A rare case of fructose-1,6-bisphosphatase deficiency: a delayed diagnosis story
-
Mahmut Cerkez Ergoren

 ,
Gulten Tuncel
,
Gulten Tuncel

Abstract
Objectives
Fructose-1,6-bisphosphatase deficiency (FBPase deficiency, OMIM 229700) is an early-onset rare genetic disorder caused by mutations in the FBP1 gene.
Case presentation
Our patient was 17-years-old when she was diagnosed with the disease. Initial sequencing analysis with Ion Torrent technology failed to detect the gross deletion that covered complete exon 2 (c.-24-26_170 + 5192del) of FBP1 gene and caused the delay in diagnosis. Deletion was then detected when sequencing was performed in an Illumina MiSeq platform.
Conclusions
This case emphasizes the importance of sequencing data analysis for precise diagnosis of rare diseases and therapy planning.
Introduction
Fructose-1,6-bisphosphatase deficiency (FBPase deficiency, OMIM 229700) is a rare, autosomal recessively inherited disorder with an estimated prevalence between 1:350.000 and 1:900.000 [1]. The disease is caused by mutations in the FBP1 gene (OMIM 611570), which encodes the FBPase enzyme involved in the gluconeogenesis pathway [2], [3]. The FBP1 gene is located on chromosome 9 and spans a region of 37,117 bases (9q22.2–q22.3) [3]. Mutations causing FBPase deficiency include a number of single nucleotide changes, indels and duplications [4].
FBPase is a key enzyme that cleaves fructose-1,6-bisphosphate into fructose-6-phosphate, so its deficiency causes defects in gluconeogenesis, which results in impaired glucose production from all gluconeogenic precursors [5]. If not managed correctly, high intake of fructose, prolonged fasting and intercurrent infections can trigger acute attacks of ketotic hypoglycaemia and lactic acidosis in patients, usually in the first months of life [2]. When preventive treatment is applied, the prognosis is usually quite good. However, non-specific presentation of the disease and patients being asymptomatic between acute attacks can lead to cases of FBPase deficiency being confused with other metabolic disorders [1]. Consequently, precise diagnosis is critical as the disorder may become fatal with episodes of severe hypoglycaemia and is often possible by measuring FBPase enzymatic activity or by genetic testing [5], [6].
Patient and methods
The current study presents the case of a 17-year-old female from a consanguineous marriage with clinical manifestations suggestive of both FBPase deficiency and a mild form of Becker disease including acute attacks, vomiting, hypoglycaemia, hyperventilation, coma, hepatomegaly and myopia.
According to the medical history of the patient, the first hypoglycaemic attack was reported when she was 18-months old and nutrition therapy was given with the pre-diagnosis of glycogen storage disease. However, acute attacks continued, which were accompanied by vomiting and hyperventilation at least once a year. Hepatomegaly and myopia were other symptoms indicative of FBPase deficiency. Previously, the genes that are associated with glycogen storage disease (G6PC, AGL (exon 2), GBE1 (exon 1 and 11), PYGL, SLC37A4, PHKA2 (exon 19 and 21), PHKG2, GYS1, GYS2, PHKB (exons 6, 11, 12, 14), SLCA2, PHKA1, PHKA2, FBP1 (exon 1), GYG1, PGK1, SLC2A2, ALDOB, ALDOA, PGAM2 were analysed with Ion Torrent sequencing technologies in a genetic diagnosis centre and no mutations were detected. Her enzymatic activity in the liver (NRV=25_70 nmol/mg of protein) was 3.1 when tested.
When the patient was admitted to Near East University Hospital with these symptoms last year, clinical exome sequencing was performed. DNA was isolated from peripheral blood samples and exons of 1,124 genes present in the Clinical Exome Solution panel were amplified. Sequencing was then performed in an Illumina MiSeq platform. Confirmation of sequencing was done with exon-specific PCR. Primers were designed within exon 2, within deletion region and equal concentrations of DNA were used in PCR.
Due to the delay in diagnosis, the patient was recommended to undergo an MRI examination for detection of possible brain damage due to the high number of hypoglycaemic attacks. Genetic counselling was advised for the patient and her family.
Results
Family pedigree is represented in Figure 1. The proband’s cousin was diagnosed with epilepsy and her older uncle had recently been diagnosed with myasthenia gravis.
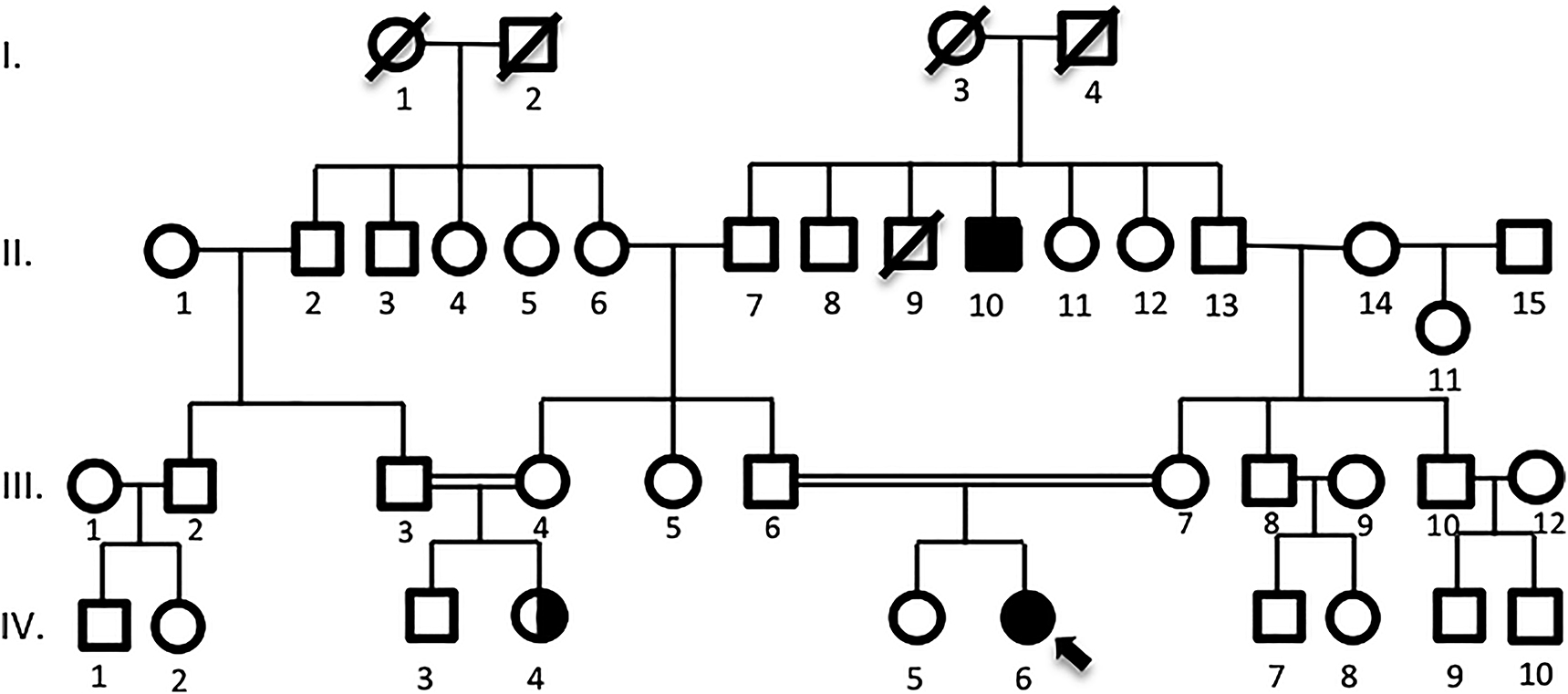
Pedigree analysis of the proband (IV-6) is shown above, who was born to a consanguineous marriage. Her cousin (IV-6) was diagnosed with epilepsy and her older uncle (II-10) was diagnosed with M. Gravis.
In clinical exome sequencing data analysis, a 5412bp-long gross deletion that covered complete exon 2 (c.-24-26_170 + 5192del) according to reference sequence NM_001127628.2, which was previously termed as exon 1 according to reference sequence NM_000507.4, in the FBP1 gene was detected in a homozygous state in the patient [7] Figure 2A. Confirmation of sequencing results with exon-specific PCR is shown in Figure 2B. No bands were visible in the proband indicating the absence of exon 2.
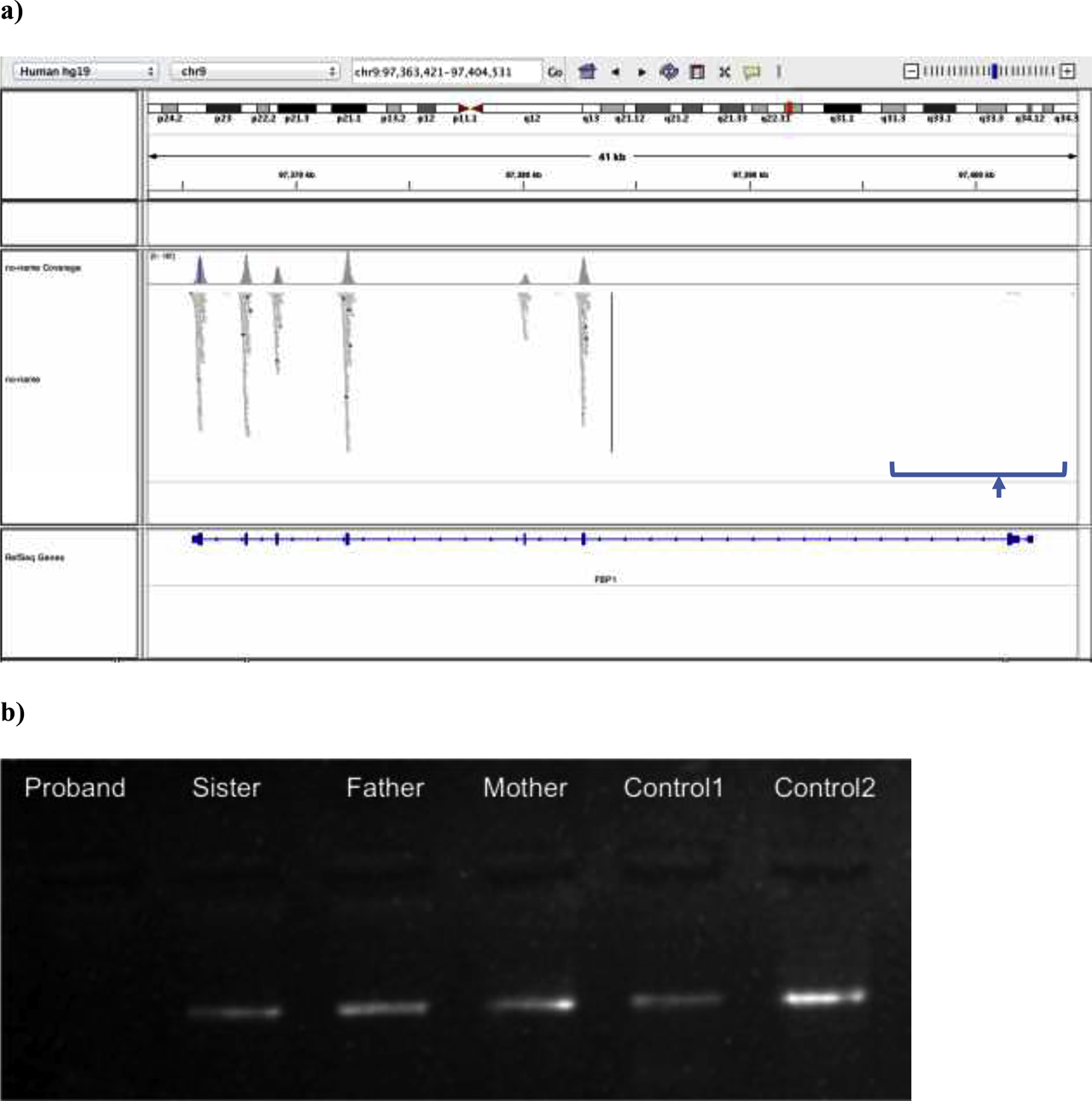
Molecular genetic analysis results of the proband. (a) Integrative Genomics Viewer (IGV) analysis of the patient’s FBP1 sequencing result is shown. Sequence is aligned to reference sequence NM_001127628.2. No reads are obtained from exon 2 of the FBP1 gene, suggesting a gross deletion of the exon. (b) The electrophoresis gel image of the exon-specific PCR analysis to confirm the exon 2 deletion of FBP1 is shown above. Primers were designed within exon 2. Homozygous deletion in the proband (no band) and heterozygous deletions in the mother, father and sister (faint bands) were confirmed by comparison of band intensities with the control bands from a heterozygous individual (Control1-faint band) and from the homozygous healthy individual (Control2-strong band). EtBr signalling under ultraviolet light was used for gel imaging.
No significant pathological change was reported in the brain MRI examination of the patient, indicating no brain damage due to the high number of hypoglycaemic attacks that the patient has experienced before diagnosis.
Discussion
The rare genetic disease, fructose 1,6-bisphosphatase (FBPase) deficiency, is caused by mutations in the FBP1 gene and manifests itself in the first months of life with episodes of hypoglycaemia and lactic acidosis. Diagnosis is important, as patients should be given a fructose- and sucrose-restricted diet, exogenous glucose administration and should avoid prolonged fasting to prevent severe hypoglycaemic attacks, which can cause brain damage and can even be fatal [1], [8], [9].
In our case, symptoms of the FBPase deficiency were initially reported in the first 18-months of life but diagnosis was not possible until she was 17. The condition was confused with glycogen storage disease. When sequencing was performed, even though it is the most common FBP1 variation in the Turkish population, homozygous exon 2 deletion (c.-24-26_170 + 5192del) could not be detected until the second clinical exome sequencing performed by our group [10]. There are several explanations for the delayed diagnosis in our patient’s case. The first is that different sequencing technologies were used in two different centres. Unfortunately, copy number variations and gross insertion/deletions can be missed in Ion Torrent sequencing, so MLPA is mostly suggested to detect these variations. Also, the first analysis with Ion Torrent was performed around 10 years before the second one. Therefore, information about the variations causing the syndrome was quite limited. Hence, analysis of bulk sequencing data coming from the whole genome, exome and targeted sequencing is of great importance for the evaluation of testing results.
As the hypoglycaemic attacks continued for so long and repeated at least once a year, brain damage was a risk. After the diagnosis was determined, the patient was given preventive treatment and the sister who is heterozygote for the mutation was advised to have genetic counselling before marriage due to the risk of giving birth to affected children if her husband has heterozygosity for the same mutation. Thus, it is imperative that a rapid and accurate diagnostic method for FBPase deficiency is established.
Conclusion
There is no doubt that next generation sequencing has revolutionized the clinical diagnostic field and increased our understanding of the rarest diseases. However, examples such as our case indicate the importance of data analysis after sequencing is performed. Bias can occur at several stages of bioinformatics analysis, especially during filtering. Therefore, use of correct parameters and repeating the analysis a few times are of high importance.
Learning Points
High-throughput sequencing technology has dramatically changed the nature of molecular diagnosis. When applied to individuals with rare-genetic syndromes, this technology allows scientist to identify genetic variants and also helps to understand the genome structure and function better. In the current study, a patient with rare FBPase deficiency was diagnosed after 15 years of initial testing using new technologies and planned therapy will improve her life quality. Additionally, this finding showed the importance of genetic counselling and patient management. Counselling will be important for the patient and her family, especially for her sister who is heterozygous for the mutation. The aim is to provide a practical guide for the effective use of this technology which will aid in clinical diagnosis, genetic counselling and patient management.
Research funding: There is no financial interest to report.
Author contributions: All co-authors have seen and agree with the contents of the manuscript.
Conflict of Interest: The authors have no conflicts of interest to declare.
Informed consent: Informed consent was obtained from the parents of the patient.
Ethical approval: The study protocol was approved by ethical committee (NEU/2019/74/923).
References
1. Li, N, Chang, G, Xu, Y, Ding, Y, Li, G, Yu, T, et al. Clinical and molecular characterization of patients with fructose 1,6-bisphosphatase deficiency (in eng). Int J Mol Sci 2017;18:857. https://doi.org/10.3390/ijms18040857.Search in Google Scholar
2. Lebigot, E, Brassier, A, Zater, M, Imanci, D, Feillet, F, Thérond, P, et al. Fructose 1,6-bisphosphatase deficiency: clinical, biochemical and genetic features in French patients (in eng). J Inherit Metab Dis 2015;38:881–7. https://doi.org/10.1007/s10545-014-9804-6.Search in Google Scholar
3. el-Maghrabi, MR, Lange, AJ, Jiang, W, Yamagata, K, Stoffel, M, Takeda, J, et al. Human fructose-1,6-bisphosphatase gene (FBP1): exon-intron organization, localization to chromosome bands 9q22.2–q22.3, and mutation screening in subjects with fructose-1,6-bisphosphatase deficiency (in eng). Genomics 1995;27:520–5. https://doi.org/10.1006/geno.1995.1085.Search in Google Scholar
4. Santer, R, du Moulin, M, Shahinyan, T, Vater, I, Maier, E, Muntau, AC, et al. A summary of molecular genetic findings in fructose-1,6-bisphosphatase deficiency with a focus on a common long-range deletion and the role of MLPA analysis (in eng). Orphanet J Rare Dis, OriginalPaper 2016;11:1–7, 2016-04-21. https://doi.org/10.1186/s13023-016-0415-1.Search in Google Scholar
5. Pinheiro, FC, Sperb-Ludwig, F, Ligabue-Braun, R, Schüler-Faccini, L, de Souza, CF, Vairo, F, et al. Genetic analysis of patients with fructose-1,6-bisphosphatase deficiency (in eng). Gene 2019;699:102–9. https://doi.org/10.1016/j.gene.2019.03.007.Search in Google Scholar
6. Kikawa, Y, Shin, YS, Inuzuka, M, Zammarchi, E, Mayumi, M. Diagnosis of fructose-1,6-bisphosphatase deficiency using cultured lymphocyte fraction: a secure and noninvasive alternative to liver biopsy (in eng). J Inherit Metab Dis 2002;25:41–6. https://doi.org/10.1023/a:1015129616599.10.1023/A:1015129616599Search in Google Scholar
7. NCBI, SG. Homo sapiens fructose-bisphosphatase 1 (FBP1), transcript variant 1, m – Nucleotide – NCBI. Available from: https://www.ncbi.nlm.nih.gov/pubmed/ [Accessed 2020].Search in Google Scholar
8. Tran, C. Inborn errors of fructose metabolism. What can we learn from them? (in eng). Nutrients 2017;9. https://doi.org/10.3390/nu9040356.Search in Google Scholar
9. Pinto, A, Alfadhel, M, Akroyd, R, Altınok, YA, Bernabei, SM, Bernstein, L, et al. International practices in the dietary management of fructose 1-6 biphosphatase deficiency (in eng). Orphanet J Rare Dis 2018;13:21. https://doi.org/10.1186/s13023-018-0760-3.Search in Google Scholar
10. Kilic, M, Kasapkara, CS, Yilmaz, DY, Ozgul, RK. Exon 2 deletion represents a common mutation in Turkish patients with fructose-1,6-bisphosphatase deficiency (in eng). Metab Brain Dis 2019;34:1487–91. https://doi.org/10.1007/s11011-019-00455-8.Search in Google Scholar
© 2020 Walter de Gruyter GmbH, Berlin/Boston
Articles in the same Issue
- Frontmatter
- Review Article
- Newly developed diagnostic methods for SARS-CoV-2 detection
- Short Communication
- Effect of hemolysis on prealbumin assay
- Research Articles
- BioVar: an online biological variation analysis tool
- High dose ascorbic acid treatment in COVID-19 patients raised some problems in clinical chemistry testing
- Immunoassay biomarkers of first and second trimesters: a comparison between pregnant Syrian refugees and Turkish women
- Association of maternal serum trace elements with newborn screening-thyroid stimulating hormone
- PIK3CA and TP53 MUTATIONS and SALL4, PTEN and PIK3R1 GENE EXPRESSION LEVELS in BREAST CANCER
- Evaluation of E2F3 and survivin expression in peripheral blood as potential diagnostic markers of prostate cancer
- Age, gender and season dependent 25(OH)D levels in children and adults living in Istanbul
- Original Article
- Fractional excretion of magnesium as an early indicator of renal tubular damage in normotensive diabetic nephropathy
- Research Articles
- Diagnostic value of laboratory results in children with acute appendicitis
- Evaluation of thiol disulphide levels in patients with pulmonary embolism
- Relationship between renal tubulointerstitial fibrosis and serum prolidase enzyme activity
- Comparison of test results obtained from lithium heparin gel tubes and serum gel tubes
- MHC Class I related chain A (MICA), Human Leukocyte Antigen (HLA)-DRB1, HLA-DQB1 genotypes in Turkish patients with ulcerative colitis
- An overview of procalcitonin in Crimean-Congo hemorrhagic fever: clinical diagnosis, follow-up, prognosis and survival rates
- Comparison of different equations for estimation of low-density lipoprotein (LDL) – cholesterol
- Case-Report
- A rare case of fructose-1,6-bisphosphatase deficiency: a delayed diagnosis story
- Research Articles
- Atypical cells in sysmex UN automated urine particle analyzer: a case report and pitfalls for future studies
- Investigation of the relationship cellular and physiological degeneration in the mandible with AQP1 and AQP3 membrane proteins
- In vitro assessment of food-derived-glucose bioaccessibility and bioavailability in bicameral cell culture system
- Letter to the Editor
- The weighting factor of exponentially weighted moving average chart
Articles in the same Issue
- Frontmatter
- Review Article
- Newly developed diagnostic methods for SARS-CoV-2 detection
- Short Communication
- Effect of hemolysis on prealbumin assay
- Research Articles
- BioVar: an online biological variation analysis tool
- High dose ascorbic acid treatment in COVID-19 patients raised some problems in clinical chemistry testing
- Immunoassay biomarkers of first and second trimesters: a comparison between pregnant Syrian refugees and Turkish women
- Association of maternal serum trace elements with newborn screening-thyroid stimulating hormone
- PIK3CA and TP53 MUTATIONS and SALL4, PTEN and PIK3R1 GENE EXPRESSION LEVELS in BREAST CANCER
- Evaluation of E2F3 and survivin expression in peripheral blood as potential diagnostic markers of prostate cancer
- Age, gender and season dependent 25(OH)D levels in children and adults living in Istanbul
- Original Article
- Fractional excretion of magnesium as an early indicator of renal tubular damage in normotensive diabetic nephropathy
- Research Articles
- Diagnostic value of laboratory results in children with acute appendicitis
- Evaluation of thiol disulphide levels in patients with pulmonary embolism
- Relationship between renal tubulointerstitial fibrosis and serum prolidase enzyme activity
- Comparison of test results obtained from lithium heparin gel tubes and serum gel tubes
- MHC Class I related chain A (MICA), Human Leukocyte Antigen (HLA)-DRB1, HLA-DQB1 genotypes in Turkish patients with ulcerative colitis
- An overview of procalcitonin in Crimean-Congo hemorrhagic fever: clinical diagnosis, follow-up, prognosis and survival rates
- Comparison of different equations for estimation of low-density lipoprotein (LDL) – cholesterol
- Case-Report
- A rare case of fructose-1,6-bisphosphatase deficiency: a delayed diagnosis story
- Research Articles
- Atypical cells in sysmex UN automated urine particle analyzer: a case report and pitfalls for future studies
- Investigation of the relationship cellular and physiological degeneration in the mandible with AQP1 and AQP3 membrane proteins
- In vitro assessment of food-derived-glucose bioaccessibility and bioavailability in bicameral cell culture system
- Letter to the Editor
- The weighting factor of exponentially weighted moving average chart