Orthoamide und Iminiumsalze, CIV. Umsetzungen von Orthoamiden der Alkincarbonsäuren mit enolisierbaren Carbonylverbindungen – Cyclisierung der Kondensationsprodukte zu Pyran-Derivaten
-
Willi Kantlehner
 ,
Kai Edelmann
,
Kai Edelmann

Abstract
Orthoamides of alkynecarboxylic acid 15 condense with enolisable β-dicarbonyl compounds and as well with acetophenones to give 3-acryl-1,1-bis(dimethyl-amino)-1,3-butadienes. Some acylbutadienes cyclize affording 2-pyranon-derivatives 33 upon heating with aqueous ethanol. 2H-pyranes are accessible from acetone dicarboxylic acid ester and orthoamides 15. The constitution of one 4-acyl-1,1-bis(dimethylamino)-1,3-butadiene (16f) and one 2H-pyrane (44b) was confirmed by crystal structure determinations.
1 Einleitung
Orthoamid-Derivate der Carbonsäuren, insbesondere der Ameisensäure 1, 2, 3 und der Kohlensäure 4, 5, 6, reagieren mit CH2-aciden Verbindungen 7 unter Kondensation. Bei Verwendung der Orthoamid-Derivate 1–3 resultieren dabei Enamine 8 [2], während die Umsetzungen mit den Orthoamiden 4 und 6 einheitlich zu Keten-O,N-acetalen 9 bzw. Ketenaminalen 10 führt. Aus den CH2-aciden 7 und dem Harnstoffderivat 5 entstehen häufig Gemische aus den Ketenderivaten 9 und 10 [3, 4]. In Gegenwart von Aktivatoren werden jedoch bevorzugt Ketenaminale erhalten [5] (Schema 1).

Orthoamid-Derivate der Ameisensäure 1–3 und Orthoamid-Derivate der Kohlensäure 4–6. Verlauf von Kondensationsreaktionen der Orthoamid-Derivate 1–6 mit CH2-aciden Verbindungen 7.
Mit einer Reihe enolisierbarer CH2-acider Verbindungen 12 wie z. B. Acetessigester, Acetylaceton, Dimedon wurden Kondensationsreaktionen mit den Orthoamiden 1–6, die Enamine 8 [6] bzw. Ketenacetale 9, 10 liefern [3], durchgeführt. Aber auch Umsetzungen von Acetophenonen [7], [8], [9] Heteroarylmethylketene [10], [11], [12], [13] und Alkylmethylketone [3] mit Acetalen des Dimethylformamids 1 sind beschrieben. Mit Aminalestern 2 wurden entsprechende Kondensationen durchgeführt [14], [15], [16], [17], [18] auch mit Tetrakis(dimethylamino)methan sind analoge Reaktionen beschrieben [19]. Während das Orthoamid-Derivat 3 CH2-acide Verbindungen glatt aminomethyleniert [6], wurde bei Umsetzungen der Orthoamide von Benzoesäure 11a bzw. Thiophencarbonsäure 11b mit Acetessigsäureester 12 bzw. Acetylaceton keine C,C-Verknüpfung beobachtet, vielmehr wurden die Dicarbonylverbindungen in Enamine 13 umgewandelt [20]. Aus den Orthoamiden 11 und nicht enolisierbaren CH2-aciden Verbindungen 7 bilden sich Enamine 14 [20] (Schema 2).

Umsetzung von enolisierbaren und nicht enolisierbaren CH2-Aciden 12 bzw. 7 mit Orthoamiden 11.
Aus Orthoamiden 6 und enolisierbaren sowie nicht enolisierbaren CH2-aciden Verbindungen wurden Ketenaminale 10 erhalten [21, 22]. Analoge Umsetzungen von enolisierbaren β-Dicarbonylverbindungen 12 (Acetylaceton, Acetessigester, Dimedon und Dimethylbarbitursäure) mit Orthoamiden von Alkincarbonsäuren 15 liefern 1,1-Bis-(dimethylamino)-1,3-butadiene 16 [23, 24] (Schema 3).

Orthoamide von Alkincarbonsäuren 15. Ketenaminale 16 aus enolisierbaren CH2-aciden Verbindungen 12 und Orthoamiden 15.
Stark enolisierte 1,3-Dicarbonylverbindungen können auch als vinyloge Carbonsäuren aufgefasst werden. Da Essigsäure mit dem Orthoamid 15c in komplexer Weise reagiert [25], haben wir das Verhalten von 1,3-Indandion 17 und 4-Hydroxycumarin 19 gegenüber den Orthoamiden 15a–d untersucht. Bei den Umsetzungen werden die Ketenaminale 18a–d bzw. 20a–c erhalten (Schema 4).

Ketenaminale 18, 20 aus stark enolisierte Carbonylverbindungen 17, 19 und Orthoamiden 15.
Die Ergebnisse stellen eine Parallele zum Verlauf der Umsetzungen von Orthoamiden 1 mit Tetronsäuren 21 dar. Hier erfolgt ebenfalls eine Aminomethylenierung der „Ketoform“ von 21 unter Bildung der Enamine 22 (Schema 5) [26].
![Schema 5:
Aminomethylenierung von Tetronsäuren 21 mit N,N-Dimethylformamidacetal (1) [26].](/document/doi/10.1515/znb-2021-0005/asset/graphic/j_znb-2021-0005_scheme_005.jpg)
Aminomethylenierung von Tetronsäuren 21 mit N,N-Dimethylformamidacetal (1) [26].
Bei den Verbindungen 18c, d sind NMR-spektroskopisch Stereoisomere nachweisbar. Es dürfte sich dabei um s-cis- und s-trans-Isomere 18A, 18B handeln, da die Bindung C2–C3 des Diensystems hohen Doppelbindungscharakter besitzt, denn im Allgemeinen werden push-pull-substituierte 1,3-Diene am richtigsten durch dipolare Grenzstrukturen beschrieben (Abbildung 1).

S-cis- und s-trans-Isomere 18A, B des Ketenaminals 18.
Die C,C-Knüpfungen, die bei der Umsetzung stark enolisierter Carbonylverbindungen mit Orthoamiden 15 beobachtet werden, erfolgen möglicherweise nach konkrrierenden Mechanismen, was am Beispiel der Umsetzungen der Orthoamide 15 mit den CH2-aciden Carbonylverbindungen 12 erläutert sei. Zunächst deprotoniert das Orthoamid 15 die CH2-acide Carbonylverbindung 12, wobei Enolationen 24 und Propiolamidiniumionen 23 entstehen. Addiert sich das Enolation mit dem Kohlenstoff am C-3 des Propiolamidiniumions, entsteht ein Allen 25 das durch Protonenwanderung in das Ketenaminal 16 übergeht (Schema 6).

Mechanismen bei der Bildung von Ketenaminalen 16 aus enolisierbaren Carbonylverbindungen 12 und Orthoamiden 15.
Lagert sich das Enolat 24 mit dem Sauerstoff am C1 das Kation 23 an, so kann sich das Addukt 26 durch eine Claisenumlagerung in 25 umwandeln, das dann in 16 übergeht. Eine analog verlaufende Claisenumlagerung, die zu 2,4-Pentadiensäureamiden 29 führt, wurde bei der Umsetzung von N,N-Dimethylacetamidacetal (27) mit Propargylalkoholen 28 beobachtet [27] (Schema 7).
![Schema 7:
Claisen Umlagerung bei der Umsetzung des N,N-Dimethylacetamidacetals 27 mit Propargylalkohol [27].](/document/doi/10.1515/znb-2021-0005/asset/graphic/j_znb-2021-0005_scheme_007.jpg)
Claisen Umlagerung bei der Umsetzung des N,N-Dimethylacetamidacetals 27 mit Propargylalkohol [27].
Stark acide Benzoylacetonitrile reagieren mit Orthoamiden 15 zu den entsprechenden Ketenaminalen [23, 24]. Aber auch aus den erheblich weniger aciden Acetophenonen 30 und Orthoamiden 15 lassen sich entsprechende Kondensationsprodukte 31 gewinnen, wenn unter Boresterkatalyse gearbeitet wird [28, 29] (Schema 8).

Ketenaminale 31 aus Acetophenonen 30 und Orthoamiden 15.
Die Reaktionsprodukte 31 fallen häufig als orangerot gefärbte, zähe Öle an, die sich destillativ nicht reinigen lassen. Bei früheren Untersuchungen haben wir unter anderem ein Orthoamid-Derivat der Benzoesäure mit Cyclohexanon umgesetzt. Das dabei gebildete Enamin 32 (Schema 9) wird dabei als zähes Öl erhalten, das aber nach Wasseraufnahme in ein stabiles, kristallines Hydrat übergeht [24].

Kondensation von Cyclohexanon mit einem Orthoamid-Derivat der Benzoesäure zum Enamin 32.
Es lag daher nahe zu prüfen, ob auch die Verbindungen 31 stabile Hydrate bilden. Wir wählten hierzu das Ketenaminal 31c, das aus 15d und 4-Nitroacetophenon (30c) darstellbar ist. Nach längerem Stehen in wässrigem Ethanol und mehrmaligen Erhitzen der homogenen Lösung schieden sich Kristalle ab, bei denen es sich aber nicht um das erwartete Hydrat von 31c, sondern um das 2-Pyron 33c handelt. Exemplarisch wurde nun untersucht, ob sich bei dieser Pyron-Synthese generell die rohen Ketenaminale 31 verwenden lassen. Dazu wurden die Acetophenone 30a, b mit den Orthoamiden 15c bzw. 15d in Acetonitril umgesetzt. Die nach dem Entfernen des Lösungsmittels verbleibenden Rückstände, die aus rohem 31a bzw. 31b bestehen, reagieren beim Erhitzen mit Ethanol/Wasser zu den 2-Pyronen 33a, b (Schema 10).

2-Pyrone 33 aus Ketenaminalen 31.
Die Pyronsynthese ist verwandt mit der bekannten, basenkatalysierten Cyclokondensation von 1,3-Diketonen mit Alkincarbonsäureestern, die 5-Acylpyran-2-on 35 liefert [30] (Schema 11).
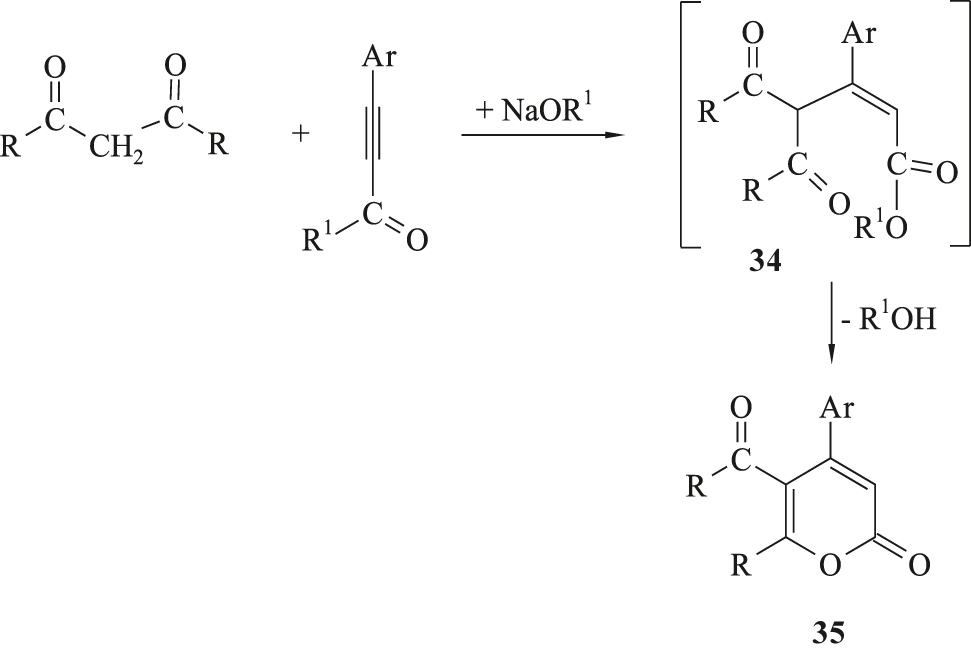
Synthese von 5-Acylpyran-2-on-Derivate 35.
Zwischenstufen sind dabei die Additionsprodukte 34, deren Enolform sich mit der Esterfunktion im Sinne einer Umesterung zu Pyran-2-on-Derivaten 35 umsetzen.
Da die Ketenaminale 31 nicht enolisierbar sind, nehmen wir an, dass die Umwandlung 31 → 33 durch eine Elektrocyclisierung der Ketenaminale 31 eingeleitet wird, wobei sich cyclische Aminalester 36 bilden (Schema 12). Durch Hydrolyse der Aminalesterfunktion in 36 entstehen die Pyrone 33.

Elektrocyclischer Ringschluss der Ketenaminale 31 zu cyclischen Aminalestern 36 und deren Hydrolyse zu Pyronen 33.
Wie die Grenzstrukturen 31B in Abbildung 2 zeigt, sind die Ketenaminale 31 deutlich polar gebaut, was die Bildung der Cycloaddukte 36 erleichtern dürfte.

Grenzstrukturen der Ketenaminale 31.
Außerdem ist bekannt, dass Aminalester außerordentlich leicht hydrolysieren.
Um nachzuweisen, dass die Pyronsynthese allgemein anwendbar ist, haben wir zunächst aus dem Orthoamid 15d und Acetylaceton 12b bzw. Dimedon 12c die Ketenaminale 16e, f dargestellt (Schema 13).

Ketenaminale 16e, f aus dem Orthoamid 15d und enolisierbaren β-Diketonen 12a, c.
Die Struktur des Ketenaminals 16f wurde durch eine Kristallstrukturanalyse gesichert. In Abbildung 3 ist die Festkörperstruktur von 16f dargestellt. In den Tabellen 1 und 2 sind ausgewählte Bindungsabstände und Bindungswinkel in 16f aufgelistet.

Molekülstruktur des Ketenaminals 16f im Kristall.
Ausgewählte Bindungsabstände im Ketenaminal 16f. Die Bezifferung der Atome entspricht der in Abbildung 3.
| Bindung | Länge [Å] | Bindung | Länge [Å] |
|---|---|---|---|
| C1–C2 | 1.456(2) | C5–C6 | 1.525(2) |
| C2–C3 | 1.354(2) | C8–C9 | 1.521(2) |
| C3–C4 | 1.469(2) | C1–N1 | 1.344(2) |
| C3–C16 | 1.489(2) | C1–N2 | 1.327(2) |
| C4–C5 | 1.424(2) | C5–O1 | 1.2481(18) |
| C4–C9 | 1.4365(19) | C9–O2 | 1.2428(19) |
Ausgewählte Bindungs- und Torsionswinkel [°] im Ketenaminal 16f.
| Bindungen | Winkel [°] | Bindungen | Winkel [°] |
|---|---|---|---|
| C1–N1–C15 | 122.12(13) | N1–C1–C2 | 120.53(13) |
| C1–N1–C14 | 121.55(13) | C1–C2–C3 | 125.51(14) |
| C15–N1–C14 | 115.04(13) | C2–C3–C4 | 125.54(13) |
| C1–N2–C12 | 120.89(13) | C2–C3–C16 | 115.10(13) |
| C1–N2–C13 | 124.20(14) | C4–C3–C16 | 118.99(12) |
| N1–C1–N2 | 120.74(13) | C5–C4–C9 | 121.01(13) |
| N2–C1–C2 | 118.60(13) | C3–C4–C5 | 120.19(13) |
| Torsionswinkel [°] | |||
| C15–N1–C1–N2 | 39.3(2) | N1–C1–C2–C3 | 49.3(2) |
| C14–N1–C1–N2 | −154.30(14) | C1–C2–C3–C4 | 10.0(2) |
| C15–N1–C1–C2 | −144.84(14) | C1–C2–C3–C16 | −177.15(14) |
| C14–N1–C1–C2 | 21.5(2) | C2–C3–C4–C5 | 39.7(2) |
| N1–C1–N2–C12 | −161.63(15) | C16–C3–C4–C5 | 132.98(14) |
| C2–C1–N2–C12 | 22.5(2) | C2–C3–C4–C9 | −142.11(16) |
| N1–C1–N2–C13 | 20.6(2) | C16–C3–C4–C9 | 45.25(19) |
| C2–C1–N2–C13 | −155.28(16) | C9–C4–C5–O1 | 179.30(14) |
| N2–C1–C2–C3 | −134.75(17) | C3–C4–C5–O1 | −2.5(2) |
Tabelle 1 entnimmt man, dass in 16f die vermeintliche C,C-Einfachbindung C2–C3 mit 1.354(2) Å wesentlich kürzer ist, als die „Doppelbindungen“ zwischen C1–C2 (1.456(2) Å) und C3–C4 (1.469(2) Å). Der Sachverhalt beweist, dass die Bindungsverhältnisse auch bei der Verbindung 16f durch die dipolare Struktur 16fB besser beschrieben werden, als durch die unpolare Struktur 16fA, denn die C2–C3-Bindung hat hohen Doppelbindungscharakter (Abbildung 4). Bezüglich der C2–C3-Bindung liegt eine „Z-Anordnung“ vor.

Grenzstrukturen des 1,3-Butadiens 16f.
Es sei angemerkt, dass sich das Ausmaß der Beteiligung polarer Grenzstrukturen wie 16f zur Beschreibung der Bindungsverhältnisse bei „Carbonylgruppen-haltigen“ Ketenaminalen 16, 18, 20 im Allgemeinen anhand der Schwingungsfrequenzen der C=O-Gruppen abschätzen lässt. Mit wachsender Polarität der Ketenaminale nimmt der Doppelbindungscharakter der C=O-Bindungen ab, d. h. die Lage der C=O-Banden im Infrarotspektrum sollten sich erniedrigen. Die Carbonylbanden von nicht-cyclischen gesättigten Ketonen liegen im Bereich 1690–1725 cm−1, in α,β-ungesättigten Ketonen findet man Absorptionen im Bereich 1665–1685 cm−1. Diese Werte sinken um weitere 20–80 cm−1, wenn Dialkylaminogruppen Bestandteile der konjugierten Systeme sind. In der folgenden Tabelle 3 ist die Lage der C=O-Banden für die Verbindungen 16a, e, f, g und 18a–d [31] zusammengestellt. Mit Ausnahme der Verbindung 18a–e beobachtet man tatsächlich erniedrigte IR-Absorptionen um 1600 und 1580 cm−1. Die scheinbar zu geringe Erniedrigung der Lage der C=O-Banden bei den Verbindungen 18a–d ist erklärbar. Bei cyclischen Ketonen bestimmt die Ringgröße die Lage der C=O-Bande im Infrarotspektrum. Diese liegen bei Cyclohexanonen zwischen 1706 und 1720 cm−1. Sie befinden sich damit im Bereich, der auch für aliphatische Ketone typisch ist. Deswegen tritt bei der Verbindung 16f eine Bandenverschiebung, die mit der bei den Verbindungen 16a, e übereinstimmt. Bei Cyclopentanonen liegt die C=O-Bande jedoch zwischen 1740 und 1750 cm−1 [31]. Geht bei den Verbindungen 18 mit steigender Polarität eine Frequenzverschiebung der C=O-Bande einher, die vergleichbar ist, mit denen, die bei den Verbindungen 16a, e, f auftreten, so sollten die Verbindungen 18 Absorptionen um 1650 cm−1 besitzen, was ja auch beobachtet wird.
Lage der C=O-Banden [cm−1] in 1,1-Diacyl-4,4-diamino-1,3-butadienen 16a, e, f, 18a–d.
| Ketenaminal | ν (C=O) [cm−1] | |
|---|---|---|

|
1600, 1580 | |

|
1590, 1580 | |

|
1600, 1580 | |

|
18a R = H 18b R = MeOCH2 18c R = Ph 18d R = 4-ClPh |
1670, 1625 1650, 1610 1650, 1610 1610, 1570 |
Das Ketenamminal 16f ist aus dem Orthoamid 15d und Dimedon (37) erhältlich. Beim Kochen mit wässrigem Ethanol wandelt es sich in das Chromenderivat 38 um (Schema 14).

Darstellung und Umwandlung des 1,1-Diamino-1,3-butadiens 16f in das Chromenderivat 37.
Bei der entsprechenden Umsetzung des Ketenaminals 16e erwarteten wir als Reaktionsprodukt das Pyron 39, erhalten wurde jedoch das deacetylierte Pyron 40. Auf welcher Stufe der Umsetzung die Deacylierung erfolgt, wurde nicht untersucht (Schema 15).

Darstellung des Pyrons 40 durch Cyclisierung des Ketenaminals 16a.
Bei der Kondensation von Acetondicarbonsäureester 41 mit dem Orthoamid 15a wurde das Ketenaminal 42a erhalten, das ausschließlich in der Ketoform vorliegt (Schema 16).

Synthese des Ketenaminals 42a. Pyranderivate 44a, b aus Acetondicarbonsäureester 41 und Orthoamiden 15c, d.
Die Umsetzung der Orthoamide 15c, d mit 41 führt unmittelbar zu den Pyranen 44a, b. Zwischenstufen dürften dabei ebenfalls Ketenaminale 42b, c sein. Offenbar cyclisiert die Enolform 43 der Ketenaminale 42b, c unter Abspaltung von Dimethylamin zu den Pyranderivaten 44a, b. Die Konstitution von 44b wurde durch eine Kristallstrukturanalyse bestätigt, deren Ergebnisse in Abbildung 5 dargestellt sind. Ausgewählte Bindungsabstände und Bindungswinkel der Verbindung 44b finden sich in den Tabellen 4 und 5.

Molekülstruktur des Pyrans 44b im Kristall.
Ausgewählte Bindungslängen [Å] in dem Pyran-3-carbonsäuremethylester 44b. Die Bezifferung der Atome entspricht der in Abbildung 5.
| Bindung | Länge [Å] | Bindung | Länge [Å] |
|---|---|---|---|
| O1–C2 | 1.3404(13) | C5–C16 | 1.4672(15) |
| O1–C6 | 1.3824(13) | C6–C19 | 1.3627(15) |
| C2–C3 | 1.3814(15) | C16–O16 | 1.2126(14) |
| C8–N7 | 1.4579(15) | O17–C16 | 1.3507(14) |
| C3–C4 | 1.3944(15) | C19–C20 | 1.4503(16) |
| C4–C5 | 1.4023(14) | O20–C20 | 1.2147(15) |
| C4–C10 | 1.4858(15) | C20–O21 | 1.3612(14) |
| C5–C6 | 1.4430(15) | O21–C22 | 1.4396(16) |
Ausgewählte Bindungs- [°] und Torsionswinkel [°] in dem Pyran-3-carbonsäuremethylester 44b.
| Bindungen | Winkel [°] | Bindungen | Winkel [°] |
|---|---|---|---|
| C2–O1–C6 | 123.75(9) | C5–C6–C19 | 128.92(10) |
| O1–C2–N7 | 112.71(10) | O1–C6–C19 | 114.29(10) |
| O1–C2–C3 | 120.68(10) | C2–N7–C9 | 119.78(10) |
| C3–C2–N7 | 126.60(10) | C2–N7–C8 | 122.48(10) |
| C2–C3–C4 | 119.01(10) | C8–N7–C9 | 117.59(10) |
| C3–C4–C10 | 115.06(9) | C5–C16–O17 | 112.43(9) |
| C3–C4–C5 | 120.84(10) | C5–C16–O16 | 126.24(10) |
| C5–C4–C10 | 123.96(9) | O16–C16–O17 | 121.29(10) |
| C4–C5–C16 | 123.34(10) | C16–O17–C18 | 115.40(9) |
| C4–C5–C6 | 118.75(9) | C6–C19–C20 | 124.60(10) |
| C6–C5–C16 | 117.87(9) | C19–C20–O21 | 109.89(10) |
| O1–C6–C5 | 116.79(9)5 | C19–C20–O20 | 128.95(11) |
| Torsionswinkel [°] | |||
| C2–O1–C6–C19 | 178.39(10) | C3–C4–C5–C6 | −4.23(15) |
| C2–O1–C6–C5 | −2.41(15) | C3–C4–C5–C16 | 173.46(10) |
| C6–O1–C2–N7 | −179.59(9) | C5–C4–C10–C11 | 136.94(11) |
| C6–O1–C2–C3 | −0.71(16) | C5–C4–C10–C15 | −48.19(16) |
| O1–C2–N7–C8 | 1.92(15) | C10–C4–C5–C6 | 171.19(9) |
| O1–C2–N7–C9 | 177.31(10) | C4–C5–C6–O1 | 4.77(14) |
| O1–C2–C3–C4 | 1.42(16) | C4–C5–C16–O16 | 151.13(12) |
| C3–C2–N7–C8 | −176.88(11) | C4–C5–C16–O17 | −31.21(14) |
| C3–C2–N7–C9 | −1.49(18) | C4–C5–C6–C19 | −176.17(11) |
| N7–C2–C3–C4 | −179.87(11) | C6–C5–C16–O16 | −31.15(16) |
| C2–C3–C4–C5 | 1.13(16) | C6–C5–C16–O17 | 146.51(10) |
| C2–C3–C4–C10 | −174.68(10) | C16–C5–C6–C19 | 6.01(17) |
| C3–C4–C10–C11 | −47.40(14) | C5–C6–C19–C20 | −177.54(11) |
| C3–C4–C10–C15 | 127.47(11) | O1–C6–C19–C20 | 1.54(16) |
| C6–C19–C20–O20 | 7.6(2) |
2 Schlussfolgerungen und Ausblick
Enolisierbare β-Dicarbonylverbindungen 12 sowie Acetophenone 30 reagieren mit Orthoamiden von Alkincarbonsäuren 15 zu 1,1-Diamino-butadienderivaten, die beim Erwärmen mit wässrigem Ethanol in 2-Pyranone übergehen. Acetondicarbonsäureester 40 setzt sich mit den Orthoamiden 15b, c unmittelbar zu 2H-Pyranen um. Von präparativen Interesse dürfte es sein, herauszufinden, ob sich diese Pyron-Synthese auch mit funktionalisierten Ketonen wie z. B. Benzoylacetonitril, Acetessigester usw. durchführen lassen und, ob α,α′-difunktionalisierte Ketone mit 15 ebenfalls zu 2H-Pyronen vom Typ 43 reagieren.
3 Experimenteller Teil
3.1 Vorbemerkung
Die Schmelzpunkte wurden mit einem Büchi Ölbad 510 bestimmt und sind unkorrigiert.
UV/VIS-Spektren wurden mit einem Spektralphotometer der ACTA-M Serie der Firma Beckmann angefertigt. Die IR-Spektren wurden mit einem Perkin-Elmer 457 und 283 Spektralphotometer aufgenommen. Die 250 MHz 1H NMR-Spektren und 62.9 MHz 13C NMR-Spektren wurden mit einem Bruker AC 250 Kernspinresonanzspektrometer aufgenommen. Die Massenspektren wurden mit einem Varian MAT 711 Massenspektrometer mit Datensystem DP 10 der Firma AMD aufgenommen. Hochauflösende Spektren wurden nach dem Peakmatching Verfahren erhalten.
Alle Umsetzungen wurden in Lösungsmitteln, die nach Standardverfahren getrocknet wurden, unter Feuchtigkeitsausschluss durchgeführt (KOH-Trockenrohr). Im Fall von Isomerengemischen werden die Protonen des einen Isomeren mit H, die des anderen mit H′ bezeichnet.
Die Orthoamide 15 wurden nach Literaturangaben hergestellt: 15a [33], 15b [23], 15c [33], 15d [33].
3.2 Umsetzung der Orthoamide 15a–d mit Indan-1,3-dion (17)
3.2.1 Allgemeine Vorschrift
Zu einer Lösung von Indan-1,3-dion (17) in 30 mL THF wird eine Lösung des Orthoamids 15 in 20 mL THF zugetropft und 20 h bei Raumtemperatur gerührt. Anschließend werden alle flüchtigen Bestandteile im Rotationsverdampfer entfernt und der Rückstand umkristallisiert.
3.2.2 Umsetzung des Orthoamids 15a mit Indan-1,3-dion
0.73 g (5 mmol) Indan-1,3-dion werden mit 0.85 g (5 mmol) 15a nach der allgemeinen Vorschrift umgesetzt. Ausb.: 0.51 g (60%) 2-[3,3-Bis(dimethylamino)-2-propenyliden]-indan-1,3-dion (18a), gelber Feststoff mit Schmp. 220–222 °C (Ethylacetat). – IR (KBr): ν = 1670, 1625 (C=O), 1580, 1560, 1550, 1535, 1515 (C=C) cm−1. – 1H NMR (250 MHz, CDCl3, TMS): δ = 3.06 (s, 12H, NMe2), 6.51 [d, J = 14.4 Hz, 1H, (Me2N)2C=CH], 7.50 [d, J = 14.4 Hz, 1H, (Me2N)2C=CH–CH], 7.49–7.64 (m, 4H, Ar) ppm. – 13C NMR (62.9 MHz, CDCl3, TMS): δ = 42.10 (NMe2), 94.87 [(Me2N)2C=CH], 110.27 [(Me2N)2C=CH–CH=C], 120.07, 120.56, 131.88, 133.12, 139.93, 141.31 (Ar), 144.43 [(Me2N)2C=CH–CH], 172.14 [(Me2N)2 C], 191.76, 192.53 (C=O) ppm. – C16H18N2O2 (270.33): ber. C 71.09, H 6.71, N 10.36; gef. C 71.01, H 6.74, N 10.36.
3.2.3 Umsetzung des Orthoamids 15b mit Indan-1,3-dion (17)
Aus 0.73 g (5 mmol) Indan-1,3-dion (17) und 1.07 g (5 mmol) 15b erhält man nach der allgemeinen Vorschrift. Ausb.: 0.64 g (41%) 2-[3,3-Bis(dimethylamino)-1-methoxymethyl-2-propenyliden]-indan-1,3-dion (18b), gelber Feststoff mit Schmp. 174–175 °C (Ethylacetat). – IR (KBr): ν = 1650, 1610 (C=O), 1590, 1565, 1540 (C=C) cm−1. – 1H NMR (250 MHz, CDCl3, TMS): δ = 3.06 (s, 12H, NMe2), 3.47 (s, 3H, O–CH 3), 4.81 (s, 2H, –CH 2–O), 5.40 [(Me2N)2C=CH], 7.35–7.46 (m, 4H, Ar) ppm. – 13C NMR (62.9 MHz, CDCl3, TMS): δ = 41.85 (NMe2), 58.74 (O–CH3), 73.08 (CH2–O), 98.85 [(Me2N)2C=CH], 106.23 [CH–C(CH2–O–CH3)=C], 119.13, 130.91, 139.79 (Ar), 156.35 (CCH2–O–CH3), 172.88 [(Me2N)2 C], 190.04 (C=O) ppm. – C18H22N2O3 (314.38): ber. C 68.77, H 7.05, N 8.91; gef. C 68.50, H 7.08, N 8.87.
3.2.4 Umsetzung des Orthoamids 15c mit Indan-1,3-dion (17)
Es werden 0.73 g (5 mmol) Indan-1,3-dion (17) und 1.23 g (5 mmol) 15c nach der allgemeinen Vorschrift umgesetzt. Ausb.: 0.90 g (52%) 2-[3,3-Bis(dimethylamino)-1-phenyl-2-propenyliden]-indan-1,3-dion (18c), gelber Feststoff mit Schmp. 238–240 °C (Ethylacetat). – IR (KBr): ν = 1650, 1610 (C=O), 1565, 1510 (C=C) cm−1. – 1H NMR (250 MHz, CDCl3, TMS): δ = 2.72 (s, 12H, NMe2), 3.07 (s, 12H′, NMe2), 5.13 [s, 1H, (Me2N)2C=CH], 6.61 [s, 1H′, (Me2N)2C=CH], 7.24–7.55 (m, 9H+9H′, Ar), H/H′=5/1 ppm. – 13C NMR (62.9 MHz, CDCl3, TMS): δ = 41.85 (NMe2), 99.40 [(Me2N)2C=CH], 103.76 [C′, (Me2N)2C=CH], 108.34 [CH–C(Ph)=C], 119.33, 127.43, 127.82, 128.71, 129.22, 131.00, 131.40, 139.60, 139.92, 140.14, 140.83 (Ar), 158.14 [CH–C(Ph)=C], 160.58 [C′, CH–C(Ph)=C], 171.66 [(Me2N)2 C], 171.96 [C′, (Me2N)2 C], 189.97, 191.64 (C=O) ppm. – C22H22N2O2 (346.43): ber. C 76.27, H 6.40, N 8.09; gef. C 76.33, H 6.38, N 8.12.
3.2.5 Umsetzung des Orthoamids 15d mit Indan-1,3-dion (17)
0.73 g (5 mmol) Indan-1,3-dion (17) werden mit 1.40 g (5 mmol) 15d entsprechend der allgemeinen Vorschrift umgesetzt. Ausb.: 1.06 g (56%) 2-[3,3-Bis(dimethylamino)-1-(p-chlorphenyl)-2-propenyliden]-indan-1,3-dion (18d), gelber Feststoff mit Schmp. 282 °C (Acetonitril). – IR (KBr): ν = 1610 (C=O), 1570, 1510 (C=C) cm−1. – 1H NMR (250 MHz, CDCl3, TMS): δ = 2.77 (s, 12H, NMe2), 3.09 (s, 12H′, NMe2), 5.08 [s, 1H, (Me2N)2C=CH], 6.62 [s, 1H′, (Me2N)2C=CH], 7.27–7.57 (m, 8H+8H′, Ar), H/H′=6/1 ppm. – 13C NMR (62.9 MHz, CDCl3, TMS): δ = 41.88 (NMe2), 99.84 [(Me2N)2C=CH], 103.32 [C′, (Me2N)2C=CH], 108.18 [C(p-Cl-Ph)=C], 119.45, 119.87, 127.68, 128.06, 130.48, 130.61, 131.16, 135.25, 138.04, 139.29, 139.82, 140.06 (Ar), 156.78 [(Me2N)2C=CH–C], 171.57 [(Me2N)2 C], 171.71 [C′, (Me2N)2 C], 189.97 (C=O) ppm. – C22H21N2O2Cl (380.87): ber. C 69.38, H 5.56, N 7.36, Cl 9.31; gef. C 69.13, H 5.60, N 7.44, Cl 9.39.
3.3 Umsetzung der Orthoamide 15a, c, d mit 4-Hydroxycumarin (19)
3.3.1 Allgemeine Vorschrift
Zu einer Lösung von 4-Hydroxycumarin (19) in 20 mL THF wird das Orthoamid 15 in 30 mL THF zugetropft und 20 h unter Rückfluss erhitzt. Anschließend werden alle flüchtigen Bestandteile im Rotationsverdampfer entfernt und der Rückstand in Aceton über Aluminiumoxid 90 (Akt. II–III) filtriert. Das Aceton wird im Rotationsverdampfer entfernt und der zurückbleibende Feststoff mit Diethylether gewaschen.
3.3.2 Umsetzung des Orthoamids 15a mit 4-Hydroxycumarin (19)
Es werden 0.81 g (5 mmol) 4-Hydroxycumarin (19) mit 0.85 g (5 mmol) 15a entsprechend der allgemeinen Vorschrift umgesetzt. Ausb.: 0.53 g (37%) 3-[3,3-Bis(dimethylamino)-2-propenyliden]-2,4-dioxo-chroman (20a), gelber Feststoff mit Schmp. 194 °C. – IR (KBr): ν = 1675 (C=O), 1610, 1600, 1535 (C=C) cm−1. – 1H NMR (250 MHz, CDCl3, TMS): δ = 3.14 (s, 12H, NMe2), 7.12 [d, J = 14.5 Hz, 1H, CH=C(NMe2)2], 7.20 [d, J = 14.5 Hz, 1H, CH–CH=C], 7.17–7.21 (m, 4H, Ar), 7.43–7.50 (m, 1H, Ar), 8.07–8.15 (m, 2H, Ar) ppm. – 13C NMR (62.9 MHz, CDCl3, TMS): δ = 42.58 (NMe2), 100.53 (C-3), 101.76 [CH=C(NMe2)2], 116.53 (C-8), 122.07 (C-4a), 122.95, 125.93, 132.31 (C-5, C-6, C-7), 151.01 [=CH–CH=C(NMe2)2], 154.40 (C-8a), 164.95 (C-2), 173.19 [C(NMe2)2], 178.21 (C-4) ppm. – C16H18N2O3 (286.33): ber. C 67.12, H 6.34, N 9.78; gef. C 67.25, H 6.40, N 9.71.
3.3.3 Umsetzung des Orthoamids 15c mit 4-Hydroxycumarin (19)
Nach der allgemeinen Vorschrift werden 0.81 g (5 mmol) 4-Hydroxycumarin (19) mit 1.23 g (5 mmol) 15c umgesetzt. Ausb.: 0.47 g (26%) 3-[3,3-Bis(dimethylamino)-1-phenyl-2-propenyliden]-2,4-dioxo-chroman (20b), gelber Feststoff mit Schmp. 215 °C erhalten. – IR (KBr): ν = 1650 (C=O), 1600, 1545 (C=C) cm−1. – 1H NMR (250 MHz, CDCl3, TMS): δ = 3.07 (s, 12H, NMe2), 5.73 [s, 1H, CH=C(NMe2)2], 7.11–7.20 (m, 2H, Ar), 7.25–7.32 (m, 3H, Ar), 7.35–7.46 (m, 1H, Ar). 7.48–7.52 (m, 2H, Ar), 8.01–8.04 (m, 1H, Ar) ppm. – 13C NMR (62.9 MHz, CDCl3, TMS): δ = 42.27 (NMe2), 98.35 (C-3), 112.59 [CH=C(NMe2)2], 116.32 (C-8), 122.31 (C-4a), 125.84, 128.97, 131.06 (C-5, C-6, C-7), 128.19, 128.97, 141.94 (Ph), 154.14 [=CH(Ph)], 160.20 (C-8a), 163.88 (C-2), 170.60 [C(NMe2)2], 174.06 (C-4) ppm. – C22H22N2O3 (362.43): ber. C 72.91, H 6.12, N 7.73; gef. C 72.82, H 6.10, N 7.66.
3.3.4 Umsetzung des Orthoamids 15d mit 4-Hydroxycumarin (19)
Es werden 0.81 g (5 mmol) 4-Hydroxycumarin (19) mit 1.40 g (5 mmol) 15d entsprechend der allgemeinen Vorschrift umgesetzt. Ausb.: 0.64 g (32%) 3-[3,3-Bis(dimethylamino)-1-(p-chlorphenyl)-2-propenyliden]-2,4-dioxo-chroman (20c), gelber Feststoff mit Schmp. 212–213 °C erhalten. – IR (KBr): ν = 1665 (C=O), 1600, 1555, 1525 (C=C) cm−1. – 1H NMR (250 MHz, CDCl3, TMS): δ = 3.09 (s, 12H, NMe2), 5.70 [s, 1H, CH=C(NMe2)2], 7.12–7.21 (m, 2H, Ar), 7.25–7.33 (m, 2H, Ar), 7.39–7.45 (m, 3H, Ar). 8.00–8.04 (m, 1H, Ar) ppm. – 13C NMR (62.9 MHz, CDCl3, TMS): δ = 42.28 (NMe2), 98.08 (C-3), 112.57 [CH=C(NMe2)2], 116.35 (C-8), 122.17 (C-4a), 122.52, 125.81, 131.23 (C-5, C-6, C-7), 128.38, 128.90, 134.71, 140.52 (p-ClPh), 154.10 [=C(p-Cl-Ph)], 158.90 (C-8a), 163.86 (C-2), 170.37 [CH=C (NMe2)2], 174.21 (C-4) ppm. – C22H21N2O3Cl (362.43): ber. C 66.58, H 5.33, N 7.06, Cl 8.93; gef. C 66.46, H 5.39, N 7.12, Cl 8.97.
3.4 2-Pyranone 33 aus Ketenaminalen 31
Allgemeine Vorschrift zur Umsetzung von Orthoamiden 15 mit Acetophenonen 30 zu Ketenaminalen 31 und deren Umwandlung in Pyrone 33.
Das Orthoamid 15 (6–15 mmol) und die entsprechende Menge des Acetophenons 30 werden in 30 mL Acetonitril gelöst. Die Lösung wird 18 h unter Rückfluss erhitzt, wobei gerührt wird. Dann wird das Lösungsmittel im Rotationsverdampfer entfernt, wobei das rohe Ketenaminal 31 zurückbleibt.
Der Rückstand wird in einem Gemisch aus 10 mL Ethanol und 20 mL Wasser aufgenommen und 6 h unter Rückfluss erhitzt und gerührt. Alle flüchtigen Bestandteile werden im Mempranpumpenvakuum abdestilliert und der Rückstand umkristallisiert.
3.4.1 Umsetzung des Orthoamids 15d mit 4-Nitroacetophenon (30c) zum Pyran-2-on 33c
Entsprechend der allgemeinen Vorschrift werden 3.49 g (12.5 mmol) des Orthoamids 15d mit 2.06 g (12.5 mmol) 4-Nitroacetophenon (30c) umgesetzt. Ausb.: 1.56 g (38%) 4-(4-Chlorphenyl)-6-(4-nitrophenyl)pyran-2-on (33c), gelber Feststoff mit Schmp. 217 °C (CH2CN). – IR (KBr): ν = 1700 (C=O), 1620, 1580 (C=C) cm−1. – 1H NMR (250 MHz, CDCl3, TMS): δ = 6.55 (d, J = 1.4 Hz, 1H, 5-H), 7.04 (d, J = 1.4 Hz, 1H, 3-H), 7.44–7.62 (m, 4H, Ar), 8.05–8.37 (m, 4H, Ar) ppm. – Aufgrund der Schwerlöslichkeit von 33c in den üblichen deuterierten Lösungsmitteln konnte kein 13C NMR-Spektrum aufgenommen werden. – C17H10ClNO4 (327.72): ber. C 62.31, H 3.08, Cl, 10.82, N 4.27; gef. C 62.23, H 3.15, Cl 10.71, N 4.45.
3.4.1.1 Umsetzung des Orthoamids 15c mit Acetophenon (30a) zum Ketenaminal 31a
Es werden 3.61 g (14.7 mmol) 15c mit 1.77 g (14.7 mmol) Acetophenon (30a) nach der allgemeinen Vorschrift Teil 1 angesetzt. Der Rückstand wird in 20 mL Diethylether gelöst. Man lässt einen Teil des Ethers verdampfen, wobei sich das Produkt abscheidet. Ausb.: 3.72 g (79%) 5,5-Bis(dimethylamino)-1,3-diphenyl-penta-2,4-dien-1-on (31a), orange-gelber Feststoff mit Schmp. 89 °C (Lit. [32]: 89 °C). IR (KBr): ν = 1620 (C=O), 1580, 1560, 1480 (C=C) cm−1. – 1H NMR (250 MHz, CDCl3, TMS): δ = 2.73 (s, 9H, NMe2), 2.92 (s, 3H, NMe2), 5.68 [d, J = 1.0 Hz, 1H, -CH=C(NMe2)2], 6.07 (d, J = 1.0 Hz, 1H, =CH–CO), 6.99–7.22 (m, 10H, Ph) ppm. – 13C NMR (62.9 MHz, CDCl3, TMS): δ = 41.22 (NMe2), 102.10, 115.18 (C-2, C-4), 127.08, 127.40, 127.67, 127.93, 128.15, 130.18, 137.81, 143.69 (Ph-C), 151.82 (C-3), 156.23 (C=O), 168.22 (C-5) ppm. – C21H24N2O (320.42): ber. C 70.72, H 7.55, N 8.74; gef. C 78.52, H 7.47, N 8.73.
3.4.1.2 Cyclisierung des Ketenaminals 31a
Nach der allgemeinen Vorschrift Teil 2 werden 1.80 g (5.6 mmol) 5,5-Bis(dimethylamino)-1,3-diphenyl-penta-2,4-dien-1-on (31a) umgesetzt. Man erhält 4,6-Diphenyl-pyran-2-on (33a) als schwach gelben Feststoff mit Schmp. 142 °C (Acetonitril). Ausb.: 0.58 g (42%). – IR (KBr): ν = 1690 (C=O), 1620, 1520, 1470 (C=C) cm−1. – 1H NMR (250 MHz, CDCl3, TMS): δ = 6.45 (d, J = 1.5 Hz, 1H, 5-H), 6.95 (d, J = 1.5 Hz, 1H, 3-H), 7.45–7.51 (m, 2H, Ph), 7.53–7.66 (m, 2H, Ph), 7.84–8.00 (m, 2H, Ph) ppm. – 13C NMR (62.9 MHz, CDCl3, TMS): δ = 101.34 (C-5), 109.22 (C-3), 125.73, 126.72, 128.95, 129.23, 130.68, 130.91, 131.51, 136.00 (Ar-Cl), 155.55 (C-4), 160.34 (C-6), 162.62 (C=O) ppm. – C17H12O2 (248.28): ber. C 82.24, H 4.87; gef. C 82.26, H 5.06.
3.4.2 Umsetzung des Orthoamids 15c mit 3-Bromacetophenon (30b) zum Pyran-2-on 33b
Es werden 1.47 g (6.0 mmol) 15c mit 1.19 g (6.0 mmol) 3-Bromacetophenon (30b) entsprechend der allgemeinen Vorschrift umgesetzt. Ausb.: 0.94 g (48%) 6-(3-Bromphenyl)-4-phenyl-pyran-2-on (33b), gelber Feststoff mit Schmp. 175 °C (Acetonitril). – IR (KBr): ν = 1690 (C=O), 1620, 1520, 1480 (C=C) cm−1. – 1H NMR (250 MHz, CDCl3, TMS): δ = 6.46 (d, J = 1.4 Hz, 1H, 5-H), 6.93 [d, J = 1.4 Hz, 1H, (C-3)-H], 7.44–7.52 (m, 5H, Ar), 7.61–7.65 (m, 2H, Ar), 7.79–7.83 (m, 2H, Ar) ppm. – 13C NMR (62.9 MHz, CDCl3, TMS): δ = 101.51 (C-5), 109.48 (C-3), 126.70, 126.97, 129.27, 129.97, 130.79, 135.82, 137.05 (Ar-C), 155.44 (C-4), 159.16 (C-6), 162.26 (C=O) ppm. – C17H11BrO2 (327.18): ber. C 62.41, H 3.39, Br 24.42; gef. C 62.32, H 3.42, Br 24.14.
3.5 Umsetzung des Orthoamids 15d mit Dimedon (37)
Es werden 2.57 g (9.2 mmol) 15d mit 1.29 g (9.2 mmol) Dimedon (37) nach der allgemeinen Vorschrift (Teil 1) umgesetzt. Die Reaktionszeit beträgt 15 h. Der ausgefallene Niederschlag wird abfiltriert. Ausb.: 3.10 g (90%) 2-[1-(4-Chlorphenyl)-3,3-bis(dimethylamino)-2-propenyliden]-5,5-dimethyl-cyclohexan-1,3-dion (16f), gelber Feststoff mit Schmp. 203 °C (Zers.). – IR (KBr): ν = 1600, 1580 (C=O), 1550, 1500, 1470 (C=C) cm−1. – 1H NMR (250 MHz, CD3OD, TMS): δ = 1.14 [s, 6H, C-5(CH3)2], 2.28 (s, 4H, CH2), 3.08 (bs, 12H, NMe2), 5.81 (s, 1H, =CH–), 7.27–7.56 (m, 4H, Ar) ppm. – 13C NMR (62.9 MHz, CD3OD, TMS): δ = 30.79 [C-5(CH3)2], 33.63 (C-5), 43.97 (NMe2), 53.06 (CH2), 113.86 (C-2), 117.83 (=CH–), 130.54, 131.52, 136.64, 143.96 (Ar-C), 159.07 [(C-2)=C(p-Cl-C6H4–)], 172.28 [–HC=C(NMe2)2], 195.23 (C=O) ppm. – C21H27ClN2O2 (374.91): ber. C 67.28, H 7.26, Cl 9.46. N 7.47; gef. C 67.29, H 7.31, Cl 9.25, N 7.45.
3.6 Cyclisierung der Ketenaminale 16f, a zu 2-Pyronen 38, 40
3.6.1 Cyclisierung von 16f
1.86 g (5.0 mmol) 2-[1-(4-Chlorphenyl)-3,3-bis(dimethylami-no)-2-propenyliden]-5,5-dimethyl-cyclohexan-1,3-dion (16f) werden nach der allgemeinen Vorschrift Teil 2 umgesetzt. Ausb.: 1.02 g (68%) 4-(4-Chlorphenyl)-7,7-dimethyl-7,8-dihydro-6H-chromen-2,5-dion (38), leicht gelblicher Feststoff mit Schmp. 164 °C (Wasser-Isopropylalkohol = 5:1). – IR (KBr): ν = 1730, 1670 (C=O), 1600, 1560, 1520 (C=C) cm−1. – 1H NMR (250 MHz, CDCl3, TMS): δ = 1.16 [s, 6H, C(CH3)2], 2.42 [s, 2H, (C-8)-H2], 2.81 [s, 2H, (C-6)-H2], 6.04 [s, 1H, (C-3)-H], 7.12 (d, J = 8.4 Hz, 2H, Ar), 7.35 (d, J = 8.4 Hz, 2H, Ar) ppm. – 13C NMR (62.9 MHz, CDCl3, TMS): δ = 28.14 (CH3), 31.99 (C-7), 42.72 (C-8), 52.18 (C-6), 112.88 (C-4a), 114.29 (C-3), 128.15, 128.58, 135.00, 135.47 (Ar-C), 155.09 (C-8a), 159.41 (C-4), 173.71 (C=O), 193.26 (C-2) ppm. – C17H15ClO3 (302.76): ber. C 67.44, H 4.99, Cl 11.71; gef. C 67.26, H 5.03, Cl 11.49.
3.6.2 Cyclisierung des Ketenaminals 16a
2.12 g (7.1 mmol) 3-Acetyl-6,6-bis(dimethylamino)-4-phenyl-2-oxo-3,5-hexadien (16a) [23] werden nach der allgemeinen Vorschrift Teil 2 umgesetzt. Ausb.: 0.75 g (57%) 6-Methyl-4-phenyl-pyran-2-on (40), leicht gelblicher Feststoff mit Schmp. 94 °C (Diethylether-Ethanol = 4:1). – IR (KBr): ν = 1700 (C=O), 1620, 1570, 1530 (C=C) cm−1. – 1H NMR (250 MHz, CDCl3, TMS): δ = 2.32 (s, 3H, CH3), 6.32, 6.35 [je s, 1H, (C-3)-H, (C-5)-H], 7.44–7.60 (m, 5H, Ph) ppm. – 13C NMR (62.9 MHz, CDCl3, TMS): δ = 20.18 (CH3), 103.48 (C-5), 108.11(C-3), 126.65, 129.16, 130.60, 135.80 (Ar-C), 155.51 (C-4), 162.16 (C-6), 163.43 (C=O) ppm. – C12H10O2 (186.21): ber. C 77.40, H 5.41; gef. C 77.16, H 5.43.
3.7 Umsetzung der Orthoamide 15a, c, d mit Acetondicarbonsäuredimethylester (41)
3.7.1 Umsetzung des Orthoamids 15a
Zu 1.74 g (10.0 mmol) Acetondicarbonsäuredimethylester (41) in 20 mL THF werden 1.69 g (10.0 mmol) 15a in 20 mL THF getropft. Nach 24 h Rühren wird die Reaktionsmischung im Rotationsverdampfer vom Lösungsmittel befreit. Das zurückbleibende Öl wird mit Diethylether zur Kristallisation gebracht und anschließend aus Methyl-tert-Butylether umkristallisiert. Ausb.: 1.10 g (37%) 7,7-Bis(dimethylamino)-4-methoxycarbonyl-3-oxo-4,6-heptadiensäuremethylester (42a), gelber Feststoff mit Schmp. 80–82 °C. – UV–VIS (CHCl3): λ max (lg ε max) = 275 (4.1847), 410 (4.7221) nm. – IR (KBr): ν = 1735, 1655 (COOMe), 1585 (CO), 1560, 1510 (C=C) cm−1. – 1H NMR (250 MHz, CDCl3, TMS): δ = 3.04 (s, 12H, NMe2), 3.70, 3.71 (je s, 3H, OMe), 3.89 (s, 2H, CH2), 6.25 [bs, 1H, (Me2N)2C=CH], 7.94 (bs, 1H, CH=C–CO) ppm. – 13C NMR (62.9 MHz, CDCl3, TMS): δ = 42.03 (NMe2), 49.74 (C-2), 50.41, 51.65 (OMe), 94.55 (C-6), 104.5 (C-4), 153.0 (C-5), 169.34 (COOMe), 170.81 (C-7), 172.91 (C-1), 189.89 (C-3) ppm. – C14H22N2O5 (298.34): ber. C 56.36, H 7.43, N 9.39; gef. C 56.23, H 7.48, N 9.24.
3.7.2 Umsetzung des Orthoamids 15c
Zu 0.78 g (4.48 mmol) Acetondicarbonsäuredimethylester (41) in 20 mL THF werden 1.10 g (4.48 mmol) 15c in 30 mL THF getropft und 18 h bei Raumtemperatur gerührt. Anschließend wird die Reaktionsmischung im Rotationsverdampfer vom Lösungsmittel befreit. Das zurückbleibende Öl wird mit etwas Ethylacetat/Petrolether (1:1) kurz zum Sieden erhitzt und über Nacht bei 0–5 °C stehengelassen. Der Niederschlag wird abfiltriert und aus Ethylacetat umkristallisiert. Ausb.: 0.91 g (62%) 5-Dimethylamino-2-methoxycarbonylmethylen-4-phenyl-2H-pyran-3-carbonsäuremethylester (44a), orangeroter Feststoff mit Schmp. 153–154 °C. – UV/VIS (CH3CN): λ max (lg ε max) = 285 (4.4206), 328 (4.0899), 450 (4.1004) nm. – IR (KBr): ν = 1680, 1630 (CO), 1540, 1505, 1485 (Ar) cm−1. – 1H NMR (250 MHz, CDCl3, TMS): δ = 3.2 (s, 6H, NMe2), 3.39, 3.65 (je s, 3H, OMe), 4.83 (s, 1H, CH=), 5.42 (s, 1H, 5-H), 7.24–7.37 (m, 5H, Ar) ppm. – 13C NMR (62.9 MHz, CDCl3, TMS): δ = 37.44 (NMe2), 50.21, 51.34 (OMe), 81.79, 84.24 (C-5, =CH), 103.41 (C-3), 126.59, 128.27, 128.57, 140.41 (Ar), 154.26 (C-4), 160.32 (s, COOMe), 166.96 (C-2, COOMe) ppm. – MS (EI, 70 eV): m/z (%) = 329.1 (100) [M]+, 298.1 (23) [M–OMe]+, 270.1 (39) [M–COOMe]+, 221.0 (14) [M–2COOMe]+, 72.0 (19) [CHCOOMe]+. – HRMS ((+)-ESI): m/z = 329.1262 (ber. 329.31264 für C18H19NO5, [M]+).
3.7.3 Umsetzung des Orthoamids 15d
Zu 0.87 g (5.0 mmol) Acetondicarbonsäuredimethylester (41) in 20 mL THF werden 1.40 g (5.0 mmol) 15d in 30 mL THF getropft. Es wird 20 h bei Raumtemperatur gerührt. Anschließend wird die Reaktionsmischung im Rotationsverdampfer vom Lösungsmittel befreit. Das zurückbleibende Öl wird mit etwas Ethylacetat/Petrolether (1:1) kurz zum Sieden erhitzt und über Nacht bei 0–5 °C stehengelassen. Der entstandene Niederschlag wird abfiltriert und aus Ethylacetat umkristallisiert. Ausb.: 1.19 g (65%) 5-Dimethylamino-2-methoxycarbonylmethylen-4-(p-chlorphenyl)-2H-pyran-3-carbonsäuremethylester (44b), roter Feststoff mit Schmp. 182–183 °C. – UV/VIS (CH3CN): λ max (lg ε max) = 287 (5.9069), 330 (5.5763), 452 (5.601) nm. – IR (KBr): ν = 1685 (CO), 1660, 1620, 1555, 1545, 1490 (C=C) cm−1. – 1H NMR (250 MHz, CDCl3, TMS): δ = 3.2 (s, 6H, NMe2), 3.42, 3.64 (je s, 3H, OMe), 4.77 (s, 1H, CH=), 5.45 (s, 1H, 3-H), 7.18, 7.32 (je d, J = 8.4 Hz, 2H, Ar) ppm. – 13C NMR (62.9 MHz, CDCl3, TMS): δ = 37.47 (NMe2), 50.24, 51.4 (OMe), 81.58, 84.87 (C-5, =CH), 102.77 (C-3), 128.07, 128.47, 134.41, 138.98 (Ar), 153.19 (C-4), 158.94 (C-6), 160.04, 166.64 (s, COOMe), 166.83 (C-2) ppm. – MS (EI, 70 eV): m/z (%) = 363.0 (100) [M]+, 332.0 (33) [M–OMe]+, 304.0 (45) [M–COOMe]+, 254.9 (10) [M–COOMe, OMe, OH]+, 323.0 (10) [M–COOMe, CHCOOMe]+, 72.1 (45) [CHCOOMe]+. – C18H18NO5Cl (363.8): ber. C 59.43, H 4.99, N 3.85, Cl 9.75; gef. C 59.39, H 5.01, N 3.65, Cl 9.59.
4 Kristallstrukturanalysen der Verbindungen 16f und 44b
Die Intensitätsdaten für 16f und 44b wurden mit einem Bruker Kappa APEXII Duo Vierkreisdiffraktometer im ω- und φ-Scanmodus bei T = 140 K (Graphit-Monochromator, MoKα-Strahlung) gemessen. Die Einkristalle wurden hierzu auf einer Teflonöse fixiert. Bei beiden Strukturen wurde eine semiempirische Absorptionskorrektur mit äquivalenten Reflexen (Multi-Scan Methode) durchgeführt.
Die Verfeinerung der Parameter wurde ohne Einschränkung nach der Methode der kleinsten Fehlerquadrate mit der vollen Matrix durchgeführt (Shelxl-2014/7). Die Ergebnisse der Kristallstrukturbestimmungen sind in den Tabellen 1, 2, 4 und 5 zusammengestellt. Tabelle 6 fasst die Kristallstrukturdaten zusammen:
Kristallografische Daten für die Verbindungen 16f und 44b.
| 16f | 44b | |
|---|---|---|
| Empirische Formel | C21H27ClN2O2 | C18H18NO5Cl |
| Molare Masse | 374.90 | 363.8 |
| Temperatur [K] | 140(2) | 140(2) |
| Kristallgröße [mm³] | 0.73 × 0.29 × 0.20 | 0.88 × 0.38 × 0.36 |
| Kristallsystem | monoklin | monoklin |
| Raumgruppe | Cc | P21/c |
| a [Å] | 12.6544(6) | 7.3644(4) |
| b [Å] | 15.1906(7) | 11.5557(6) |
| c [Å] | 11.4930(8) | 20.2541(9) |
| β [°] | 119.988(2) | 96.300(2) |
| Volumen V [Å3] | 1913.52(19) | 1713.23(15) |
| Z | 4 | 4 |
| ρ ber [g cm−3] | 1.301 | 1.41 |
| Wellenlänge λ [Å] | 0.71073 | 0.71073 |
| μ [mm−1] | 0.218 | 0.252 |
| F(000) [e] | 800 | 760 |
| θ-Bereich [°] | 2.29–30.59 | 2.02–30.5 |
| Index Bereiche hkl | −17 → 18, ±21, ±16 | ±10, ±16, ±28 |
| Gemessene Reflexe | 30,936 | 28,894 |
| Unabhängige Reflexe/R int | 5828/0.0260 | 5220/0.0269 |
| Beobachtete Reflexe [I > 2 σ(I)] | 5549 | 4424 |
| Verfeinerte Parameter | 242 | 231 |
| R1/wR2 [I > 2 σ(I)] | 0.0284/0.0694 | 0.0389/0.0997 |
| R1/wR2 (alle Daten) | 0.0312/0.0707 | 0.0488/0.1043 |
| GooF (F 2) | 1.022 | 1.023 |
| Flack x | 0.008(10) | – |
| Extinktionskoeffizient | 0.0049(7) | 0.0196(14) |
| Restelektronendichte [e Å−3] | 0.301/−0.177 | 0.428/−0.276 |
CCDC 2056034 (16f) und CCDC 2056035 (44b) enthalten die beim Cambridge Crystallographic Data Center hinterlegten Kristallstrukturdaten. Anforderung www.ccd.cam.ac.uk/data request/cif.
-
Author contributions: All the authors have accepted responsibility for the entire content of this submitted manuscript and approved submission.
-
Research funding: None declared.
-
Conflict of interest statement: The authors declare no conflicts of interest regarding this article.
Literatur
1. Kantlehner, W., Mezger, J., Tiritiris, I., Kreß, R., Frey, W. Z. Naturforsch. 2021, 76b, 133–162; https://doi.org/10.1515/znb-2020-0074.Search in Google Scholar
2. Eine Zusammenstellung von Übersichtsartikeln findet sich unter loc. cit. [2] bei: Kantlehner, W., Stieglitz, R., Hauber, M., Haug, E., Regele, C. J. Prakt. Chem. 2000, 342, 256–268.10.1002/(SICI)1521-3897(200003)342:3<256::AID-PRAC256>3.0.CO;2-GSearch in Google Scholar
3. Meerwein, H., Florian, W., Schön, N., Stopp, G. Liebigs Ann. Chem. 1961, 641, 1–39; https://doi.org/10.1002/jlac.19616410102.Search in Google Scholar
4. Kantlehner, W., Jaus, H., Kienitz, L., Bredereck, H. Liebigs Ann. Chem. 1979, 2096–2113; https://doi.org/10.1002/jlac.197919791220.Search in Google Scholar
5. Kantlehner, W., Heckel, B., Mezger, J. Z. Naturforsch. 2020, 75b, 865–880; https://doi.org/10.1515/znb-2020-0072.Search in Google Scholar
6. Bredereck, H., Effenberger, F., Brendle, T. Angew. Chem., Int. Ed. Engl. 1966, 5, 132; https://doi.org/10.1002/anie.196601321.Search in Google Scholar
7. Föhlisch, B. Chem. Ber. 1971, 104, 348–349; https://doi.org/10.1002/cber.19711040141.Search in Google Scholar
8. Lin, Y., Lang, S. A.Jr. J. Heterocycl. Chem. 1977, 14, 343–347; https://doi.org/10.1016/b978-0-08-019602-2.50020-4.Search in Google Scholar
9. Lin, Y., Lang, S. A.Jr. J. Org. Chem. 1980, 45, 4857–4860; https://doi.org/10.1021/jo01312a011.Search in Google Scholar
10. Boamah, P. J., Haider, N., Heinisch, G. J. Heterocycl. Chem. 1989, 26, 933–939; https://doi.org/10.1002/jhet.5570260410.Search in Google Scholar
11. Sarodnick, G. Chem. Ztg. 1991, 115, 217–218; https://doi.org/10.1016/0300-9572(91)90015-q.Search in Google Scholar
12. Jameson, D. L., Guise, L. E. Tetrahedron Lett. 1991, 18, 1993–2002; https://doi.org/10.1016/s0040-4039(00)78890-3.Search in Google Scholar
13. Gupton, J. T., Hicks, F. A., Wilkinson, D. R., Petrich, S. A., Sikorski, J. A. Heterocycles 1994, 37, 487–499; https://doi.org/10.3987/com-93-s38.Search in Google Scholar
14. Bredereck, H., Effenberger, F., Botsch, H. J. Chem. Ber. 1964, 97, 3397–3406; https://doi.org/10.1002/cber.19640971220.Search in Google Scholar
15. Bredereck, H., Simchen, G., Griebenow, W. Chem. Ber. 1974, 107, 1545–1554; https://doi.org/10.1002/cber.19741070515.Search in Google Scholar
16. Bennett, G. B., Simpson, W. R. J., Mason, R. B., Strohschein, R. J., Mensukhani, R. J. Org. Chem. 1977, 48, 221–225; https://doi.org/10.1021/jo00422a009.Search in Google Scholar
17. Eiden, F., Hereis, C. Arch. Pharm. (Weinheim) 1978, 311, 287–293; https://doi.org/10.1002/ardp.19783110404.Search in Google Scholar PubMed
18. Takeuchi, N., Ochi, K., Murase, M., Tobinaga, S. J. Chem. Soc., Chem. Commun. 1980, 13, 593–594; https://doi.org/10.1039/c39800000593.Search in Google Scholar
19. Weingarten, H., Edelmann, K. N. J. Org. Chem. 1967, 33, 3293–3294; https://doi.org/10.1021/jo01286a005.Search in Google Scholar
20. Kantlehner, W., Haug, E., Stieglitz, R., Frey, W., Kreß, R., Mezger, J. Z. Naturforsch. 2002, 57b, 399–419; https://doi.org/10.1515/znb-2002-0406.Search in Google Scholar
21. Kantlehner, W., Mergen, W. W., Haug, E., Speh, P., Kapasakalidis, J. J., Bräuner, H.-J. Liebigs Ann. Chem. 1985, 1804–1816; https://doi.org/10.1002/jlac.198519850908.Search in Google Scholar
22. Kantlehner, W., Mergen, W. W., Haug, E. Liebigs Ann. Chem. 1983, 290–298; https://doi.org/10.1002/jlac.198319830214.Search in Google Scholar
23. Kantlehner, W., Vettel, M., Lehmann, H., Edelmann, K., Stieglitz, R., Ivanov, I. C. J. Prakt. Chem. 1998, 340, 408–423; https://doi.org/10.1002/prac.19983400503.Search in Google Scholar
24. Kantlehner, W., Haug, E., Stieglitz, R., Frey, W., Kreß, R., Mezger, J. Z. Naturforsch. 2002, 57b, 399–419; https://doi.org/10.1515/znb-2002-0406.Search in Google Scholar
25. Weingarten, H. Tetrahedron 1968, 24, 2767–2772; https://doi.org/10.1016/s0040-4020(01)82548-5.Search in Google Scholar
26. Gelin, S., Chantegrel, B. J. Heterocycl. Chem. 1981, 18, 663–665; https://doi.org/10.1002/jhet.5570180403.Search in Google Scholar
27. Parker, K. A., Kosley, R. W. Tetrahedron Lett. 1976, 341–344; https://doi.org/10.1016/s0040-4039(00)93726-2.Search in Google Scholar
28. Kantlehner, W., Lehmann, H., Edelmann, K., Mezger, J., Ivanov, I. C. Appl. Catal. A 2008, 336, 148–154; https://doi.org/10.1016/j.apcata.2007.08.027.Search in Google Scholar
29. Kantlehner, W., Mezger, J., Lehmann, H., Edelmann, K., Frey, W. Z. Naturforsch. 2018, 73b, 689–702; https://doi.org/10.1515/znb-2018-0065.Search in Google Scholar
30. Kohler, E. P. J. Am. Chem. Soc. 1982, 44, 384–390; https://doi.org/10.1007/bf00319924.Search in Google Scholar
31. Bellamy, L. J. Ultrarot-Spektrum und chemische Konstitution, 2. Auflage; Kapitel 9, D. Steinkopf: Darmstadt, 1966; pp. 101–106.Search in Google Scholar
32. Kantlehner, W., Lehmann, H., Edekmann, K., Mezger, J., Ivanov, I. C. Appl. Catal., A 2008, 326, 148–154; https://doi.org/10.1016/j.apcata.2007.08.027.Search in Google Scholar
33. Kantlehner, W., Speh, P., Lehmann, H., Bräuner, H.-J. Chem. Ztg. 1990, 184, 176–178.Search in Google Scholar
© 2021 Walter de Gruyter GmbH, Berlin/Boston
Articles in the same Issue
- Frontmatter
- In this issue
- Research Articles
- Orthoamide und Iminiumsalze, CIV. Umsetzungen von Orthoamiden der Alkincarbonsäuren mit enolisierbaren Carbonylverbindungen – Cyclisierung der Kondensationsprodukte zu Pyran-Derivaten
- 1,3-Dimethyl-tetrakis(2-triphenylsilylethyl)dimethyldisiloxane: a new carbosilane for the preparation of high-refractive-index films
- Antibacterial activity of some chemical constituents from Trichilia prieuriana (Meliaceae)
- A cobalt-based coordination polymer with a tripodal carboxylate ligand: synthese, structure and properties
- The stannides Ca1.692Pt2Sn3.308, SrPtSn2 and EuAuSn2
- Synthesis and coordination chemistry of silver(I), gold(I) and gold(III) complexes with picoline-functionalized benzimidazolin-2-ylidene ligands
Articles in the same Issue
- Frontmatter
- In this issue
- Research Articles
- Orthoamide und Iminiumsalze, CIV. Umsetzungen von Orthoamiden der Alkincarbonsäuren mit enolisierbaren Carbonylverbindungen – Cyclisierung der Kondensationsprodukte zu Pyran-Derivaten
- 1,3-Dimethyl-tetrakis(2-triphenylsilylethyl)dimethyldisiloxane: a new carbosilane for the preparation of high-refractive-index films
- Antibacterial activity of some chemical constituents from Trichilia prieuriana (Meliaceae)
- A cobalt-based coordination polymer with a tripodal carboxylate ligand: synthese, structure and properties
- The stannides Ca1.692Pt2Sn3.308, SrPtSn2 and EuAuSn2
- Synthesis and coordination chemistry of silver(I), gold(I) and gold(III) complexes with picoline-functionalized benzimidazolin-2-ylidene ligands