Grignard Reagents and Nickel
-
Fabienne Fache


1 Introduction
Grignard reagents, discovered more than one century ago, are still widely used to promote carbon–carbon bond formation. Over the time, their reactivity has been modulated by the presence of various additives, especially transition metals salts. In this area, nickel had played a major role in the development of new and selective cross-coupling reactions between organomagnesium derivatives and C(sp2)-halides. It is generally admitted that the respective contributions of Kumada and Tamao in Japan [1] and Corriu in France [2] constitute a major breakthrough in the field of organometallic chemistry. Forty years after these two seminal contributions, the Kumada Tamao-Corriu reaction and related processes are still investigated but also applied in industrial processes and used in the synthesis of complex molecules. Thanks to the design of more and more sophisticated ligands, including pincer and NHC ligands, this coupling reaction initially performed with halogen derivatives has been successfully applied to a large number of starting materials. Nowadays, the cleavage of carbon–oxygen and carbon–sulfur bonds has considerably enhanced the potential of this reaction in term of regioselectivity. Furthermore, the challenging sp3–sp3 cross-coupling has been addressed. Finally, the control of the stereochemistry has been considered, which also allows new perspectives for the future.
The mechanism generally admitted is depicted in Figure 1. A nickel(0) species is generated in situ by reduction of a nickel(II) complex mainly by action of the Grignard species present in excess. Then, the oxidative addition results in the formation of a nickel(II) complex A which can undergo a transmetallation with the Grignard reagent leading to species B. An isomerization step via intermediate C, followed by a subsequent reductive elimination, release the coupled product D and regenerate the effective catalyst.

Generally admitted mechanism for the Kumada-Corriu reaction.
In the context of enantioselective processes, the coupling between an optically active Grignard reagent 1 and vinyl bromide was investigated and led to adduct 2 with almost complete retention of the configuration when conducted in the presence of NiCl2dppf at low temperature (Figure 2). This demonstrated that the key transmetallation step occurred surely according to a concerted polar mechanism [3]. In contrast, the same reaction promoted by iron or cobalt resulted in partial racemization of the final product which supports a radical based mechanism.

Enantioselective transformation as a proof for a concerted polar mechanism.
The success of the different Ni-catalyzed cross-couplings [4] resulted from a large number of items such as:
The ligands, especially mono- and diphosphines, which stabilize the organometallic intermediate to avoid a homodimerization process (Kharasch reaction).
The elimination which usually occurred with alkyl Grignard reagents can be strongly reduced.
In Csp2 –Csp2 cross-couplings, the initial geometry of the vinyl derivatives is maintained.
2 Coupling reactions of aryl Grignard reagents
2.1 With aryl derivatives
2.1.1 With aryl bromides and chlorides
The formation of diaryl compounds has been extensively studied. The success of the couplings depends both on the structure of the substrates (nature of the leaving group, steric hindrance, etc.) but also on the nature of the nickel catalyst and principally the type of ligands directly linked to the metal.
Use of phosphine ligands
At the very beginning of the development of the Kumada-Corriu reaction [5–6], phosphine ligands were used to stabilize the nickel catalyst, the catalytic activity being significantly dependent on the nature of the phosphine (NiCl2(dppp) > NiCl2(dppe) > NiCl2(PR3)2 = NiCl2(dppb)). The Kumada-Tamao-Corriu reaction has attracted chemical companies to access fine intermediates such as substituted styrenes and biphenyls but also, at a later stage, for the final functionalization of the target molecules [7–8]. The reactions usually are highly yielding and selective. They are also highly attractive because of the large availability of chloro-derivatives as precursors of both Grignard reagents and cheap coupling partners. Of interest, industrial applications required low charge of catalyst of common and cheap diphosphine ligands. Moreover, these reactions are usually performed under suitable conditions of temperature. However, some drawbacks should be pointed out, for example Grignard reagents require anhydrous conditions, and they are not compatible with a large variety of (reductive) functionalities. Finally, the work-up of the reaction generates a large amount of magnesium salts and thus, requires a process for waste treatment.
PDE472 4, a selective inhibitor of the phosphodiesterase PDE4D isoenzyme has been prepared on pilot-plant scale taking advantage of the Kumada-Corriu coupling (Figure 3). The coupling reaction of 4-chloropyridine in toluene, with the p-methoxyphenyl Grignard was performed to produce 3 in 74 % yield. The success of the reaction depends on the dryness of the chloropyridine [9].

Access to 4-arylpyridine 3, a key intermediate for the synthesis of PDE 472.
To produce the HIV protease inhibitor atazanavir, chemists at Novartis have reported a multi-kg synthesis of substituted p-aryl benzaldehydes based on a Ni-catalyzed coupling between 2-bromopyridine and aryl Grignard derivatives (Figure 4). The Ni(0) which is required for the oxidative addition was generated in situ by reduction of the catalyst by DiBAL-H. After removal of the protective group, aldehyde 5 was isolated in 90 % yield. When performed in the absence of DiBAL-H, the yield dropped dramatically to 68 % [10–11].

Access to aldehyde 5, precursor of atazanavir.
Catalyst NiCl2(dppp), in combination with lithium triarylmagnesiate, instead of the classical organomagnesium reagents, allowed the Kumada-Corriu cross-coupling of a variety of aryl bromides and chlorides in good yields, and less than 10 % of the homo-coupling products were formed (Figure 5) [12].

Coupling of aryl halides with lithium triarylmagnesiates catalyzed by NiCl2 and diphosphine.
Indole-derived air stable diphosphine ligands such as L1 were also used successfully for the cross-coupling of a variety of aryl and heteroaryl chlorides with aryl Grignard reagents (Figure 6) [13].

Indole-derived air-stable diphosphine ligand L1 in Kumada-Corriu reaction.
Calix[4]-diphosphine ligands were synthesized and proved to be efficient ligands, providing that the reactions were performed at 100 °C in dioxane with a PhMgBr/ArX ratio of 2/1. Compared to results obtained with dppp, which is considered in the field of diphosphines as one of the most efficient ligand [1], nickel complexes prepared from L2 gave better results. The coupling was presumably facilitated by a temporary increase of the P-Ni-P angle and thus an increase of the steric pressure of the P-substituents on the two organic frameworks (Figure 7) [14].

Formation of bis-aryls using calixarene phosphine ligands.
Nevertheless, ligand L3, the monophosphine analog of L2, turned out to be more suitable and efficient at room temperature with aryl chlorides and four times more active than triphenylphosphine [15]. The orientation of the P–Ni bond towards the calixarene axis (and not outwards) seems to increase the ligand bulk and thus favored the formation of a mono-ligand Ni complex [16]. The mono-iminophosphorane analog L4 (R = o-anisyl) allowed cross-coupling of aryl bromides in dioxane at 100 °C with a very low catalyst loading of 0.001 mol %. Thus, in 1 h, bromoanisole reacted with PhMgBr to give 46 % of the expected coupling product. In comparison with the cavityfree iminophosphorane Ph3P=N(o-anisyl), the activity was ten fold higher, due to a more highly crowded metal environment in favor of a mono-ligated intermediate, more reactive than bis-ligated complexes and endo-located metal centers.
Among monophosphine ligands, tris(t-butyl)phosphane L5 was also particularly efficient for the cross-coupling of aryl chlorides with aryl Grignard at ambient temperature [17]. The sulfur analog (t-Bu)2P(S)H L6[18] and its oxygen analog (t-Bu)2P(O)H L8, which revealed to be the best ligand [19], were also used successfully at room temperature in THF, with the advantage of being air stable and particularly low cost and accessible (Figure 8).
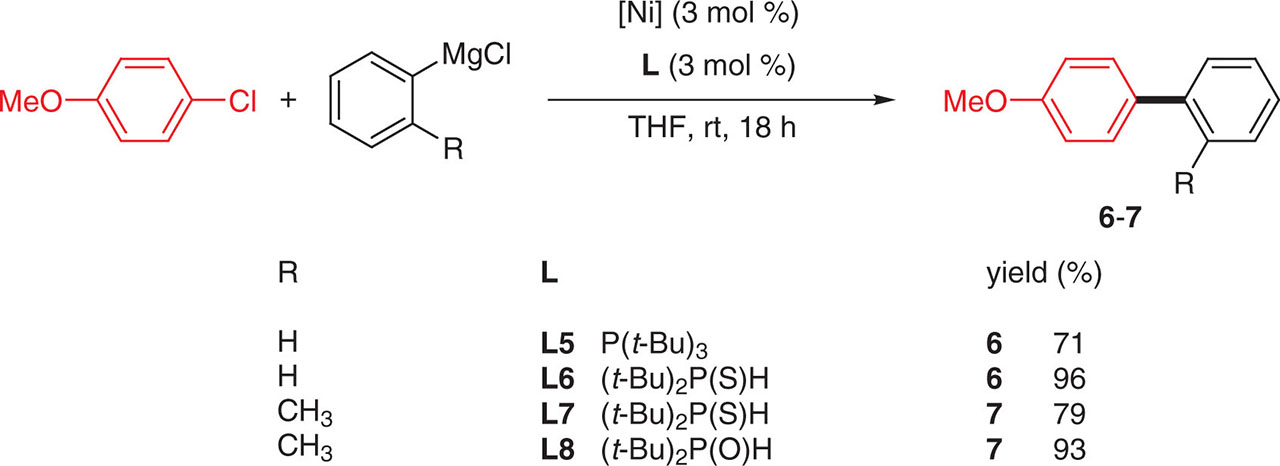
Monophosphine ligands and Kumada-Corriu coupling reaction.
Unactivated, deactivated and functionalized aryl chlorides reacted also with aryl Grignard reagents when monophosphine based catalysts C1a-b, in connection with additives such as LiCl or ZnCl2 were used (Figure 9). This significantly expanded the scope of Kumada cross-coupling reaction [20].

Influence of additives on the coupling of aryl halides with tolyl Grignard reagent.
Phosphine oxides turned out to be excellent ligands for the activation of C–Cl bonds. Thus, the particularly congested structure L9 showed one of the highest activity among the already described ligands [21]. Its sulfur analog, the diaminophosphine sulfide ligand L10, turned out to be less active and selective (Figure 10) [22].

Application of phosphine oxide L9 in the Kumada-Corriu reaction.
Binaphthyls play a major role as ligands or auxiliaries in numerous asymmetric reactions. To access to related derivatives, the asymmetric cross-coupling of two naphthyl derivatives has been successfully investigated by the group of Hayashi and Ito, in Japan. To significantly favor the formation of one of the two atropoisomers, chiral ferrocenylphosphine ligand L11 was used and gave excellent stereoselectivities (Figure 11) [23].

Stereoselective synthesis of binaphthyl derivatives via an asymmetric Kumada-Corriu cross-coupling.
Use of bidentate P,N Ligands
Bidentate diarylamido phosphine nickel chelates as C2 turned out to be involved in very active catalytic systems (Figure 12) [24].

Bidentate diarylamido phosphine C2 as ligand for the Kumada-Corriu reaction of aryl halides.
Very recently, the Sommer’s group [25] used the hybrid P,N ligand C3 for Kumada-nickel catalyst transfer polycondensation of sterically hindered thiophenes (Figure 13). They showed that such a system is highly suitable to control the polymerization of thiophene-based monomers with sterically very demanding side chain, much more efficient than the commercially available NiCl2(dppe) and NiCl2(dppp) catalysts.

Polycondensation of sterically hindered thiophenes.
Use of tridentate pincer ligands
Tridentate pincer ligands containing phosphorus were also considered (Figure 14). Thus, P,N,P ligands as C4, [26]C5, [27] or C6, P,C,N ligands as C7, C8[28] and P,C,P ligand such as C9[29] have been tested successfully in the aryl–aryl coupling, even with aryl chlorides.

Various P,N,P ligands, P,C,N and P,C,P ligands used for the aryl–aryl coupling.
As depicted in Figure 15, C6 and other catalysts such as C10, C11 or C12 are particularly relevant for the formation of biphenyl derivatives [30–32]. It has been assumed that (P,N,P) pincers in C6 and C11 are less labile than (P,N,N) ligands as in C10 and C12. This difference directly impacts the oxidative addition step and favors the C–C bond formation [33].

Comparison of (P,N,P) and (P,N,N) pincer ligands for the formation of biphenyl derivatives.
Other pincer ligands of the less widespread P,N,S and P,N,O types were examined in the Kumada-Corriu reaction [34]. If the P,N,O chelate nickel complexes showed high catalytic activity for the coupling of unactivated, or deactivated aryl-, heteroaryl- and vinyl chlorides with aryl Grignard reagents, the P,N,S analogs were far less active (Figure 16).

Comparison of (P,N,S) and (P,N,O) pincer ligands for the formation of biphenyl derivatives.
Use of NHC carbenes as ligands
N-Heterocyclic carbenes are strong σ-donor ligands especially effective when the oxidative addition of an aryl halide to the catalyst is the rate-determining step. The main interest of N-heterocyclic carbene ligands resides in the robustness of the associated catalysts in comparison to the phosphorus ones, avoiding decomposition or deactivation. The first described air-stable efficient carbene ligand L12 (Figure 17) [17] combined with Ni(acac)2, allowed the cross-coupling of aryl chlorides with aryl Grignard at room temperature, in THF with a catalyst loading of 3 mol %.

First efficient air-stable NHC ligand for the Kumada-Corriu reaction.
Nakamura et al. successfully used NHC carbenes such as L12 for the coupling of aryl halides with aryl Grignard reagents [35]. The formation of homocoupling byproducts was mainly suppressed by using a nickel halide salt (Figure 18).

Inhibition of the homocoupling process induced by L12 and nickel(II) fluoride.
Only one or two NHC ligands could be coordinated to the metal, depending on the steric hindrance on the carbene ring. With ligands L13 and L14 (Figure 19), the metal can be coordinated to two NHC subunits leading to a stabilized nickel complex which is efficient for the cross-coupling between an aryl Grignard reagent with an aryl halide [36–37].

Other NHC ligands used in Kumada-Corriu reaction.
Heterogeneous version of N-heterocyclic carbene precursors was proposed using copolymer-embedded nickel nanoparticles C15[38]. In this case, no additional coordination site was necessary, the nickel particles being stabilized by the structure of the polymer. After 10 recyclings of the catalyst, no loss of activity and selectivity was observed in the coupling. This catalyst is efficient for the coupling of aryl halides with aryl Grignard reagents (Figure 20).

Heterogeneous NHC ligand.
Other NHC-ligands containing additional coordinating functions which should provide a good catalyst efficiency have been successfully developed for the Kumada-Tamao-Corriu reaction involving aryl chlorides and aryl Grignard reagents. Thus, at room temperature in THF with 3 mol % catalyst loading, the ligand L15 with the phosphane-NHC bidentate ligand [39–40] turned out to be far more active than L12, and remained the best nickel-NHC catalyst to date for the coupling of 4-chloroanisole with phenyl magnesium chloride (Figure 21).

Cross-coupling catalyzed by a NHC-ligand containing an additional phosphane group.
Nickel catalyst possessing six-, seven- or eight-membered ring N-heterocyclic carbenes C16–18 (Figure 22) have also been tested [41]. At room temperature in THF, the best results were obtained in the coupling of aryl chlorides in the presence of C16 as catalyst.

Six-, seven- and eight-membered ring NHC ligands.
NHC-carbene ligand L16 combined with Ni(acac)2[42] or a diaza function [CNN]-pincer nickel complexes such as C19[43], C20[44], and C21[45], were synthesized and successfully involved in Kumada-Corriu coupling between phenyl chloride and tolyl Grignard reagent (Figure 23).

NHC ligands with additional coordinating sites used in the Kumada-Corriu reaction.
Bridged di-NHC complexes, C22a-c and C23, were also developed and tested for the synthesis of 6 as a model reaction [46–48]. The bridge length influences the formation of monochelate complex versus less reactive dichelate species. The 1,3-propanediyl-bridged dicarbene ligand C22c turned out to be the most active of the series for the coupling of aryl Grignard reagents with aryl halides (Figure 24).

Bridged di-NHC complexes.
Tridentate bis(carbene)-derived nickel(II)-pincer complexes were also developed. Thus, Inamoto et al. tested successfully catalyst C24 on a wide range of aryl halides as electrophiles [49]. The reaction was performed in THF, at room temperature, with a catalyst loading of 5 mol % (Figure 25).

Tridentate bis(carbene)-derived nickel (II)-pincer complexes.
Heterogeneous catalysis
Several heterogeneous systems have been developed for the Kumada-Corriu reaction using Salen ligands bound either on a Merrifield resin, such as C25[50–51] or aminomethylpyrrolidine framework attached on silica, such as C26[52] for the aryl–aryl coupling (Figure 26). Isolated yields of cross-coupling reaction were quiet good (around 70 % e.g. for the reaction of 4-bromoanisole and phenyl magnesium bromide). In addition, in both cases, the catalytic systems remained active even after 5 runs with almost no leaching (<1 %), which could be interesting for the development of green industrial processes.

Heterogeneous systems using N containing ligands for the Kumada-Corriu reaction.
As a source of nickel, Ni/C can be used for the Kumada-Corriu reaction of aryl chlorides and aryl Grignard reagents (Figure 27). The best conditions required refluxing THF, the presence of a phosphine (generally PPh3) to minimize the homocoupling side reaction. While this catalytic system allowed the scale-up, no recycling of the catalyst was reported [53–55].

Kumada-Corriu reaction of aryl chlorides and aryl Grignard reagents catalyzed by Ni/C.
2.1.2 With aryl fluorides
For a long time, fluorinated compounds have been considered as inert materials in cross-coupling reactions. Due to the design of new catalysts NHC-Ni(0) C27, the Kumada-Corriu reaction has been successfully achieved between fluoroarenes and aryl magnesium bromides. The reaction was efficient in a large number of cases even with electron-deficient substrates. Steric hindrance induced by substitution on the ortho position considerably reduced the yields of the couplings. Mechanistic studies revealed that the process occurred probably via a polar process, while reaction promoted by NiCl2 took place via a radical process [17]. Air stable phosphine oxide ligands such as L17[56] and P,N,N-pincer nickel complexes such as C28[57] have also been tested to be efficient even for the coupling of electron-enriched substrates which are usually more reluctant to cross-couplings (Figs. 28 and 29).

Kumada-Corriu reaction between fluoroarenes and aryl magnesium bromides.

Coupling between fluoroanisoale and aryl magnesium bromides.
In order to reach mono- or difluorinated compounds, the activation of di- and trifluoroarenes has been deeply investigated [58–60]. However, no significant selectivities were observed when reactions were realized in the presence of nickel salts. In contrast, palladium catalysis employing PdCl2(dppf) gave only the monoarylated adduct 14 in 91 % yield (Figure 30).

Selectivities of nickel-catalyzed Kumada-Corriu reactions starting from difluoroarenes.
2.1.3 With aryl thioethers
In 1979, Wenkert et al. demonstrated that arylthiols, sulfides underwent coupling with phenyl- or p-tolyl-magnesium bromides in the presence of 10 mol % of NiCl2(PPh3)2 catalyst (Figure 31) [61]. The process required higher temperatures and longer reaction times than with halides as the carbon-sulfide bond is stronger and therefore more reluctant to oxidative addition [62].

Nickel-catalyzed Kumada-Corriu reaction with arylthiol derivatives.
The scope was expanded to a great variety of heterocyclic aryl sulfides (Figure 32) [63].

Nickel-catalyzed Kumada-Corriu reaction with heterocyclic aryl sulfides.
The electron-rich NiCl2(PPh3)(IPr) catalyst C29 was successfully employed in the cross-coupling of bulky 2-methylthio-substituted benzofurans with aryl Grignard reagents, yielding highly fluorescent benzofurans such as 25 (Figure 33) [64].

Access to highly fluorescent benzofurans via the nickel-catalyzed Kumada-Corriu reaction.
Taking advantage of this coupling, a flexible access to eupomatenoids, a class of natural products has been designed [64]. A Kumada-Corriu coupling between 2-methylthiobenzofuran 26 and the Grignard reagent 27, prepared from 5-bromo-1,3-benzodioxole, led the expected compound 28 in high yield (Figure 34).

Access to eupomatenoids using the Kumada-Corriu reaction.
2.1.4 With aryl sulfones, sulfonates, sulfonamides
The use of aryl tert-butyl sulfones as electrophilic coupling partners was described by the group of Julia. The ability of the tert-butyl sulfonyl group to direct ortho-lithiation was later applied to the synthesis of ortho-substituted unsymmetrical biaryls (Figure 35) [65].

Aryl tert-butyl sulfones as electrophilic coupling partners.
Arenesulfonates can undergo nickel-catalyzed cross-coupling with aryl Grignard nucleophiles to give unsymmetrical biaryls. Park et al. studied the C–C bond formation from neopentyl arylsulfonates with arylmagnesium halides and showed that the NiCl2(dppf) catalyst inserts in the C(sp2)-S rather than in the C(sp3)-O bond (Figure 36) [66].

Arenesulfonate used for the synthesis of unsymmetrical biaryls.
Similarly, Snieckus et al. reported the coupling of a variety of arenesulfonamides with phenylmagnesium halides in refluxing toluene under classical nickel catalysis (Figure 37) [67].

Arenesulfonamides as partners for the synthesis of unsymmetrical biaryls.
2.1.5 With aryl ethers
Cross-couplings with oxygenated derivatives represent new perspectives in Kumada-Corriu reactions. Due to their large availability from natural biosources, they represent an interesting pool of starting materials. Their use has been recently reviewed [68] and will probably increase in the near future.
2.1.5.1 Aryl alkylethers and aryl silylethers
Aryl ethers constitute more attractive electrophiles than sulfonates as they are more readily available and more environmentally benign as they generate less waste. However, the C-OMe bond is more reluctant towards oxidative addition and, thus, less reactive in cross-coupling reactions due to the higher activation energy for its cleavage. In a pioneering work, Wenkert and co-workers described the cross-coupling of aryl methyl ethers with phenyl magnesium bromide in the presence of 10 mol % of NiCl2(PPh3)2 in refluxing benzene [69]. Two decades later, Dankwardt found that nickel complexes with electron-rich phosphine ligands possessing a larger cone angle (e.g., PCy3 or PhPCy2) were efficient at 60 °C for the arylation of diversely substituted methoxyarenes (Figure 38) [70].

Fonctionalized arylethers as efficient electrophiles for the Kumada-Corriu reaction.
More recently, several contributions reported the design of new ligands to improve the efficiency and the selectivity of nickel-catalyzed Kumada-Corriu coupling reactions of aryl ethers. In 2011, Wang and Xie reported that electron-rich and sterically hindered pyrazolyl amino phosphines C1b (cf. Figure 9) allowed for the coupling of π-extended aromatic and alkenyl ethers under mild conditions (THF, room temperature or 50 °C) [71]. The more challenging aryl methyl ethers still required high temperatures to react (toluene, 120 °C). In 2012, and for the first time, Nicasio et al. reported the use of N-heterocyclic carbene ligands as an alternative to phosphine ligands in the nickel-catalyzed Kumada-Tamao-Corriu coupling of π-extended and regular aryl methyl ethers under relatively mild conditions (THF, 60 °C) [72]. The scope was again limited to non-functionalized coupling partners due to the high reactivity of Grignard reagents (Figure 39).

NHC ligand for the coupling of arylethers.
An initial study on the nickel-catalyzed alkenyl C-OSi bond activation for C-C bond formation with aryl and alkyl Grignard reagents was reported in 1980 by Kumada et al. [73]. Although a few aromatic silyl ethers were mentioned to react under the conditions optimized for the Kumada-Corriu coupling of aromatic alkyl ethers, no systematic study had been carried out on these substrates until 2011. Different silyloxy groups were tested and led to the cross-coupling products in similar yields compared to the OTMS derivatives whatever the steric hindrance of the silyl protecting group. It is worth to note that the C-OSi bond can be activated in the presence of a methoxy group (Figure 40) [74].

C–C bond formation using silyloxyaryl derivatives.
2.1.5.2 Aryl and alkenyl carbamates and phosphates
In 1992, the group of Snieckus showed that aryl carbamates and triflates could be coupled to aryl Grignard in similar conditions [75].

Carbamates as electrophiles in nickel-catalyzed Kumada-Corriu reaction.
Compared to other O-based electrophiles, the carbamate moiety offers the ability to effect directed ortho-metallation, thus providing a potential entry to complex poly-substituted aromatics. The use of hydroxyphosphine ligand L18 proved efficient in the rate acceleration of the nickel-catalyzed coupling of aryl carbamates (Figure 42) [76].

Hydroxyphosphine L18 as an efficient ligand for the nickel-catalyzed coupling of aryl carbamates.
The intermediate nickel phosphine/magnesium alkoxide bimetallic catalytic species induces the C–O bond cleavage by a synergetic push-pull action. These conditions were also effective with aryl phosphates, which were initially considered in 1981 by Kumada et al. using Ni(acac)2 as the catalyst [77–78].
2.1.5.3 Diaryl sulfates and aryl sulfamates
In the search of environmentally benign cross-coupling transformations, diaryl sulfates were found by Shi et al. to be efficient green organic electrophiles [79]. Both of the aryl groups can be inserted into the product, while the process generates harmless inorganic salts (e.g. MgSO4 and MgX2) as by-products. Interestingly, the reaction conditions are mild enough to leave C-F and C-OMe bonds unreactive. The proposed mechanism involves a monosulfate magnesium salt which proved to be a reactive intermediate in the catalytic cycle (Figure 43). In 2005, Snieckus et al. described the use of more electron-rich IMesNHC ligands for the nickel-catalyzed cross-coupling of O-sulfamates electrophiles in moderate to good yields [80].

Diaryl sulfates as efficient green organic electrophiles for the Kumada-Corriu reaction.
2.1.5.4 Magnesium aryloates
The direct transformation of readily available phenol derivatives would represent a major breakthrough in nickel-catalyzed cross-coupling reactions as the use of free alcohols would be step- and atom-economic as well as environmentally benign. The process would indeed generate less organic waste and would avoid additional transformations of the alcohol to good leaving groups (e.g., phosphonates or sulfonates) or the use of toxic organic halides as electrophiles. The first achievement towards this goal was reported by Shi et al. using magnesium naphtholates [81]. The NiF2/PCy3-catalyzed cross-coupling of aryl Grignard reagents with different naphthols was performed in non-polar solvents after in situ-preformation of the corresponding magnesium salts with methylmagnesium bromide. The process was believed to proceed through the Ni(0)/Ni(II) catalytic cycle and the C-O naphthoxide bond cleavage was supposedly activated by Mg2+ in the oxidative addition step to the Ni(0) species. Unfortunately, the parent phenols could not be converted to biphenyls under these conditions (Figure 44).

Cross-coupling of aryl Grignard reagents with functionalized 2-naphthols.
2.2 With vinyl derivatives
2.2.1 Vinyl halides
Vinyl chloride is one of the most reactive C(sp2)-halides and has been widely used in the industry for the synthesis of styrene derivatives in multi-tons scale. Chemists at Hokko Chemical industry in Japan have used inexpensive NiCl2(dppp) as catalyst. For security reasons, they have replaced highly flammable ether by a mixture of benzene/ THF or toluene/THF (Figure 45) [82].
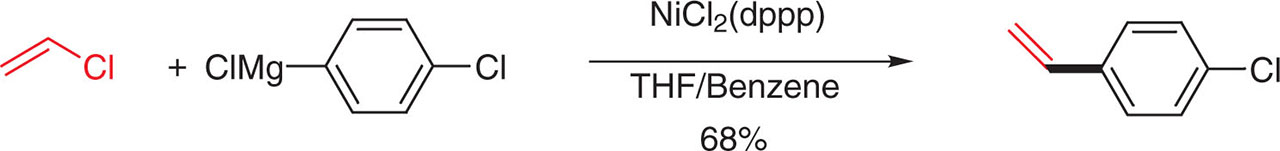
Cross-coupling of vinyl chloride with aryl Grignard reagents.
Dppp can be replaced by 4,6-bis(3-diisopropylphosphinophenyl) dibenzofuran L19, a related disphosphine ligand, which coordinate to Ni(I) to produce the [(iPrDPDB-Fphos)NiCl] complex and allowed the synthesis of styrene from vinyl chloride and phenyl Grignard reagent in nearly 85 % isolated yield (Figure 46) [83].

Diphosphine ligand L19 designed for the synthesis of styrenes from vinyl chloride.
Tetradentate nickel catalyst C31 was also particularly efficient for the coupling of unactivated vinyl chlorides (Figure 47) [84].

Tetradentate nickel complex C31 for the coupling of unactivated vinyl chlorides.
In 2014, Pugh et al. used the same NiCl2(dppp)system for the synthesis of a polymerizable 1-ethoxyvinylbenzocyclobutenes. These latter could undergo radical polymerization at 100–150 °C leading to cross-linked polymers (Figure 48) [85].

Access to polymerizable cyclobutene from vinylbromide.
2.2.2 With vinyl thioethers
Following the early results obtained by Wenkert et al., Takei et al. reported the reaction between alkenyl and aryl sulfides in nickel-catalyzed cross-coupling reactions with phenyl Grignard reagents [86]. The olefinic products were obtained with retention of configuration suggesting a high stereospecificity for each step of the process (Figure 49).

Stereoselective cross-coupling of alkenyl sulfides.
2.2.3 With cyclic enol ethers
The group of Martin found that a combination of Ni(cod)2 with L20. HCl, a NHC precursor, and LiCl as the additive allowed the cross-coupling of cyclic vinyl ethers when performed at low temperatures [87]. Under these conditions, the use of functionalized Grignard reagents furnished the corresponding homoallylic alcohols with complete Z-stereoselectivity (Figure 50).

Stereoselective access to homoallylic alcohols from dihydrofuran.
2.3 With alkynyl halides
1-Bromoalkynes have also been used as electrophiles to achieve Ni-catalyzed cross-coupling reactions with aryl magnesium bromides [88]. Among the different catalysts tested, the combination of NiCl2 with a triarylphosphine appeared as the most promising system to obtain high yields in the coupling product (Figure 51).

1,2-Disubstituted acetylenes from bromo(ethynyl)benzene.
2.4 Miscellaneous
2.4.1 With propargyl bromide
The coupling of aryl Grignard with propargyl bromide led to an arylallene derivative 29 contaminated by small amounts of the corresponding propargyl arene 30 (Figure 52) [89].

Allene derivatives from propargyl bromide.
2.4.2 With α-bromoketones
α-Bromopropiophenones can be easily substituted by an aryl group when treated with an aryl Grignard and a small amount of a nickel catalyst. This reaction has been extensively studied in the presence of chiral ligands. Of interest, the reaction is stereoconvergent: both enantiomers of the starting material led to the final chiral product with the same configuration and a similar level of induction. The best results were obtained when dimethyl bis-oxazoline ligand L21, derived from arylglycine, was used. To reach similar ee with dialkylketones, the process needed however to be conducted in the presence of the less common and more hindered ligand L22 and with this ligand, the ee values culminate to the range of 85–90 % (Figure 53) [90].

Enantioselective formation of a-arylketones.
We have to point out that, recently, the group of Walsh has shown that the Kumada-Corriu reaction gave even better results when performed in the presence of cobalt salts associated to the same bis-oxazolidine ligands [91].
2.4.3 Three component reaction
To extend the scope of the Kumada-Corriu coupling reaction, nickel-catalyzed three component process has been designed [92]. The reaction was carried out in the presence of the two classical partners, an alkyl halide and a phenyl Grignard reagent, and also in the presence of 2,3-dialkyl-1,3-butadienes. Interestingly, functionalizations occurred selectively on the two opposite terminal positions of the diene and delivered polysubstituted alkenes in reasonable yields but with a poor E/Z ratio (Figure 54). It was assumed that alkyl radical species were generated by a Single Electron Transfer (SET) process from a nickelate complex intermediate.

Cross-coupling of alkyl halides, 1,3-butadienes and aryl Grignard reagents.
A three-component reaction leading to tetrasubstituted alkenes has been devised by Zhao, Hayashi et al. [93]. By adding a very small amount of a nickel salt to a mixture of an internal alkyl(phenyl)acetylene, an aryl Grignard reagent and an aryl iodide, an arylmagnesiation occured rapidly, followed by an efficient Kumada-Corriu coupling. The Z-selectivity observed for the first step was essential for the stereoselective synthesis of pharmaceutical compounds such as tamoxifen 33 (Figure 55).

Tetrasubstituted alkenes by a three-component reaction.
3 Coupling reactions of alkenyl Grignard reagents
3.1 With aryl derivatives
3.1.1 With aryl halides
In contrast to the Kumada-Tamao-Corriu reaction with aryl Grignard reagents, the nickel-catalyzed coupling of alkenyl magnesium reagents received much less attention since a first report in 1975 by the group of Kumada [94]. The cross-coupling of alkenyl Grignard reagents with aryl or vinyl halides was found to occur with cis/trans isomerization of the olefinic bond depending on the reactivity of the halide, the bulkyness of the Grignard reagent and the ArX/R’MgX ratio (Figure 56). The vinyl Grignard reagents appeared far less reactive than their aryl counterparts [95].

Unselective access to styrene derivatives from vinyl Grignard reagents.
The group of Bach studied the functionalization of dibromobenzofurans 35 and observed a selective alkenylation at the C-5 position in high yields under Kumada-Corriu conditions. A further Negishi coupling reaction led to different eupomatenoids 37 in high yields (Figure 57) [96].

Selective alkenylation of dibromobenzofurans with vinyl Grignard reagents.
3.1.2 With aryl sulfides
The group of Wenkert reported the selective cross-coupling of methyl aryl sulfides over iso-propyl aryl sulfides with alkenyl Grignard reagents, albeit in moderate yields [97]. The coupling conditions using NiCl2(PPh3)2 as catalyst also led to the reduction product of the thiomethyl substituent (Figure 58).

Regioselective coupling of aryl sulfides with vinyl Grignard reagents.
3.1.3 With aryl carbamates
The coupling of a phenanthrenyl (N,N-diethyl)carbamate was performed with vinyl magnesium bromide and Ni(acac)2 as the catalyst to deliver a styrene derivative however, in moderate yield [75].

Coupling of arylcarbamates with vinyl magnesium bromide.
3.2 With vinyl derivatives
3.2.1 With vinyl sulfones
Conjugated dienes can be obtained with complete retention of configuration by the Ni-catalyzed coupling of vinyl sulfones with vinylmagnesium bromide (Figure 60) [98]. The outcome of the reaction is highly dependent on the nickel catalyst (40 % yield with Ni(acac)2 and 23 % yield with NiCl2/2PPh3) leading, in all cases, to the formation of the reduction product along with the coupling product.

Synthesis of 1,3-dienes from vinylsulfones and vinyl Grignard reagent.
3.2.2 With vinyl carbamates
The coupling of (Z)-vinyl (N,N-diisopropyl)carbamate 38 with different vinyl magnesium bromides in the presence of 10 mol % of Ni(acac)2 led to (Z)-alkenyl derivatives with high stereoselectivity (Figure 61) [99].

Coupling of (Z)-vinyl carbamates with vinyl magnesium bromides.
3.3 With alkyl derivatives
The coupling of vinyl magnesium chloride with alkyl fluorides in the presence of a nickel catalyst gave rise to an alkylative dimerization of the vinyl Grignard reagent to form a 2-alkyl-3-butenyl Grignard intermediate which is hydrolyzed to the corresponding butene (Figure 62) [100]. The nickelate complex C32 was suggested by Kambe et al. to be a reactive intermediate in the different plausible reaction pathways.

Coupling of alkylfluorides with vinyl Grignard reagent.
3.4 Miscellaneous
3.4.1 Three component reactions
A novel nickel-catalyzed coupling reaction between alkenyl magnesium reagents and methylenecyclopropanes in the presence of a chlorosilane, as the terminal electrophile, was developed by Kambe et al. [101]. The corresponding carbomagnesation products were obtained by the selective cleavage of the distal C–C bond of methylenecyclopropanes through nickel oxidative addition (Figure 63).

Selective cleavage of methylenecyclopropanes with vinyl Grignard reagents.
3.4.2 Ring-opening of cyclic ethers
Kambe et al. have explored the nickel-catalyzed coupling of vinyl magnesium chloride with 3- or 4-membered cyclic ethers [102]. As for alkyl fluorides, the vinyl Grignard reagent underwent a dimerization prior to the coupling with the electrophile giving access to pentenol or hexenol derivatives 40 (Figure 62). The postulated mechanism involves the nickelate complex C47 which can either react directly with cyclic ethers to form the coupling product 40 or undergo a transmetallation step to yield (2-butene1,4-diyl)magnesium 39, followed by electrophilic trapping with a cyclic ether. Interestingly, no reaction took place when using substituted vinyl Grignard reagents (Figure 64). Chlorosilanes as electrophilic partners led to 1,4-disilyl-2-butenes under the same reaction conditions.
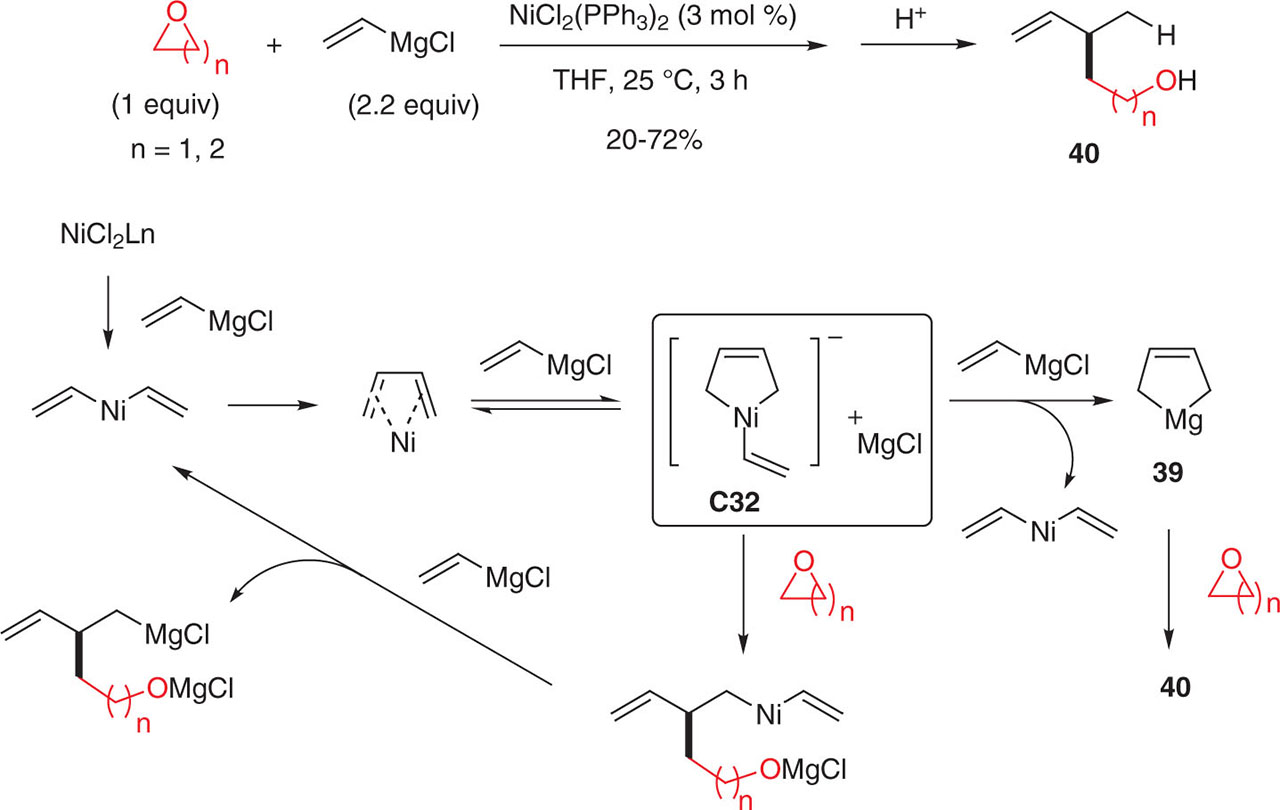
Catalyzed dimerization of vinyl Grignard with 3- or 4-membered cyclic ethers.
4 Coupling reactions of Alkynyl Grignard Reagents
4.1 With alkyl halides
The functionalization of terminal alkynes is usually achieved by well-established and high yielding Sonogashira reaction conditions. In order to avoid the use of expensive palladium salts, the development of reactions between alkynyl Grignard reagents and primary alkyl halides are still challenging, especially due to the lower nucleophilic character of metal alkynylides and their propensity to dimerize. Very recently, successful processes have been published (Figure 65). For example coupling between acetylenic Grignard reagents and primary alkyl iodides or bromides have been reported in the presence of Nickamine C33 and diamine O-TMEDA as the additive [103]. From a mechanistic point a view, this additive plays an important role: it could coordinate to the magnesium salt favouring the transmetallation and the formation of an alkynyl complex with Nickamine, but it could also minimize the homocoupling side-reaction. Moreover, radical intermediates are probably involved as suggested by the ring-opening observed with cyclopropylmethyl bromide used in this reaction as the cross-coupling partner.

Disubstituted alkynes from alkynyl Grignard reagents using Nickamine complex C33.
Interestingly, the reaction is compatible with sulfur-containing groups. Attempts to perform the same process with secondary halides are however unsuccessful.
4.2 With alkynyl sulfones
Diyne derivatives have also been prepared at room temperature by condensation of readily available alkynyl Grignard reagents onto alkynyl sulfones (Figure 66). The process is an extension of the Ni-catalyzed cross-coupling of aryl sulfones with Grignard reagents previously discovered by Julia et al. [65], in which the catalyst Ni(acac)2 was reduced in situ to Ni(0) by the acetylide present in excess. The yields are in the range from 42 % to 83 %. The best results were observed for the formation of bisaryldiynes (R = Ar) [104].

1,3-Diyne derivatives from alkynyl sulfones.
4.3 With ethers
4.3.1 Aryl ethers
The functionalization of terminal alkynes has also been extended to anisole derivatives [105]. In the presence of Ni(cod)2 and a NHC ligand bearing bulky groups, the reaction of triisopropylsilylacetylide with electron-enriched anisoles delivered the cross-coupling adducts in high yields (Figure 67). The presence of sterically hindered substituents on the acetylide has also a major impact on the success of the process (Figure 68). Under these conditions, a selective cleavage of the C-F bond of fluoroanisole was observed and this difference of reactivity could be promising for multi- and sequential cross-coupling reactions.

Influence of the NHC ligand on the coupling of TIPS-alkynyl-MgBr with p-tert-butylanisole.

Influence of the alkynyl nucleophile on the NHC-Ni-catalyzed coupling of p-tert-butylanisole.
This coupling reaction found application for the ethynylation of a large number of methoxyaryl derivatives and tolerate numerous functionalities (alcohol, phenol, amine, etc.) without significant decrease of the yields.
4.3.2 Enol derivatives
Enol ethers
Silyl enol ether 42 underwent a smooth coupling with an alkynyl Grignard reagent leading to enyne 43, while dihydrofuran 44 underwent a ring-opening leading to compound 45 (Figure 69) [105].

1,3-Enynes by Ni-catalyzed alkynylation of TMS enol ethers and dihydrofurans.
Vinyl carbamates
We have to point out that (Z)-vinylcarbamates 46 were also implicated in reaction with alkynyl Grignard reagents to deliver enynes 47 which could find application in the total synthesis of complex molecules such as avermectins [106]. The Z/E ratio was affected by both the nature of the catalyst but also by the substitution of the oxygen atom present at the homoallylic position (Figure 70).

(Z)-Enynes from (Z)-vinyl carbamates and hexynyl magnesium bromide.
5 Coupling reactions of Alkyl Grignard Reagents
5.1 With aryl derivatives
5.1.1 Aryl halides
As an example, the Kumada-Corriu reaction between alkylmagnesium reagents and aryl halides has been advantageously applied to the synthesis of (S)-macrostomine 48 by Comins et al. in only five steps (Figure 71). After optimization of the reaction conditions, three equivalents of a benzyl Grignard and a large amount of nickel catalyst were required to achieve the final step in 63 % yield [107].

Key Kumada cross-coupling of a benzyl Grignard fragment in the synthesis of (S)-macrostomine.
Similarly, as reported for aryl Grignard reagents in Section 2.2, the coupling between aryl fluorides and alkyl magnesium reagents can be also efficiently achieved. As depicted in Figure 72, the reaction can be performed in the presence of catalyst C34, with alkyl Grignard without observation of β-hydride elimination. It is anticipated that magnesium phenolate could be the key element for the success of the reaction. Moreover, cleavage of C-F bond, under these conditions, was observed in the presence of weaker carbon-halogen bonds [108].

Selective coupling of aryl fluorides with alkyl-MgX using catalyst C34.
5.1.2 Aryl sulfides
As already pointed out with aryl Grignard, efficient coupling reactions were noticed between methyl Grignard and aryl thiols, thioethers, sulfoxides or sulfones when performed in the presence of 10 mol % of NiCl2(PPh3)2 (Figure 73) [61].

Coupling of methyl magnesium bromide with different thioaryl derivatives.
In the presence of this additive, the reductive cleavage of C(sp2)-sulfide bonds by secondary alkyl Grignard reagents has been reported. Fortunately, the use of bidentate phosphine ligands allowed a higher selectivity for the cross-coupling product [109]. For example, the synthesis of 4-cyclohexyl-2,6-diphenylpyridine via the coupling of cyclohexylmagnesium bromide and the corresponding 4-thiomethyl derivative occurred when NiCl2(dppp) was present in the reaction media (Figure 74) [63].

Coupling of aryl thioethers with primary and secondary alkyl magnesium reagents.
A combination of Ni(acac)2 and (Z)-1,2-diphosphinoalkene L23 was also described for the cross-coupling of arylsulfides with primary and secondary alkyl Grignard reagents (Figure 75) [110]. Nevertheless, the coupling reactions with iPrMgX led to partial isomerization of the alkyl nucleophile.

Coupling of aryl thioethers with Ni(acac)2 and (Z)-1,2-diphosphinoalkene L23 as an additive.
More recently, the NiCl2/butadiene catalytic system developped by Kambe et al. proved efficient in the coupling of 2-thiomethyl-substituted 1,3-benzothiazoles with secondary alkyl Grignard reagents [111]. Under these conditions, the formation of isomerized products and the possible reduction of thioethers are minimized. However, non π-extended systems such as thiazoles and thiazolines are not reactive. In the case of arylsulfonates, alkylmagnesium reagents bearing no β-hydrogen reacted efficiently at room temperature, when the more electron-rich NHC ligand IPr.HBF4 was used as the ligand (Figure 76) [112].

Coupling of aryl sulfonates with alkyl Grignard reagents using the electron-rich NHC ligand.
5.1.3 Aryl methyl ethers
In 2008, Shi et al. reported the methylation of different alkoxy naphthalenes including alkyl-, aryl- and silyl ethers by using 5 mol % of NiCl2(PCy3)2 in toluene [113]. Comparative experiments showed that π-extended aryl methyl ethers reacted faster than phenyl methyl ethers (Figure 77).

Ni-catalyzed methylation of aryl or naphthyl methyl ethers with MeMgBr.
5.1.4 Aryl phosphonates
Functionalization of 49 by using a similar coupling allowed the introduction of a prenylated chain onto 1,5-naphthalenediol derivatives (Figure 78) [114]. The resulting compound was transformed in few steps into (+)-cryptotanshinone 51.

Selective coupling of aryl phosphate with an alkyl Grignard in the synthesis of 51.
5.2 With alkenyl derivatives
5.2.1 Alkenyl halides
The access to chiral 2-aryl propanoic acids is of great interest due to their pharmaceutical properties. In this context, the synthesis of precursors such as 3-aryl-1-butenes, has been intensively studied. Enantioselective Kumada-Tamao-Corriu reaction between benzyl Grignard reagents and vinyl bromide has been carried out in the presence of various chiral phosphine ligands [115–119]. Enantiomeric excesses up to 88 % were recorded with the readily available ligands L27 and L28 (Figure 79) [118–119]. However, the process cannot actually compete in terms of selectivities with other procedures to access α-aryl propionic acids as hydrogenation or hydroformylation of parent styrenes are extremely efficient.

Enantioselective coupling of secondary benzyl Grignard reagents with vinyl bromide.
The coupling of gem-difluoroalkenes has also been considered (Figure 80). The Nicatalyzed reaction was attractive with alkyl Grignard reagents compared to the process performed in the presence of palladium salts for which lower yields were recorded, probably due to side processes such as β-elimination [120].

Trisubstituted alkenes from gem-difluoroalkenes and ethyl magnesium chloride.
5.2.2 Enol carbamates and phosphates
The reaction of enol carbamates 54 with alkyl Grignard reagents initially reported by Kocienski et al. occurred with retention of configuration (Figure 81) [121].

Coupling of (Z)-enol carbamates and alkyl Grignard nucleophiles.
Kumada-Corriu reactions involving the cleavage of a C–O bond has been advantageously achieved to prepare different natural products. For example, phosphate enol ether 56 was easily generated from the corresponding bicyclic ketone. When this phosphate enol ether was involved in a nickel-catalyzed coupling with methyl Grignard reagent, it was transformed to the corresponding alkene in 73 % yield and was further transformed into the marine diterpene fuscol (Figure 82) [122].

Synthesis of fuscol involving the coupling of intermediate enol phosphate 56.
A key intermediate in the total synthesis of ∆1-trans-tetrahydrocannabinol 58 was also prepared in excellent yield by a 1,4-addition/regioselective phosphorylation/ Kumada-Corriu coupling sequence (Figure 83) [123].

Methylation of enol phosphates 56 in the total synthesis of 58.
5.3 With alkyl derivatives
5.3.1 Alkyl halides
The major challenge in the alkyl–alkyl Kumada cross-coupling reactions is to suppress the β-hydride elimination step that may occur from the intermediate alkyl– nickel complex, leading to olefins as the side products. Therefore, the reductive elimination step has to occur rapidly to avoid this competing process. The first Ni-catalyzed alkyl–alkyl cross-coupling between primary alkyl halides (Br, Cl) and tosylates and primary or secondary Grignard reagents was reported by Kambe et al. in 2002 using NiCl2/1,3-butadiene as a unique catalytic system [4, 124]. Interestingly, the Csp3-Br bond reacted faster than the Csp2-Br bond yielding p-bromohexylbenzene in quantitative GC yield even if Csp3-X bonds are more electron-rich than Csp2-X bonds and thus more reluctant to oxidative addition. Moreover, (bromomethyl)cyclopropane led to the corresponding alkylcyclopropane in 87 % yield which seems to preclude the formation of an alkyl radical as intermediate (Figure 84).

Alkyl–alkyl coupling in the presence of Ni(acac) and 1,3-butadiene.
Based on these results, a non-radical mechanism involving Ni(II)/Ni(IV) species in the catalytic cycle was proposed by Kambe et al. (Figure 85). The 1,3-butadiene additive converts Ni(0) to a bis-π-allyl nickel(II) complex E which reacts first with R’-MgX to form an anionic complex F with enhanced nucleophilicity as a key intermediate. The oxidative addition of alkyl halides to complex F then yields Ni(IV) complex G which undergoes reductive elimination to the coupled product, regenerating the bis-π-allyl nickel complex E. The activation of the alkyl halide may also be due to a strong interaction with the magnesium salt.

Postulated mechanism for the alkyl–alkyl coupling using Ni/1,3-butadiene system.
This catalytic system also proved to be efficient for the coupling of primary alkyl fluorides albeit in lower yields due to the formation of elimination products [125]. In this case, CuCl2 was found to be superior to NiCl2 for the coupling. It was later shown that the combination of Ni(acac)2 (0.6 mol %) and a 1,3,8,10-tetraene as an additive improved the efficiency of the coupling of alkyl fluorides [126]. Functionalized alkyl halides and tosylates could also be coupled by using these conditions [127].
The use of 3 mol % of a pincer (NN2)-Ni catalyst C33 synthesized from an amidobis(amine) tridentate ligand was described by Hu et al. for the coupling of functionalized primary and secondary alkyl iodides and bromides with alkylmagnesium nucleophiles [128–129]. At low temperature (–35 °C) and, using dimethylacetamide as solvent, a slow addition of an alkyl Grignard reagent (1 equiv) on a large range of unactivated alkyl halides yielded the coupled product in moderate to high yields (Figure 86). Notably, alkyl triflates and alkyl tosylates were inert. A structure-activity study demonstrated that bidentate nickel catalysts C35 and C36 gave better yields for the coupling of secondary alkyl halides, which was attributed to a higher structural flexibility around the nickel center [130].

Alkyl–alkyl Kumada-Corriu coupling by using pincer C33-nickel complex.
Under these catalytic conditions, a diastereoselective alkyl–alkyl Kumada coupling was also reported by Hu et al. for the formation of the conformationally most stable 1,4-trans-dialkyl cyclohexanes and 1,3-cis-dialkyl cyclohexanes and tetrahydropyrans, whatever the diastereomeric ratio of the corresponding cyclic halide substrates [131]. The alkyl–alkyl cross-coupling was believed to occur via the transmetallation of C33 by RMgX in a first step to form a Ni–alkyl intermediate, followed by oxidative addition of the alkyl halide and reductive elimination step to produce the coupled product. An extensive mechanistic investigation revealed that the key intermediate for the activation of the alkyl halide was a bimetallic [(N2N)Ni-R](RMgCl) complex and that the oxidative addition step involved radical alkyl intermediates [132].
More recently, the group of Cardenas reported the use of tetramethylethylenediamine (TMEDA) as additive in the Ni(acac)2-catalyzed coupling of non-activated primary and secondary iodides with alkylmagnesium reagents [133]. On the basis of experimental and computational studies evidencing the intermediacy of an alkyl radical from the alkyl iodide, a cascade cyclization-coupling was applied to a variety of iodoalkene substrates for the sequential formation of two alkyl–alkyl bonds (Figure 87).

Ni-catalyzed sequential radical cyclization Kumada-Tamao-Corriu coupling of iodoalkenes.
The catalytic system was also efficient in the cross-coupling of functionalized benzyl chlorides with alkyl Grignard reagents [134].
5.3.2 Sulfones
Li and co-workers reported the Kumada coupling of activated alkyl sulfones with Grignard reagents with the NiI2(PPh3)2/PCy3 system (Figure 88) [135]. A range of aryl ketones were synthesized in good yields (61–77 %) from 1-aryl-2-tosylethanones without affecting the carbonyl group. Interestingly, the outcome of the coupling of 1-(1-tosylethyl)arenes was completely reversed by changing the solvent: the coupled product 62 was obtained in toluene or cyclohexane whereas the use of THF yielded the methylstyrene derivatives 64.

Ni(II)-catalyzed reactions of ketosulfones and sulfones with methyl Grignard reagent.
5.3.3 Alkyl ethers
In 2008, Shi et al. reported the first C(sp3)-O(ether) activation in the coupling of benzyl alkyl ethers with alkyl Grignard reagents [136]. Primary and secondary benzyl ethers reacted in the presence of NiCl2(dppf)2 as a catalyst to give similar yields in the cross-coupling products (Figure 89). The use of iPrMgX and n-BuMgBr led to lower yields in the coupled products, probably due to a competitive α-β-H elimination pathway.

Methylation of benzyl methyl ether using NiCl2(dppf)2 as catalyst.
Cross-coupling of non racemic bis-aryl ethers 65 with methyl Grignard reagent has been applied to prepare enantio-enriched diarylethanes 66, precursors of anti-insomnia drugs (Figure 90) [137–138] It is worth noting that the transfer of chirality is sizeable.

Diastereoselective Ni-catalyzed methylation of chiral bis-aryl ether 65.
A similar coupling has been also realized starting from both racemic chiral cyclic ethers 67 and 69. Introduction of a methyl group occured in a stereospecific manner with a complete inversion of configuration at the electrophilic center (Figure 91). It is noteworthy that the same authors reported the ring-opening of the corresponding lactones with organozinc reagents (Negishi coupling) which also occured nicely and with very high stereoselectivities [139]. The reactions were conducted in the presence of DPEphos L29 and of (R)- or (S)-BINAP, L30. No match/mismatch effects were observed in the later case demonstrating that the chirality of the ligand has no influence on the stereoselectivity of the reaction.
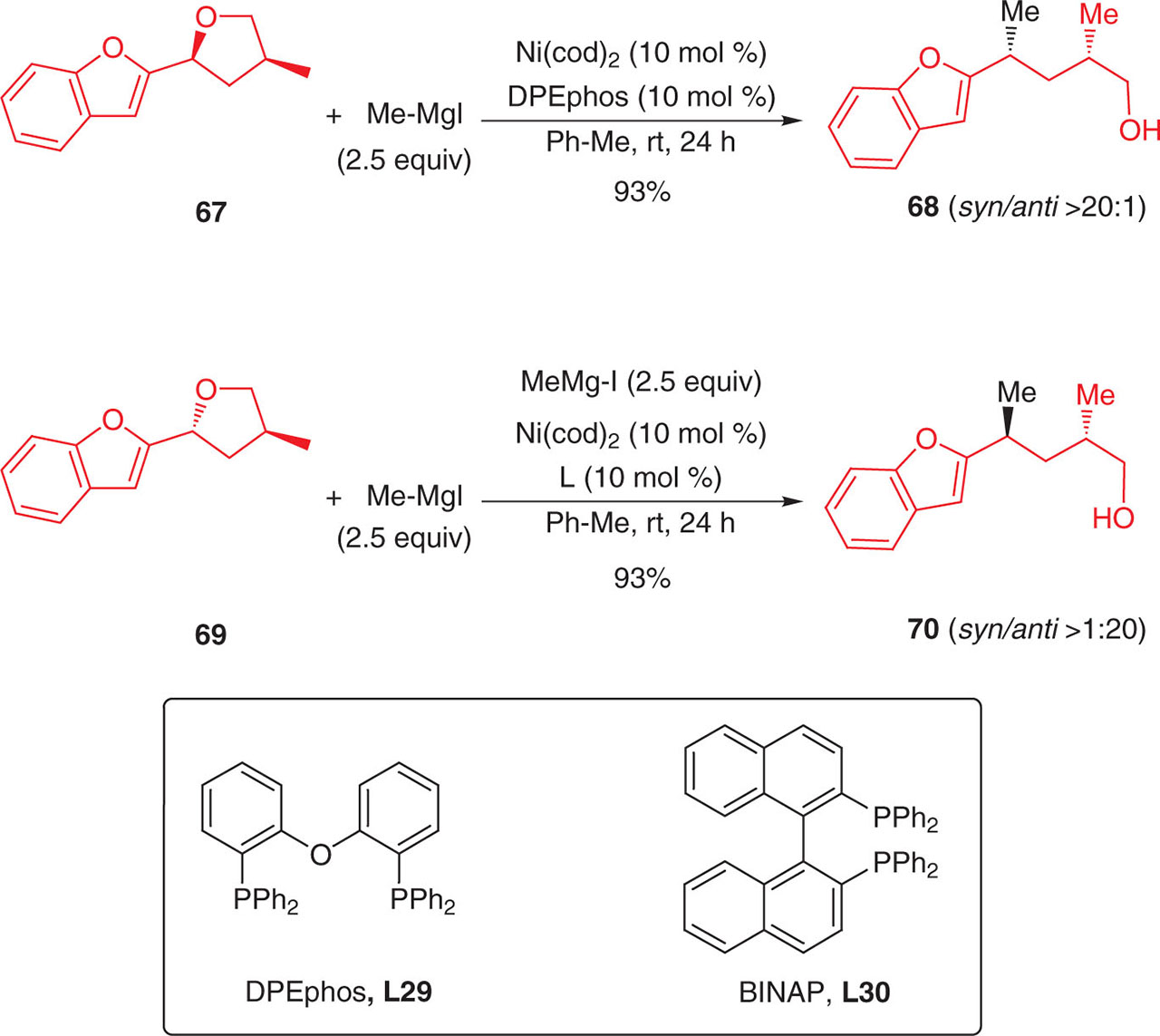
Diastereoselective ring-opening of chiral cyclic benzyl ethers by Kumada-Corriu reaction.
5.3.4 Benzyl alcohols
The cross-coupling of benzylic C(sp3)-OH bond was also successfully achieved with methyl Grignard reagent (Figure 92) [140]. Starting from naphthylmethanol, 2-ethylnaphthalene was obtained in 68 % yield by using bidentate 1, 2-bis(dicylohexylphosphino)ethane (DCyPE) L31 as ligand. Unfortunately, other alkyl Grignard reagents led mainly to reductive products.

Methylation of benzyl alcohols with Ni(acac)2 and bidentate phosphine L31.
6 Conclusion
In parallel to the well-established reactivity of Grignard reagents towards electrophiles, a complementary set of reactions has been designed when these organomagnesium derivatives are associated with appropriate nickel catalysts. Due to the lower cost of nickel salts, the coupling reactions depicted in this Chapter seems of particular interest when compared to procedures using palladium salts. The major drawbacks are still the use of substoechiometric amounts of Grignard reagents and the β-hydride elimination frequently observed with alkyl derivatives.
Acknowledgment
This article is also available in: Cossy, Grignard Reagents and Transition Metal Catalysts. De Gruyter (2016), isbn 978–3–11–035266–5.
References
[1] Tamao K, Sumitami M, Kumada M, J Am Chem Soc 1972, 94, 4374–76.10.1021/ja00767a075Search in Google Scholar
[2] Corriu RJP, Masse J, J Chem Soc, Chem Commun 1972, 144.10.1039/c3972000144aSearch in Google Scholar
[3] Hölzer B, Hoffmann RW, Chem Commun 2003, 732–3.10.1039/b300033hSearch in Google Scholar
[4] Terao J, Kambe N, Acc Chem Res 2008, 41, 1545–54.10.1021/ar800138aSearch in Google Scholar
[5] Tamao K, J Organomet Chem 2002, 653, 23–6.10.1016/S0022-328X(02)01159-2Search in Google Scholar
[6] Kiso Y, Tamao K, Kumada M, J Organomet Chem 1973, 50, C12–C14.10.1016/S0022-328X(00)95063-0Search in Google Scholar
[7] Picquet M, Platinum Metals Rev 2013, 57, 272–80.10.1595/147106713X672320Search in Google Scholar
[8] Magano J, Dunetz JR, Chem Rev 2011, 111, 2177–250.10.1021/cr100346gSearch in Google Scholar PubMed
[9] Manley PW, Acemoglu M, Marterer W, Pachinger W, Org Process Res Dev 2003, 7, 436–45.10.1021/op025615qSearch in Google Scholar
[10] Bold G, Fässler A, Capraro H-G, Cozens R, Klimkait T, Lazdins J, Mestan J, Poncioni B, Rösel J, Stover D, Tintelnot-Blomley M, Acemoglu F, Beck W, Boss E, Eschbach M, Hürlimann T, Masso E, Roussel S, Ucci-Stoll K, Wyss D, Lang M, J Med Chem 1998, 41, 3387–401.10.1021/jm970873cSearch in Google Scholar PubMed
[11] Fan X, Song Y-L, Long Y-Q, Org Process Res Dev 2008, 7, 69–75.10.1021/op7001563Search in Google Scholar
[12] Lau SYW, Hughes G, O’Shea PD, Davies IW, Org Lett 2007, 9, 2239–42.10.1021/ol070841bSearch in Google Scholar
[13] Ghosh R, Sarkar A, J Org Chem 2010, 75, 8283–6.10.1021/jo1016458Search in Google Scholar
[14] Monnereau L, Semeril D, Matt D, Toupet L, Mota AJ, Adv Synth Catal 2009, 351, 1383–9.10.1002/adsc.200800759Search in Google Scholar
[15] Monnereau L, Semeril D, Matt D, Chem Commun 2011, 47, 6626–8.10.1039/c0cc05805jSearch in Google Scholar
[16] Monnereau L, Semeril D, Matt D, Adv Synth Catal 2013, 355, 1351–60.10.1002/adsc.201300091Search in Google Scholar
[17] Bohm VPW, Weskamp T, Gstöttmayr CWK, Herrmann WA, Angew Chem Int Ed 2000, 39, 1602–4.10.1002/(SICI)1521-3773(20000502)39:9<1602::AID-ANIE1602>3.0.CO;2-NSearch in Google Scholar
[18] Li GY, Marshall WJ, Organometallics 2002, 21, 590–1.10.1021/om010828+Search in Google Scholar
[19] Li GY, Angew Chem Int Ed 2001, 40, 1513–151610.1002/1521-3773(20010417)40:8<1513::AID-ANIE1513>3.0.CO;2-CSearch in Google Scholar
[20] Xie L-G, Wang Z-X, Chem Eur J 2010, 16, 10332–6.10.1002/chem.201001022Search in Google Scholar
[21] Ackermann L, Born R, Spatz JH, Meyer D, Angew Chem Int Ed 2005, 44, 7216–9.10.1002/anie.200501860Search in Google Scholar PubMed
[22] Ackermann L, Wechsler C, Kapdi AR, Althammer A, Synlett 2010, 294–8.10.1055/s-0029-1219166Search in Google Scholar
[23] Hayashi T, Hayashizaki K, Kiyoi T, Ito Y, J Am Chem Soc 1988, 110, 8153–6.10.1021/ja00232a030Search in Google Scholar
[24] Chen M-T, Lee W-Y, Tsai T-L, Liang L-C, Organometallics 2014, 33, 5852–62.10.1021/om5006353Search in Google Scholar
[25] Schiefer D, Wen T, Wang Y, Goursot P, Komber H, Hanselmann R, Braunstein P, Reiter G, Sommer M, ACS Macro Lett 2014, 3, 617–21.10.1021/mz500282jSearch in Google Scholar
[26] Liang L-C, Chien P-S, Lin J-M, Huang M-H, Huang Y-L, Liao J-H, Organometallics 2006, 25, 1399–411.10.1021/om050943aSearch in Google Scholar
[27] Venkanna GT, Tammineni S, Arman HD, Tonzetich ZJ, Organometallics 2013, 32, 4656–63.10.1021/om400630qSearch in Google Scholar PubMed PubMed Central
[28] Sanford J, Dent C, Masuda JD, Xia A, Polyhedron 2011, 30, 1091–4.10.1016/j.poly.2011.01.007Search in Google Scholar
[29] Sun Y, Li X, Sun H, Inorg Chim Acta 2014, 415, 95–7.10.1016/j.ica.2014.02.030Search in Google Scholar
[30] Sun K, Wang L, Wang Z-X, Organometallics 2008, 27, 5649–56.10.1021/om800580pSearch in Google Scholar
[31] Liang L-C, Lee W-Y, Hung Y-T, Hsiao Y-C, Cheng L-C, Chen W-C, Dalton Trans 2012, 41, 1381–8.10.1039/C1DT11338KSearch in Google Scholar
[32] Wang Z-X, Wang L, Chem Commun 2007, 2423–5.10.1039/b702027aSearch in Google Scholar PubMed
[33] Zhang X-Q, Wang Z-X, Synlett 2013, 24, 2081–4.10.1055/s-0033-1339653Search in Google Scholar
[34] Liu N, Wang Z-X, J Org Chem 2011, 76, 10031–8.10.1021/jo201821gSearch in Google Scholar PubMed
[35] Hatakeyama T, Hashimoto S, Ishizuka K, Nakamura M, J Am Chem Soc 2009, 131, 11949–63.10.1021/ja9039289Search in Google Scholar PubMed
[36] Tennyson AG, Lynch VM, Bielawski CW, J Am Chem Soc 2010, 132, 9420–9.10.1021/ja102686uSearch in Google Scholar PubMed
[37] Jothibasu R, Huang K-W, Huynh HV, Organometallics 2010, 29, 3746–52.10.1021/om100241vSearch in Google Scholar
[38] Soulé J-F, Miyamura H, Kobayashi S, J Am Chem Soc 2013, 135, 10602–5.10.1021/ja404006wSearch in Google Scholar PubMed
[39] Wolf J, Labande A, Daran J-C, Poli R, J Organomet Chem 2006, 691, 433–43.10.1016/j.jorganchem.2005.09.010Search in Google Scholar
[40] Wolf J, Labande A, Natella M, Daran J-C, Poli R, J Mol Catal A: Chem 2006, 259, 205–12.10.1016/j.molcata.2006.06.004Search in Google Scholar
[41] Page MJ, Lu WY, Poulten RC, Carter E, Algarra AG, Kariuki BM, MacGregor SA, Mahon MF, Cavell KJ, Murphy DM, Whittlesey MK, Chem Eur J 2013, 19, 2158–67.10.1002/chem.201202950Search in Google Scholar PubMed
[42] Mitsudo K, Doi Y, Sakamoto S, Murakami H, Mandai H, Suga S, Chem Lett 2011, 40, 936–8.10.1246/cl.2011.936Search in Google Scholar
[43] Zhang C, Wang Z-X, Organometallics 2009, 28, 6507–14.10.1021/om9006399Search in Google Scholar
[44] Guo W-J, Wang Z-X, J Org Chem 2013, 78, 1054–61.10.1021/jo302425xSearch in Google Scholar PubMed
[45] Sun Y, Li X, Sun H, Dalton Trans 2014, 43, 9410–3.10.1039/C4DT00461BSearch in Google Scholar
[46] Guo J, Lv L, Wang X, Cao C, Pang G, Shi Y, Inorg Chem Commun 2013, 31, 74–8.10.1016/j.inoche.2013.03.011Search in Google Scholar
[47] Berding J, Lutz M, Spek AL, Bouwman E, Organometallics 2009, 28, 1845–54.10.1021/om8010596Search in Google Scholar
[48] Vinh Huynh H, Jothibasu R, Eur J Inorg Chem 2009, 1926–31.10.1002/ejic.200801149Search in Google Scholar
[49] Inamoto K, Kuroda J-i, Sakamoto T, Hiroya K, Synthesis 2007, 2853–61.10.1055/s-2007-983845Search in Google Scholar
[50] Styring P, Grindon C, Fisher CM, Catal Lett 2001, 77, 219–25.10.1023/A:1013209202418Search in Google Scholar
[51] Phan NTS, Brown DH, Adams H, Spey SE, Styring P, Dalton Trans 2004, 33, 1348–57.10.1039/b316553cSearch in Google Scholar
[52] Chen X, Wang L, Liu J, Synthesis 2009, 2408–12.10.1055/s-0029-1216832Search in Google Scholar
[53] Lipshutz BH, Tomioka T, Blomgren PA, Sclafani JA, Inorg Chim Acta 1999, 296, 164–9.10.1016/S0020-1693(99)00388-6Search in Google Scholar
[54] Tasler S, Lipshutz BH, J Org Chem 2003, 68, 1190–9.10.1021/jo020297eSearch in Google Scholar PubMed
[55] Frieman BA, Taft BR, Lee C-T, Butler T, Lipshutz BH, Synthesis 2005, 2989–93.10.1055/s-2005-872145Search in Google Scholar
[56] Ackermann L, Born R, Spatz JH, Meyer D, Angew Chem, Int Ed 2005, 44, 7216–9.10.1002/anie.200501860Search in Google Scholar PubMed
[57] Wu D, Wang Z-X, Org Biomol Chem 2014, 12, 6414–24.10.1039/C4OB01041HSearch in Google Scholar
[58] Ahrens T, Kohlmann J, Ahrens M, Braun T, Chem Rev 2015, 115, 931–72.10.1021/cr500257cSearch in Google Scholar PubMed
[59] Saeki T, Takashima T, Tamao K, Synlett 2005, 1771–4.10.1055/s-2005-871571Search in Google Scholar
[60] Guo W-J, Wang Z-X, J Org Chem 2013, 78, 1054–61.10.1021/jo302425xSearch in Google Scholar PubMed
[61] Wenkert E, Ferreira TW, Michelotti EL, J Chem Soc, Chem Commun 1979, 637–8.10.1039/C39790000637Search in Google Scholar
[62] Dubbaka SR, Vogel P, Angew Chem Int Ed 2005, 44, 7674–84.10.1002/anie.200463007Search in Google Scholar PubMed
[63] Wenkert E, Hanna JM Jr, Leftin MH, Michelotti EL, Potts KT, Usifer D, J Org Chem 1985, 50, 1125–6.10.1021/jo00207a044Search in Google Scholar
[64] Murakami K, Yorimitsu H, Osuka A, Angew Chem Int Ed 2014, 53, 7510–3.10.1002/anie.201403288Search in Google Scholar PubMed
[65] Clayden J, Cooney JJA, Julia M, J Chem Soc, Perkin Trans 1, 1995, 7–14.10.1039/p19950000007Search in Google Scholar
[66] Cho C-H, Yun H-S, Park K, J Org Chem 2003, 68, 3017–25.10.1021/jo026449nSearch in Google Scholar PubMed
[67] Milburn RR, Snieckus V, Angew Chem Int Ed 2004, 43, 888–91.10.1002/anie.200352633Search in Google Scholar PubMed
[68] Li W-N, Wang Z-L, RSC Advances 2013, 3, 25565–75.10.1039/c3ra44884cSearch in Google Scholar
[69] Wenkert E, Michelotti EL, Swindell CS, J Am Chem Soc 1979, 101, 2246–7.10.1021/ja00502a074Search in Google Scholar
[70] Dankwardt JW, Angew Chem Int Ed 2004, 43, 2428–32.10.1002/anie.200453765Search in Google Scholar PubMed
[71] Xie L-G, Wang Z-X, Chem Eur J 2011, 17, 4972–5.10.1002/chem.201003731Search in Google Scholar
[72] Iglesias MJ, Prieto A, Nicasio C, Org Lett 2012, 14, 4318–21.10.1021/ol302112qSearch in Google Scholar
[73] Hayashi T, Katsuro Y, Kumada H, Tetrahedron Lett 1980, 21, 3915–18.10.1016/0040-4039(80)80215-2Search in Google Scholar
[74] Zhao F, Yu D-G, Zhu R-Y, Xi Z, Shi Z-J, Chem Lett 2011, 40, 1001–3.10.1246/cl.2011.1001Search in Google Scholar
[75] Sengupta S, Leite M, Raslan DS, Quesnelle C, Snieckus V J Org Chem 1992, 57, 4066–8.10.1021/jo00041a004Search in Google Scholar
[76] Yoshikai N, Matsuda H, Nakamura E, J Am Chem Soc 2009, 131, 9590–9.10.1021/ja903091gSearch in Google Scholar
[77] Hayashi T, Fujiwa T, Okamoto Y, Katsuro Y, Kumada M, Synthesis 1981, 1001–3.10.1055/s-1981-29680Search in Google Scholar
[78] Hayashi T, Katsuro Y, Okamoto Y, Kumada M, Tetrahedron Lett 1981, 22, 4449–52.10.1016/S0040-4039(01)82980-4Search in Google Scholar
[79] Guan B-T, Lu X-Y, Zheng Y, Yu D-G, Wu T, Li K-L, Li B-J, Shi Z-J, Org Lett 2010, 12, 396–9.10.1021/ol9028308Search in Google Scholar PubMed
[80] Macklin TK, Snieckus V, Org Lett 2005, 7, 2519–22.10.1021/ol050393cSearch in Google Scholar PubMed
[81] Yu D-G, Li B-J, Zheng S-F, Guan B-T, Wang B-Q, Shi Z-J, Angew Chem Int Ed 2010, 49, 4566–70.10.1002/anie.200907359Search in Google Scholar
[82] Banno T, Hayakawa Y, Umeno M J Organomet Chem 2002, 653, 288–291.10.1016/S0022-328X(02)01165-8Search in Google Scholar
[83] Marlier EE, Tereniak SJ, Ding K, Milliken JE, Lu CC, Inorg Chem 2011, 50, 9290–9.10.1021/ic200589eSearch in Google Scholar
[84] Xi Z, Liu B, Chen W, J Org Chem 2008, 73, 3954–7.10.1021/jo800197uSearch in Google Scholar
[85] Pugh C, Baker J, Storms WK, Synlett 2014, 25, 148–52.10.1055/s-0033-1339925Search in Google Scholar
[86] Okumara H, Miura M, Takei H, Tetrahedron Lett 1979, 43–6.10.1016/S0040-4039(01)85876-7Search in Google Scholar
[87] Cornella J, Martin R, Org Lett 2013, 6298–301.10.1021/ol4031815Search in Google Scholar PubMed
[88] Li Q-H, Ding Y, Yang X-J, Chin Chem Lett 2014, 25, 1296–1300.10.1016/j.cclet.2014.04.019Search in Google Scholar
[89] Li Q, Gau H, Synlett 2012, 747–50.10.1055/s-0031-1290365Search in Google Scholar
[90] Lou S, Fu GC, J Am Chem Soc 2010, 132, 1264–6.10.1021/ja909689tSearch in Google Scholar PubMed PubMed Central
[91] Mao J, Liu F, Wang M, Wu L, Zheng B, Liu S, Zhong J, Bian Q, Walsh PJ, J Am Chem Soc 2014, 136, 17662–8.10.1021/ja5109084Search in Google Scholar
[92] Terao J, Nii S, Chowdhury FA, Nakamura A, Kambe N, Adv Synth Catal 2004, 346, 905–8.10.1002/adsc.200404041Search in Google Scholar
[93] Xue F, Zhao J, Hor TSA, Hayashi T, J Am Chem Soc 2015, 137, 3189–92.10.1021/ja513166wSearch in Google Scholar
[94] Zembayashi M, Tamao K, Kumada M, Tetrahedron Lett 1975, 21, 1719–22.10.1016/S0040-4039(00)72242-8Search in Google Scholar
[95] Tamao K, Sumitani K, Kiso Y, Zembayashi M, Fujioka A, Kodama S, Nakajima I, Minato A, Kumada M, Bull Chem Soc Jpn 1976, 49, 1958–69.10.1246/bcsj.49.1958Search in Google Scholar
[96] Bach T, Bartels M, Synthesis 2003, 925–939.10.1055/s-2003-38695Search in Google Scholar
[97] Tiecco M, Testaferri L, Tingoli M, Wenkert E, Tetrahedron 1983, 39, 2289–94.10.1016/S0040-4020(01)91955-6Search in Google Scholar
[98] Fabre J-L, Julia M, Verpeaux J-N, Bull Soc Chim Fr 1985, 762–71.Search in Google Scholar
[99] Porée F-H, Clavel A, Betzer J-F, Pancrazi A, Ardisson J, Tetrahedron Lett 2003, 44, 7553–6.10.1016/S0040-4039(03)01804-5Search in Google Scholar
[100] Terao J, Watabe H, Kambe N, J Am Chem Soc 2005, 127, 3656–7.10.1021/ja042565rSearch in Google Scholar PubMed
[101] Terao J, Tomita M, Singh SP, Kambe N, Angew Chem Int Ed 2010, 49, 144–7.10.1002/anie.200904721Search in Google Scholar PubMed
[102] Fujii Y, Terao J, Watabe H, Watanabe H, Kambe N, Tetrahedron 2007, 63, 6635–41.10.1016/j.tet.2007.04.013Search in Google Scholar
[103] Vechorkin O, Godinat A, Scopelliti R, Hu X, Angew Chem Int Ed 2011, 50, 11777–81.10.1002/anie.201105964Search in Google Scholar PubMed
[104] Fang K, Xie M, Zhang Z, Ning P, Shu G, Tetrahedron Lett 2013, 54, 3819–21.10.1016/j.tetlet.2013.05.049Search in Google Scholar
[105] Tobisu M, Takahira T, Ohtsuki A, Chatani N, Org Lett 2015, 17, 680–3.10.1021/ol503707mSearch in Google Scholar PubMed
[106] Madec D, Pujol S, Henryon V, Férézou JP, Synlett 1995, 435–8.10.1055/s-1995-4980Search in Google Scholar
[107] Enamorado MF, Ondacchi PW, Comins DL, Org Lett 2010, 12, 4513–5.10.1021/ol101887bSearch in Google Scholar PubMed
[108] Wang J-R, Manabe K, Org Lett 2009, 11, 741–4.10.1021/ol802824bSearch in Google Scholar PubMed
[109] Luh T-Y, Ni Z-J, Synthesis 1990, 89–103.10.1055/s-1990-26798Search in Google Scholar
[110] Kanemura S, Kondoh A, Yorimitsu H, Oshima K, Synthesis 2008, 2659–64.10.1055/s-2008-1067193Search in Google Scholar
[111] Ghaderi A, Iwasaki T, Fukuoka A, Terao J, Kambe N, Chem Eur J 2013, 19, 2951–5.10.1002/chem.201203413Search in Google Scholar PubMed
[112] Kim C-B, Jo H, Ahn B-K, Kim CK, Park K, J Org Chem 2009, 74, 9566–9.10.1021/jo902151hSearch in Google Scholar
[113] Guan B-T, Xiang S-K, Wu T, Sun Z-P, Wang B-Q, Zhao K-Q, Shi Z-J, Chem Comm 2008, 1437–9.10.1039/b718998bSearch in Google Scholar
[114] Jiang Y-Y, Li Q, Lu W, Cai J-C, Tetrahedron Lett 2003, 44, 2073–5.10.1016/S0040-4039(03)00191-6Search in Google Scholar
[115] Chelucci G, Cabras MA, Botteghi C, Marchetti M, Tetrahedron: Asymmetry 1994, 5, 299–302.10.1016/S0957-4166(00)86190-XSearch in Google Scholar
[116] Wimmer P, Widhaim M, Tetrahedron: Asymmetry 1995, 6, 657–60.10.1016/0957-4166(95)00053-RSearch in Google Scholar
[117] Terfort A, Brunner H, J Chem Soc, Perkin Trans 1, 1996, 1467–79.10.1039/p19960001467Search in Google Scholar
[118] Cross G, Vriesema BK, Boven G, Kellogg RM, van Bolhuis F, J. Organomet. Chem. 1989, 370, 357–81.10.1016/0022-328X(89)87299-7Search in Google Scholar
[119] Pellet-Rostaing S, Saluzzo C, Ter Halle R, Breuzard J, Vial L, Le Guyader F, Lemaire M, Tetrahedron: Asymmetry 2001, 12, 1983–5.10.1016/S0957-4166(01)00342-1Search in Google Scholar
[120] Dai W, Xiao J, Jin G, Wu J, Cao S, J Org Chem 2014, 79, 10537–46.10.1021/jo5022234Search in Google Scholar PubMed
[121] Kocienski P, Dixon NJ, Synlett 1989, 52–4.10.1055/s-1989-34703Search in Google Scholar
[122] Iwashima M, Nagaoka H, Kobayashi K, Yamada Y, Tetrahedron Lett 1992, 33, 81–2.10.1016/S0040-4039(00)77678-7Search in Google Scholar
[123] William AD, Kobayashi Y, Org Lett 2001, 3, 2017–20.10.1021/ol010071iSearch in Google Scholar PubMed
[124] Terao J, Watanabe H, Ikumi A, Kuniyasu H, Kambe N, J Am Chem Soc 2002, 124, 4222–3.10.1021/ja025828vSearch in Google Scholar PubMed
[125] Terao J, Ikumi A, Kuniyasu H, Kambe N, J Am Chem Soc 2003, 125, 5646–7.10.1021/ja034201pSearch in Google Scholar PubMed
[126] Terao J, Todo H, Watanabe H, Ikumi A, Kambe N, Angew Chem Int Ed 2004, 43, 6180–3.10.1002/anie.200460246Search in Google Scholar PubMed
[127] Singh SP, Terao J, Kambe N, Tetrahedron Lett 2009, 50, 5644–6.10.1016/j.tetlet.2009.07.094Search in Google Scholar
[128] Vechorkin O, Csok Z, Scopelliti R, Hu X, Chem Eur J 2009, 15, 3889–99.10.1002/chem.200802059Search in Google Scholar PubMed
[129] Vechorkin O, Hu X, Angew Chem Int Ed 2009, 48, 2937–40.10.1002/anie.200806138Search in Google Scholar PubMed
[130] Ren P, Vechorkin O, von Allmen K, Scopelliti R, Hu X, J Am Chem Soc 2011, 133, 7084–95.10.1021/ja200270kSearch in Google Scholar PubMed
[131] Perez Garcia PM, Di Franco T, Orsino A, Ren P, Hu X, Org Lett 2012, 14, 4286–9.10.1021/ol302067bSearch in Google Scholar PubMed
[132] Breitenfeld J, Ruiz J, Wodrich MD, Hu X, J Am Chem Soc 2013, 135, 12004–12.10.1021/ja4051923Search in Google Scholar PubMed
[133] Guisan-Ceinos M, Soler-Yanes R, Collado-Sanz D, Phapale VB, Bunuel E, Cardenas DJ, Chem Eur J 2013, 19, 8405–10.10.1002/chem.201300882Search in Google Scholar PubMed
[134] Soler-Yanes R, Guisan-Ceinos M, Bunuel E, Cardenas DJ, Eur J Org Chem 2014, 6625–9.10.1002/ejoc.201403007Search in Google Scholar
[135] Wu J-C, Gong L-B, Xia Y, Song R-J, Xie Y-X, Li J-H, Angew Chem Int Ed 2012, 51, 9909–13.10.1002/anie.201205969Search in Google Scholar PubMed
[136] Guan B-T, Xiang S-K, Wang B-Q, Sun Z-P, Wang Y, Zhao K-Q, Shi Z-J, J Am Chem Soc 2008, 130, 3268–9.10.1021/ja710944jSearch in Google Scholar PubMed
[137] Taylor BLH, Swift EC, Waetzig JD, Jarvo ER, J Am Chem Soc 2011, 133, 389–91.10.1021/ja108547uSearch in Google Scholar PubMed
[138] Taylor BLH, Jarvo ER, Synlett 2011, 2761–5.10.1055/s-0031-1289871Search in Google Scholar
[139] Tollefson EJ, Dawson DD, Osborne CA, Jarvo ER, J Am Chem Soc 2014, 136, 14951–8.10.1021/ja5076426Search in Google Scholar PubMed PubMed Central
[140] Yu D-G, Wang X, Zhu R-Y, Luo S, Zhang X-B, Wang B-Q, Wang L, Shi Z-J, J Am Chem Soc 2012, 134, 14638–41.10.1021/ja307045rSearch in Google Scholar PubMed
© 2016 by Walter de Gruyter Berlin/Boston