
Exploring inherited vitamin B responsive disorders in the Moroccan population: cutting-edge diagnosis via GC-MS profiling
-
Samira Najeh
 ,
Es-Said Sabir
,
Imane Assiri
,
Miloud Hammoud
,
Karima Lafhal
,
Maroua Jakani
,
Abdelaati Berrachid
,
Hanane Choubbane
,
Zakaria Baki
,
Safya Sbyea
,
Salwa Baki
,
Hajar Qorchi
,
Mina Aallam
,
Najib Kissani
,
Lhoucine Gebrati
,
My Ahmed El Amiri
,
Abdelkader Outzourhit
and
Naima Fdil
,
Es-Said Sabir
,
Imane Assiri
,
Miloud Hammoud
,
Karima Lafhal
,
Maroua Jakani
,
Abdelaati Berrachid
,
Hanane Choubbane
,
Zakaria Baki
,
Safya Sbyea
,
Salwa Baki
,
Hajar Qorchi
,
Mina Aallam
,
Najib Kissani
,
Lhoucine Gebrati
,
My Ahmed El Amiri
,
Abdelkader Outzourhit
and
Naima Fdil


Abstract
Objectives
B vitamins are a group of eight essential nutrients involved in various biological processes, including energy production and the proper functioning of the nervous system. They play a critical role in certain inherited metabolic diseases (IMDs). Deficiencies in the metabolism of thiamine (B1), biotin (B7), and cobalamin (B12) can lead to serious health issues, including confusion, seizures, respiratory problems, encephalopathy, coma, hearing and vision loss, ataxia, and more. The main objective of our study is to highlight the role of Gas Chromatography-Mass Spectrometry (GC/MS) in detecting vitamin B-responsive IMDs in Moroccan patients.
Case presentation
Urine samples from symptomatic individuals suspected of having intermediate metabolism deficiencies, excluding aminoacidopathies, were analyzed using GC/MS. Three cases of IMDs were identified: pyruvate dehydrogenase complex deficiency, biotinidase deficiency, and methylmalonic aciduria. Each patient presented with neurological symptoms and metabolic abnormalities. Following targeted vitamin supplementation (B1, B7, or B12), significant clinical improvement was observed, with resolution of neurological manifestations and normalization of brain CT findings.
Conclusions
Urinary metabolite screening using GC/MS in patients with neurological and other symptoms indicative of IMDs can facilitate practical, reliable diagnosis and accessible management. GC/MS analysis is a highly sensitive, rapid, and precise technique for quantifying a wide range of metabolites in human urine. We recommend considering such analysis as a routine test for specific symptoms in the context of personalized and functional medicine practice.
Introduction
The identification and quantification of small molecules involved in metabolism within biological systems play a crucial role in diagnosing various diseases, thus improving the prognosis and quality of patient’s life. Since early 1980s, the combination of chromatography and mass spectrometry is utilized for precise and reproducible analysis of metabolites in biological fluids or tissue samples [1]. Currently, Urine is a hopeful sample for qualitative and quantitative determination of hundreds of hydroxylated molecules, which derive from various sources, including both normal and abnormal metabolism [2], 3].
Morocco is a developing country, recently known for the diversity and high prevalence of inherited metabolic diseases due to consanguineous marriages and the lack of reference laboratories [4]. Research on organic acidurias started in 2018 at our laboratory with great support by Spanish experts [5], 6].
The organic acidemias or organic acidurias (OA), are a class of inborn errors of metabolism, usually caused by a deficiency in an enzymatic activity of amino acid, carbohydrate or lipid metabolism [7]. Among these diseases are inherited metabolic disorders responsive to B vitamins [8], 9].
B vitamins play a crucial role in several metabolic processes, including the metabolism of carbohydrates, fats, and proteins. They act as cofactors or coenzymes in reactions that release energy from food, synthesize neurotransmitters, and maintain neurological function. These vitamins are also essential for the synthesis and repair of DNA, as well as the regulation of gene expression [10], 11]. Disruption in these pathways can lead to the accumulation of harmful metabolites, which are detectable in biological samples. Thus, in some patients, abnormal organic acids detected through gas chromatography-mass spectrometry (GC/MS), often point to specific deficiencies of B vitamins.
The B vitamins group consists of eight vitamins; vitamin B1 (thiamine), vitamin B2 (riboflavin), vitamin B3 (niacin), vitamin B5 (pantothenic acid), vitamin B6 (pyridoxine), vitamin B7 (biotin), vitamin B9 (folic acid or folate) and vitamin B12 (cobalamin or cyanocobalamin) [12]. Each of these vitamins plays distinct but interconnected roles in metabolic pathways. Vitamin B1 (thiamine) is critical for the oxidative decarboxylation of pyruvate to acetyl-CoA, a key step in carbohydrate metabolism. It acts as a coenzyme for the pyruvate dehydrogenase complex (PDHC), linking glycolysis to the citric acid cycle [13]. Vitamin B7 (biotin) is a coenzyme for carboxylation reactions essential in fatty acid synthesis, amino acid catabolism, and gluconeogenesis [14]. Vitamin B12 (cobalamin) is involved in two critical enzymatic reactions: the conversion of homocysteine to methionine via methionine synthase and the conversion of methylmalonyl-CoA to succinyl-CoA via methylmalonyl-CoA mutase [15]. Disruptions in these vitamin-dependent pathways result in specific metabolic disorders, which can be detected by the accumulation of abnormal metabolites in biological samples.
In this study, we focus on three metabolic disorders associated with deficiencies in thiamine, biotin, and cobalamin, each leading to the buildup of characteristic metabolites:
Pyruvate dehydrogenase complex deficiency, which affects carbohydrate metabolism, leads to elevated levels of lactate and pyruvate. The PDHC enzyme relies on thiamine (vitamin B1) as a cofactor to convert pyruvate into Acetyl-CoA. In its absence, pyruvate accumulates and is diverted towards lactate, resulting in lactic acidosis [16].
Biotinidase deficiency is a metabolic disorder characterized by insufficient activity of the Biotinidase enzyme, which is responsible for recycling biotin by releasing it from biotin-dependent carboxylases. The resulting shortage of free biotin impairs the function of biotin-dependent carboxylases, leading to the accumulation of potentially toxic metabolites, including 3-hydroxyisovaleryl-carnitine in plasma and 3-hydroxyisovaleric acid in urine [17], 18].
Methylmalonic aciduria (cobalamin disorders), the condition is characterized by elevated levels of methylmalonic acid (MMA) due to a deficiency in methylmalonyl-CoA mutase, an enzyme that requires vitamin B12 for its activity [19]. The accumulation of MMA leads to metabolic acidosis and various neurological symptoms, including confusion, seizures, and coma [20].
This study reports the cases of three patient’s vitamin B-responsive metabolic disorders identified by analyzing abnormal profiles of urinary organic acids using GC/MS. These initial findings were subsequently confirmed through specific enzymatic assays, emphasizing the critical role of early diagnosis and appropriate vitamin supplementation in improving patient outcomes.
Case presentation
In this study, we report three cases of patients with rare metabolic disorders diagnosed through biochemical analyses and confirmed by enzymatic and/or genetic testing. The patients, aged between 10 and 24 years, presented with a range of symptoms, from intermittent neurological episodes to severe neuromuscular impairments and hematological abnormalities.
These patients were managed according to established protocols for hereditary metabolic diseases, and biochemical investigations led to the identification of three distinct conditions: thiamine-responsive pyruvate dehydrogenase complex (PDHC) deficiency, biotinidase deficiency, and vitamin B12-responsive methylmalonic aciduria.
Samples
During the patient’s decompensation, samples for basic and specific biochemical analysis, as well as for special studies, were collected prior to the initiation of any supportive measures. This was done in accordance with the established protocols for handling suspected hereditary metabolic diseases at our hospital. The samples for specific biochemical analyses, were collected as follows:
Urine: first-void morning urine samples (2–4 mL)
Blood: venous blood collected in tubes containing EDTA and heparin (Improvacuter®, Guangzhou Improve Medical Instruments Co. Ltd), and 60–70 μL of whole blood spotted on Whatman filter paper (Whatman-GE no. 903, Sigma-Aldrich, Saint-Louis, MO, USA), blood samples are dedicated to specific enzyme assays and/or genetic analysis to confirm diagnoses.
Sample preparation for gas chromatography analysis (GC/MS)
The procedure used to extract urinary organic acids was carried out following Nakagawa et al. and Prieto, J. A., et al. with some slight modifications [5], 21]. In a 10 mL centrifuge tube with a glass stopper, 25 µl of internal standard solution (tropic acid, 0.5 mg/mL) was added to 2 mL of urine. The mixture was acidified with 100 µL of HCl (6 N) and stirred for 30 s with a vortex mixer. The mixture was centrifuged at 4,000×g for 10 min, the organic acids were extracted twice with 6 mL of ethyl acetate. After centrifugation, the organic phase was collected and evaporated at 50 °C under a gentle nitrogen flow. The final dry residue was derivatized using 80 µL of N, O-bis (trimethylsilyl) trifluoroacetamide at 60 °C for 1 h, then, 1 µL were injected into the GC column.
GC/MS analysis
GC/MS analysis was performed using a Thermo Scientific Trace 1300 Gas Chromatography coupled with an ISQ LT Single Quadruple Mass Spectrometer on a GC column TG-5MS; 30 m × 0.25 mm × 0.25 µm. The GC was equipped with a split-mode capillary injection port held at 275 °C with a split flow 19.5 mL/min. The chromatographic Temperature program was as follows: initial Temperature was 70 °C, held for 5 min, increased to 300 °C a rest of 4.5 °C/min and a held for 5 min with a helium flow of 1 mL/min. Transfer line temperature was 280 °C, and ion source temperature was 230 °C. The mass spectrometer operated in scan mode into a scan range from 50 to 480 m/z, using a 4 min solvent delay. The metabolites were identified by the NIST, mainlib and replib libraries. Sample metabolites were determined by quantifying the areas of chromatographic peaks corresponding to total ions. The area of each peak, representing a specific compound, is compared with the area of the corresponding peak in a reference sample (normal).
Pathway analysis
Two combined ways can be used to interpret results. Scientific literature is the initial approach to find links between the identified metabolites and known metabolic diseases based on previous published studies, researchers can associate the detected metabolites in their samples with abnormal metabolic pathways or specific biological processes related to the diseases. The second approach involves analysis tools like MetaboAnalyst 4.0, which is a free online platform specifically designed for metabolomic data analysis and interpretation. This platform offers a wide range of powerful tools and algorithms that enable the analysis of metabolomics data generated by various techniques, such as mass spectrometry and chromatography.
Results
Validation methodology for metabolomics identification
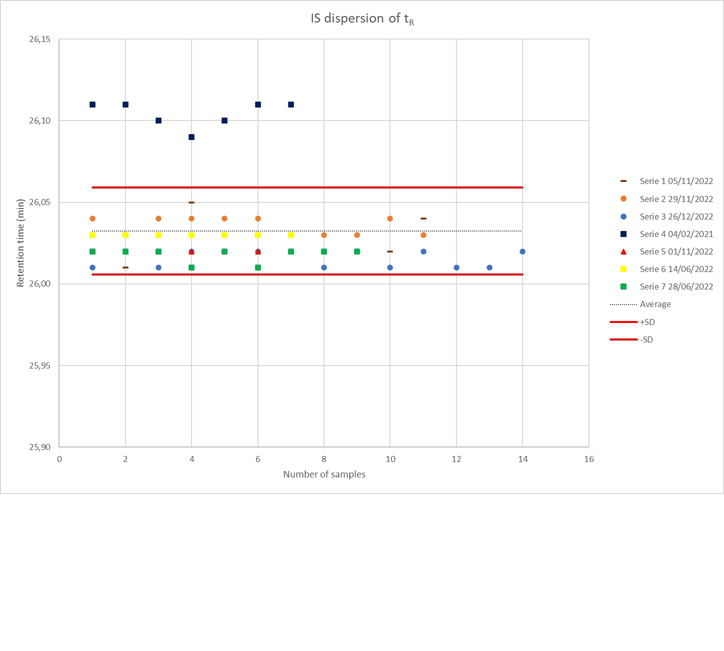
We used tropic acid as an internal standard (IS) in each urine sample (from both control and patient groups) at a consistent volume. The IS helps assess the repeatability of the analytical run and allows for data filtering prior to analysis by accounting for accurate mass-to-charge ratios, retention time, and signal drift variations.
The use of an IS offers several benefits: First, it ensures reproducibility by allowing us to “condition” or equilibrate the analytical system with matrix injections before starting the main analytical run. Second, it provides information necessary for calculating technical and analytical precision. Lastly, it corrects signal variations both within and between analytical runs or batch analyses.
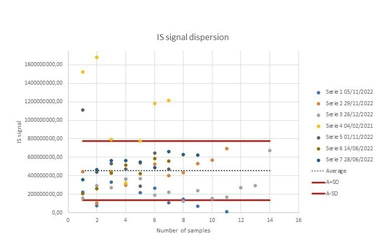
To assess instrumental repeatability before data processing, we conducted statistical analyses on three parameters: peak retention time, intensity, and mass-to-charge ratio (m/z). For this purpose, the total signal from each chromatogram was plotted against each sample (in sequence/run order) (Supplementary Figures 1–2). The dispersion of the IS in the plot reflects the instrumental repeatability.
We also evaluated selectivity by comparing a blank sample to spiked samples containing tropic acid (IS) (Supplementary Figure 3). This comparison demonstrated good selectivity of the method.
Patients
Patient one
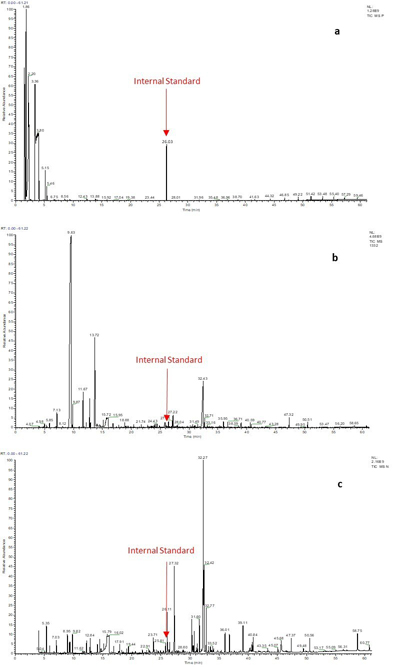
A 10-year-old female, born from a first-degree consanguineous marriage, presented with no similar conditions among her family members. Over the past four years, she has experienced two acute episodes of muscular weakness, ataxia, and neuroimaging abnormalities, all of which were reversible. She was referred by the neurology department with the same symptoms, now accompanied by optic atrophy and alterations in visual potential. Laboratory examinations, including complete blood count, renal function, lipid profile, and liver function, were in the normal ranges, a moderated lactic acid elevation was found in plasma; 3.20 mmol/L (0.5–2.20) mmol/L, brain MRI shows sequential bi-pallidal lacunar lesions of ischemia associated with bilateral and symmetrical pallidal and cerebral peduncle signal abnormalities related to anoxo-ischemic injury. Urinary GC/MS analysis highlights elevated amount of lactic acid, pyruvic acid and hydroxybutiric acid (Figure 1, Table S1) evoking intermittent-relapsing Pyruvate dehydrogenase complex (PDHC) deficiency, PDHC activity in cultured skin fibroblasts was reduced to 0.543 nM/min/mg protein (normal: 1.13–6.67 nM/min/mg protein). The patient was treated with thiamine supplementation and showed almost complete clinical recovery within one month, except for a persistent tremor in her left hand. One month after starting thiamine supplementation, follow-up tests were performed. These tests revealed normalization of lactic acid levels and no presence of molecules indicating a deficiency in internal metabolism.
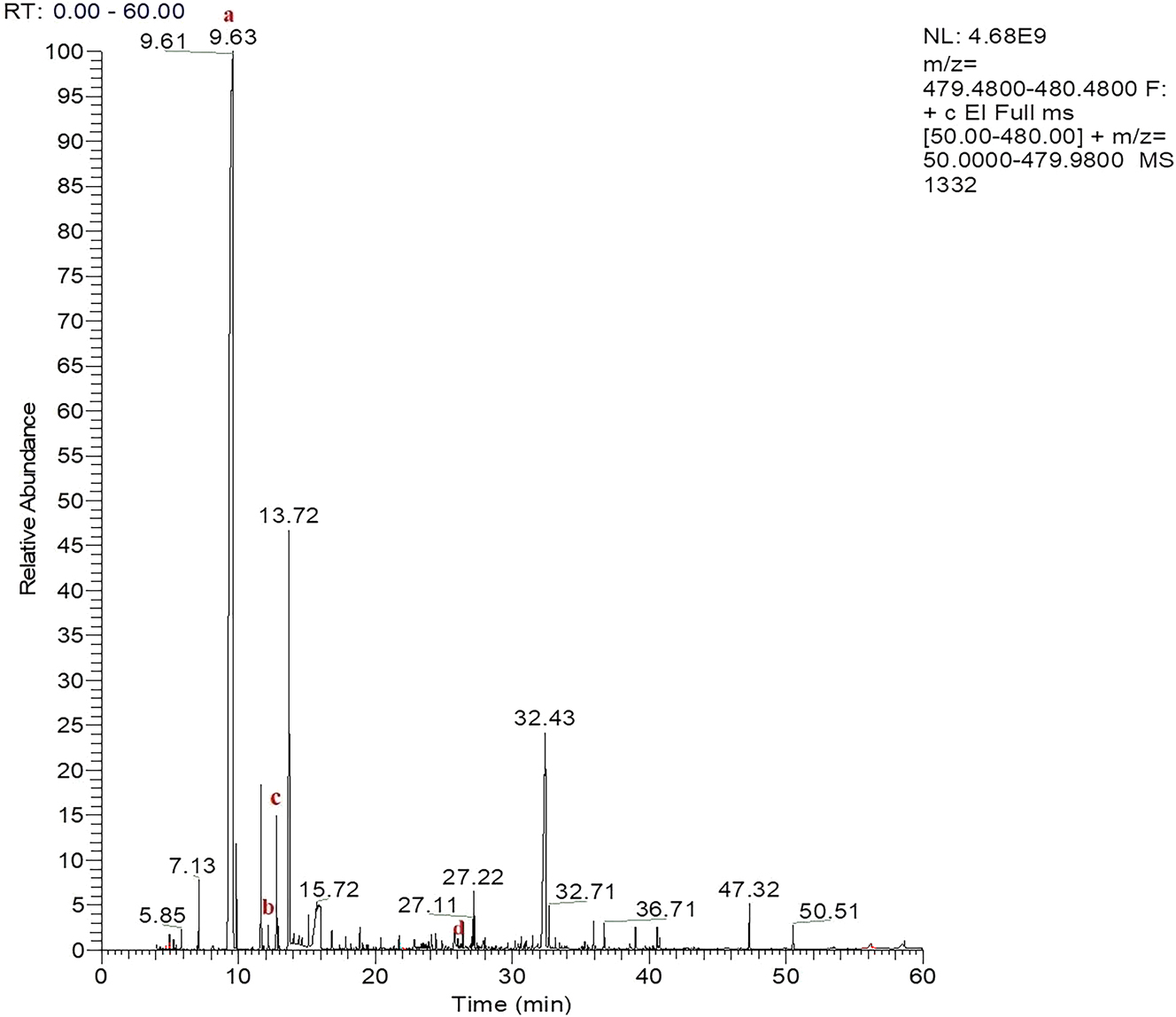
Chromatogram of organic acids of the first case obtained with the GC-MS method. (a) Lactic acid, (b) hydroxybutyric acid, (c) pyruvic acid, (d) Tropic acid.
Patient two
A 24-year-old male, born of a first-degree consanguineous marriage, has a sister with similar but more severe conditions. The patient presented with seizures, myoclonic jerks, spasms, and severe hypotonia, initially suggesting adult-onset metachromatic leukodystrophy. However, this was ruled out by the absence of urinary sulfatides. MRI imaging indicated a late-onset central nervous system demyelinating disorder. Urinary GC-MS analysis revealed a high rate of 3-Hydroxyisovaleric acid (Figure 2, Supplementary Table 1), a metabolite indicative of Biotinidase deficiency.
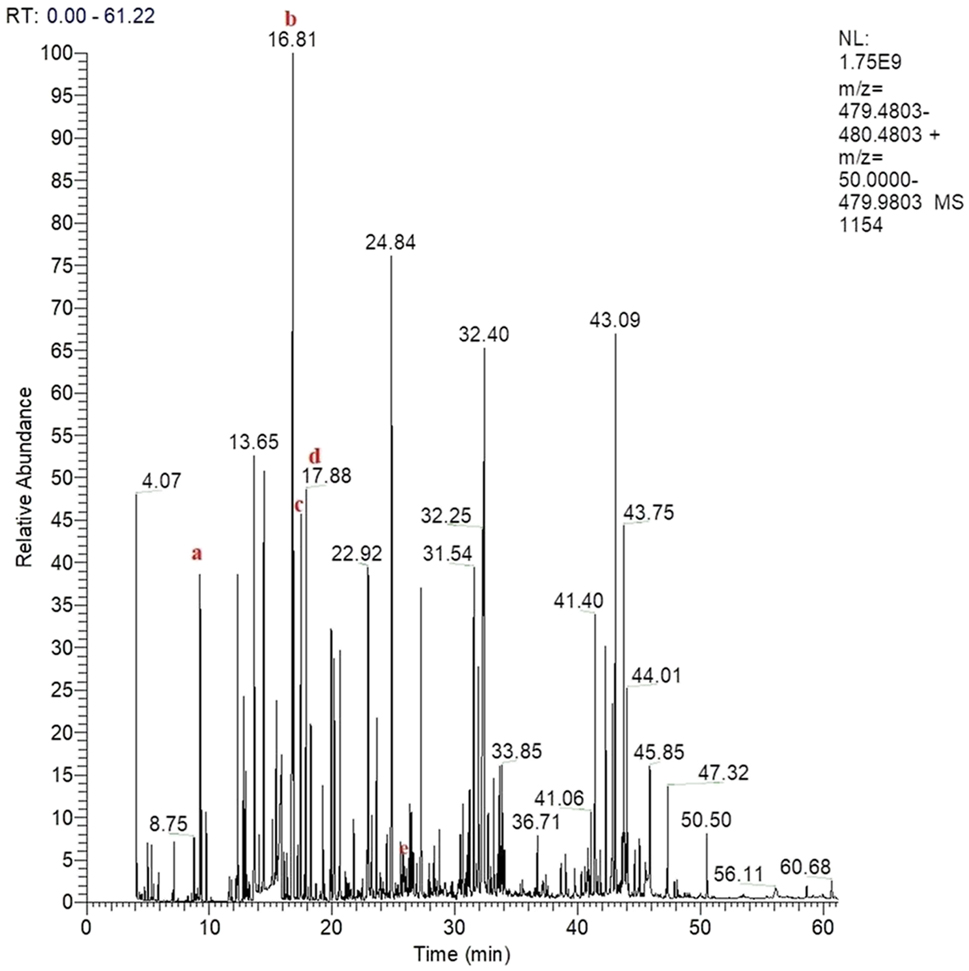
Chromatogram of organic acids of the second case obtained with the GC-MS method. (a) Lactic acid, (b) 3-hydroxyisovaleric acid, (c) valproic acid, (d) butanedioic acid, (e) Tropic acid.
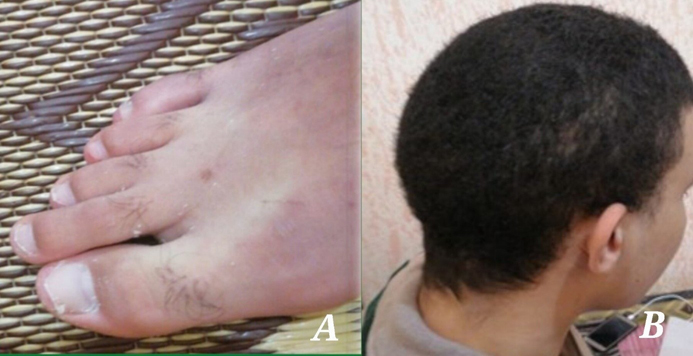
A meticulous reexamination of the patient revealed peeling of the fingertips (Supplementary Figure 4A) and sparse hair (Supplementary Figure 4B), classic symptoms of biotin deficiency. The patient’s Biotinidase activity was low at 1.4 IU/L (normal range >3.5 IU/L). Despite improvement in his general condition after biotin supplementation, the mental retardation remained irreversible.
Patient three
A 13-year-old male, with no history of consanguinity, was hospitalized in the oncology department for pancytopenia. A bone marrow biopsy revealed eosinophilia, hypoplastic marrow without fibrosis and blasts, and significant hepatosplenomegaly. Several biochemical tests were performed. The plasma amino acid profile showed an elevated tyrosine level of 368.09 umol/L (normal range: 19–119 umol/L), while phenylalanine and leucine were within normal ranges. The serum zinc level was normal at 125.95 ug/dL (normal range: 60–150 ug/dL), and serum B12 level was 1,500 pg/mL (normal range: 180–914).
Chromatography of the urinary lipid extract revealed semultaneous excretion of several sphingolipids (Globotriaosylceramide, glycosylceramides, lactosylceramides, non-hydroxylated sulfatides), indicating a deterioration in general condition. GC-MS analysis of organic acids detected an extremely high level of methylmalonic acid (Figure 3, Table S1), indicating methylmalonic aciduria. Methylmalonyl-CoA mutase activity was not detectable, consistent with the diagnosis of methylmalonic aciduria. The patient recovered within two months following B12 supplementation.
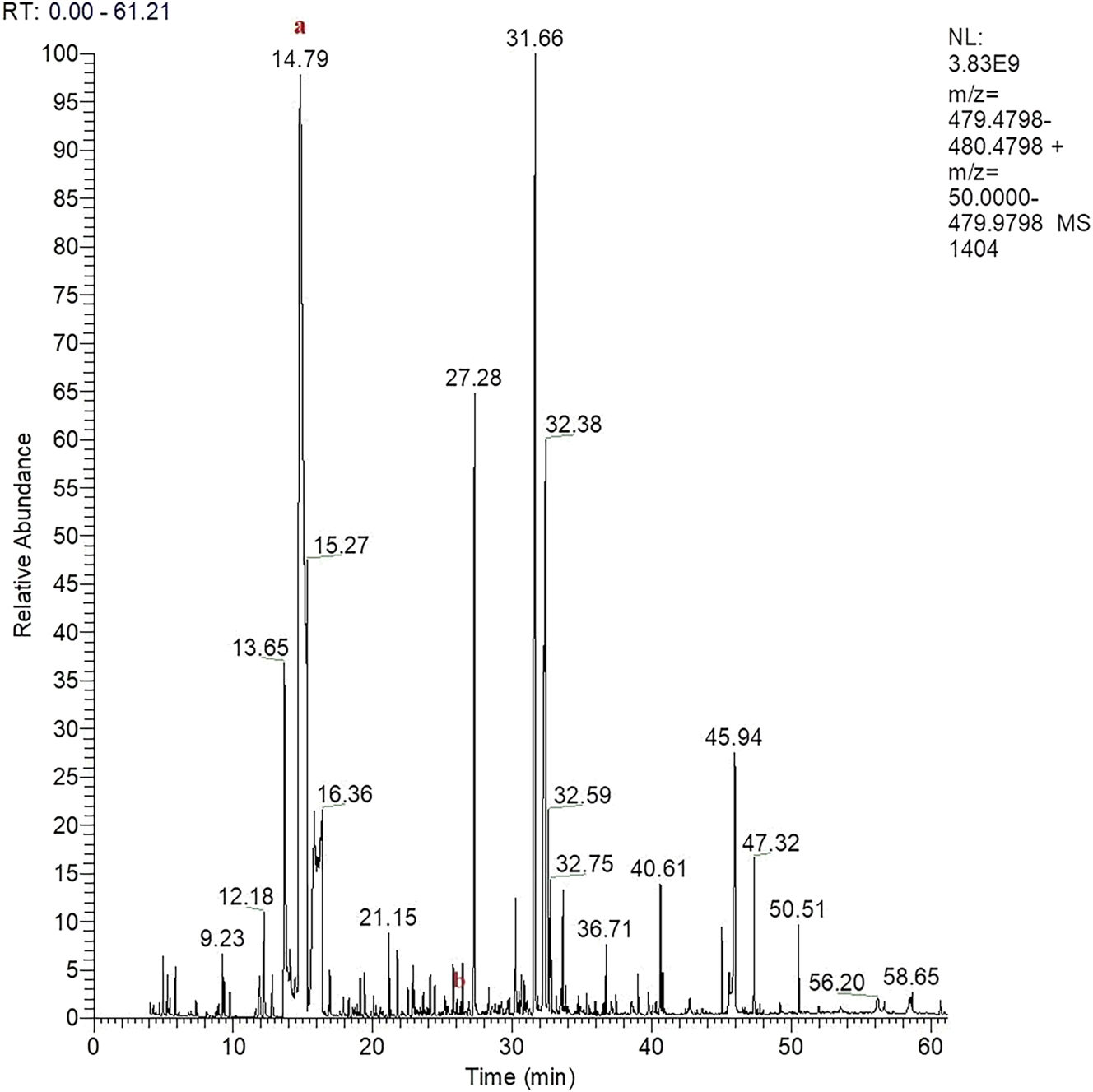
Chromatogram of organic acids of the third case obtained with the GC-MS method. (a) Methylmalonic aciduria, (b) Tropic acid.
Discussion
B vitamins are a group of water-soluble vitamins that play an important role in various metabolic processes in the body, such as energy production, DNA synthesis, nervous system regulation, and red blood cell formation [12]. Deficiencies or impaired utilization of Thiamine (B1), Biotin (B7), and Cobalamin (B12) can lead to severe metabolic disorders, [10], 22]. The present study investigated the metabolic profiles of three of our patients suspected with inherited metabolic diseases using GC/MS to assess urinary organic acids levels, we were able to detect distinct biochemical signatures associated with these deficiencies. These findings underscore the importance of metabolic profiling in the diagnosis of rare inherited metabolic disorders, particularly in resource-limited settings, where advanced biochemical and genetic testing is not readily accessible.
In patients with pyruvate dehydrogenase complex (PDHC) deficiency, clinical presentations can exhibit significant heterogeneity, The severity of symptoms can range from lactic acidosis and malformations of the central nervous system to other postnatal abnormalities such as cystic lesions of the cerebral cortex, brainstem, and basal ganglia, ataxia, and psychomotor delay [23], 24].
This is exemplified by case 1, whose elevated lactic acid levels, and increased excretion of pyruvic and hydroxybutyric acids, strongly indicated a metabolic defect in energy production [25]. These findings are consistent with previous studies, which reported similar metabolic imbalances in PDHC deficiency cases [26].
Moreover, the observed accumulation of pyruvic acid, coupled with the urinary hydroxybutyric acid excretion, suggests a potential shift towards anaerobic metabolism [27], which aligns with the known impact of PDHC deficiency on aerobic energy pathways. Similar metabolic profiles have been reported by Sofou, Kalliopi et al. in a cohort of patients exhibiting lactic acidosis and neurological deterioration [28]. These findings reinforce the importance of early detection and targeted therapeutic interventions, to manage energy production defects in PDHC deficiency [26].
In our case, the significantly reduced PDHC activity in cultured fibroblasts confirmed the presence of a PDHC defect, explaining the patient’s metabolic profile. The decrease in PDHC activity leads to impaired conversion of pyruvate to acetyl-CoA, contributing to the accumulation of pyruvate and the subsequent rise in lactate and hydroxybutyrate levels. Our findings are in line with existing literature, where low enzymatic activity is often associated with intermittent or severe forms of the disease [29], 30].
What distinguishes our case is the rapid and nearly complete recovery following thiamine supplementation, suggesting a thiamine-responsive variant of PDHC deficiency. This phenomenon has been documented in the literature, several studies, reported similar positive responses to thiamine therapy in patients with comparable biochemical profiles [16]. Such responsiveness underscores the critical need for early and targeted treatment to avert irreversible neurological damage. After one month of thiamine supplementation, the patient showed significant clinical improvement, with almost complete resolution of her symptoms except for a mild tremor in her left hand. Follow-up biochemical analysis showed normalization of lactate levels and the absence of previously detected abnormal organic acids, further supporting the therapeutic efficacy of thiamine in this case.
These results highlight the importance of early and targeted therapeutic interventions in PDHC deficiency, particularly in patients with thiamine-responsive variants. Early supplementation can prevent irreversible neurological damage, as observed in our patient, where timely intervention led to substantial clinical improvement. However, it is important to note that while the clinical recovery was remarkable, the persistence of a tremor suggests that further investigation is needed to fully understand the long-term outcomes and potential residual effects of the disease.
Biotinidase deficiency (BD), a rare autosomal recessive disorder, manifests with neurological, respiratory, and cutaneous symptoms, including hypotonia, ataxia, alopecia, and metabolic acidosis [31]. In our study, the patient (case 2) presented with characteristic clinical symptoms, including seizures, severe hypotonia, as well as skin abnormalities such as alopecia and scaly nails. The organic acid analysis revealed a significant elevation of 3-hydroxyisovaleric acid, that strongly suggests the presence of a profound Biotinidase deficiency [32]. However, it’s important to stress that in most patients, analysis of organic acids in the urine may not reveal pathology, and enzyme activity should be measured in patients with clinical signs related to the disease [33]. This highlights the importance of thorough metabolic evaluation in the assessment of atypical neurological symptoms. The measurement of Biotinidase activity revealed significantly reduced levels (1.4 IU/L) compared to normal values (>3.5 IU/L), thus corroborating the severity of the patient’s condition.
Biotin treatment, when administered early, can lead to rapid clinical and biochemical improvements. Data show that biotin supplementation often improves neurological and cutaneous symptoms [17], 34], 35]. However, in our case, despite a favorable response to treatment, neurological sequelae, including mental retardation, remain irreversible. This observation aligns with previous studies reporting that even with early intervention, some patients may experience permanent cognitive deficits [17], 36].
An essential aspect to consider is the clinical variability of BD, which can make early diagnosis challenging. Indeed, some patients may not exhibit classic symptoms until adolescence or adulthood, delaying diagnosis. Therefore, it is imperative to raise awareness among healthcare professionals about the need to consider Biotinidase deficiency in the differential diagnoses of neurological disorders, particularly in patients with a family history of hereditary disorders or unexplained neurological symptoms.
The case 3 highlights several critical aspects of methylmalonic acidemia (MMA), a severe and heterogeneous disorder affecting the metabolism of methylmalonate and cobalamin [19]. Although this condition typically presents in infancy with severe symptoms such as poor feeding, hypotonia, seizures, and, tragically, death in some cases [17], our patient, a 13-years-old male, exhibited a significant elevation in methylmalonic acid levels detected by GC-MS, suggesting a later or atypical form of MMA.
Although this disorder is often responsive to vitamin B12 supplementation, leading to significant clinical improvements [37], Some previous studies have shown that 15–30 % of individuals with high serum vitamin B12 levels also have elevated methylmalonic acid concentrations, which may reflect renal dysfunction rather than an actual vitamin B12 deficiency [38], 39]. However, in our case, the patient initially had high levels of vitamin B12 with no renal issues but still responded positively to B12 supplementation. This highlights the importance of distinguishing methylmalonic acidemia as a vitamin B12-responsive inherited metabolic disorder rather than a simple vitamin B12 deficiency [40]. It is important to emphasize the extent of this deficit which severely affected the metabolism of sphingolipids (urinary excretion of sulfatides, sphingomyelin, hexosylceramides,…) associating our patient’s disease with a poor prognosis; if the confirmatory diagnosis, inducing good management, was not made using GC/MS. For a wide range of our patients, those described in this paper, and a larger number suffering from other pathologies that we detected by GC/MS analysis and who were treated in our University hospital with dietary restriction, vitamins and/or L-carnitine supplementation, we observed a more favorable prognosis for most of them, with rapid improvement in symptomatology, indicating the importance of urinary OA screening using GC/MS to diagnose these diseases even after the onset of symptoms (selective screening).
We have tested the contribution of this analytical method, taking into account its affordability, the time invested, its realization on fresh urine and/or dried urine drops and the accessible interpretation of the obtained results.
Conclusions
The screening of organic acids by GC/MS represents a valuable resource in managing of inherited vitamin deficiencies, particularly those responsive to vitamin B. This technique provides a highly sensitive diagnostic approach, allowing for the detection of abnormal metabolites associated with genetically based vitamin metabolism deficiencies. The GC/MS is a valuable support that plays a vital role in early diagnosis, therefore in preventing irreversible sequelae, it helps clinicians to design personalized treatment plans, providing vitamin supplements tailored to the specific needs of patients. Thus, highlighting the contribution of this analysis for the establishment of personalized medicine for tailored prevention and treatment.
Acknowledgments
The authors would like to express their gratitude for the collaborative efforts that contributed to the successful completion of this research. The authors sincerely thank all the patients and their families for accepting to contribute to this study.
-
Research ethics: The local Institutional Review Board deemed the study exempt from review.
-
Informed consent: Informed consent was obtained from all individuals included in this study.
-
Author contributions: All authors have accepted responsibility for the entire content of this manuscript and approved its submission.
-
Use of Large Language Models, AI and Machine Learning Tools: ChatGPT (OpenAI) was used to improve the language and clarity of the manuscript. No content was generated that replaces original scientific analysis or interpretation.
-
Conflict of interest: Authors state no conflict of interest.
-
Research funding: None declared.
-
Data availability: None declared.
References
1. Nagana Gowda, GA, Djukovic, D. Overview of mass spectrometry-based metabolomics: opportunities and challenges. Methods Mol Biol 2014;1198:3–12. https://doi.org/10.1007/978-1-4939-1258-2_1.Search in Google Scholar PubMed PubMed Central
2. Caterino, M, Ruoppolo, M, Villani, GRD, Marchese, E, Costanzo, M, Sotgiu, G, et al.. Influence of sex on urinary organic acids: a cross‐sectional study in children. Int J Mol Sci 2020;21:582. https://doi.org/10.3390/ijms21020582.Search in Google Scholar PubMed PubMed Central
3. Tsoukalas, D, Alegakis, A, Fragkiadaki, P, Papakonstantinou, E, Nikitovic, D, Karataraki, A, et al.. Application of metabolomics: focus on the quantification of organic acids in healthy adults. Int J Mol Med 2017;40:112–20. https://doi.org/10.3892/ijmm.2017.2983.Search in Google Scholar PubMed PubMed Central
4. Fdil, N, Sabir, ES, Ezoubeiri, A, Elqadiry, R, Daoudi, A, Lalaoui, A, et al.. Implementation of an affordable method for MPS diagnosis from urine screening to enzymatic confirmation: results of a pilot study in Morocco. Clin Lab 2020;66:391–9. https://doi.org/10.7754/clin.lab.2019.190720.Search in Google Scholar
5. Prieto, JA, Andrade, F, Aldámiz-Echevarría, L, Sanjurjo, P. Análisis de ácidos orgánicos en orina mediante cromatografía de gases-espectrometría de masas. Quim Clin 2007;26:66–72.Search in Google Scholar
6. García-Villoria, J, Navarro-Sastre, A, Fons, C, Pérez-Cerdá, C, Baldellou, A, Fuentes-Castelló, MÁ, et al.. Study of patients and carriers with 2-methyl-3-hydroxybutyryl-CoA dehydrogenase (MHBD) deficiency: difficulties in the diagnosis. Clin Biochem 2009;42:27–33. https://doi.org/10.1016/j.clinbiochem.2008.10.006.Search in Google Scholar PubMed
7. Wajner, M, de Moura Coelho, D, Ingrassia, R, de Oliveira, AB, Busanello, EN, Raymond, K, et al.. Selective screening for organic acidemias by urine organic acid GC–MS analysis in Brazil: fifteen-year experience. Clin Chim Acta 2009;400:77–81. https://doi.org/10.1016/j.cca.2008.10.007.Search in Google Scholar PubMed
8. Dhir, S, Tarasenko, M, Napoli, E, Giulivi, C. Neurological, psychiatric, and biochemical aspects of thiamine deficiency in children and adults. Front Psychiatry 2019;10:1–15. https://doi.org/10.3389/fpsyt.2019.00207.Search in Google Scholar PubMed PubMed Central
9. Watanabe, T, Oguchi, KI, Ebara, S, Fukui, T. Measurement of 3-hydroxyisovaleric acid in urine of biotin-deficient infants and mice by HPLC. J Nutr 2005;135:615–8. https://doi.org/10.1093/jn/135.3.615.Search in Google Scholar PubMed
10. Kennedy, DO. B vitamins and the brain: mechanisms, dose and efficacy–a review. Nutrients 2016;8. https://doi.org/10.3390/nu8020068.Search in Google Scholar PubMed PubMed Central
11. Hanna, M, Jaqua, E, Nguyen, V, Clay, J. B vitamins: functions and uses in medicine. Perm J 2022;26:89–97. https://doi.org/10.7812/tpp/21.204.Search in Google Scholar
12. Mikkelsen, K, Apostolopoulos, V. B Vitamins and ageing. Subcell Biochem 2018;90:451–70. https://doi.org/10.1007/978-981-13-2835-0_15.Search in Google Scholar PubMed
13. Mrowicka, M, Mrowicki, J, Dragan, G, Majsterek, I. The importance of thiamine (vitamin B1) in humans. Biosci Rep 2023;43:1–18. https://doi.org/10.1042/bsr20230374.Search in Google Scholar
14. Mardhiah, M, Azize, NAA, Yakob, Y, Affandi, O, Hock, NL, Rowani, MR, et al.. Clinical, biochemical and mutational findings in biotinidase deficiency among Malaysian population. Mol Genet Metab Reports 2020;22:1–5. https://doi.org/10.1016/j.ymgmr.2019.100548.Search in Google Scholar PubMed PubMed Central
15. Riphagen, IJ, Minović, I, Groothof, D, Post, A, Eggersdorfer, ML, Kootstra-Ros, JE, et al.. Methylmalonic acid, vitamin B12, renal function, and risk of all-cause mortality in the general population: results from the prospective Lifelines-MINUTHE study. BMC Med 2020;18:1–9. https://doi.org/10.1186/s12916-020-01853-x.Search in Google Scholar PubMed PubMed Central
16. Pavlu-Pereira, H, Silva, MJ, Florindo, C, Sequeira, S, Ferreira, AC, Duarte, S, et al.. Pyruvate dehydrogenase complex deficiency: updating the clinical, metabolic and mutational landscapes in a cohort of Portuguese patients. Orphanet J Rare Dis 2020;15:1–14. https://doi.org/10.1186/s13023-020-01586-3.Search in Google Scholar PubMed PubMed Central
17. Canda, E, Kalkan Uçar, S, Çoker, M. Biotinidase deficiency: prevalence, impact and management strategies. Pediatr Heal Med Ther 2020. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32440248.Search in Google Scholar
18. Liu, S, Zhang, Y, Deng, Z, He, H, Zheng, X, Hong, Q, et al.. Delayed biotin therapy in a child with atypical profound biotinidase deficiency: late arrival of the truth and a lesson worth thinking. Int J Mol Sci 2023;24:10239. https://doi.org/10.3390/ijms241210239.Search in Google Scholar PubMed PubMed Central
19. Zhou, X, Cui, Y, Han, J. Methylmalonic acidemia: current status and research priorities. Intractable Rare Dis Res 2018;7:73–8. https://doi.org/10.5582/irdr.2018.01026.Search in Google Scholar PubMed PubMed Central
20. Forny, P, Plessl, T, Frei, C, Bürer, C, Froese, DS, Baumgartner, MR. Spectrum and characterization of bi-allelic variants in MMAB causing cblB-type methylmalonic aciduria. Hum Genet 2022;141:1253–67. https://doi.org/10.1007/s00439-021-02398-6.Search in Google Scholar PubMed PubMed Central
21. Nakagawa, K, Kawana, S, Hasegawa, Y, Yamaguchi, S. Simplified method for the chemical diagnosis of organic aciduria using GC/MS. J Chromatogr, B: Anal Technol Biomed Life Sci 2010;878:942–8. https://doi.org/10.1016/j.jchromb.2010.02.020.Search in Google Scholar PubMed
22. Hanna, M, Jaqua, E, Nguyen, V, Clay, J. B vitamins: functions and uses in medicine. Perm J 2022;29:89–97. https://doi.org/10.7812/tpp/21.204.Search in Google Scholar PubMed PubMed Central
23. Meldau, S, Fratter, C, Bhengu, LN, Sergeant, K, Khan, K, Riordan, GT, et al.. Pitfalls of relying on genetic testing only to diagnose inherited metabolic disorders in non-western populations - 5 cases of pyruvate dehydrogenase deficiency from South Africa. Mol Genet Metab Reports 2020;24:100629. https://doi.org/10.1016/j.ymgmr.2020.100629.Search in Google Scholar PubMed PubMed Central
24. Natarajan, N, Tully, HM, Chapman, T. Prenatal presentation of pyruvate dehydrogenase complex deficiency. Pediatr Radiol 2016;46:1354–7. https://doi.org/10.1007/s00247-016-3585-z.Search in Google Scholar PubMed PubMed Central
25. Giribaldi, G, Doria-Lamba, L, Biancheri, R, Severino, M, Rossi, A, Santorelli, FM, et al.. Intermittent-relapsing pyruvate dehydrogenase complex deficiency: a case with clinical, biochemical, and neuroradiological reversibility. Dev Med Child Neurol 2012;54:472–6. https://doi.org/10.1111/j.1469-8749.2011.04151.x.Search in Google Scholar PubMed
26. Patel, KP, O’Brien, TW, Subramony, SH, Shuster, J, Stacpoole, PW. The spectrum of pyruvate dehydrogenase complex deficiency: clinical, biochemical and genetic features in 371 patients. Mol Genet Metab 2012;106:385–94. https://doi.org/10.1016/j.ymgme.2012.03.017.Search in Google Scholar PubMed PubMed Central
27. Gonçalves, ÁC, Vieira, JF, Rodrigues, ACN, Murta, EFC, Marchini, JS, Michelin, MA, et al.. Benfotiamine supplementation increases thiamine in muscle of endurance-trained mice and affects the energy metabolism. J Nutr Metab 2024;2024:6102611. https://doi.org/10.1155/2024/6102611.Search in Google Scholar PubMed PubMed Central
28. Sofou, K, Dahlin, M, Hallböök, T, Lindefeldt, M, Viggedal, G, Darin, N. Ketogenic diet in pyruvate dehydrogenase complex deficiency: short- and long-term outcomes. J Inherit Metab Dis 2017;40:237–45. https://doi.org/10.1007/s10545-016-0011-5.Search in Google Scholar PubMed PubMed Central
29. DeBrosse, SD, Okajima, K, Zhang, S, Nakouzi, G, Schmotzer, CL, Lusk-Kopp, M, et al.. Spectrum of neurological and survival outcomes in pyruvate dehydrogenase complex (PDC) deficiency: lack of correlation with genotype. Mol Genet Metab 2012;107:394–402. https://doi.org/10.1016/j.ymgme.2012.09.001.Search in Google Scholar PubMed
30. Kim, JH, Kim, HR, Jang, JH, Jo, HS, Lee, KH. A case of early diagnosis of pyruvate dehydrogenase complex deficiency: the use of next-generation sequencing. Iran J Pediatr 2019;29:0–4. https://doi.org/10.5812/ijp.84965.Search in Google Scholar
31. Demirtürk, Z, Şentürk, E, Köse, A, Özcan, PE, Telci, L. A case of biotinidase deficiency in an adult with respiratory failure in the intensive care unit. Balkan Med J 2016;33:563–5. https://doi.org/10.5152/balkanmedj.2016.150359.Search in Google Scholar PubMed PubMed Central
32. Kim, YN, Yum, MS, Kim, MJ, Ko, TS. Profound biotinidase deficiency: a rare but treatable inborn error of metabolism in a neonate with recurrent seizures. Ann Child Neurol 2023:1–5.10.26815/acn.2023.00192Search in Google Scholar
33. Devanapalli, B, Sze Hui Wong, R, Lim, N, Ian Andrews, P, Vijayan, K, Kim, WT, et al.. Biotinidase deficiency: a treatable neurometabolic disorder. Brain Dev Case Reports 2024;2:100021. https://doi.org/10.1016/j.bdcasr.2024.100021.Search in Google Scholar
34. Szymańska, E, ͆redzińska, M, Ługowska, A, Pajdowska, M, Rokicki, D, Tylki-Szymańska, A. Outcomes of oral biotin treatment in patients with biotinidase deficiency - twenty years follow-up. Mol Genet Metab Reports 2015;5:33–5. https://doi.org/10.1016/j.ymgmr.2015.09.004.Search in Google Scholar PubMed PubMed Central
35. León-Del-Río, A. Biotin in metabolism, gene expression, and human disease. J Inherit Metab Dis 2019;42:647–54. https://doi.org/10.1002/jimd.12073.Search in Google Scholar PubMed
36. Wolf, B. Clinical issues and frequent questions about biotinidase deficiency. Mol Genet Metab 2010;100:6–13. https://doi.org/10.1016/j.ymgme.2010.01.003.Search in Google Scholar PubMed
37. Held, PK, Singh, E, Schwoerer, JS. Screening for methylmalonic and propionic acidemia: clinical outcomes and follow-up recommendations. Int J Neonatal Screen 2022;8. https://doi.org/10.3390/ijns8010013.Search in Google Scholar PubMed PubMed Central
38. Hao, Q, Jiang, B, Zhao, Y, Hu, Z. Adult-onset combined methylmalonic acidemia and hyperhomocysteinemia, cblC type with aortic dissection and acute kidney injury: a case report. BMC Nephrol 2024;25:1–5. https://doi.org/10.1186/s12882-023-03414-9.Search in Google Scholar PubMed PubMed Central
39. Clarke, R, Refsum, H, Birks, J, Evans, JG, Johnston, C, Sherliker, P, et al.. Screening for vitamin B-12 and folate deficiency in older persons. Am J Clin Nutr 2003;77:1241–7. https://doi.org/10.1093/ajcn/77.5.1241.Search in Google Scholar PubMed
40. Froese, DS, Gravel, RA. Genetic disorders of vitamin B 12 metabolism: eight complementation groups - eight genes. Expert Rev Mol Med 2010;12:1–20. https://doi.org/10.1017/s1462399410001651.Search in Google Scholar PubMed PubMed Central
Supplementary Material
This article contains supplementary material (https://doi.org/10.1515/tjb-2025-0081).
© 2025 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution 4.0 International License.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Articles in the same Issue
- Frontmatter
- Review Article
- Unveiling the hidden clinical and economic impact of preanalytical errors
- Research Articles
- To explore the role of hsa_circ_0053004/hsa-miR-646/CBX2 in diabetic retinopathy based on bioinformatics analysis and experimental verification
- Study on the LINC00578/miR-495-3p/RNF8 axis regulating breast cancer progression
- Comparison of two different anti-mullerian hormone measurement methods and evaluation of anti-mullerian hormone in polycystic ovary syndrome
- The evaluation of the relationship between anti angiotensin type I antibodies in hypertensive patients undergoing kidney transplantation
- Evaluation of neopterin, oxidative stress, and immune system in silicosis
- Assessment of lipocalin-1, resistin, cathepsin-D, neurokinin A, agmatine, NGF, and BDNF serum levels in children with Autism Spectrum Disorder
- Regulatory nexus in inflammation, tissue repair and immune modulation in Crimean-Congo hemorrhagic fever: PTX3, FGF2 and TNFAIP6
- Pasteur effect in leukocyte energy metabolism of patients with mild, moderate, and severe COVID-19
- Thiol-disulfide homeostasis and ischemia-modified albumin in patients with sepsis
- Myotonic dystrophy type 1 and oxidative imbalance: evaluation of ischemia-modified albumin and oxidant stress
- Antioxidant and alpha-glucosidase inhibitory activities of flavonoids isolated from fermented leaves of Camellia chrysantha (Hu) Tuyama
- Examination of the apelin signaling pathway in acetaminophen-induced hepatotoxicity in rats
- Integrating network pharmacology, in silico molecular docking and experimental validation to explain the anticancer, apoptotic, and anti-metastatic effects of cosmosiin natural product against human lung carcinoma
- Validation of Protein A chromatography: orthogonal method with size exclusion chromatography validation for mAb titer analysis
- The evaluation of the efficiency of Atellica UAS800 in detecting pathogens (rod, cocci) causing urinary tract infection
- Case Report
- Exploring inherited vitamin B responsive disorders in the Moroccan population: cutting-edge diagnosis via GC-MS profiling
- Letter to the Editor
- Letter to the Editor: “Gene mining, recombinant expression and enzymatic characterization of N-acetylglucosamine deacetylase”
Articles in the same Issue
- Frontmatter
- Review Article
- Unveiling the hidden clinical and economic impact of preanalytical errors
- Research Articles
- To explore the role of hsa_circ_0053004/hsa-miR-646/CBX2 in diabetic retinopathy based on bioinformatics analysis and experimental verification
- Study on the LINC00578/miR-495-3p/RNF8 axis regulating breast cancer progression
- Comparison of two different anti-mullerian hormone measurement methods and evaluation of anti-mullerian hormone in polycystic ovary syndrome
- The evaluation of the relationship between anti angiotensin type I antibodies in hypertensive patients undergoing kidney transplantation
- Evaluation of neopterin, oxidative stress, and immune system in silicosis
- Assessment of lipocalin-1, resistin, cathepsin-D, neurokinin A, agmatine, NGF, and BDNF serum levels in children with Autism Spectrum Disorder
- Regulatory nexus in inflammation, tissue repair and immune modulation in Crimean-Congo hemorrhagic fever: PTX3, FGF2 and TNFAIP6
- Pasteur effect in leukocyte energy metabolism of patients with mild, moderate, and severe COVID-19
- Thiol-disulfide homeostasis and ischemia-modified albumin in patients with sepsis
- Myotonic dystrophy type 1 and oxidative imbalance: evaluation of ischemia-modified albumin and oxidant stress
- Antioxidant and alpha-glucosidase inhibitory activities of flavonoids isolated from fermented leaves of Camellia chrysantha (Hu) Tuyama
- Examination of the apelin signaling pathway in acetaminophen-induced hepatotoxicity in rats
- Integrating network pharmacology, in silico molecular docking and experimental validation to explain the anticancer, apoptotic, and anti-metastatic effects of cosmosiin natural product against human lung carcinoma
- Validation of Protein A chromatography: orthogonal method with size exclusion chromatography validation for mAb titer analysis
- The evaluation of the efficiency of Atellica UAS800 in detecting pathogens (rod, cocci) causing urinary tract infection
- Case Report
- Exploring inherited vitamin B responsive disorders in the Moroccan population: cutting-edge diagnosis via GC-MS profiling
- Letter to the Editor
- Letter to the Editor: “Gene mining, recombinant expression and enzymatic characterization of N-acetylglucosamine deacetylase”