Genetic dissection of the Mastl-Arpp19/Ensa-PP2A-B55δ pathway in mammalian cells
-
Muhammed Kasim Diril

 and
Mehmet Erguven
and
Mehmet Erguven

Abstract
Objectives
Mastl is an essential kinase required for inhibition of the phosphatase activity directed toward Cdk1/cyclin B substrates during mitosis. Mastl phosphorylates two small evolutionarily conserved proteins, Arpp19 and Ensa converting them into strong inhibitors of PP2A-B55δ. Mastl-Arpp19/Ensa-PP2A regulatory pathway has been mainly studied in Xenopus egg extracts and Drosophila using biochemical and genetic approaches. Studies in mammalian cells and genetically modified mouse models have suggested distinct but important functions for Arpp19 and Ensa, in mitosis and S-phase, respectively. A detailed comparative analysis of the Arpp19 and Ensa functions in mammalian cells has not been performed.
Methods
We utilized Mastl conditional knockout (CKO) mouse embryonic fibroblasts (MEF), to investigate the roles of Mastl-Arpp19/Ensa-PP2A pathway components in mitosis and cellular proliferation. We used viral transduction for overexpression or silencing of these genes in conjunction with inducible genetic Mastl knockout to assess their roles in relation to each other.
Results
We show that, Arpp19 is expressed at significantly higher levels in MEFs in comparison to Ensa. Silencing of Arpp19, but not Ensa, results in reduced cellular proliferation. Overexpression of WT Arpp19 or its phosphomimetic mutant (S67D) partially restores mitosis arrest duration in Mastl knockout MEFs, however cellular proliferation block cannot be rescued. Silencing of B55δ expression has a similar outcome as Arpp19 overexpression, underscoring the opposite roles of these genes in mitosis.
Conclusions
Our results show that Arpp19 is the major Mastl substrate during mitotic division of MEFs. Ensa expression is low and it is not essential for cell cycle.
Introduction
Cyclin-dependent kinases (Cdk), in complex with specific cyclins, regulate progression through phases of the cell cycle by phosphorylating different sets of substrates during quiescence to G1 transition, S-phase and mitosis [1]. Cdk1 is the only essential cyclin-dependent kinase for cell division [2, 3] and in complex with cyclin B1, it governs dramatic mitotic events including nuclear envelope breakdown, chromosome condensation and spindle formation [4] by phosphorylating a large number of mitotic substrates [5].
In order to maintain the mitotic proteins at a phosphorylated state, counteracting phosphatase activity of PP2A-B55 needs to be suppressed during mitosis [6]. Greatwall/Mastl kinase, first identified in Drosophila as a mutation that causes chromosome decondensation [7], was later found to be responsible for suppression of PP2A-B55 in Xenopus egg extracts [8]. During mitotic entry Cdk1 activates Mastl kinase by phosphorylation, which, in turn phosphorylates Arpp19 and Ensa, two small proteins with highly conserved sequence. Upon phosphorylation, these proteins become potent and competitive inhibitors of PP2A-B55 [9], [10], [11], [12], [13]. Biochemical studies in Xenopus egg extracts suggested that Mastl is essential for mitotic entry [14]. Although this finding was confirmed in HeLa cells where Mastl expression was silenced by siRNAs [15], subsequent studies using MEFs derived from Mastl conditional knockout (CKO) mice, showed that it is not required for mitotic entry [16, 17]. Therefore, in contrast to deletion of Cdk1 [3], Mastl knockout cell can enter mitosis however due to a weakening of the spindle assembly checkpoint, they prematurely exit with chromosome segregation defects [16].
After identification of the two specific substrates of Mastl kinase, Ensa and Arpp19 [10, 11], several studies performed in different model organisms further characterized their distinct functions in development and cell cycle: mitosis, meiosis and S-phase. Notably, Arpp19 but not Ensa was found to be required and sufficient for meiosis in porcine oocytes which don’t express Ensa due to a point mutation [18]. Similarly, gene knockout studies showed that mouse embryogenesis and development can occur in absence of Ensa but not Arpp19 [19]. On the other hand, Ensa depletion in human cell lines resulted in extension of S-phase, due to PP2A-B55-dependent dephosphorylation and subsequent degradation of the replication factor treslin [20]. Although Mastl knockout had a moderately similar outcome, Arpp19 deletion did not affect treslin stability, different from Ensa [19].
In this study, we have utilized Mastl CKO MEFs and performed additional genetic interventions to Mastl-Arpp19/Ensa-PP2A pathway via viral transduction. Our results demonstrate that overexpression of Arpp19 or knockdown of the gene coding for PP2A-B55δ subunit does not rescue Mastl loss. Nevertheless, reduced mitotic entry defect observed in Mastl KO MEFs can be partially restored. On the other hand, silencing of Arpp19, but not Ensa, significantly reduced cellular proliferation rate although cells could still divide. Therefore, Mastl is the only crucial component of the Mastl-Arpp19/Ensa-PP2A pathway.
Materials and methods
Mastl CKO immortalized MEF lines and cell cycle synchronization
Generation of immortalized Mastl FLOX/FLOX conditional knockout MEF lines was described before [16]. MEFs were synchronized at the quiescent state G0 by growing them to 100% confluence (contact inhibition) and culturing in reduced serum media (0.2% fetal calf serum) for three days. Genetic Mastl knockout was achieved by Cre-mediated recombination during the last 24 h when almost all of the cells were quiescent. Experiments were done in pairs comprising control and Mastl deficient conditions, where CKO MEFs were treated with DMSO (Control) or 20 ng/mL 4-OHT (Mastl knockout or null). To release cells into cell cycle entry synchronously, they were trypsinized and replated at lower density (30–40% confluent). Cells were arrested in mitosis by the addition of 500 ng/mL nocodazole between 24–28 or 24–30 h after release.
qPCR expression analysis
Total RNA was extracted using MN NucleoSpin RNA II kit according the manufacturer’s protocol. For each RT-PCR reaction, first strand cDNA was synthesized from 1 μg total RNA using the Maxima First Strand cDNA synthesis kit (Thermo Fisher, K1642). Arpp19 and Ensa mRNA levels at different time points after release of control or 4-OHT induced synchronized CKO MEFs were determined by RT-PCR using cyclophilin A, Mastl, Arpp19, Ensa primers (Supplementary Table S1). PCR amplification was carried out using the Maxima SYBR Green qPCR Master Mix (Fermentas, K0252) and the appropriate primer pair (Supplementary Table S1). The reactions were monitored continuously in a RotorGene thermal cycler (Corbett Research) using the following program: 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s, 55 °C for 30 s, and 72 °C for 30 s. All data were normalized to the expression levels of cyclophilin A housekeeping gene using the (2−ΔΔcT) method.
Cell culture, retroviral transduction, and stable cell line engineering
PLAT-E cells (Cell Biolabs), 293FT cells (Invitrogen) and Mastl conditional knockout immortalized MEFs [16] were grown in DMEM supplemented with 10% FBS, and antibiotics. Cells were cultured in a 95% humidified incubator with 5% CO2 at 37 °C. Lipofectamine 3000 transfection reagent (L3000015; Thermo Scientific) was used for transfection of PLAT-E and 293FT cells. PLAT-E and 293FT culture and virus production were carried out according to the manufacturer’s instructions. The viral supernatant was harvested on second and third days post-transfection, and stored at 4 °C up to a week until use.
Polybrene was mixed with retroviral medium to 8 μg/mL final concentration, immediately before infection of MEFs. MEFs were transduced when they are 40% confluent. Viral media was collected two days after the day of transfection to be used in the first round of viral transduction. One day post-transduction, the used viral media was removed and a second round of transduction was performed by using the viral media that was harvested at the third day of transfection. 8 h after the second round of transduction, the viral media was removed. Cells were cultured further in normal growth medium overnight for recovery, prior to antibiotic selection. The stable cell lines were generated under 2–4 μg/mL puromycin selection. Cells were selected for a week and negative control cells died within the first two days of selection. Mastl knockout was induced by adding 4-OHT to the culture medium at 20 ng/mL final concentration. The control cells were treated with an equal volume of DMSO. The knockout-induced stable cell lines were subjected to limited dilution approximately 6 days post-induction.
Cell viability and proliferation assays
The alamarBlue proliferation assay was carried out in 96-well plate format in three replicates. The daily measurements were initiated one day after seeding the cells. Cells were incubated in 150 µL of assay medium for 4 h. The assay medium was prepared by diluting 1 volume of alamarBlue dye reagent (BUF012A; Bio-Rad) in 9 volumes of growth medium. The metabolic activity was quantified fluorometrically by using 560 nm excitation wavelength and recording the emission at 590 nm. The assay was performed for 6 successive days.
The proliferation rate was also measured by a modified 3T3 assay in 6-well plates [3]. 20,000 cells were seeded per well and cells were counted after 3 days and 20,000 cells were plated again. Counts were repeated for four successive passages.
Antibodies and plasmids
The rabbit anti-Mastl polyclonal antibody was self-made in Kaldis laboratory [16]. The rabbit anti-ArppEnsa sera were raised against a N-terminal GST-tagged mouse Arpp19 C-terminal region (25–112 a.a.). Commercially available primary antibodies used in immunoblots were mouse anti-HA tag (clone 12CA5; Roche), rabbit anti-Ensa (Cell Signaling Technology, clone 8770) and mouse anti-HSP90 (610419; BD Biosciences). For FACS analysis of mitotic and S-phase cells, AlexaFluor 488 conjugated Phospho-Histone H3 (Ser10) antibody (Cell Signaling Technology, 9708), CDK phosphorylated substrate set two rabbit monoclonal antibody (Cell Signaling Technology, 9477) and APC conjugated anti BrdU antibody (BD-Pharmingen, 623551) were used.
For expression of shRNAs, pSUPER.retro.puro (OligoEngine) plasmid was used [3]. For lentiviral expression of HA-tagged Arpp19 and Ensa, pBOBI vector backbone, together with helper plasmids pMDLg/pRRE, pRSV-Rev and pMD2.G (VSV-G) were used. All plasmids were prepared at transfection quality using PureLink™ HiPure Plasmid Miniprep Kit (Invitrogen) and following manufacturer’s instructions.
FACS analysis
For BrdU labeling and FACS analysis, MEFs synchronized by serum starvation were released into cell cycle for 18 h and pulse labeled with 100 μM BrdU (BD Pharmingen, #550891) for 1 h. Cells were recovered by trypsinization and fixed in −20 °C cold 70% EtOH, stained with APC conjugated anti-BrdU antibodies (BD Pharmingen, #623551) and propidium iodide (Sigma, #81845). S-phase analysis was done using FACSCalibur flow cytometer (BD Biosciences) and data were analyzed by FlowJo 8 software.
For detection of mitotic index in synchronized but not arrested cells, cells were fixed 24 h after release. For mitotic arrest, cells were incubated in 500 ng/mL nocodazole for four or 6 h after 24 h following release. Cells were processed and analyzed by FACS as described earlier [16].
Results
Mouse embryonic fibroblasts predominantly express Arpp19
In our previous work, we have created a conditional knockout mouse model to investigate functions of mammalian Mastl kinase in a physiological setting. Although Mastl deficient embryos could be detected as late as 7.5 dpc, these were much smaller in size and most likely survived until this stage thanks to the presence of maternally deposited Mastl mRNA in eggs. Tamoxifen induced genetic deletion of Mastl in midgestation stage embryos, resulted in a block of cell division in all organs and tissues analyzed [16]. Therefore, Mastl is essential for mitotic division in all cells of the developing embryo.
A recent study used CKO mouse models to study the in vivo functions of Mastl substrates Ensa and Arpp19 using genetically modified mouse models [19]. Genetic deletion of Arpp19 resulted in early embryonic lethality and lack of cellular proliferation in MEFs, with phenotypes reminiscent of Mastl KO. Ensa deficient mice were born albeit at a lower ratio, suggesting that it does not have an essential function in cell division. On the other hand, as reported earlier [20], treslin was found to be degraded in Ensa and Mastl deficient MEFs but Arpp19 loss had no effect. Both Arpp19 and Ensa genes were shown to be expressed at comparable rates in early embryos [19].
Before studying Mastl-Arpp19/Ensa-PP2A pathway in our Mastl CKO MEFs, we sought to investigate the expression levels of Arpp19 and Ensa in MEFs. We characterized the similarity of protein and DNA sequences (Supplementary Figure S1). These proteins are evolutionarily well conserved and display 76% identity in their peptide sequences. Especially the C-terminal 88 residues are 89% identical which we used to develop antibodies recognizing both proteins (Supplementary Figure S1A).
Alignment of the coding nucleotide sequences showed 69% identity with several long stretches with 100% identity. We made use of the identical DNA sequences to design primers that would amplify same length amplicons from both genes, here after we name as ArppEnsa. We designed gene specific forward primers with similar DNA sequences that differed at the 3′ four nucleotides to distinguish and specifically measure expression levels of either (Supplementary Figure S1B). Amplification of cDNA derived from primary MEFs by Real-Time PCR showed that cycle threshold value for Arpp19 was seven cycles earlier than Ensa. This was seen as a good indication that Arpp19 expression is significantly higher than Ensa at RNA level (Figure 1A).

Arpp19 is the predominantly expressed Mastl substrate in MEFs. (A) cDNA prepared from reverse transcription of primary MEF total RNA extract was amplified using cyclophilin A, Arpp19 and Ensa specific primers. Realtime analysis of fluorescence increase was measured by instrument software (RotorGene thermal cycler, Corbett Research). (B) Mastl CKO MEFs were transduced with 1X and 10X titers of Arpp19 or Ensa lentiviral overexpression vectors. mRNA levels were quantified by real-time qPCR analysis. Arpp19 and Ensa expression levels were normalized to cyclophilin A, a house keeping gene. Y-axis represents folds change in expression in comparison to mock (transduced with empty vector). Ensa and Arpp19 charts were derived from samples amplified with gene specific primers. ArppEnsa chart shows the combined expression levels of Arpp19 and Ensa, amplified with primers common to both genes. (C) WT MEFs (con) were infected with pSUPER.retro.puro vectos with two different shRNAs against each of Arpp19 and Ensa. Cells were selected with puromycin and expression levels cyclophilin A (20 cycles), Ensa, Arpp19 and ArppEnsa (30 cycles) were analyzed by gel electrophoresis. Mock (empty vector). (D) Expression levels in the cells as in (C) were quantified by real-time qPCR analysis and plotted after normalization as in (B) Y-axis represents the change in expression level compared to mock.
Next, we used lentiviral vectors to overexpress Arpp19 or Ensa in MEFs, and monitored the change of expression. Real-time qPCR analysis of the common ArppEnsa fragment after transduction of cells with either HA-Arpp19 or HA-Ensa showed a comparable increase (Figure 1B). Immunofluorescence and immunoblot analysis of the cells using anti-HA antibodies confirmed that the expression levels were similar (Supplementary Figure S3A, B). These data indicate equal levels of ectopic gene expression efficiency for both constructs. Next we analyzed the mRNA levels of Arpp19 and Ensa using gene specific primers. Viral transduction with 1X and 10X viral titers resulted in an 80–180 fold increase in Ensa mRNA level, respectively. By contrast, Arpp19 mRNA level increased by only 3–9 folds depending on the titers used. Taken together, these results show, expression of endogenous Arpp19 RNA is more than twenty folds higher than Ensa in MEFs.
To compare the expression levels by a different method, we used shRNA-mediated gene silencing. We designed shRNAs targeting specifically Arpp19 or Ensa (Supplementary Figure S1B; Supplementary Table S1) and cloned them into pSUPER.retro.puro vector (OligoEngine). After retroviral transduction and puromycin selection of MEFs [3], RNA levels were analyzed by agarose gel electrophoresis and real-time qPCR (Figure 1C, D). Although, all used shRNAs specifically silenced their target sequences to variable efficiencies, only Arpp19 shRNAs could decrease the levels of the ArppEnsa amplicon. This findings substantiate our above results and confirm that Arpp19 is the predominantly expressed Mastl kinase substrate in MEFs.
Finally, we analyzed the tissue specific expression of Arpp19 and Ensa by qPCR. Arpp19 was expressed in all tissues and midgestation stage embryos. Ensa had a similar expression pattern except brain tissue, where it was expressed at significantly lower levels (Supplementary Figure S2A). Unlike cyclin A2 and Mastl, expression of Arpp19 and Ensa did not change during cell cycle entry and progression. Mastl loss did not have any effect on the expression levels (Supplementary Figure S2B).
Development of an Arpp19-Ensa antibody with dual specificity
Arpp19 and Ensa specific antibodies have been developed and described in literature [19, 21]. These antibodies were generally raised against N-terminal non-conserved sequences of either protein or C-terminal unique sequence of Ensa. We aimed to develop antibodies that will recognize both proteins with equal efficiency.
C-terminal domains of mouse Arpp19 (residues 25–112) and Ensa (residues 30–117) share 89% sequence identity (Supplementary Figure S1A). We reasoned that an antisera developed against Arpp19 C-terminal would recognize Ensa as efficiently. We expressed and purified a recombinant GST-Arpp19 (25–112) fusion protein and used it as an antigen for raising antisera in rabbits, hereafter referred to as ArppEnsa antibody. ArppEnsa antibodies recognized mouse Arpp19 and Ensa with equal efficiency and sensitivity (Supplementary Figure S3A–C).
Previous work suggested that Ensa is the major Mastl substrate in HeLa cells and Arpp19 expression is at insignificant levels [22]. We wanted to compare Arpp19 and Ensa protein levels in MEFs. Protein extracts were prepared from MEFs where Arpp19 or Ensa expression was silenced by shRNAs (Figure 1C, D). Immunoblotting with ArppEnsa antibodies revealed that, only Arpp19 knockdown resulted in loss of the recognized band, corresponding to both substrates (Supplementary Figure S3D). Silencing of Ensa did not have a noticeable effect on ArppEnsa band intensity. Our results confirm that Arpp19 is the main Mastl substrate in MEFs and Ensa expression is low.
Silencing of Arpp19 results in reduced proliferation and motility in MEFs
Roles of Arpp19 and Ensa in mammalian cell cycle and carcinogenesis have been investigated in several studies, with differing and sometimes conflicting findings, in different cell lines and cancers. Ensa but not Arpp19, was proposed to be the essential Mastl substrate in mitotic cell cycle in HeLa cells [22]. Increased Ensa expression was shown to be a poor prognostic marker in triple-negative breast cancer [23]. On the other hand, in a knockout mouse study, Arpp19 was found to be the more important Mastl substrate in embryogenesis and cellular proliferation [19]. Arpp19 was also found to promote proliferation and metastasis of human gliomas [24]. Therefore, we sought to answer which of the two substrates is important in cell division and migration in MEFs.
We designed shRNAs specifically targeting Arpp19 or Ensa (Supplementary Figure S1B). We found that these shRNAs effectively and specifically silence the expression of their intended targets in MEFs (Figure 1C, D). Silencing of Arpp19, but not Ensa led to loss of the ArppEnsa antibody recognized band in immunoblots (Supplementary Figure S3D). Knockdown of Arpp19 but not Ensa, decreased the proliferation rate of these MEFs (data not shown).
Next, we generated clonal cell lines from Arpp19 or Ensa knockdown MEF pools, using limited dilution method we described previously [25]. In Ensa shRNA-expressing clones, the intensity of the ArppEnsa band remained unchanged (Figure 2A, lower panel) and these clones proliferated well. However, Arpp19 shRNA expressing clones displayed a marked decrease in ArppEnsa band intensity and they proliferated slower (Figure 2A–C). We selected clones 1 and 3 for further characterization as they had undetectable or significantly reduced levels of Arpp19, respectively. Clone 1 and 3 had a markedly slower proliferation rate according to AlamarBlue and 3T3 assays. Nevertheless, both clones could proliferate and they were maintained in cell culture without difficulty for more than ten passages. We then asked if Arpp19 loss affects cell motility and migration. In wound healing assays, we found that clone 1 and 3 motility was reduced compared to control cells (Figure 2C). Therefore, silencing of Arpp19 expression in MEFs results in decreased motility as previously reported for human glioma cell line A172 [24].

Arpp19 knockdown causes inhibition of proliferation and cell migration. (A) WT MEFs were infected with retroviral shRNA constructs against Ensa (Ensa-2) and Arpp19 (Arpp-1) and selected with 2.5 µg/mL puromycin for 14 days. Clonal cell lines were derived from the puromycin resistant cell pools via limited dilution. Protein extracts from the selected clones, as well as puromycin resistant pools (pool) and empty vector infected MEFs (con), were analyzed by immunoblotting using ArppEnsa antibody. Arpp19 shRNA clones 1 and 3 displayed the highest decrease in ArppEnsa band intensity. Ensa knockdown did not have an effect on ArppEnsa band intensity. (B) Proliferation rates of Arpp19 shRNA clones 1 and 3 were analyzed by alamarBlue proliferation assay (left) and 3T3 assay (right). AFU:Arbitrary fluorescence units. (C) Cells were grown to confluence in u-dish culture inserts (Ibidi) prior to wound healing/migration assay. Gap closure was monitored at 24 and 48 h after formation of the physical gaps.
Overexpression of Arpp19 does not rescue general proliferation defect of Mastl deficiency
Phosphorylation of Arpp19 by Mastl kinase during mitosis potentiates its binding to PP2A-B55δ. Upon phosphorylation, its affinity to PP2A increases and it acts as a competitive inhibitor by occupying the binding site and blocking the access of Cdk1-phosphorylated substrates [13]. Phosphorylated Endos, which is the single orthologue of Arpp19 and Ensa in Drosophila, binds to PP2A and gets steadily dephosphorylated during mitosis. However, this so called anti-Endos activity is very slow compared to other phosphorylated Cdk substrates, resulting in maintenance of mitotic phosphorylated state [12]. In the absence of phosphorylation by Mastl kinase, Xenopus Ensa can still interact and partly inhibit PP2A [26], and Drosophila Endos can bind to it [27, 28]. A phosphomimetic point mutation (S68D) in Drosophila Endos has been shown to effectively interact with PP2A and inhibit its phosphatase activity [28]. We reasoned that, if the stoichiometric ratio of Arpp19 to PP2A-B55δ is increased, this could result in suppression of the phosphatase activity during mitosis and rescue the phenotypes attributed to Mastl loss.
Mastl CKO MEFs were infected with lentiviral constructs for overexpression of HA-tagged WT mouse Arpp19 or its phosphomimetic (PM) mutant S62D. Lentiviral expression of the ectopic constructs resulted in a 7–8 fold increase in Arpp19 mRNA, whereas ArppEnsa common fragment was increased by 5–6 folds (Supplementary Figure S4A). Ectopic HA-Arpp19 constructs localized to both cytoplasm and nucleus (Supplementary Figure S4B) as reported earlier for Endos [28]. We used limited dilution method to isolate monoclonal cell lines that expressed HA-tagged Arpp19 at much higher levels than endogenous protein (Supplementary Figure S4C, D). We selected one clonal cell line stably overexpressing WT (#6) or phosphomimetic mutant (#5) of Arpp19 for further analysis (Figure 3A).
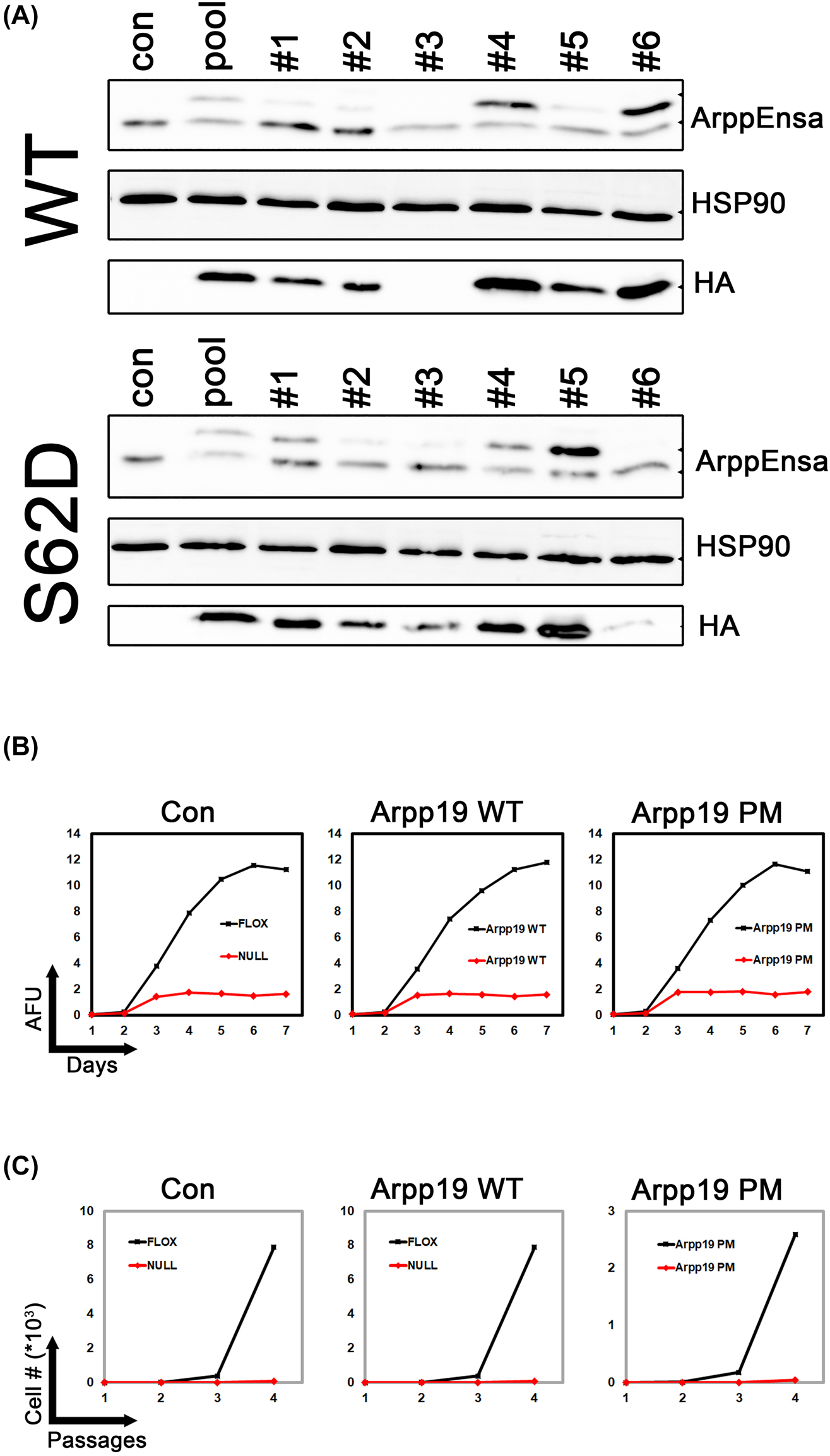
Overexpression of Arpp19 does not rescure Mastl loss. (A) Mastl CKO MEFs (con) were transduced with lentiviral constructs for overexpression of HA-tagged Arpp19 WT or phosphomimetic mutant S62D (PM). Seven days after viral transduction (pool), cells were seeded by limited dilution into 96-well plates. Single clonal cell lines were derived. Overexpression of Arpp19 was analyzed by immunoblotting using antibodies against ArppEnsa or HA-tag. (B) Mastl CKO MEF clonal cell lines overexpressing Arpp19 WT (clone #6) and S62D (clone #5) were treated with 20 ng/mL 4′OHT to induce Cre-mediated knockout in Mastl gene. Cellular proliferation was monitored by alamarBlue proliferation assay. AFU, Arbitrary fluorescence units. (C) Cells treated as in (B) were analyzed for proliferation by 3T3 assay for four passages.
We used 4′OHT to induce recombination mediated genetic loss of Mastl in the CKO MEFs as described in our earlier work [16]. Mastl knockout blocked cellular proliferation as expected. Overexpression of WT or phosphomimetic App19 did not rescue this phenotype (Figure 3B). Therefore, increased levels of Arpp19 or its phosphomimetic mutant cannot compensate for Mastl loss in mitotic cell division.
Overexpression of Arpp19 restores the duration of mitotic arrest in Mastl KO MEFs
MEFs can be synchronized to a quiescent state, also referred as G0, by serum starvation (culturing in media with 0.2% serum) and contact inhibition (100% confluent) for three days. During this stage, expression of most cell cycle regulatory genes including Mastl kinase is downregulated to undetectable levels in immunoblots. After three days, cells can be made to reenter cell cycle synchronously by plating them at a lower density in full (10% serum) medium [3].
In our past work, we have developed a protocol that allows genetic deletion of Mastl during the last 24 h of serum starvation (see Materials and Methods). Under these conditions, genetically WT and Mastl KO cells enter cell cycle in the absence of Mastl protein. As cells progress through cell cycle, Mastl kinase can be produced in WT but not in KO cells, [16]. This method allows us to compare mitotic progression in WT and Mastl deficient cells under otherwise identical states.
In our previous work, we have shown that all Mastl deficient MEFs can enter mitosis, albeit, at a slower rate than WT cells. Mitotic index (percentage of cells in mitosis) is increased in Mastl KO cells as duration of mitosis (time spent between nuclear envelope breakdown and degradation of Geminin) is longer. Mastl KO results in reduced phosphorylation of Mps1 and a weakened spindle assembly checkpoint (SAC) signal. SAC weakening results in mitotic exit without correcting chromosome attachment errors. Weakening of the SAC signal can be monitored by arresting the cells in mitosis using spindle poisons (nocodazole or EG5 kinesin inhibitor). Mastl KO MEFs cannot maintain the mitotic state as long as the controls [16].
We wanted to test if Arpp19 WT or its phosphomimetic mutant’s overexpression could rescue any of these phenotypes associated with Mastl loss. Control and Arpp19 overexpressing clonal cell lines entered cell cycle with the same efficiency, whether they expressed Mastl or not (Supplementary Figure S5A). Analysis of the cells progressing through cell cycle revealed that, Mastl KO cells had a higher mitotic index at 24 h post cell cycle entry as expected. Arpp19 WT or PM overexpression reduced the percentage of Mastl KO mitotic cells to Mastl WT levels (Figure 4A, B). Our results suggest that, Arpp19 overexpression rescues the long mitosis phenotype of Mastl deficient cells.

Overexpression of Arpp19 restores weak SAC in Mastl KO MEFs. (A) Mastl CKO MEF clones overexpressing Arpp19 WT and PM (Figure 3) were synchronized to enter cell cycle (see materials and methods). 24 h after release, the mitotic index was calculated by FACS analysis of Phospho-Histone H3 (Ser10) antibody labelled cells (H3P). Propidium iodide (PI) was used to measure the DNA content. Percentage of mitotic cells were determined by gating analysis of the H3P positive population with 4 N DNA content. FACS data were analyzed by FlowJo 8 software. (B) Mitotic index values in (A) were plotted as histograms. (C) Cells treated as in (A) were arrested in mitosis for 4 h after 24 h release by adding 500 ng/mL nocodazole in their growth medium. Cells that remain arrested in mitosis were analyzed by FACS as in (A). (D) Values in (C) were plotted as histograms.
Next, we analyzed the strength of the SAC signaling by arresting the cells in mitosis for different periods of time and monitoring the ratio of mitotic cells by FACS analysis, using antibodies directed against serine10 phosphorylated histone H3, a mitotic marker. 24 h after cell cycle entry, nocodazole was added to the culture medium. After fours of further incubation, cells were fixed and analyzed. This results in arrest of the cells in metaphase after they enter mitosis. The percentage of mitotic cells in Mastl KO population is lower since they cannot maintain the mitotic state as good as WT cells. This is attributed to the reduced phosphorylation of Mps1, a kinase crucial for maintenance of spindle assembly checkpoint [16]. Overexpression of Arpp19 WT or PM resulted in partial rescue of this phenotype (Figure 4C, D). This did not change if the duration of mitotic arrest was increased to 6 h (Supplementary Figure S5B). Therefore, Arpp19 overexpression partly restores the SAC signal strength.
Silencing of B55δ has a similar affect as Arpp19 overexpression
PP2A is a heterotrimeric protein complex with three subunits. These are structural A, catalytic C, and regulatory B subunits. PP2A with B55δ subunit is responsible for dephosphorylation of Cdk substrates during mitotic exit [6]. Mastl phosphorylated Arpp19 and Ensa can interact with PP2A at the interface between C and B55 subunits [26]. Presence of a molar excess of Endos, the Drosophila orthologue of Arpp19, and its phosphorylation during mitosis by Mastl ensures efficient inhibition of protein phosphatase activity [12]. As discussed above, increasing the WT or phosphomimetic Arpp19 protein levels in the cells by overexpression restored some of the phenotypes associated with Mastl loss. In a similar fashion, PP2A inhibitor okadaic acid extended the time Mastl KO cells can maintain their mitotic arrested state upon nocodazole arrest [16]. Therefore, we reasoned that silencing of the B55δ subunit expression may have a similar outcome, by increasing the Arpp19:PP2A stoichiometric ratio.
We designed shRNAs targeted against mouse B55δ to knockdown its expression in Mastl CKO MEFs. Retroviral expression of the shRNA vector resulted in efficient silencing of the B55δ mRNA (Figure 5A, B). Induction of Mastl deletion by addition of 4′OHT to the culture medium resulted in a halt of cellular proliferation in both control and B55δ shRNA expressing cells (Figure 5C, D). These are similar to what we found for Arpp19 overexpression in Mastl CKO MEFs.

Knockdown of PP2A B55δ subunit does not rescure Mastl loss. (A) Mastl CKO MEFs were infected with empty (con) or B55δ shRNA expressing retroviral vectors (Figure 2). After selection with puromycin (2.5 µg/mL), RNA extracts were prepared and B55δ expression was analyzed by agarose gel electrophoresis of the RT-PCR products. (B) Real-time qPCR analysis of the samples in (A) were performed as described in Figure 1D. (C) AlamarBlue proliferation assay was performed in Mastl CKO MEFs selected with puromycin after transduced with empty (shCon) or B55δ shRNA (shB55 δ) expressing retroviral constructs (pSUPER.retro.puro). Cells were untreated (black, FLOX) or induced to undergo Mastl gene deletion (red, NULL). AFU, Arbitrary fluorescence units. (D) Cells treated as in (C) were analyzed for proliferation by 3T3 assay for three passages.
Next, we tested whether B55δ knockdown would display a similar effect as Arpp19 overexpression (WT or PM) in nocodazole arrested Mastl KO MEFs. Silencing of B55δ expression partly restored the percentage of Mastl KO cells that can stay arrested in mitosis (Figure 6). When we used antibodies that can detect phosphorylated substrates of Cdk1, we found that the mitotic phosphorylations in absence of Mastl kinase were partly restored (Figure 6C, D). Therefore, B55δ knockdown has a similar effect as Arpp19 overexpression in Mastl deficient cells.

Knockdown of PP2A B55δ subunit restores phosphorylation of Cdk substrates in mitosis. (A) Control or B55δ knockdown Mastl CKO MEFs (Figure 5) were synchronized to enter cell cycle (see materials and methods). 24 h after release, cells were treated with 500 ng/mL nocodazole for another 4 h to arrest them in mitosis. Percentage of cells that remained arrested in mitosis was calculated by FACS analysis of Phospho-Histone H3 (Ser10) antibody labelled cells (H3P). Propidium iodide (PI) was used to measure the DNA content. Percentage of mitotic cells were determined by gating analysis of the H3P positive population with 4 N DNA content. (B) Values in (A) were plotted as histograms. (C) Cells in (A) were immunostained using antibodies that recognize phosphorylated substrates of Cdk1 (CDKPS). (D) Values in (C) were plotted as histograms.
Discussion
Studies on the Cdk1-Mastl-Arpp19/ENSA-PP2A-B55δ axis as a central mitotic regulatory pathway took a start with the discovery of the greatwall mutation in Drosophila, which caused chromosome condensation and mitotic exit defects in mutant cells [7]. Subsequent in vitro work using Xenopus egg extracts demonstrated the importance of Mastl kinase in regulation of the PP2A-B55δ phosphatase activity directed against Cdk1-phosphorylated mitotic substrates [8, 29]. Finally, the discovery of Arpp19 and Ensa as the Mastl’s key substrates that bind to PP2A and inhibit its activity during mitosis, completed the missing link in the pathway [10, 11].
Physiological roles of Mastl kinase, Arpp19 and Ensa have been investigated by conditional knockout mouse models [16, 19]. A detailed study of the Mastl-Arpp19/Ensa-PP2A pathway in mammalian cells, by inquisition of the relationship of the components, has not been done yet. In this study, we aimed to address this.
We have shown that, Arpp19 is the predominantly expressed and relevant Mastl kinase substrate in MEFs. Although, both genes are ubiquitously expressed in a wide array of tissues we tested, it is not clear whether Ensa contributes to the suppression of PP2A activity during mitosis. Our work here and other published studies [19, 20] demonstrated that these two proteins have distinct functions in S-phase and mitosis. However, additional work needs to be done to confirm this in different cell types.
Silencing of Arpp19 but not Ensa expression results in a significant reduction in cellular proliferation rate. Similar results have been obtained using Arpp19 CKO MEFs [19]. Nevertheless, unlike Mastl deletion which results in a complete block of cellular proliferation [16], Arpp19 knockout or knockdown MEFs could still proliferate at a slower pace. This could be due to compensation by Ensa. However, we did not test whether Ensa overexpression can rescue the Arpp19 loss, or whether simultaneous knockdown of Ensa and Arpp19 would lead to a more pronounced, Mastl loss-like phenotype.
In an attempt to rescue Mastl loss phenotypes in MEFs, we tried suppressing PP2A phosphatase activity in two alternate ways: (i) by overexpressing Arpp19 (WT or PM mutant), (ii) or by silencing the expression of B55δ subunit. In both cases we had the same outcome: weakening of the SAC signal was restored (percentage of mitotically arrested MEFs is increased) but cells could not divide (lack of proliferation). Since PP2A phosphatase activity can be suppressed in Mastl deficient MEFs, it is worth investigating why the cells cannot proliferate. This could be due to the disturbance of timing of PP2A suppression, as overexpression of Arpp19 or reduction of B55δ levels are continuous throughout the cell cycle. An alternative explanation would be, Mastl has other, yet to be discovered, mitotic substrates independent of the Arpp19/Ensa-PP2A pathway, and Mastl-dependent phosphorylation of these proteins is essential for mitosis.
Although, the phosphomimetic mutant of Arpp19 was shown to inhibit PP2A-B55δ phosphatase activity more efficient than WT [28], we did not observe a difference between the two variants in our assays. This could be due to high overexpression of WT Arpp19 being sufficient to block PP2A activity effectively.
Funding source: Dokuz Eylül University Scientific Research Projects Grant
Award Identifier / Grant number: DEU-BAP Project No: 2016.KB.SAG.014
Funding source: TUBITAK (The Scientific and Technological Research Council of Turkey)
Award Identifier / Grant number: 1002 program Project No. 217Z248
Funding source: Turkish Academy of Sciences and The Science Academy, Turkey (Additional support received by MKD)
Award Identifier / Grant number: GEBIP award
Award Identifier / Grant number: BAGEP award
Acknowledgments
We thank Dr. Philipp Kaldis for sharing the plasmids, cell lines and antibodies used in this study.
-
Research funding: This work was supported by Dokuz Eylül University Scientific Research Projects grant (DEU-BAP Project No: 2016.KB.SAG.014) and TUBITAK (The Scientific and Technological Research Council of Turkey) 1002 program Project No. 217Z248. MKD received additional support from Turkish Academy of Sciences (GEBIP award) and The Science Academy, Turkey (BAGEP award). The funding organizations played no role in the study design, in the collection, analysis, and interpretation of data, in the writing of the report, or in the decision to submit the report for publication.
-
Author contributions: MKD conceived and designed the project, ME and MKD acquired the data, ME and MKD analyzed and interpreted the data, MKD wrote the paper. Both authors have accepted responsibility for the entire content of this manuscript and approved its submission.
-
Competing interests: Authors state no conflict of interest.
-
Informed consent: Not applicable.
-
Ethical approval: The local Institutional Review Board approved the study. Izmir Biyotıp ve Genom Merkezi, GOEK, 2022-024, 27 Aug 2022.
References
1. Satyanarayana, A, Kaldis, P. Mammalian cell-cycle regulation: several cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009;28:2925–39. https://doi.org/10.1038/onc.2009.170.Search in Google Scholar PubMed
2. Santamaría, D, Barrière, C, Cerqueira, A, Hunt, S, Tardy, C, Newton, K, et al.. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 2007;448:811–5. https://doi.org/10.1038/nature06046.Search in Google Scholar PubMed
3. Diril, MK, Ratnacaram, CK, Padmakumar, VC, Du, T, Wasser, M, Coppola, V, et al.. Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc Natl Acad Sci USA 2012;109:3826–31. https://doi.org/10.1073/pnas.1115201109.Search in Google Scholar PubMed PubMed Central
4. Morgan, DO. The cell cycle: principles of control. London: New Science Press; 2007.Search in Google Scholar
5. Petrone, A, Adamo, ME, Cheng, C, Kettenbach, AN. Identification of candidate cyclin-dependent kinase 1 (Cdk1) substrates in mitosis by quantitative phosphoproteomics. Mol Cell Proteomics 2016;15:2448–61. https://doi.org/10.1074/mcp.m116.059394.Search in Google Scholar PubMed PubMed Central
6. Mochida, S, Ikeo, S, Gannon, J, Hunt, T. Regulated activity of PP2A-B55 is crucial for controlling entry into and exit from mitosis in Xenopus egg extracts. EMBO J 2009;28:2777–85. https://doi.org/10.1038/emboj.2009.238.Search in Google Scholar PubMed PubMed Central
7. Yu, J, Fleming, SL, Williams, B, Williams, Ev., Li, ZX, Somma, P, et al.. Greatwall kinase: a nuclear protein required for proper chromosome condensation and mitotic progression in Drosophila. JCB (J Cell Biol) 2004;164:487–92. https://doi.org/10.1083/jcb.200310059.Search in Google Scholar PubMed PubMed Central
8. Castilho, PV, Williams, BC, Mochida, S, Zhao, Y, Goldberg, ML. The M phase kinase greatwall (Gwl) promotes inactivation of PP2A/B55, a phosphatase directed against CDK phosphosites. Mol Biol Cell 2009;20:4777–89. https://doi.org/10.1091/mbc.e09-07-0643.Search in Google Scholar PubMed PubMed Central
9. Virshup, DM, Kaldis, P. Cell biology. Enforcing the Greatwall in mitosis. Science 2010;330:1638–9. https://doi.org/10.1126/science.1199898.Search in Google Scholar PubMed
10. Gharbi-Ayachi, A, Labbé, JC, Burgess, A, Vigneron, S, Strub, JM, Brioudes, E, et al.. The substrate of Greatwall kinase, Arpp19, controls mitosis by inhibiting protein phosphatase 2A. Science 2010;330:1673–7. https://doi.org/10.1126/science.1197048.Search in Google Scholar PubMed
11. Mochida, S, Maslen, SL, Skehel, M, Hunt, T. Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science 2010;330:1670–3. https://doi.org/10.1126/science.1195689.Search in Google Scholar PubMed
12. Williams, BC, Filter, JJ, Blake-Hodek, KA, Wadzinski, BE, Fuda, NJ, Shalloway, D, et al.. Greatwall-phosphorylated Endosulfine is both an inhibitor and a substrate of PP2A-B55 heterotrimers. Elife 2014;2014:1–34. https://doi.org/10.7554/elife.01695.Search in Google Scholar PubMed PubMed Central
13. Labbé, JC, Vigneron, S, Méchali, F, Robert, P, Roque, S, Genoud, C, et al.. The study of the determinants controlling Arpp19 phosphatase-inhibitory activity reveals an Arpp19/PP2A-B55 feedback loop. Nat Commun 2021;12:1–19. https://doi.org/10.1038/s41467-021-23657-0.Search in Google Scholar PubMed PubMed Central
14. Yu, J, Zhao, Y, Li, ZX, Galas, S, Goldberg, ML. Greatwall kinase participates in the Cdc2 autoregulatory loop in Xenopus egg extracts. Mol Cell 2006;22:83–91. https://doi.org/10.1016/j.molcel.2006.02.022.Search in Google Scholar PubMed
15. Burgess, A, Vigneron, S, Brioudes, E, Labbé, JC, Lorca, T, Castro, A. Loss of human Greatwall results in G2 arrest and multiple mitotic defects due to deregulation of the cyclin B-Cdc2/PP2A balance. Proc Natl Acad Sci USA 2010;107:12564–9. https://doi.org/10.1073/pnas.0914191107.Search in Google Scholar PubMed PubMed Central
16. Diril, MK, Bisteau, X, Kitagawa, M, Caldez, MJ, Wee, S, Gunaratne, J, et al.. Loss of the greatwall kinase weakens the spindle assembly checkpoint. PLoS Genet 2016;12:1–26. https://doi.org/10.1371/journal.pgen.1006310.Search in Google Scholar PubMed PubMed Central
17. Álvarez-Fernández, M, Sánchez-Martínez, R, Sanz-Castillo, B, Gan, PP, Sanz-Flores, M, Trakala, M, et al.. Greatwall is essential to prevent mitotic collapse after nuclear envelope breakdown in mammals. Proc Natl Acad Sci USA 2013;110:17374–9. https://doi.org/10.1073/pnas.1310745110.Search in Google Scholar PubMed PubMed Central
18. Li, YH, Kang, H, Xu, YN, Heo, YT, Cui, XS, Kim, NH, et al.. Greatwall kinase is required for meiotic maturation in porcine Oocytes1. Biol Reprod 2013;89:1–7. https://doi.org/10.1095/biolreprod.113.109850.Search in Google Scholar PubMed
19. Hached, K, Goguet, P, Charrasse, S, Vigneron, S, Sacristan, MP, Lorca, T, et al.. ENSA and ARPP19 differentially control cell cycle progression and development. JCB (J Cell Biol) 2019;218:541–58. https://doi.org/10.1083/jcb.201708105.Search in Google Scholar PubMed PubMed Central
20. Charrasse, S, Gharbi-Ayachi, A, Burgess, A, Vera, J, Hached, K, Raynaud, P, et al.. Ensa controls S-phase length by modulating Treslin levels. Nat Commun 2017;8:1–15. https://doi.org/10.1038/s41467-017-00339-4.Search in Google Scholar PubMed PubMed Central
21. Adhikari, D, Diril, MK, Busayavalasa, K, Risal, S, Nakagawa, S, Lindkvist, R, et al.. Mastl is required for timely activation of APC/C in meiosis I and Cdk1 reactivation in meiosis II. JCB (J Cell Biol) 2014;206:843–53. https://doi.org/10.1083/jcb.201406033.Search in Google Scholar PubMed PubMed Central
22. Cundell, MJ, Bastos, RN, Zhang, T, Holder, J, Gruneberg, U, Novak, B, et al.. The BEG (PP2A-B55/ENSA/greatwall) pathway ensures cytokinesis follows chromosome separation. Mol Cell 2013;52:393–405. https://doi.org/10.1016/j.molcel.2013.09.005.Search in Google Scholar PubMed PubMed Central
23. Chen, YY, Ge, JY, Zhu, SY, Shao, ZM, Yu, KD. Copy number amplification of ENSA promotes the progression of triple-negative breast cancer via cholesterol biosynthesis. Nat Commun 2022;13:791. https://doi.org/10.1038/s41467-022-28452-z.Search in Google Scholar PubMed PubMed Central
24. Jiang, T, Zhao, B, Li, X, Wan, J. ARPP-19 promotes proliferation and metastasis of human glioma. Neuroreport 2016;27:960–6. https://doi.org/10.1097/wnr.0000000000000638.Search in Google Scholar
25. Erguven, M, Kilic, S, Karaca, E, Diril, MK. Genetic complementation screening and molecular docking give new insight on phosphorylation-dependent Mastl kinase activation. J Biomol Struct Dyn 2022;1–13. https://doi.org/10.1080/07391102.2022.2131627. 36270968.Search in Google Scholar PubMed
26. Mochida, S. Regulation of α-endosulfine, an inhibitor of protein phosphatase 2A, by multisite phosphorylation. FEBS J 2014;281:1159–69. https://doi.org/10.1111/febs.12685.Search in Google Scholar PubMed
27. Kim, MY, Bucciarelli, E, Morton, DG, Williams, BC, Blake-Hodek, K, Pellacani, C, et al.. Bypassing the Greatwall-Endosulfine pathway: plasticity of a pivotal cell-cycle regulatory module in Drosophila melanogaster and caenorhabditis elegans. Genetics 2012;191:1181–97. https://doi.org/10.1534/genetics.112.140574.Search in Google Scholar PubMed PubMed Central
28. Larouche, M, Kachaner, D, Wang, P, Normandin, K, Garrido, D, Yao, C, et al.. Spatiotemporal coordination of Greatwall-Endos-PP2A promotes mitotic progression. JCB (J Cell Biol) 2021;220. https://doi.org/10.1083/jcb.202008145.Search in Google Scholar PubMed PubMed Central
29. Vigneron, S, Brioudes, E, Burgess, A, Labbé, JC, Lorca, T, Castro, A. Greatwall maintains mitosis through regulation of PP2A. EMBO J 2009;28:2786–93. https://doi.org/10.1038/emboj.2009.228.Search in Google Scholar PubMed PubMed Central
Supplementary Material
This article contains supplementary material (https://doi.org/10.1515/tjb-2022-0191).
© 2023 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Frontmatter
- Review
- A dual role of proton pump inhibition on cancer: a critical review
- Opinion Paper
- The consumption of vegetable oils and their possible participation as a neuroprotector
- Research Articles
- Analysis of inappropriate repeated laboratory testing
- SNP based analysis depicts phenotypic variability in heme oxygenase-1 protein
- The D allele of angiotensin-converting enzyme gene insertion/deletion polymorphism is associated with the lung involvement in COVID-19
- The effect of survivin gene in breast cancer risk and prognosis
- Increase of Claudin-5, ICAM-1 and eNOS expressions in human brain endothelial cells by ammonium chloride
- The effect of different anti-inflammatory treatment strategies on process of atherosclerosis in ankylosing spondylitis patients
- Genetic dissection of the Mastl-Arpp19/Ensa-PP2A-B55δ pathway in mammalian cells
- Investigation into miRNA profile in patient groups with and without ST elevation
- Prognostic value of total thiol and D-dimer in patients hospitalized with COVID-19
- Case Report
- The effect of heterophilic antibody interference in thyroglobulin measurement on different immunoassay devices
Articles in the same Issue
- Frontmatter
- Review
- A dual role of proton pump inhibition on cancer: a critical review
- Opinion Paper
- The consumption of vegetable oils and their possible participation as a neuroprotector
- Research Articles
- Analysis of inappropriate repeated laboratory testing
- SNP based analysis depicts phenotypic variability in heme oxygenase-1 protein
- The D allele of angiotensin-converting enzyme gene insertion/deletion polymorphism is associated with the lung involvement in COVID-19
- The effect of survivin gene in breast cancer risk and prognosis
- Increase of Claudin-5, ICAM-1 and eNOS expressions in human brain endothelial cells by ammonium chloride
- The effect of different anti-inflammatory treatment strategies on process of atherosclerosis in ankylosing spondylitis patients
- Genetic dissection of the Mastl-Arpp19/Ensa-PP2A-B55δ pathway in mammalian cells
- Investigation into miRNA profile in patient groups with and without ST elevation
- Prognostic value of total thiol and D-dimer in patients hospitalized with COVID-19
- Case Report
- The effect of heterophilic antibody interference in thyroglobulin measurement on different immunoassay devices