Investigating the performance of AIC in selecting phylogenetic models
-
Dwueng-Chwuan Jhwueng


Abstract
The popular likelihood-based model selection criterion, Akaike’s Information Criterion (AIC), is a breakthrough mathematical result derived from information theory. AIC is an approximation to Kullback-Leibler (KL) divergence with the derivation relying on the assumption that the likelihood function has finite second derivatives. However, for phylogenetic estimation, given that tree space is discrete with respect to tree topology, the assumption of a continuous likelihood function with finite second derivatives is violated. In this paper, we investigate the relationship between the expected log likelihood of a candidate model, and the expected KL divergence in the context of phylogenetic tree estimation. We find that given the tree topology, AIC is an unbiased estimator of the expected KL divergence. However, when the tree topology is unknown, AIC tends to underestimate the expected KL divergence for phylogenetic models. Simulation results suggest that the degree of underestimation varies across phylogenetic models so that even for large sample sizes, the bias of AIC can result in selecting a wrong model. As the choice of phylogenetic models is essential for statistical phylogenetic inference, it is important to improve the accuracy of model selection criteria in the context of phylogenetics.
Acknowledgments
We thank David Posada for the helpful discussion on phylogenetic model selection. We thank Diego Darriba for his generous help with implementing the phylogenetic model selection program jModelTest. Jhwueng’s research was supported by the National Science Council Award #NSC-101-2118-M-035-001 Taiwan and Postdoctoral Fellowship at the National Institute for Mathematical and Biological Synthesis (NIMBioS), an Institute sponsored by the National Science Foundation, the U.S. Department of Homeland Security, and the U.S. Department of Agriculture through NSF Awards #EF-0832858 and #DBI-1300426, with additional support from The University of Tennessee, Knoxville. We also thank NIMBioS Working Group-Gene Tree/Species Tree Reconciliation for providing the opportunity that the authors of this paper could meet to discuss this project. This research was partially supported by the National Science Foundation (DMS-1222745 to LL). Huzurbazar’s research was supported by a grant to the University of Wyoming from the National Science Foundation under grant DMS-1100615. Huzurbazar’s contribution was based upon work partially supported by the National Science Foundation under Grant DMS-1127914 to the Statistical and Applied Mathematical Sciences Institute. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
Appendix A
A1 The likelihood function of general substitution models
Given the DNA sequence data D of size N×M where each element of D, Duv represents the vth (v=1, …, M) nucleotide of sequence u (u=1, …, N). There are 4N possible nucleotide patterns for each column in data D. Let pi be the probability of observing the ith nucleotide pattern. The probability pi is the summation of the product of transition probabilities {P(t)=Pij(t); i, j=A, C, G, T} (see Figure A1). Thus, pi=pi(ϕ) is a function of model parameters ϕ=(θ, τ, b) where θ represents the parameters in the substitution model (typically the rate matrix Q, and the equilibrium vector π=(πA, πC, πG, πT)). When substitution rates are variable over sites, the heterogeneity of rates such as invariant parameter I, and the gamma rate parameter γ will be included in the substitution models. The tree topology τ and its branch lengths b are also treated as parameters for phylogenetic tree estimation. The rate matrix, Q, describes the rate at which bases of one type change into bases of another type. The rate matrix has the following structure
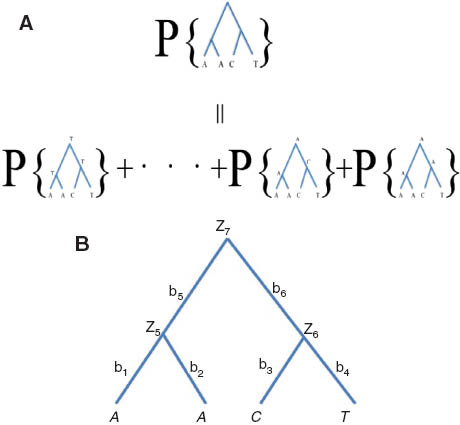
The probability density function p(·) of a column of nucleotides given the phylogenetic tree. (A) The column of nucleotides (A, A, C, T) are at the tips of the tree. The probability of (A, A, C, T) given the tree (on the top) is equal to the sum of the probabilities at the bottom, in which the nucleotides at the internal nodes of the tree are given. Each probability at the bottom is the product of Pij(t)s, the probabilities for individual branches on the tree. (B) A four taxa rooted phylogenetic tree. The ith site observed at tip has pattern AACT. The internal nodes zk∈{A, C, G, T}, 5≤k≤7 are ancestral status; and bj≥0, 1≤j≤6 are branches lengths.
where μxy represents the transition rate from base x to base y and the diagonals of the matrix are chosen so that the rows sum to zero: μx=–Σ{y|y≠x}μxy. The equilibrium row vector π must satisfy πQ=0. Let P(t) be the transition probability matrix, in which Pxy(t) is the probability of base x changing to y after a period of time t. Since this is a continuous time Markov Chain, the transition probability matrix satisfies a first order ordinary system differential equation P′(t)=QP(t), to which the solution is P(t∣Q)=eQt. The substitution models considered in this paper are time reversible. Thus, the rate matrix Q can be diagonalized and has real eigenvalues. It is straightforward that all elements Pxy(t) in the transition probability matrix P(t) have a continuous second order partial derivative with respect to t and parameters in the rate matrix Q.
As an example, we express probability pi for a particular pattern {AACT} observed at the tips of a 4-taxon rooted tree (Figure A1B). Let zk∈{A, C, G, T}, 5≤k≤7 be the ancestor status, let bj>0,1≤j≤6 be the branch lengths. Let π be the equilibrium base frequencies, and we assume that the nucleotides at the root of the tree have reached the equilibrium frequencies π. The probability of observing pattern AACT at the tips of the tree is a sum over all possible assignments of nucleotides to internal nodes, i.e.,
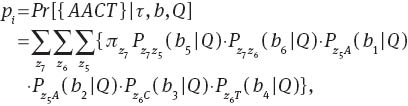
where Pxy(bj∣Q) is the transition probability that nucleotide y is substituted for x over a branch length bj. Equation (A1) has 43=64 terms. In general the expression for k species will have 22k–2 terms. As pi is the sum of multiplications of equilibrium frequencies π and transition probabilities Pxy(b), pi has a continuous second-order partial derivative with respect to branch lengths b and parameters in the rate matrix Q and equilibrium frequency vector π.
Let
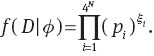
It follows from (2) that the log likelihood function L is
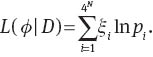
The log likelihood function for a fixed model m∈Ω is denoted by Lm(ϕm∣D), or simply by Lm. If the tree topology τ is given, pi has continuous second-order partial derivatives with respect to parameters ϕ. It follows immediately that the likelihood function Lm(ϕm∣D) has continuous second-order partial derivatives with respect to ϕ as Lm is the sum of the weighted logarithm of pi shown in (A3).
A2 The flow chart for estimating the expected KL divergence
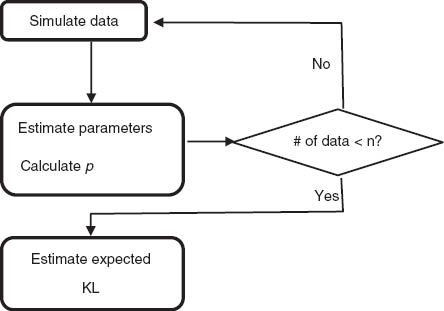
The flow chart for estimating the expected KL divergence.
References
Abdo, Z., V. Minin, P. Joyce and J. Sullivan (2005): “Accounting uncertainty in the tree topology has little effect on the decision theoretic approach on model selection in phylogenetic estimation.” Mol. Biol. Evol., 22, 691–703.Search in Google Scholar
Akaike, H. (1974): “A new look at the statistical model identification,” IEEE Trans. Aut. Control, 19, 716–723.Search in Google Scholar
Alfaro, M. and J. Huelsenbeck (2006): “Comparative performance of bayesian and aicbased measures of phylogenetic model uncertainty,” Syst. Biol., 55, 89–96.Search in Google Scholar
Anisimova, M. and O. Gascuel (2006): “Approximate likelihood-ratio test for branches: a fast, accurate and powerful alternative,” Syst. Biol., 55, 539–552.Search in Google Scholar
Boettiger, C., G. Coop and P. Ralph (2012): “Is your phylogeny informative? Measuring the power of comparative methods,” Evolution, 66, 2240–2251.10.1111/j.1558-5646.2011.01574.xSearch in Google Scholar PubMed PubMed Central
Bos, D. and D. Posada (2005): “Using models of nucleotide evolution to build phylogenetic trees,” Developmental and Comparative Immunolology, 29, 211–227.10.1016/j.dci.2004.07.007Search in Google Scholar PubMed
Buckley, T. and C. Cunningham (2002): “The effects of nucleotide substituion model assumptions on estimates of nonparametric bootstrap support,” Mol. Biol. Evol., 19, 394–405.Search in Google Scholar
Burham, K. and D. Anderson (2004): Model selection and multimodel inference, Springer-Verlag: New York.10.1007/b97636Search in Google Scholar
Cunningham, C., H. Zhu and D. Hillis (1998): “Best-fit maximum likelihood models for phylogenetic inference: empirical tests with known phylogenies,” Evolution, 52, 978–987.10.1111/j.1558-5646.1998.tb01827.xSearch in Google Scholar PubMed
Darriba, D., G. Taboada, R. Doallo and D. Posada (2012): “Jmodeltest 2: more models, new heuristics and parallel computing,” Nature Methods, 9, 772.10.1038/nmeth.2109Search in Google Scholar PubMed PubMed Central
Davison, A. (2003): Statistical models, Cambridge University Press: New York.10.1017/CBO9780511815850Search in Google Scholar
Evans, J. and J. Sullivan (2010): “Approximation model probabilities in bic and dt approaches to model selection in phylogenetics,” Mol. Biol. Evol., 28, 343–349.Search in Google Scholar
Felsenstein, J. (1981): “Evolutionary trees from dna sequences: a maximum likelihood approach,” J. Mol. Evol., 17, 368–376.Search in Google Scholar
Frati, F. (1997): “Evolution of the mitochondrial coii gene in collembola,” J. Mol. Evol., 44, 145–158.Search in Google Scholar
Guindon, S. and O. Gascuel (2003): “A simple, fast and accurate method to estimate large phylogenies by maximum-likelihood,” Syst. Biol., 52, 696–704.Search in Google Scholar
Hayasaka, K., T. Gojobori and S. Horai (1988): “Molecular phylogeny and evolution of primate mitochondrial DNA,” Mol. Biol. Evol., 5, 626–644.Search in Google Scholar
Holder, M., P. Lewis and D. Swofford (2010): “The akaike information criterion will not choose the no common mechanism model,” Syst. Biol., 59, 477–485.Search in Google Scholar
Huelsenbeck, J. and K. Crandall (1997): “Phylogeny estimation and hypothesis testing using maximum likelihood,” Annu. Rev. Ecol. Evol. Syst., 42, 247–264.Search in Google Scholar
Huelsenbeck, J., B. Larget and M. Alfaro (2004): “Bayesian phylogenetic model selection using reversible jump markov chain monte carlo,” Mol. Biol. Evol., 21, 1123–1133.Search in Google Scholar
Hurvich, C. and C.-L. Tsai (1989): “Regression and time series model selection in small samples,” Biometrika, 76, 297–307.10.1093/biomet/76.2.297Search in Google Scholar
Ishiguro, M., Y. Sakamoto and G. Kitagawa (1997): “Bootstrapping log likelihood and eic, an extension of aic,” Ann. I. Stat. Math., 49, 411–434.Search in Google Scholar
Jermiin, L., V. Jayaswal, F. Ababneh and J. Robinson (2008): “Phylogenetic model evaluation,” Methods Mol. Biol., 452, 31–64.Search in Google Scholar
Johnson, J. and K. Omland (2004): “Model selection in ecology and evolution,” Trends Ecol. Evol., 19, 101–108.Search in Google Scholar
Jukes, T. and C. Cantor (1969): “Evolution of protein molecules,” In: Munro, H.N. (Eds.), Mammalian protein metabolism. Academic Press: New York. 21–132.10.1016/B978-1-4832-3211-9.50009-7Search in Google Scholar
Kelchner, S. (2009): “Phylogenetic models and model selection for noncoding Dna,” Plant Syst. Evol., 282, 109–126.Search in Google Scholar
Kelchner, S. and M. Thomas (2007): “Model use in phylogenetics: nine key questions,” Trends Ecol. Evol., 282, 109–126.Search in Google Scholar
Kimura, M. (1980): “A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences,” J. Mol. Evol., 16, 111–120.Search in Google Scholar
Luo, A., H. Qiao, Y. Zhang, W. Shi, Y. Ho, W. Xu, A. Zhang and C. Zhu (2010): “Performance of crtiteria for selecting evolutionary models in phylogenetics: a comprehensive study based on simulated datasets,” BMC Evol. Biol., 10, 242.Search in Google Scholar
Minin, V., Z. Abdo, P. Joyce and J. Sullivan (2003): “Performance-based selection of likelihood models for phylogeny estimation,” Syst. Biol., 52, 674–683.Search in Google Scholar
Pol, D. (2004): “Empirical problems of the hierarchical likelihood ratio test for model selection,” Syst. Biol., 53, 949–962.Search in Google Scholar
Posada, D. (2008): “Jmodeltest: phylogenetic model averaging,” Mol. Biol. Evol., 25, 1253–1256.Search in Google Scholar
Posada, D. and T. Buckley (2004): “Model selection and model averaging in phylogenetics: advantage of akaike information criterion and baysian approaches over likelihood ratio tests,” Syst. Biol., 53, 793–808.Search in Google Scholar
Posada, D. and K. Crandall (1998): “Modeltest: testing the model of DNA substitution,” Bioinformatics, 14, 817–818.10.1093/bioinformatics/14.9.817Search in Google Scholar PubMed
Posada, D. and K. Crandall (2001): “Selecting the best-fit model of nucleotide substitution,” Syst. Biol., 50, 580–601.Search in Google Scholar
Rambaut, A. and N. Grassly (1997): “Seq-gen: an application for the monte carlo simulation of dna sequence evolution along phylogenetic tree,” Comput. Appl. Biosci., 13, 235–238.Search in Google Scholar
Rippinger, J. and J. Sullivan (2008): “Does choice in model selection affect maximum likelihood analysis?” Syst. Biol., 57, 76–85.Search in Google Scholar
Schwarz, G. (1978): “Estimating the dimension of a model,” Ann. Stat., 6, 461–464.Search in Google Scholar
Self, S. and K.-Y. Liang (1987): “Asymptotic properties of maximum likelihood estimators and likelihood ratio tests under nonstandard conditions,” J. Am. Stat. Assoc., 82, 605–610.Search in Google Scholar
Shapiro, B., A. Rambaut and A. Drummond (2006): “Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences,” Mol. Biol. Evol., 23, 7–9.Search in Google Scholar
Sullivan, J. and P. Joyce (2005): “Model selection in phylogenetics,” Annu. Rev. Ecol. Evol. Syst., 36, 445–466.Search in Google Scholar
Sullivan, J. and D. Swofford (1997): “Are guinea pigs rodents? The importance of adequate models in molecular phylogenetics,” J. Mamm. Evol., 4, 77–86.Search in Google Scholar
Tavaré, S. (1986): “Some probabilistic and statistical problems in the analysis of dna sequences,” Lect. Math. Life Sci. (American Mathematical Society), 17, 57–86.Search in Google Scholar
Wu, C., M. Suchard and A. Drummond (2013): “Bayesian selection of nucleotide substitution models and their site assignments,” Mol. Biol. Evol., 30, 669–688.Search in Google Scholar
Yang, Z. (1994): “Maximum likelihood phylogenetic estimation from dna sequeces with variable rates over sites: approximate methods,” J. Mol. Evol., 39, 306–314.Search in Google Scholar
Zharkikh, A. (1994): “Estimation of evolutionary distances between nucleotide sequences,” J. Mol. Evol., 39, 315–329.Search in Google Scholar
© 2014 by De Gruyter
Articles in the same Issue
- Frontmatter
- Review
- Comparison of algorithms to infer genetic population structure from unlinked molecular markers
- Research Articles
- Comparison of statistical methods for finding network motifs
- A sequential naïve Bayes classifier for DNA barcodes
- Biological pathway selection through Bayesian integrative modeling
- Investigating the performance of AIC in selecting phylogenetic models
- Multiclass cancer classification based on gene expression comparison
- Protein domain hierarchy Gibbs sampling strategies
Articles in the same Issue
- Frontmatter
- Review
- Comparison of algorithms to infer genetic population structure from unlinked molecular markers
- Research Articles
- Comparison of statistical methods for finding network motifs
- A sequential naïve Bayes classifier for DNA barcodes
- Biological pathway selection through Bayesian integrative modeling
- Investigating the performance of AIC in selecting phylogenetic models
- Multiclass cancer classification based on gene expression comparison
- Protein domain hierarchy Gibbs sampling strategies