A 16-year-old boy with arthritis, rash, and hemoptysis: Beyond “undifferentiated connective tissue disease”?
-
 ,
und
,
und

Type I interferonopathy is a group of autoinflammatory disorders characterized by activation of type I interferon (IFN-I) signaling and upregulated expression of IFN-stimulated genes (ISGs). It can present both “autoinflammation” and “autoimmunity” phenomenon, and therefore has overlap manifestations with rheumatic diseases such as systemic lupus erythematosus (SLE). Since IFN-I exerts its functions via JAK/STAT (Janus kinase/signal transduction and activator of transcription) pathways, JAK inhibitors showed efficacy in the treatment of type I interferonopathy. The study reports a 16-year-old patient who suffered from intermittent anemia, rash, arthritis, and hemoptysis and was diagnosed with type I interferonopathy.
A 16-year-old boy was admitted in Peking Union Medical College hospital because of fatigue, intermittent cough, and fever for 13 years, and dyspnea on exertion for 3 years. The boy had been suffering from intermittent severe anemia with a minimum hemoglobin 30 g/L associated with dark urine, jaundice of skin and sclera for the past 13 years. The examination of thalassemia and other congenital hemolytic anemia was normal. Meanwhile, he had been suffering from relapsing fever, cough, and sputum, which gradually worsened. He also presented with relapsing arthritis involving metacarpophalangeal joints, facial rash, and fingertip and toe erythema for the last 10 years (Figure 1A,B). At the age of 13, he had fever, cough, and hemoptysis. Since then, he began to feel dyspnea on exertion. Pedigree analysis (Figure 2A) revealed that his mother (II-6) had polyarthritis, and frequently had fingertip and toe erythema since her teenage (Figure 1C,D), with positive rheumatic factor (RF), antinuclear antibody (ANA), and anti-SSA and anti-cyclic citrullinated peptide (CCP) antibodies. His aunt (II-9) had intermittent arthralgia. His grandmother (I-4) had also been suffering from polyarthritis mainly involving proximal interphalangeal joints for years. His elder sister (III-5) occasionally had facial rash, and fingertip and toe erythema (Figure 1E,F).
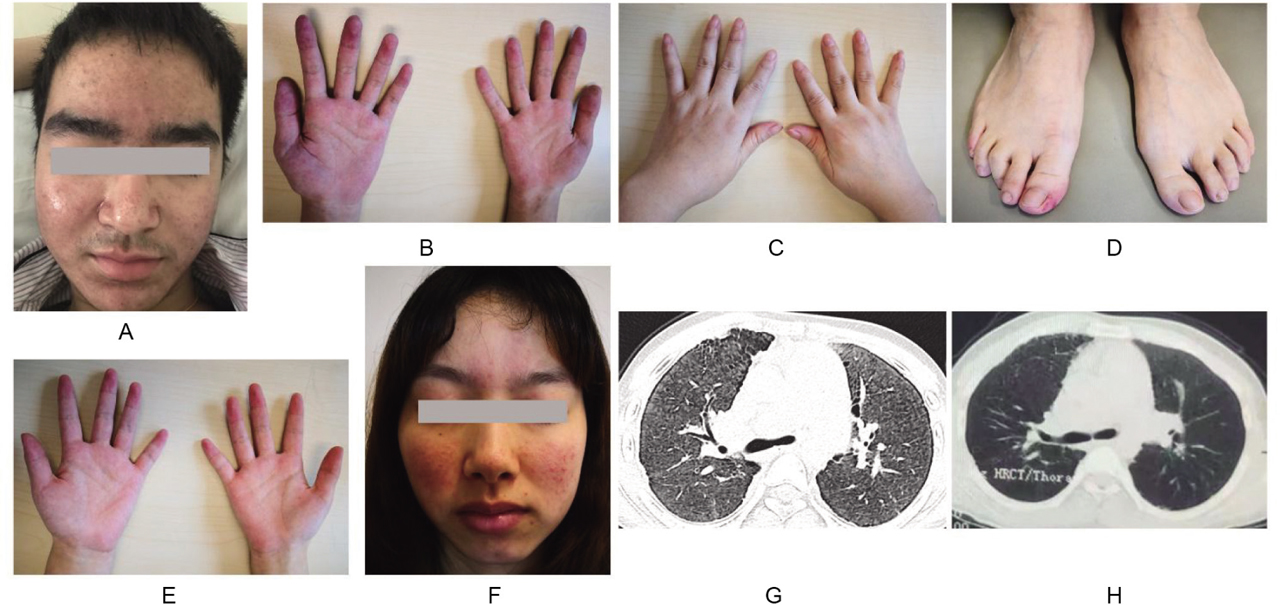
(A, B) The facial rash and fingertip erythema of the patient. (C, D) The polyarthritis and toe erythema of the patient's mother. (E, F) The fingertip erythema and facial rash of the patient's elder sister. (G) The chest HRCT of the patient showed diffused severe ILD with ground glass opacity and subpleural small cysts in both lungs at baseline. The radiologic changes in HRCT were in accordance with chronic recurrent DAH and pulmonary hemosiderosis. (H) The ILD in HRCT scan was improved after 1-year treatment of tofacitinib and HCQ. DAH, diffuse alveolar hemorrhage; HCQ, hydroxychloroquine; HRCT, high-resolution computed tomography; ILD, interstitial lung disease.
The complete blood count was normal with hemoglobin 144 g/L. The complete biochemistry panel and urine analysis were within the normal range. The erythrocyte sedimentation rate (ESR) level was elevated (34 mm/h), as well as C-reactive protein (CRP) (27.43 mg/L) and immunoglobulin G (IgG) (18.75 g/L). Serological testing for autoantibodies showed that ANA was positive (1:80, speckle pattern) with positive anticentromere antibody. He also had low-titer lupus anticoagulant, anti-beta2-glycoprotein I (β2GPI), RF, and anti-CCP antibody. His brain natriuretic peptide (BNP) and cardiac troponin I (cTnI) levels were within the normal range. Peripheral vascular ultrasound found no sign of thrombosis. His brain magnetic resonance imaging (MRI) and X-ray of both hands were normal. The exams showed severe hypoxemia (percutaneous oxygen saturation [SpO2] 85% at room air and pO2 48 mmHg in arterial blood gas [ABG] analysis). Pulmonary function test revealed severe restrictive ventilation dysfunction with decreased diffusion function (forced expiratory volume in 1 s [FEV1]/forced volume vital capacity [FVC] 90.37%, FVC 40.4%, diffusing capacity of the lung for carbon monoxide [DLCO] 20.3%). Chest high-resolution computed tomography (HRCT) scan showed diffused severe ground glass opacity in both lungs (Figure 1G), which were in accordance with chronic recurrent diffuse alveolar hemorrhage (DAH) and pulmonary hemosiderosis. Severe tricuspid regurgitation and pulmonary hypertension (evaluated pulmonary artery systolic pressure [PASP] 97 mmHg) was detected by echocardiography. The right heart catheter (RHC) examination confirmed the precapillary pulmonary artery hypertension (pulmonary artery pressure [PAP] 108/46 (70) mmHg, pulmonary arterial wedge pressure [PAWP] 13 mmHg).
Regarding multiple organ involvements including arthritis, rash, intermittent anemia, severe pulmonary hypertension, interstitial lung disease (ILD), positive ANA, and other autoantibodies, and elevated inflammatory parameters, he was suspected of having undifferentiated connective tissue disease at first. However, many of his manifestations were inexplicable by connective tissue disease, for example, the disease-onset age of 3 years, the normal levels of complements, the low-titer autoantibodies, and the family history of similar symptoms. Therefore, the whole-exome sequencing by next-generation sequencing was performed. Although the genetic testing for existing autoinflammatory diseases, especially type I interferonopathies, was normal, the blood tests of his family members showed that the expression of ISGs was significantly upregulated in the patient and his mother, compared with his father and health control (Figure 2B–G). Hence, he was finally diagnosed with type I interferonopathy.

(A) Pedigree analysis of the patient. Arrow, proband; black symbols, affected individuals; open symbols, unaffected individuals. (B) The expression of ISGs was significantly upregulated in the patient, compared with his father. (C–G) Real-time quantitative PCR for 5 ISG genes of the proband compared to his family members and the healthy control. For all RT-qPCR (Reverse transcription quantitative real-time polymerase chain reaction) assays, β-ACTIN was used as an endogenous control. Data shown were mean ± standard deviation (n ≥ 3). β-ACTIN, beta-actin; ISGs, IFN-stimulated genes.
The patient was treated with tofacitinib 5 mg twice a day and hydroxychloroquine (HCQ) 200 mg twice a day. At 6-month follow-up, his symptom of dyspnea on exertion was relieved and hypoxemia was improved (SpO2 95% at room air and pO2 79 mmHg in ABG test). The ESR, CRP, and IgG levels returned to normal. The evaluated PASP by echocardiography was decreased (50 mmHg). There was no significant improvement in chest HRCT and mild improvement in pulmonary function test (FEV1/FVC 94.04%, FVC 47.8%, and DLCO 26.9%). At 1-year follow-up, he was stable with normal ESR, CRP, and IgG levels. The ILD in HRCT scan was improved (Figure 1H). The echocardiography only showed mild tricuspid regurgitation and normal evaluated PASP (34 mmHg). Unfortunately, due to the COVID-19 pandemic, he did not come back for a regular follow-up at 1.5 years and died due to severe pneumonia and rapidly progressive respiratory failure.
Type I interferonopathy is a group of rare genetic autoinflammatory disorders characterized by prominent activation of IFN-I signaling.[1] Since IFN-I, including IFN-α and IFN-β, affects not only innate immunity, but also adaptive immunity,[2] type I interferonopathy may be characterized by autoinflammation manifestations such as fever, cutaneous rash, or intracranial calcifications, and varying autoimmune features such as positive autoantibodies, systemic involvement of pulmonary or kidney, or even SLE-like manifestations.[3] It used to be considered as monogenic lupus or lupus-like syndromes years ago.[4] Since our patient, as well as his mother, had prominent upregulated expression of ISGs, which meant abnormal activation of IFN-I signaling, atypical manifestation of systemic involvement, positive autoantibodies, and elevated inflammatory parameters, we considered the diagnosis of type I interferonopathy instead of undifferentiated connective tissue disease.
Until now, there have been many reported kinds of type I interferonopathy including Aicardi-Goutières syndrome, STING (Stimulator of interferon genes) -associated vasculopathy with onset in infancy (SAVI), spondyloenchondro-dysplasia with immune dysregulation (SPENCD),[5] and coatomer protein subunit alpha (COPA) syndrome, among others.[6] Patients with SAVI usually present with systemic inflammation, cutaneous rash, ILD, and pulmonary hypertension.[7, 8] As a recently described disease, COPA syndrome is characterized by activation of IFN-I, recurrent DAH, and/or ILD, associated with arthritis, lupus-like nephropathy, and positive antibodies.[6, 9] Since our patient had systemic inflammation, facial rash, intermittent anemia due to recurrent DAH, ILD, and pulmonary hypertension, SAVI and COPA syndrome were the most likely clinical diagnoses. Unfortunately, we did not find any genetic mutations related to type I interferonopathies including SAVI and COPA syndrome. Nevertheless, given the significantly upregulated ISGs, which were detected in the proband and his mother, they could be diagnosed as type I interferonopathy with a combination of their manifestations.
As IFN-I exerts its functions via JAK/STAT pathways, JAK inhibitors were used in type I interferonopathies including SAVI and COPA syndrome and showed good efficacy.[10, 11] The efficacy of tofacitinib in the patient might due to his upregulated ISGs. Furthermore, JAK inhibitors are also a novel therapy for rheumatic diseases such as SLE[12, 13] because of the overactivity of IFN-I pathway in these diseases.[2, 14] It is inspiring that the progress of type I interferonopathy may provide more insights into the treatment strategies in other rheumatic diseases.
Acknowledgments
We appreciate all support from the patient and his family.
Conflict of Interest
Xiaofeng Zeng is the Editor-in-Chief of the journal, and Mengtao Li is an Associate Editor-in-Chief. The article was subject to the journal's standard procedures, with peer review handled independently of these members and their research groups.
Informed Consent
The authors certify that they have obtained all appropriate patient consent documents. In the documents, the patient has given his consent for his images and other clinical information to be published in the journal.
Ethical Statement
We have obtained the patients’ written informed consent form for participation in the study and for publication of their data.
Funding
This work was supported by the Natural Science Foundation of Beijing (Grant No. 7192170); the Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (CIFMS) (Grant No. 2017-I2M-3-001); the National Key Research and Development Program of China (Grant Nos 2016YFC0901500; 2016YFC0901501); and CAMS Innovation Fund for Medical Sciences (CIFMS) (Grand No. 2021-I2M-1-005).
References
[1] Yu ZX, Song HM. Toward a Better Understanding of Type I Interferonopathies: A Brief Summary, Update and Beyond. World J Pediatr, 2020;16(1):44–51.10.1007/s12519-019-00273-zSuche in Google Scholar PubMed
[2] Kretschmer S, Lee-Kirsch MA. Type I Interferon-Mediated Autoinflammation and Autoimmunity. Curr Opin Immunol, 2017;49:96–102.10.1016/j.coi.2017.09.003Suche in Google Scholar PubMed
[3] Crow YJ, Manel N. Aicardi-Goutieres Syndrome and the Type I Interferonopathies. Nat Rev Immunol, 2015;15(7):429–440.10.1038/nri3850Suche in Google Scholar PubMed
[4] Alperin JM, Ortiz-Fernandez L, Sawalha AH. Monogenic Lupus: A Developing Paradigm of Disease. Front Immunol, 2018;9:2496.10.3389/fimmu.2018.02496Suche in Google Scholar PubMed PubMed Central
[5] Bousfiha A, Jeddane L, Picard C, et al. The 2017 IUIS Phenotypic Classification for Primary Immunodeficiencies. J Clin Immunol, 2018;38(1):129–143.10.1007/s10875-017-0465-8Suche in Google Scholar PubMed PubMed Central
[6] Frémond ML, Nathan N. COPA Syndrome, 5 years After: Where are we? Joint Bone Spine, 2021;88(2):105070.10.1016/j.jbspin.2020.09.002Suche in Google Scholar PubMed
[7] Liu Y, Jesus AA, Marrero B, et al. Activated STING in a Vascular and Pulmonary Syndrome. N Engl J Med, 2014;371(6):507–518.10.1056/NEJMoa1312625Suche in Google Scholar PubMed PubMed Central
[8] Saldanha RG, Balka KR, Davidson S, et al. A Mutation Outside the Dimerization Domain Causing Atypical STING-Associated Vasculopathy With Onset in Infancy. Front Immunol, 2018;9:1535.10.3389/fimmu.2018.01535Suche in Google Scholar PubMed PubMed Central
[9] Vece TJ, Watkin LB, Nicholas S, et al. Copa Syndrome: A Novel Autosomal Dominant Immune Dysregulatory Disease. J Clin Immunol, 2016;36(4):377–387.10.1007/s10875-016-0271-8Suche in Google Scholar PubMed PubMed Central
[10] Gómez-Arias PJ, Gómez-García F, Hernández-Parada J, et al. Efficacy and Safety of Janus Kinase Inhibitors in Type I Interferon-Mediated Monogenic Autoinflammatory Disorders: A Scoping Review. Dermatol Ther (Heidelb), 2021;11(3):733–750.10.1007/s13555-021-00517-9Suche in Google Scholar PubMed PubMed Central
[11] Krutzke S, Rietschel C, Horneff G. Baricitinib in Therapy of COPA Syndrome in a 15-year-old Girl. Eur J Rheumatol. 2019:7 (Suppl 1):1–4.10.5152/eurjrheum.2019.18177Suche in Google Scholar PubMed PubMed Central
[12] Wallace DJ, Furie RA, Tanaka Y, et al. Baricitinib for Systemic Lupus Erythematosus: A Double-Blind, Randomised, Placebo-Controlled, Phase 2 Trial. Lancet, 2018;392(10143):222–231.10.1016/S0140-6736(18)31363-1Suche in Google Scholar PubMed
[13] You H, Zhang G, Wang Q, et al. Successful Treatment of Arthritis and Rash with Tofacitinib in Systemic Lupus Erythematosus: The Experience from a Single Centre. Ann Rheum Dis, 2019;78(10): 1441–1443.10.1136/annrheumdis-2019-215455Suche in Google Scholar PubMed
[14] Muskardin TLW, Niewold TB. Type I Interferon in Rheumatic Diseases. Nat Rev Rheumatol, 2018;14(4):214–228.10.1038/nrrheum.2018.31Suche in Google Scholar PubMed PubMed Central
© 2022 Wei Bai et al., published by Sciendo
This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
Artikel in diesem Heft
- Review
- Myositis-specific antibodies: Overview and clinical utilization
- Interleukin-6 blocking therapy for COVID-19: From immune pathogenesis to clinical outcomes
- Contribution of impaired DNASE1L3 activity to anti-DNA autoantibody production in systemic lupus erythematosus
- Original Article
- Effects of conventional rehabilitative and aerobic training in patients with idiopathic inflammatory myopathy
- Effectiveness of SB4 transition from originator etanercept in rheumatoid arthritis and axial spondyloarthritis: A subgroup analysis from the BENEFIT study
- Special Report
- The challenges and future perspective for the management of systemic lupus erythematosus in China: A concise annual report of 2020
- Images
- Andersson lesion in ankylosing spondylitis
- Letter
- A 16-year-old boy with arthritis, rash, and hemoptysis: Beyond “undifferentiated connective tissue disease”?
Artikel in diesem Heft
- Review
- Myositis-specific antibodies: Overview and clinical utilization
- Interleukin-6 blocking therapy for COVID-19: From immune pathogenesis to clinical outcomes
- Contribution of impaired DNASE1L3 activity to anti-DNA autoantibody production in systemic lupus erythematosus
- Original Article
- Effects of conventional rehabilitative and aerobic training in patients with idiopathic inflammatory myopathy
- Effectiveness of SB4 transition from originator etanercept in rheumatoid arthritis and axial spondyloarthritis: A subgroup analysis from the BENEFIT study
- Special Report
- The challenges and future perspective for the management of systemic lupus erythematosus in China: A concise annual report of 2020
- Images
- Andersson lesion in ankylosing spondylitis
- Letter
- A 16-year-old boy with arthritis, rash, and hemoptysis: Beyond “undifferentiated connective tissue disease”?