, Kalra VK. Ethanol-induced expression of ET-1 and ET-BR in liver sinusoidal endothelial cells and human endothelial cells involves hypoxia-inducible factor-1α and microrNA-199. J Immunol 2009; 183: 5232–43.10.4049/jimmunol.0901084Search in Google Scholar PubMed PubMed Central
34. Andresen J, Shafi NI, Bryan RM Jr. Endothelial influences on cerebrovascular tone. J Appl Physiol 2006; 100: 318–27.10.1152/japplphysiol.00937.2005Search in Google Scholar PubMed
35. Rodríguez-Pascual F, Busnadiego O, Lagares D, Lamas S. Role of endothelin in the cardiovascular system. Pharmacol Res 2011; 63: 463–72.10.1016/j.phrs.2011.01.014Search in Google Scholar PubMed
36. Bouallegue A, Daou GB, Srivastava AK. Endothelin-1-induced signaling pathways in vascular smooth muscle cells. Curr Vasc Pharmacol 2007; 5: 45–52.10.2174/157016107779317161Search in Google Scholar PubMed
37. Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 1996; 379: 557–60.10.1038/379557a0Search in Google Scholar PubMed
38. Chahdi A, Sorokin A. Endothelin-1 induces p66Shc activation through EGF receptor transactivation: role of β1 Pix/Gαi3 interaction. Cell Signal 2010; 22: 325–9.10.1016/j.cellsig.2009.09.039Search in Google Scholar PubMed PubMed Central
39. Grantcharova E, Reusch HP, Grossmann S, Eichhorst J, Krell HW, Beyermann M, Rosenthal W, Oksche A. N-terminal proteolysis of the endothelin B receptor abolishes its ability to induce EGF receptor transactivation and contractile protein expression in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 2006; 6: 1288–96.10.1161/01.ATV.0000220377.51354.30Search in Google Scholar PubMed
40. Vacca F, Bagnato A, Catt KJ, Tecce R. Transactivation of the epidermal growth factor receptor in endothelin-1-induced mitogenic signaling in human ovarian carcinoma cells. Cancer Res 2000; 60: 5310–7.Search in Google Scholar
41. Giaid A, Gibson SJ, Ibrahim BN, Legon S, Bloom SR, Yanagisawa M, Masaki T, Varndell IM, Polak JM. Endothelin-1, an endothelium-derived peptide, is expressed in neurons of the human spinal cord and dorsal root ganglia. Proc Natl Acad Sci USA 1989; 86: 7634–8.10.1073/pnas.86.19.7634Search in Google Scholar PubMed PubMed Central
42. Loesch A, Milner P, Burnstock G. Endothelin in perivascular nerves. An electron-immunocytochemical study of rat basilar artery. Neuroreport 1998; 9: 3903–6.10.1097/00001756-199812010-00025Search in Google Scholar PubMed
43. Nakamura S, Naruse M, Naruse K, Shioda S, Nakai Y, Uemura H. Colocalization of immunoreactive endothelin-1 and neurohypophysial hormones in the axons of the neural lobe of the rat pituitary. Endocrinology 1993; 132: 530–3.10.1210/endo.132.2.8425473Search in Google Scholar PubMed
44. Yoshizawa T, Kimura S, Kanazawa I, Uchiyama Y, Yanagisawa M, Masaki T. Endothelin localizes in the dorsal horn and acts on the spinal neurons: possible involvement of dihydropyridine-sensitive calcium channels and substance P release. Neurosci Lett 1989; 102: 179–84.10.1016/0304-3940(89)90075-XSearch in Google Scholar
45. Chichorro JG, Zampronio AR, Cabrini DA, Franco CR, Rae GA. Mechanisms operated by endothelin ETA and ETB receptors in the trigeminal ganglion contribute to orofacial thermal hyperalgesia induced by infraorbital nerve constriction in rats. Neuropeptides 2009; 43: 133–42.10.1016/j.npep.2008.12.001Search in Google Scholar
46. Dashwood MR, Loesch A. Endothelin-1 as a neuropeptide: neurotransmitter or neurovascular effects? J Cell Commun Signal 2010; 4: 51–62.10.1007/s12079-009-0073-3Search in Google Scholar
47. Matsuo Y, Mihara S, Ninomiya M, Fujimoto M. Protective effect of endothelin type A receptor antagonist on brain edema and injury after transient middle cerebral artery occlusion in rats. Stroke 2001; 32: 2143–8.10.1161/hs0901.94259Search in Google Scholar
48. Barone FC, Globus MY, Price WJ, White RF, Storer BL, Feuerstein GZ, Busto R, Ohlstein EH. Endothelin levels increase in rat focal and global ischemia. J Cereb Blood Flow Metab 1994; 14: 337–42.10.1038/jcbfm.1994.41Search in Google Scholar
49. Lampl Y, Fleminger G, Gilad R, Galron R, Sarova-Pinhas I, Sokolovsky M. Endothelin in cerebrospinal fluid and plasma of patients in the early stage of ischemic stroke. Stroke 1997; 28: 1951–5.10.1161/01.STR.28.10.1951Search in Google Scholar
50. Ziv I, Fleminger G, Djaldetti R, Achiron A, Melamed E, Sokolovsky M. Increased plasma endothelin-1 in acute ischemic stroke. Stroke 1992; 23: 1014–6.10.1161/01.STR.23.7.1014Search in Google Scholar
51. Skopál J, Turbucz P, Vastag M, Bori Z, Pék M, deChâtel R, Nagy Z, Tóth M, Karádi I. Regulation of endothelin release from human brain microvessel endothelial cells. J Cardiovasc Pharmacol 1998; 31(Suppl 1): S370–2.10.1097/00005344-199800001-00104Search in Google Scholar
52. Desai D, He S, Yorio T, Krishnamoorthy RR, Prasanna G. Hypoxia augments TNF-α-mediated endothelin-1 release and cell proliferation in human optic nerve head astrocytes. Biochem Biophys Res Commun 2004; 318: 642–8.10.1016/j.bbrc.2004.04.073Search in Google Scholar
53. Ehrenreich H, Costa T, Clouse KA, Pluta RM, Ogino Y, Coligan JE, Burd PR. Thrombin is a regulator of astrocytic endothelin-1. Brain Res 1993; 600: 201–7.10.1016/0006-8993(93)91374-2Search in Google Scholar
54. Didier N, Romero IA, Créminon C, Wijkhuisen A, Grassi J, Mabondzo A. Secretion of interleukin-1β by astrocytes mediates endothelin-1 and tumour necrosis factor-α effects on human brain microvascular endothelial cell permeability. J Neurochem 2003; 86: 246–54.10.1046/j.1471-4159.2003.01829.xSearch in Google Scholar
55. Ehrenreich H, Anderson RW, Ogino Y, Rieckmann P, Costa T, Wood GP, Coligan JE, Kehrl JH, Fauci AS. Selective autoregulation of endothelins in primary astrocyte cultures: endothelin receptor-mediated potentiation of endothelin-1 secretion. New Biol 1991; 3: 135–341.Search in Google Scholar
56. Turner AJ, Murphy LJ. Molecular pharmacology of endothelin converting enzymes. Biochem Pharmacol 1996; 51: 91–102.10.1016/0006-2952(95)02036-5Search in Google Scholar
57. Rodriguiz RM, Gadnidze K, Ragnauth A, Dorr N, Yanagisawa M, Wetsel WC, Devi LA. Animals lacking endothelin-converting enzyme-2 are deficient in learning and memory. Genes Brain Behav 2008; 7: 418–26.10.1111/j.1601-183X.2007.00365.xSearch in Google Scholar PubMed PubMed Central
58. Eckman EA, Watson M, Marlow L, Sambamurti K, Eckman CB. Alzheimer’s disease β-amyloid peptide is increased in mice deficient in endothelin-converting enzyme. J Biol Chem 2003; 278: 2081–4.10.1074/jbc.C200642200Search in Google Scholar PubMed
59. Palmer JC, Baig S, Kehoe PG, Love S. Endothelin-converting enzyme-2 is increased in Alzheimer’s disease and up-regulated by Aβ. Am J Pathol 2009; 175: 262–70.10.2353/ajpath.2009.081054Search in Google Scholar PubMed PubMed Central
60. Palmer J, Love S. Endothelin receptor antagonists: potential in Alzheimer’s disease. Pharmacol Res 2011; 63: 525–31.10.1016/j.phrs.2010.12.008Search in Google Scholar PubMed
61. Roosterman D, Cottrell GS, Padilla BE, Muller L, Eckman CB, Bunnett NW, Steinhoff M. Endothelin-converting enzyme 1 degrades neuropeptides in endosomes to control receptor recycling. Proc Natl Acad Sci USA 2007; 104: 11838–43.10.1073/pnas.0701910104Search in Google Scholar PubMed PubMed Central
62. Cattaruzza F, Cottrell GS, Vaksman N, Bunnett NW. Endothelin-converting enzyme 1 promotes re-sensitization of neurokinin 1 receptor-dependent neurogenic inflammation. Br J Pharmacol 2009; 156: 730–9.10.1111/j.1476-5381.2008.00039.xSearch in Google Scholar PubMed PubMed Central
63. Sharkey J, Ritchie IM, Kelly PA. Perivascular microapplication of endothelin-1: a new model of focal cerebral ischaemia in the rat. J Cereb Blood Flow Metab 1993; 13: 865–71.10.1038/jcbfm.1993.108Search in Google Scholar PubMed
64. Macrae IM, Robinson MJ, Graham DI, Reid JL, McCulloch J. Endothelin-1-induced reductions in cerebral blood flow: dose dependency, time course, and neuropathological consequences. J Cereb Blood Flow Metab 1993; 13: 276–84.10.1038/jcbfm.1993.34Search in Google Scholar PubMed
65. Zhou QL, Strichartz G, Davar G. Endothelin-1 activates ETA receptors to increase intracellular calcium in model sensory neurons. Neuroreport 2001; 12: 3853–7.10.1097/00001756-200112040-00050Search in Google Scholar
66. Gokin AP, Fareed MU, Pan HL, Hans G, Strichartz GR, Davar G. Local injection of endothelin-1 produces pain-like behavior and excitation of nociceptors in rats. J Neurosci 2001; 21: 5358–66.10.1523/JNEUROSCI.21-14-05358.2001Search in Google Scholar
67. Khodorova A, Montmayeur JP, Strichartz G. Endothelin receptors and pain. J Pain 2009; 10: 4–28.10.1016/j.jpain.2008.09.009Search in Google Scholar
68. Barr TP, Kam S, Khodorova A, Montmayeur JP, Strichartz GR. New perspectives on the endothelin axis in pain. Pharmacol Res 2011; 63: 532–40.10.1016/j.phrs.2011.02.002Search in Google Scholar
69. Rogers SD, Peters CM, Pomonis JD, Hagiwara H, Ghilardi JR, Mantyh PW. Endothelin B receptors are expressed by astrocytes and regulate astrocyte hypertrophy in the normal and injured CNS. Glia 2003; 41: 180–90.10.1002/glia.10173Search in Google Scholar
70. Wilhelmsson U, Li L, Pekna M, Berthold CH, Blom S, Eliasson C, Renner O, Bushong E, Ellisman M, Morgan TE, Pekny M. Absence of glial fibrillary acidic protein and vimentin prevents hypertrophy of astrocytic processes and improves post-traumatic regeneration. J Neurosci 2004; 24: 5016–21.10.1523/JNEUROSCI.0820-04.2004Search in Google Scholar
71. Peters CM, Rogers SD, Pomonis JD, Egnaczyk GF, Keyser CP, Schmidt JA, Ghilardi JR, Maggio JE, Mantyh PW. Endothelin receptor expression in the normal and injured spinal cord: potential involvement in injury-induced ischemia and gliosis. Exp Neurol 2003; 180: 1–13.10.1016/S0014-4886(02)00023-7Search in Google Scholar
72. Cazaubon S, Chaverot N, Romero IA, Girault JA, Adamson P, Strosberg AD, Couraud PO. Growth factor activity of endothelin-1 in primary astrocytes mediated by adhesion-dependent and -independent pathways. J Neurosci 1997; 17: 6203–12.10.1523/JNEUROSCI.17-16-06203.1997Search in Google Scholar
73. Gadea A, Schinelli S, Gallo V. Endothelin-1 regulates astrocyte proliferation and reactive gliosis via a JNK/c-Jun signaling pathway. J Neurosci 2008; 28: 2394–408.10.1523/JNEUROSCI.5652-07.2008Search in Google Scholar
74. Koyama Y, Baba A. Endothelins are extracellular signals modulating cytoskeletal actin organization in rat cultured astrocytes. Neuroscience 1994; 61: 1007–16.10.1016/0306-4522(94)90420-0Search in Google Scholar
75. Koyama Y, Baba A. Endothelin-induced cytoskeletal actin re-organization in cultured astrocytes: inhibition by C3 ADP-ribosyltransferase. Glia 1996; 16: 342–50.10.1002/(SICI)1098-1136(199604)16:4<342::AID-GLIA6>3.0.CO;2-1Search in Google Scholar
76. Giaume C, Cordier J, Glowinski J. Endothelins inhibit junctional permeability in cultured mouse astrocytes. Eur J Neurosci 1992; 4: 877–81.10.1111/j.1460-9568.1992.tb00198.xSearch in Google Scholar
77. Tabernero A, Sánchez-Alvarez R, Medina JM. Increased levels of cyclins D1 and D3 after inhibition of gap junctional communication in astrocytes. J Neurochem 2006; 96: 973–82.10.1111/j.1471-4159.2005.03623.xSearch in Google Scholar
78. Hamby ME, Sofroniew MV. Reactive astrocytes as therapeutic targets for CNS disorders. Neurotherapeutics 2010; 7: 494–506.10.1016/j.nurt.2010.07.003Search in Google Scholar
79. Koyama Y, Michinaga S. Regulations of astrocytic functions by endothelins: roles in the pathophysiological responses of damaged brains. J Pharmacol Sci 2012; 118: 401–7.10.1254/jphs.11R13CPSearch in Google Scholar
80. Gadea A, Aguirre A, Haydar TF, Gallo V. Endothelin-1 regulates oligodendrocyte development. J Neurosci 2009; 29: 10047–62.10.1523/JNEUROSCI.0822-09.2009Search in Google Scholar
81. Vidovic M, Chen MM, Lu QY, Kalloniatis KF, Martin BM, Tan AH, Lynch C, Croaker GD, Cass DT, Song ZM. Deficiency in endothelin receptor B reduces proliferation of neuronal progenitors and increases apoptosis in postnatal rat cerebellum. Cell Mol Neurobiol 2008; 8: 1129–38.10.1007/s10571-008-9292-zSearch in Google Scholar
82. Nishikawa K, Ayukawa K, Hara Y, Wada K, Aoki S. Endothelin/endothelin-B receptor signals regulate ventricle-directed interkinetic nuclear migration of cerebral cortical neural progenitors. Neurochem Int 2011; 58: 261–72.10.1016/j.neuint.2010.11.013Search in Google Scholar
83. Zimmermann M, Seifert V. Endothelin and subarachnoid hemorrhage: an overview. Neurosurgery 1998; 43: 863–75.10.1097/00006123-199810000-00083Search in Google Scholar
84. Franceschini R, Gandolfo C, Cataldi A, Del Sette M, Rolandi A, Corsini G, Rolandi E, Barreca T. Twenty-four-hour endothelin-1 secretory pattern in stroke patients. Biomed Pharmacother 2001; 5: 272–6.10.1016/S0753-3322(01)00059-2Search in Google Scholar
85. Barone FC, White RF, Elliott JD, Feuerstein GZ, Ohlstein EH. The endothelin receptor antagonist SB 217242 reduces cerebral focal ischemic brain injury. J Cardiovasc Pharmacol 1995; 26(Suppl 3): S404–7.10.1097/00005344-199506263-00119Search in Google Scholar
86. Patel TR, Galbraith SL, McAuley MA, Doherty AM, Graham DI, McCulloch J. Therapeutic potential of endothelin receptor antagonists in experimental stroke. J Cardiovasc Pharmacol 1995; 26(Suppl 3): S412–5.10.1097/00005344-199506263-00121Search in Google Scholar
87. Bhardwaj A, Wu Y, Hurn PD, Kirsch JR, Traystman RJ. Administration of selective endothelin receptor type A antagonist Ro 61-1790 does not improve outcome in focal cerebral ischemia in cat. J Cereb Blood Flow Metab 2000; 20: 499–504.10.1097/00004647-200003000-00008Search in Google Scholar PubMed
88. Kaundal RK, Deshpande TA, Gulati A, Sharma SS. Targeting endothelin receptors for pharmacotherapy of ischemic stroke: current scenario and future perspectives. Drug Discov Today 2012; 17: 793–804.10.1016/j.drudis.2012.02.017Search in Google Scholar PubMed
89. Nag S, Manias JL, Stewart DJ. Pathology and new players in the pathogenesis of brain edema. Acta Neuropathol 2009; 118: 197–217.10.1007/s00401-009-0541-0Search in Google Scholar PubMed
90. Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci 2009; 32: 638–47.10.1016/j.tins.2009.08.002Search in Google Scholar PubMed PubMed Central
91. Koyama Y, Tanaka K. Endothelins stimulate the production of stromelysin-1 in cultured rat astrocytes. Biochem Biophys Res Commun 2008; 371: 659–63.10.1016/j.bbrc.2008.04.064Search in Google Scholar PubMed
92. Koyama Y, Tanaka K. Intracerebroventricular administration of an endothelin ETB-receptor agonist increases expression of matrix metalloproteinase-2 and -9 in rat brain. J Pharmacol Sci 2010; 114: 433–43.10.1254/jphs.10195FPSearch in Google Scholar
93. Koyama Y, Nagae R, Tokuyama S, Tanaka K. I.c.v administration of an endothelin ETB receptor agonist stimulates vascular endothelial growth factor-A production and activates vascular endothelial growth factor receptors in rat brain. Neuroscience 2011; 192: 689–98.10.1016/j.neuroscience.2011.05.058Search in Google Scholar PubMed
94. Koyama Y, Maebara Y, Hayashi M, Nagae R, Tokuyama S, Michinaga S. Endothelins reciprocally regulate VEGF-A and angiopoietin-1 production in cultured rat astrocytes: implications on astrocytic proliferation. Glia 2012; 60: 1954–63.10.1002/glia.22411Search in Google Scholar PubMed
95. Barone FC, Ohlstein EH, Hunter AJ, Campbell CA, Hadingham SH, Parsons AA, Yang Y, Shohami E. Selective antagonism of endothelin-A-receptors improves outcome in both head trauma and focal stroke in rat. J Cardiovasc Pharmacol 2000; 36 (5 Suppl 1): S357–61.10.1097/00005344-200036051-00104Search in Google Scholar
96. Vajkoczy P, Meyer B, Weidauer S, Raabe A, Thome C, Ringel F, Breu V, Schmiedek PJ. Clazosentan (AXV-034343), a selective endothelin A receptor antagonist, in the prevention of cerebral vasospasm following severe aneurysmal subarachnoid hemorrhage: results of a randomized, double-blind, placebo-controlled, multicenter phase IIa study. Neurosurgery 2005; 103: 9–17.10.3171/jns.2005.103.1.0009Search in Google Scholar
97. Macdonald RL, Kassell NF, Mayer S, Ruefenacht D, Schmiedek P, Weidauer S, Frey A, Roux S, Pasqualin A, CONSCIOUS-1 Investigators. Clazosentan to overcome neurological ischemia and infarction occurring after subarachnoid hemorrhage (CONSCIOUS-1): randomized, double-blind, placebo-controlled phase 2 dose-finding trial. Stroke 2008; 39: 3015–21.10.1161/STROKEAHA.108.519942Search in Google Scholar
98. Shaw MD, Vermeulen M, Murray GD, Pickard JD, Bell BA, Teasdale GM. Efficacy and safety of the endothelin, receptor antagonist TAK-044 in treating subarachnoid hemorrhage: a report by the Steering Committee on behalf of the UK/Netherlands/Eire TAK-044 Subarachnoid Haemorrhage Study Group. J Neurosurg 2000; 93: 992–7.10.3171/jns.2000.93.6.0992Search in Google Scholar
99. Ishikawa N, Takemura M, Koyama Y, Shigenaga Y, Okada T, Baba A. Endothelins promote the activation of astrocytes in rat neostriatum through ETB receptors. Eur J Neurosci 1997; 9: 895–901.10.1111/j.1460-9568.1997.tb01440.xSearch in Google Scholar
100. Koyama Y, Tsujikawa K, Matsuda T, Baba A. Intracerebroventricular administration of an endothelin ETB receptor agonist increases expressions of GDNF and BDNF in rat brain. Eur J Neurosci 2003; 18: 887–94.10.1046/j.1460-9568.2003.02797.xSearch in Google Scholar
101. Koyama Y, Takemura M, Fujiki K, Ishikawa N, Shigenaga Y, Baba A. BQ788, an endothelin ETB receptor antagonist attenuates stab wound injury-induced reactive astrocytes in rat brain. Glia 1999; 26: 268–71.10.1002/(SICI)1098-1136(199905)26:3<268::AID-GLIA8>3.0.CO;2-GSearch in Google Scholar
102. Koyama Y, Yoshioka Y, Shinde M, Matsuda T, Baba A. Focal adhesion kinase mediates endothelin-induced cyclin D3 expression in rat cultured astrocytes. J Neurochem 2004; 90: 904–12.10.1111/j.1471-4159.2004.02546.xSearch in Google Scholar
103. Koyama Y, Yoshioka Y, Hashimoto H, Matsuda T, Baba A. Endothelins increase tyrosine phosphorylation of astrocytic focal adhesion kinase and paxillin accompanied with their association with cytoskeletal components. Neuroscience 2000; 101: 219–27.10.1016/S0306-4522(00)00330-4Search in Google Scholar
104. Koyama Y, Yoshioka Y, Matsuda T, Baba A. Focal adhesion kinase is required for endothelin-induced cell cycle progression of astrocytes. Glia 2003; 43: 185–9.10.1002/glia.10240Search in Google Scholar
105. Ehrenreich H, Nau TR, Dembowski C, Hasselblatt M, Barth M, Hahn A, Schilling L, Sirén A-L, Brück W. Endothelin B receptor deficiency is associated with an increased rate of neuronal apoptosis in the dentate gyrus. Neuroscience 2000; 95: 993–1001.10.1016/S0306-4522(99)00507-2Search in Google Scholar
106. Laziz I, Larbi A, Grebert D, Sautel M, Congar P, Lacroix MC, Salesse R, Meunier N. Endothelin as a neuroprotective factor in the olfactory epithelium. Neuroscience 2011; 172: 20–9.10.1016/j.neuroscience.2010.10.063Search in Google Scholar
107. Yagami T, Ueda K, Sakaeda T, Okamura N, Nakazato H, Kuroda T, Hata S, Sakaguchi G, Itoh N, Hashimoto Y, Fujimoto M. Effects of an endothelin B receptor agonist on secretory phospholipase A2-IIA-induced apoptosis in cortical neurons. Neuropharmacology 2005; 48: 291–300.10.1016/j.neuropharm.2004.09.011Search in Google Scholar
108. Yagami T, Ueda K, Asakura K, Kuroda T, Hata S, Sakaeda T, Kambayashi Y, Fujimoto M. Effects of endothelin B receptor agonists on amyloid β protein (25–35)-induced neuronal cell death. Brain Res 2002; 948: 72–81.10.1016/S0006-8993(02)02951-7Search in Google Scholar
109. Duan X, Kang E, Liu CY, Ming GL, Song H. Development of neural stem cell in the adult brain. Curr Opin Neurobiol 2008; 8: 108–15.10.1016/j.conb.2008.04.001Search in Google Scholar
110. Blesch A. Neurotrophic factors in neurodegeneration. Brain Pathol 2006; 16: 295–303.10.1111/j.1750-3639.2006.00036.xSearch in Google Scholar
111. Bath KG, Lee FS. Neurotrophic factor control of adult SVZ neurogenesis. Dev Neurobiol 2010; 70: 339–49.Search in Google Scholar
112. Koyama Y, Baba A, Matsuda T. Endothelins stimulate the expression of neurotrophin-3 in rat brain and rat cultured astrocytes. Neuroscience 2005; 136: 425–33.10.1016/j.neuroscience.2005.08.004Search in Google Scholar
113. Koyama Y, Tsujikawa K, Matsuda T, Baba A. Endothelin-1 stimulates GDNF expression in cultured rat astrocytes. Biochem Biophys Res Commun 2003; 303: 1101–5.10.1016/S0006-291X(03)00491-1Search in Google Scholar
114. Koyama Y, Tsujikawa K, Matsuda T, Baba A. Endothelin increases expression of exon III- and exon IV-containing brain-derived neurotrophic factor transcripts in cultured astrocytes and rat brain. J Neurosci Res 2005; 80: 809–16.10.1002/jnr.20512Search in Google Scholar PubMed
115. Ladenheim RG, Lacroix I, Foignant-Chaverot N, Strosberg AD, Couraud PO. Endothelins stimulate c-fos and nerve growth factor expression in astrocytes and astrocytoma. J Neurochem 1993; 60: 260–6.10.1111/j.1471-4159.1993.tb05846.xSearch in Google Scholar PubMed
116. Leonard MG, Briyal S, Gulati A. Endothelin B receptor agonist, IRL-1620, provides long-term neuroprotection in cerebral ischemia in rats. Brain Res 2012; 1464: 14–23.10.1016/j.brainres.2012.05.005Search in Google Scholar PubMed
©2013 by Walter de Gruyter Berlin Boston
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Articles in the same Issue
- Masthead
- Masthead
- Reviews
- Human neuronal cells: epigenetic aspects
- Endothelin systems in the brain: involvement in pathophysiological responses of damaged nerve tissues
- The molecular biology of selenocysteine
- MicroRNA biogenesis and variability
- Short Conceptual Overviews
- Progress in mitochondrial epigenetics
- Raf kinases in signal transduction and interaction with translation machinery
- Deoxyadenosine family: improved synthesis, DNA damage and repair, analogs as drugs
- Long antisense non-coding RNAs and the epigenetic regulation of gene expression
- New insights into the activation of sterol regulatory element-binding proteins by proteolytic processing
- Amyloid-like fibrils labeled with magnetic nanoparticles
Articles in the same Issue
- Masthead
- Masthead
- Reviews
- Human neuronal cells: epigenetic aspects
- Endothelin systems in the brain: involvement in pathophysiological responses of damaged nerve tissues
- The molecular biology of selenocysteine
- MicroRNA biogenesis and variability
- Short Conceptual Overviews
- Progress in mitochondrial epigenetics
- Raf kinases in signal transduction and interaction with translation machinery
- Deoxyadenosine family: improved synthesis, DNA damage and repair, analogs as drugs
- Long antisense non-coding RNAs and the epigenetic regulation of gene expression
- New insights into the activation of sterol regulatory element-binding proteins by proteolytic processing
- Amyloid-like fibrils labeled with magnetic nanoparticles