Glossary of terms used in physical organic chemistry (IUPAC Recommendations 2021)
-
Charles L. Perrin

 ,
Israel Agranat
,
Israel Agranat

Abstract
This Glossary contains definitions, explanatory notes, and sources for terms used in physical organic chemistry. Its aim is to provide guidance on the terminology of physical organic chemistry, with a view to achieving a consensus on the meaning and applicability of useful terms and the abandonment of unsatisfactory ones. Owing to the substantial progress in the field, this 2021 revision of the Glossary is much expanded relative to the previous edition, and it includes terms from cognate fields.
Introduction to the 2021 revision
General remarks
The first Glossary of Terms Used In Physical Organic Chemistry was published in provisional form in 1979 and in revised form in 1983, incorporating modifications agreed to by IUPAC Commission III.2 (Physical Organic Chemistry) [1].
A further revision was undertaken under the chairmanship of Paul Müller, which was published in 1994 [2]. The work was coordinated with that of other Commissions within the Division of Organic Chemistry. In 1999, Gerard P. Moss, with the assistance of Charles L. Perrin, converted this glossary to a World Wide Web version [3]. The Compendium of Chemical Terminology [4] (Gold Book) incorporated many of the terms in the later version.
This Glossary has now been thoroughly revised and updated, to be made available as a Web document. The general criterion adopted for the inclusion of a term in this Glossary has been its wide use in the present or past literature of physical–organic chemistry and related fields, with particular attention to those terms that have been ambiguous. It is expected that the terms in this Glossary will be incorporated within the online version of the IUPAC Gold Book, which is the merged compendium of all glossaries [4].
The aim of this Glossary is to provide guidance on the terminology of physical-organic chemistry, with a view to achieving a far-reaching consensus on the definitions of useful terms and the abandonment of unsatisfactory ones. According to Antoine Lavoisier, “Comme ce sont les mots qui conservent les idées et qui les transmettent, il en résulte qu’on ne peut perfectionner le langage sans perfectionner la science, ni la science sans le langage,” (“As it is the words that preserve the ideas and convey them, it follows that one cannot improve the language without improving science, nor improve science without improving the language.”) [5]. Our approach has been to take or update entries from the previous glossary, whereas new terms were added by virtue of their usage in the current literature and the diverse knowledge of the members of the Task Force.
The Task Force is pleased to acknowledge the generous contributions of many scientists who helped by proposing or defining new terms or by criticizing or modifying existing ones.
We specially note the able technical assistance of Gerard P. Moss (Queen Mary University of London) and the advice of Christian Reichardt (Marburg). We also thank the several reviewers whose comments improved the presentation, especially Jan Kaiser (University of East Anglia).
Arrangement, Abbreviations, and Symbols
The arrangement is alphabetical, with terms with Greek letters following those in the corresponding Latin ones. Italicized words in the body of a definition, as well as those cited at the end, point to relevant cross-references. Literature references direct the reader either to the original literature where the term was originally defined or to pertinent references where it is used, including other IUPAC glossaries, where definitions may differ from those here.
Definitions of techniques not directly used for measurements in physical-organic chemistry are not included here but may be consulted in specialized IUPAC texts, including the IUPAC Recommendations for Mass Spectrometry [6], the Glossary of Terms Used In Theoretical Organic Chemistry [7], the Glossary of Terms Used in Photochemistry 3rd edition [8], the Glossary of Terms Used in Photocatalysis and Radiation Catalysis [9], and the Basic Terminology of Stereochemistry [10].
In accordance with IUPAC recommendations [11], the symbol ‡ to indicate transition state (“double dagger”) is used as a prefix to the appropriate quantities, e.g., ∆‡G rather than ∆G‡. In equations including a logarithmic function, the procedure recommended by the IUPAC was adopted, i.e., to divide each dimensioned quantity by its unit. Because this procedure often introduces a cluttering of the equations, we have, in some cases, chosen a short-hand notation, such as ln {k(T)}, where the curly brackets indicate an argument of dimension one, corresponding to k(T)/[k(T)], where the square brackets indicate that the quantity is divided by its unit, as recommended in [11].
Note on the identification of new and/or revised terms
Terms that are found in the previous version of this Glossary [2, 3] and currently incorporated into the IUPAC “Gold Book” [4] are identified with a reference to the Glossary of Terms used in Physical Organic Chemistry (1994), i.e., [3], whereas revised terms are designated as rev[3]. Minor changes such as better wording, additional cross-referencing, or reorganization of the text without changing the concept are, in general, not considered revisions. However, the improved version should replace the older one in the “Gold Book”. New terms and terms from other IUPAC documents are not identified as such. In many cases, new references have been added in the definitions.
Alphabetical entries
A factor
Arrhenius factor
(SI unit same as rate constant: s−1 for first-order reaction).
Pre-exponential factor in the Arrhenius equation for the temperature dependence of a reaction rate constant.
Note 1: According to collision theory, A is the frequency of collisions with the correct orientation for reaction.
Note 2: The common unit of A for second-order reactions is dm3 mol−1 s−1.
See also A value, energy of activation, entropy of activation.
rev[3]
A value
Steric substituent parameter expressing the conformational preference of an equatorial substituent relative to an axial one in a monosubstituted cyclohexane.
Note 1: This parameter equals ΔrG° for the equatorial to axial equilibration, in kJ mol−1. For example, ACH3 is 7.28 kJ mol−1, a positive value because an axial methyl group is destabilized by a steric effect.
Note 2: The values are also known as Winstein-Holness A values.
abstraction
Chemical reaction or transformation, the main feature of which is the bimolecular removal of an atom (neutral or charged) from a molecular entity.
Examples:
See detachment.
[3]
acceptor parameter (A)
acceptor number (deprecated).
Unit and dimension 1.
Quantitative measure, devised by Gutmann [15] of the Lewis acidity of a solvent, based on the 31P chemical shift of dissolved triethylphosphine oxide (triethylphosphane oxide).
Note: The term acceptor number, designated by AN, is a misnomer and ought to be called acceptor parameter, A, because it is an experimental value [16].
rev[3]
acid
Molecular entity or chemical species capable of donating a hydron (proton) or capable of forming a bond with the electron pair of a Lewis base.
See Brønsted acid, Lewis acid, Lewis base.
See also hard acid.
[3]
acidity
(1) Of a compound:
Tendency of a Brønsted acid to act as a hydron (proton) donor or tendency of a Lewis acid to form Lewis adducts and π-adducts.
Note: Acidity can be quantified by the acidity constant, by association constants for formation of Lewis adducts and π-adducts, or by the enthalpy or Gibbs energy of deprotonation in the gas phase. In water the acidity constant Ka of acid HA is {H3O+}{A–}/{HA}.
(2) Of a medium, usually one containing Brønsted acids:
Tendency of the medium to hydronate a specific reference base.
Note 1: The acidity of a medium is quantitatively expressed by the appropriate acidity function.
Note 2: Media having an acidity greater than that of 100 % H2SO4 are often called superacids.
See [17].
rev[3]
acidity function
Measure of the thermodynamic hydron-donating or -accepting ability of a solvent system or a closely related thermodynamic property, such as the tendency of the lyate ion of the solvent system to form Lewis adducts.
Note 1: Acidity functions are not unique properties of the solvent system alone but depend on the solute (or family of closely related solutes) with respect to which the thermodynamic tendency is measured.
Note 2: Commonly used acidity functions are extensions of pH to concentrated acidic or basic solutions. Acidity functions are usually established over a range of compositions of such a system by UV/Vis spectrophotometric or NMR measurements of the degree of hydronation (or Lewis adduct formation) for the members of a series of structurally similar indicator bases (or acids) of different strength: the best known of these is the Hammett acidity function Ho (for primary aromatic amines as indicator bases).
For detailed information on other acidity functions, on excess acidity, on the evaluation of acidity functions, and on the limitations of the concept, see [18], [19], [20], [21].
[3]
activated complex
See activated state.
rev[3]
activated state
In theories of unimolecular reactions an energized chemical species, often characterized by the superscript ‡, where the excitation is specific and the molecule is poised for reaction.
Note 1: Often used as a synonym for activated complex or transition state, but not restricted to transition-state theory.
Note 2: This is distinct from an energized molecule, often characterized by the superscript *, in which excitation energy is dispersed among internal degrees of freedom.
Note 3: This is not a complex according to the definition in this Glossary.
See also transition state, transition structure.
activation energy
See energy of activation.
See [11], section 2.12.
[3]
activation strain model
See distortion interaction model.
addition reaction
Chemical reaction of two or more reacting molecular entities, resulting in a product containing all atoms of all components, with formation of two chemical bonds and a net reduction in bond multiplicity in at least one of the reactants.
Note 1: The addition to an entity may occur at only one site (1,1-addition, insertion), at two adjacent sites (1,2-addition), or at two non-adjacent sites (1,3- or 1,4-addition, etc.). Examples
Note 2: This is distinguished from adduct formation, which is less specific about bonding changes.
Note 3: The reverse process is called an elimination reaction.
See also cheletropic reaction, cycloaddition, insertion.
rev[3]
additivity principle
Hypothesis that each of several structural features of a molecular entity makes an independent, transferable, and additive contribution to a property of the substance concerned.
Note 1: More specifically, it is the hypothesis that each of the several substituent groups in a parent molecule makes a separate and additive contribution to the standard Gibbs energy change or Gibbs energy of activation corresponding to a particular chemical reaction.
Note 2: The enthalpies of formation of series of compounds can be described by additivity schemes [22].
Note 3: Deviations from additivity may be remedied by including terms describing interactions between atoms or groups.
rev[3]
adduct
New chemical species AB, each molecular entity of which is formed by direct combination of two (or more) separate molecular entities A and B in such a way that there is no loss of atoms from A or B.
Example: adduct formed by interaction of a Lewis acid with a Lewis base:
Note 1: Stoichiometries other than 1:1 are also possible, e.g., a bis-adduct (2:1).
Note 2: An intramolecular adduct can be formed when A and B are groups contained within the same molecular entity.
Note 3: If adduct formation is prevented by steric hindrance, frustrated Lewis pairs may result.
See also addition reaction, frustrated Lewis pair, Lewis adduct, Meisenheimer adduct, π-adduct.
rev[3]
agostic
Feature of a structure in which a hydrogen atom is bonded to both a main-group atom and a metal atom.
Example, [(1–3-η)-but-2-en-1-yl-η2-C4,H4](η5-cyclopentadienyl)cobalt(+1), where the agostic bond is denoted by a half arrow.
Note: The expression η-hydrido-bridged is also used to describe the bonding arrangement with a bridging hydrogen, but this usage is deprecated.
rev[3]
alcoholysis
Reaction with an alcohol solvent.
Examples:
See solvolysis.
rev[3]
allotropes
Different structural modifications of an element, with different bonding arrangements of the atoms.
Examples for carbon include diamond, fullerenes, graphite, and graphene.
See [28].
allylic substitution reaction
Substitution reaction on an allylic system with leaving group (nucleofuge) at position 1 and double bond between positions 2 and 3. The incoming group may become attached to atom 1, or else, the incoming group may become attached at position 3, with movement of the double bond from 2,3 to 1,2.
Example:
Note: This term can be extended to systems such as:
and also to propargylic substitution, with a triple bond between positions 2 and 3 and possible rearrangement to an allenic product.
rev[3]
alternant
Property of a conjugated system of π electrons whose atoms can be divided into two sets (marked as “starred” and “unstarred”) so that no atom of either set is directly linked to any other atom of the same set.
Examples:
Note 1: According to several approximate theories (including HMO theory), the π MOs for an alternant hydrocarbon are paired, such that for an orbital of energy α + xβ there is another of energy α − xβ. The coefficients of paired molecular orbitals at each atom are the same, but with opposite sign for the unstarred atoms, and the π electron density at each atom in a neutral alternant hydrocarbon is unity.
rev[3]
ambident
ambidentate, bidentate
Characteristic of a chemical species whose molecular entities each possess two alternative, distinguishable, and strongly interacting reactive centers, to either of which a bond may be made.
Note 1: Term most commonly applied to conjugated nucleophiles, for example, an enolate ion (which may react with electrophiles at either the α-carbon atom or the oxygen) or a 4-pyridone, and also to the vicinally ambident cyanide ion and to the cyanate ion, thiocyanate ion, sulfinate ions, nitrite ion, and unsymmetrical hydrazines.
Note 2: Ambident electrophiles are exemplified by carboxylic esters RC(O)OR′, which react with nucleophiles at either the carbonyl carbon or the alkoxy carbon, and by Michael acceptors, such as enones, that can react at either the carbonyl or the β-carbon.
Note 3: Molecular entities containing two non-interacting (or feebly interacting) reactive centers, such as dianions of dicarboxylic acids, are not generally considered to be ambident or bidentate and are better described as bifunctional.
Note 4: The Latin root of the word implies two reactive centers, but the term has also been applied to chemical species with more than two reactive centers, such as an acyl thiourea, RCONHCSNHR′, with nucleophilic O, S, and N. For such species, the term polydentate (or multidentate) is more appropriate.
[3]
ambiphilic
Both nucleophilic and electrophilic.
Example:
See also amphiphilic, but the distinction between ambi (Latin: both) and amphi (Greek: both) and the application to hydrophilic and lipophilic or to nucleophilic and electrophilic is arbitrary.
aminoxyl
Compound having the structure
Note: The synonymous term ‘nitroxyl radical’ erroneously suggests the presence of a nitro group; its use is deprecated.
amphiphilic
Both hydrophilic and lipophilic, owing to the presence in the molecule of a large organic cation or anion and also a long hydrocarbon chain (or other combination of polar and nonpolar groups, as in nonionic surfactants)
Examples:
Note: The presence of distinct polar (hydrophilic) and nonpolar (hydrophobic) regions promotes the formation of micelles in dilute aqueous solution.
See also ambiphilic, hydrophilicity, hydrophobicity.
[3]
amphiprotic (solvent)
Feature of a self-ionizing solvent possessing characteristics of both Brønsted acid and base.
Examples: H2O, CH3OH.
[3]
amphoteric
Property of a chemical species that can behave as either an acid or as a base.
Examples: H2O, HCO3− (hydrogen carbonate)
Note: This property depends upon the medium in which the species is investigated. For example HNO3 is an acid in water but becomes a base in H2SO4.
rev[3]
anchimeric assistance
neighboring group participation
[3]
anionotropy
Rearrangement or tautomerization in which the migrating group moves with its electron pair.
See [36].
[3]
annelation
Alternative, but less desirable term for annulation.
Note: The term is widely used in German and French languages.
[3]
annulation
Transformation involving fusion of a new ring to a molecule via two new bonds.
Note: Some authors use the term annelation for the fusion of an additional ring to an already existing one and annulation for the formation of a ring from an acyclic precursor.
See also cyclization.
[3]
annulene
Conjugated monocyclic hydrocarbon of the general formula CnHn (n even) with the maximum number of noncumulative double bonds and without side chains.
Note: In systematic nomenclature, an annulene may be named [n]annulene, where n is the number of carbon atoms, e.g., [8] annulene for cycloocta-1,3,5,7-tetraene.
See [39].
See aromatic, Hückel (4n + 2) rule.
[3]
anomeric effect
Tendency of an electronegative substituent alpha to a heteroatom in a six-membered ring to prefer the axial position, as in the anomers of glucopyranose.
Note 1: The effect can be generalized to the conformational preference of an electronegative substituent X to be antiperiplanar to a lone pair of atom Y in a system R–Y–C–X with geminal substituents RY and X.
Note 2: The effect can be attributed, at least in part, to n-σ* delocalization of the lone pair on Y into the C–X σ* orbital.
anomers
The two stereoisomers (epimers) of a cyclic sugar or glycoside that differ only in the configuration at C1 of aldoses or C2 of ketoses (the anomeric or acetal/ketal carbon).
Example: 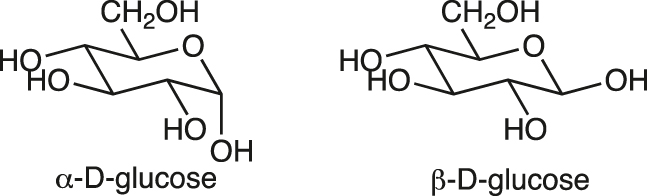
See [10].
[3]
antarafacial, suprafacial
Two spatially different ways whereby bonding changes can occur when a part of a molecule undergoes two changes in bonding (bond-making or bond-breaking), either to a common center or to two related centers external to itself. These are designated as antarafacial if opposite faces of the molecule are involved and suprafacial if both changes occur at the same face. The concept of face is clear from the diagrams in the cases of planar (or approximately planar) frameworks with interacting π orbitals.
For examples of the use of these terms, see cycloaddition, sigmatropic rearrangement.
See also anti, σ, π.
rev[3]
anti
Stereochemical relationship of two substituents that are on opposite sides of a reference plane, in contrast to syn, which means “on the same side”.
Note 1: Two substituents attached to atoms joined by a single bond are anti if the torsion angle (dihedral angle) between the bonds to the substituents is greater than 90°, in contrast to syn if it is less than 90°.
Note 2: A further distinction is made between antiperiplanar, synperiplanar, anticlinal, and synclinal.
Note 3: When the terms are used in the context of chemical reactions or transformations, they designate the relative orientation of substituents in the substrate or product:
Addition to a carbon–carbon double bond:

Alkene-forming elimination:

Aldol reaction (where syn and anti designate the relative orientation of CH3 and OH in the product)

Note 4: In examples (1) and (2), anti processes are always antarafacial and syn processes are suprafacial.
Note 5: In the older literature, the terms anti and syn were used to designate stereoisomers of oximes and related compounds. That usage was superseded by the terms trans and cis or E and Z.
See [10].
rev[3]
anti-Hammond effect
If a structure lying off the minimum-energy reaction path (MERP) is stabilized, the position of the transition state moves toward that structure.
See Hammond Postulate, More O’Ferrall–Jencks diagram, perpendicular effect.
rev[3]
aprotic (solvent)
non-HBD solvent (non-Hydrogen-Bond Donating solvent)
Solvent that is not capable of acting as a hydrogen-bond donor.
Note: Although this definition applies to both polar and nonpolar solvents, the distinction between HBD and non-HBD (or between protic and aprotic) is relevant only for polar solvents.
See HBD solvent.
rev[3]
aquation
Incorporation of one or more integral molecules of water into a species, with or without displacement of one or more other atoms or groups.
Example: The incorporation of water into the inner ligand sphere of an inorganic complex.
See [47].
See also hydration.
[3]
aromatic (adj.), aromaticity (n.)
Having a chemistry typified by benzene (traditionally).
Feature of a cyclically conjugated molecular entity whose electronic energy is significantly lower or whose stability is significantly greater (owing to delocalization) than that of a hypothetical localized structure (e.g., Kekulé structure).
Note 1: If the molecular entity is of higher energy or less stable than a hypothetical localized structure, the entity is said to be antiaromatic.
Note 2: A geometric parameter indicating bond-length equalization has been used as a measure of aromatic character, as expressed in the harmonic oscillator model of aromaticity [48].
Note 3: The magnitude of the magnetically induced ring current, as observed experimentally by NMR spectroscopy or by the calculated nucleus-independent chemical shift (NICS) value, is another measure of aromaticity [49].
Note 4: The terms aromatic and antiaromatic have been extended to describe the stabilization or destabilization of transition states of pericyclic reactions. The hypothetical reference structure is here less clearly defined, and use of the term is based on application of the Hückel (4n + 2) rule and on consideration of the topology of orbital overlap in the transition state, whereby a cycle with (4n + 2) electrons and a Möbius cycle with 4n electrons are aromatic. Reactions of molecules in the ground state involving antiaromatic transition states proceed much less easily than those involving aromatic transition states.
See [50], [51], [52]. See 19 articles in [53].
See also Hückel (4n + 2) rule, Möbius aromaticity.
[3]
Arrhenius equation
Empirical expression for the temperature dependence of a reaction rate constant k as
with A being the pre-exponential factor (Arrhenius A factor) and EA being the Arrhenius energy of activation, both considered to be temperature-independent.
rev[3]
aryne
Hydrocarbon derived from an arene by formal removal of two vicinal hydrogen atoms.
Example: 1,2-didehydrobenzene (benzyne)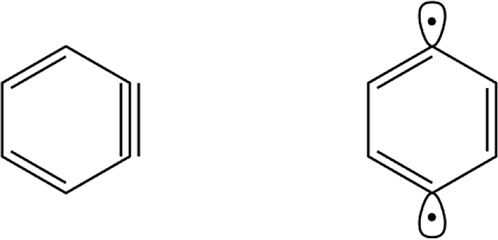
Note 1: 1,4-Didehydrobenzene (“p-benzyne diradical”, structure above right). Despite common usage, this is not an aryne because there is no triple bond, and the usage is deprecated.
Note 2: Arynes are usually transient species.
Note 3: The analogous heterocyclic compounds are called heteroarynes or hetarynes.
rev[3]
association
Assembling of separate molecular entities into any aggregate, especially of oppositely charged free ions into ion pairs or larger and not necessarily well-defined clusters of ions held together by electrostatic attraction.
Note: The term signifies the reverse of dissociation, but is not commonly used for the formation of definite adducts by colligation or coordination.
[3]
asymmetric induction
Preferential formation in a chemical reaction of one enantiomer or diastereoisomer over the other as a result of the influence of a chiral center (stereogenic center, chiral feature) in the substrate, reagent, catalyst, or environment.
Note: The term also refers to the formation of a new chiral center or chiral feature preferentially in one configuration under such an influence.
See [10].
rev[3]
atomic charge
Net charge due to the nucleus and the average electronic distribution in a given region of space. This region is considered to correspond to an atom in a molecular entity.
Note 1: The boundary limits of an atom in a polyatomic molecular entity cannot be defined, as they are not a quantum-mechanical observable. Therefore, different conceptual schemes of dividing a molecule into individual atoms will result in different atomic charges.
Note 2: The atomic charge on an atom should not be confused with its formal charge. For example, the N in NH4+ is calculated to carry a net negative charge even though its formal charge is +1, and each H is calculated to carry a net positive charge even though its formal charge is 0.
See [7].
atomic orbital
Wavefunction that depends explicitly on the spatial coordinates of only one electron around a single nucleus.
See also molecular orbital.
See [7].
[3]
atropisomers
Stereoisomers that are enantiomeric owing to hindered rotation about a single bond.
Example (2,2′-bis(diphenylphosphino)-1,1′-binaphthyl, BINAP):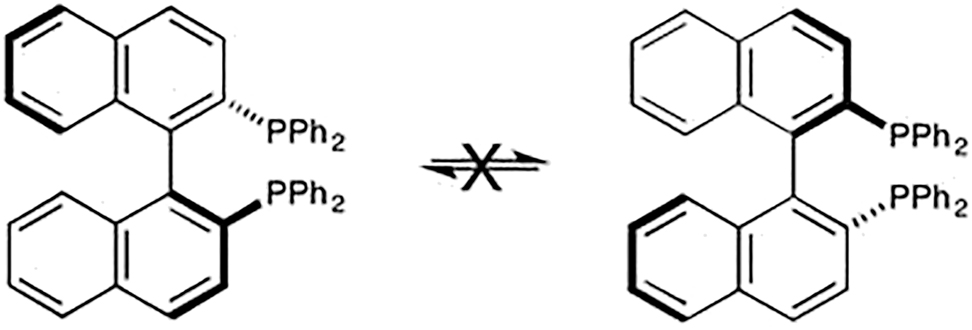
See [10].
attachment
Transformation by which one molecular entity (the substrate) is converted into another by the formation of one (and only one) two-center bond between the substrate and another molecular entity and which involves no other changes in connectivity in the substrate.
Example: formation of an acyl cation by attachment of carbon monoxide to a carbenium ion (R+):
See also colligation.
rev[3]
autocatalytic reaction
Chemical reaction in which a product (or a reaction intermediate) also functions as a catalyst.
Note: In such a reaction, the observed rate of reaction is often found to increase with time from its initial value.
Example: acid-catalyzed bromination of acetophenone, PhCOCH3, because the reaction generates HBr, which functions as a catalyst.
rev[3]
automerization
degenerate rearrangement
[3]
autoprotolysis
Proton transfer reaction (hydron transfer reaction) between two identical amphoteric molecules (usually of a solvent), one acting as a Brønsted acid and the other as a Brønsted base.
Example:
[3]
autoprotolysis constant
Product of the activities of the species produced as the result of autoprotolysis.
Example: The autoprotolysis constant for water, Kw (or KAP), is equal to the product of the relative activities of the hydronium and hydroxide ions at equilibrium in pure water.
Note: Because the relative activity a(H2O) of water at equilibrium is imperceptibly different from unity (with mole fraction as the activity scale and pure un-ionized water as the standard state), the denominator in the expression for the thermodynamic equilibrium constant Kw° for autoprotolysis has a value very close to 1.
Furthermore, owing to the low equilibrium extent of dissociation, and if infinite dilution is selected as the standard state, the activity coefficients of the hydronium and hydroxide ions in pure water are very close to unity. This leads to the relative activities of H3O+ and HO− being virtually identical with the numerical values of their molar concentrations, if the molar scale of activity is used and 1 mol dm−3 is chosen as the standard state. Thus,
where {H3O+} and {HO−} are the reduced concentrations. The autoprotolysis constant has the unit (and dimension) 1 because each relative activity has dimension 1.
rev[3]
α (alpha)
Designation applied to the carbon to which a functional group is attached.
In carbohydrate nomenclature, a stereochemical designation of the configuration at the anomeric carbon.
Parameter in a Brønsted relation expressing the sensitivity of the rate of protonation to acidity.
Parameter in Leffler’s relation expressing the sensitivity of changes in Gibbs activation energy to changes in overall Gibbs energy for an elementary reaction.
See [10].
rev[3]
α-effect
Positive deviation of an α nucleophile (one bearing an unshared pair of electrons on an atom adjacent to the nucleophilic site) from a Brønsted-type plot of lg {knuc} vs. pKa. The argument in the lg function should be of dimension 1. Thus, reduced rate coefficients should be used. Here {knuc} = knuc/[knuc] is the reduced knuc.
Note 1: More generally, it is the influence on the reactivity at the site adjacent to the atom bearing a lone pair of electrons.
Note 2: The term has been extended to include the effect of any substituent on an adjacent reactive center, e.g., the α-silicon effect.
See also Brønsted relation.
rev[3]
α-elimination
1,1-elimination
Transformation of the general type
where the central atom Z is commonly carbon.
See also elimination.
rev[3]
Baldwin’s rules
Set of empirical rules for closures of 3- to 7-membered rings.
Note: The favored pathways are those in which the length and nature of the linking chain enable the terminal atoms to achieve the proper geometry and orbital overlaps for reaction.
Example: Hex-5-en-1-yl radical undergoes 5-exo-trig cyclization to cyclopentylmethyl radical rather than 6-endo-trig cyclization to cyclohexyl radical.
rev[3]
base
Chemical species or molecular entity having an available pair of electrons capable of forming a bond with a hydron (proton) (see Brønsted base) or with the vacant orbital of some other species (see Lewis base).
See also hard base, superbase.
[3]
basicity
Tendency of a Brønsted base to act as a hydron (proton) acceptor.
Note 1: The basicity of a chemical species is normally expressed by the acidity or acid-dissociation constant of its conjugate acid (see conjugate acid–base pair).
Note 2: To avoid ambiguity, the term pKaH should be used when expressing basicity by the acid-dissociation constant of its conjugate acid. Thus, the pKaH of NH3 is 9.2, while its pKa, expressing its acidity, is 38.
Tendency of a Lewis base to act as a Lewis acid acceptor.
Note 3: For Lewis bases, basicity is expressed by the association constants of Lewis adducts and π-adducts or by the enthalpy of an acid/base reaction.
Note 4: Spectroscopic shifts induced by acid/base adduct formation can also be used as a measure of the strength of interaction.
See [63].
rev[3]
bathochromic shift (effect)
Shift of a spectral band to lower frequencies (longer wavelengths).
Note: This is informally referred to as a red shift and is opposite to a hypsochromic shift (“blue shift”), but these historical terms are discouraged because they apply only to visible transitions.
See [8].
[3]
Bell–Evans–Polanyi principle
Linear relation between energy of activation (EA) and enthalpy of reaction (ΔrH), sometimes observed within a series of closely related reactions.
benzyne
1,2-Didehydrobenzene (a C6H4aryne derived from benzene) and its derivatives formed by substitution.
Note: The terms m- and p-benzyne are occasionally used for 1,3- and 1,4-didehydrobenzene, respectively, but these are incorrect because there is no triple bond.
[3]
bidentate
Feature of a ligand with two potential binding sites.
bifunctional catalysis
Catalysis (usually of hydron transfer) by a chemical species involving a mechanism in which two functional groups are implicated in the rate-limiting step, so that the corresponding catalytic coefficient is larger than that expected for catalysis by a chemical species containing only one of these functional groups.
Example: Hydrogen carbonate is a particularly effective catalyst for the hydrolysis of 4-hydroxybutyranilide (N-phenyl-4-hydroxybutanamide) because it catalyzes the breakdown of the tetrahedral intermediate to expel aniline:
Note: The term should not be used to describe a concerted process involving the action of two different catalysts.
See [68], [69], [70], [71], [72].
rev[3]
bifurcation
Feature on a potential-energy surface whereby a minimum-energy reaction path (MERP) emanating from a saddle point (corresponding to a transition structure) splits in two and leads to alternative products without intervening minima or secondary barriers to overcome. A bifurcation arises when the curvature of the surface in a direction perpendicular to the MERP becomes zero and then negative; it implies the existence of a lower-energy transition structure with a transition vector orthogonal to the original MERP.
bimolecular
See molecularity.
[3]
rev[3]
binding site
Specific region (or atom) in a molecular entity that is capable of entering into a stabilizing interaction with another atomic or molecular entity.
Example: an active site in an enzyme that interacts with its substrate.
Note 1: Typical modes of interaction are by covalent bonding, hydrogen bonding, coordination, and ion-pair formation, as well as by dipole-dipole interactions, dispersion forces, hydrophobic interactions, and desolvation.
Note 2: Two binding sites in different molecular entities are said to be complementary if their interaction is stabilizing.
[3]
biradical
diradical
See [8].
rev[3]
blue shift
Informal expression for hypsochromic shift, but this historical term is discouraged because it applies only to visible transitions.
rev[3]
Bodenstein approximation
See steady state.
[3]
bond
Balance of attractive and repulsive forces between two atoms or groups of atoms, resulting in sufficient net stabilization to lead to the formation of an aggregate conveniently considered as an independent molecular entity.
Note: The term usually refers to the covalent bond.
See [75].
See also agostic, coordination, hydrogen bond, multi-center bond.
rev[3]
bond dissociation
See heterolysis, homolysis.
Note: In ordinary usage the term refers to homolysis. If not, it should be specified as heterolytic.
[3]
bond-dissociation energy
(derived SI unit: kJ mol−1)
Energy required to break a given bond of some specific molecular entity by homolysis from its potential-energy minimum.
Note: This is the quantity that appears in the Morse potential.
See also bond-dissociation enthalpy.
rev[3]
bond-dissociation enthalpy
(derived SI unit: kJ mol−1)
Standard molar enthalpy required to break a given bond of some specific molecular entity by homolysis.
Example: For
Note: Although DH° is commonly used,
See also bond-dissociation energy, bond energy, heterolytic bond-dissociation enthalpy.
bond energy
(derived SI unit: kJ mol−1)
Enthalpy of bond dissociation at 0 K.
See bond-dissociation enthalpy.
rev[3]
bond enthalpy (mean bond enthalpy, mean bond energy)
Average value of the gas-phase bond-dissociation enthalpies (usually at a temperature of 298 K) for all bonds of the same type within the same chemical species.
Example: For methane, the mean bond enthalpy is 415.9 kJ mol−1, one-fourth the enthalpy of reaction for
Note: More commonly, tabulated mean bond energies (which are really enthalpies) are values of bond enthalpies averaged over a number of selected chemical species containing that type of bond, such as 414 kJ mol−1 for C–H bonds in a group of R3CH (R = H, alkyl).
See [76].
bond order
Theoretical index of the degree of bonding between two atoms, relative to that of a normal single bond, i.e., the bond provided by one localized electron pair.
Example: In ethene, the C–C bond order is 2, and the C–H bond order is 1.
Note 1: In valence-bond theory, it is a weighted average of the bond orders between the respective atoms in the various resonance forms. In molecular-orbital theory, it is calculated from the weights of the atomic orbitals in each of the occupied molecular orbitals. For example, in valence-bond theory, the bond order between adjacent carbon atoms in benzene is 1.5; in Hückel molecular orbital theory, it is 1.67.
Note 2: Bond order is often derived from the electron distribution.
Note 3: The Pauling bond order n (as often used in the bond-energy-bond-order model) is a simple function of change in bond length d, where the value of the coefficient c is often 0.3 Å (for n > 1) or 0.6 Å (for n < 1).
rev[3]
bond-energy-bond-order model (BEBO):
Empirical procedure for estimating activation energy, involving relationships among bond length, bond-dissociation energy, and bond order.
bond-stretch isomers
Two (or more) molecules with the same spin multiplicity but with different lengths for one or more bonds.
Note: This feature arises because the potential-energy surface, which describes how the energy of the molecule depends on geometry, shows two (or more) minima that are not merely symmetry-related.
borderline mechanism
Mechanism intermediate between two extremes, for example, a nucleophilic substitution intermediate between SN1 and SN2 or intermediate between electron transfer and SN2.
[3]
Born–Oppenheimer approximation
Representation of the complete wavefunction as a product of electronic and nuclear parts,
See [7].
Bredt’s rule
Prohibition of placing a double bond with one terminus at the bridgehead atom of a polycyclic system unless the rings are large enough to accommodate the double bond without excessive strain.
Example: Bicyclo[2.2.1]hept-1-ene (A), which is capable of existence only as a transient species, although its higher homologues, bicyclo[3.3.1]non-1-ene (B) and bicyclo[4.2.1]non-1(8)-ene (C), with double bond at the bridgeheads, have been isolated.
For limitations, see [86].
Note: For an alternative formulation, based on the instability of a trans double bond in a small ring (fewer than 8 atoms), see [87].
rev[3]
bridged carbocation
Carbocation (real or hypothetical) in which there are two (or more) carbon atoms that could in alternative Lewis formulas be designated as carbenium centers but which is instead represented by a structure in which a group (a hydrogen atom or a hydrocarbon residue, possibly with substituents in non-involved positions) bridges these potential carbenium centers.
Note: Electron-sufficient bridged carbocations are distinguished from electron-deficient bridged carbocations. Examples of the former, where the bridging uses π electrons, are phenyl-bridged ions (for which the trivial name phenonium ion has been used), such as A. These ions are straightforwardly classified as carbenium ions. The latter type of ion necessarily involves three-center bonding because the bridging uses σ electrons. The hydrogen-bridged carbocation B contains a two-coordinate hydrogen atom, whereas structures C, D, and E (the 2-norbornyl cation) contain five-coordinate carbon atoms.
See [88].
See also carbonium ion, multi-center bond, neighboring group participation.
For the definitive X-ray structure of the norbornyl cation, see [89].
rev[3]
bridgehead (atom)
Atom that is part of two or more rings in a polycyclic molecule and that is separated from another bridgehead atom by bridges all of which contain at least one other atom.
Example: C1 and C4 in bicyclo[2.2.1]heptane, but not C4a and C8a in decahydronaphthalene.
bridging ligand
Ligand attached to two or more, usually metallic, central atoms.
See [28].
[3]
Brønsted acid
Molecular entity capable of donating a hydron (proton) to a base (i.e., a hydron donor) or the corresponding chemical species.
Examples: H2O, H3O+, CH3COOH, H2SO4,
See also conjugate acid–base pair.
[3]
Brønsted base
Molecular entity capable of accepting a hydron (proton) from a Brønsted acid (i.e., a hydron acceptor) or the corresponding chemical species.
Examples: HO−, H2O,
See also conjugate acid–base pair.
[3]
Brønsted relation
Either of the equations
where α, β, and C are constants for a given reaction series (α and β are called Brønsted exponents or Brønsted parameters). The arguments in the lg functions should be of dimension 1. Thus, reduced rate coefficients should be used: {kA} = kA/[kA] and {kHA} = kHA/[kHA], which are the reduced catalytic coefficients of reactions whose rates depend on the concentrations of acid HA or of its conjugate base A–, {KHA} = KHA/[KHA] is the reduced acid dissociation constant of HA, p is the number of equivalent acidic protons in HA, and q is the number of equivalent basic sites in A–. The chosen values of p and q should always be specified. (The charge designations of HA and A– are only illustrative.).
Note 1: The equations are often written without reduced variables, whereupon the slope α or β, obtained from a graph or least-squares analysis, is correct because it is the derivative of a logarithmic quantity, of dimension 1.
Note 2: The Brønsted relation is often termed the Brønsted catalysis law. Although justifiable on historical grounds, use of this name is not recommended, since Brønsted relations are known to apply to many uncatalyzed and pseudo-catalyzed reactions (such as simple proton [hydron] transfer reactions).
Note 3: The term pseudo-Brønsted relation is sometimes used for reactions that involve nucleophilic catalysis instead of acid–base catalysis. Various types of Brønsted parameters have been proposed such as βnuc or βlg for nucleophile or leaving group, respectively.
See also linear free-energy relation (linear Gibbs energy relation).
rev[3]
Bunnett–Olsen equations
Relations between lg([SH+]/[S]) + Ho and Ho + lg{[H+]} for base S in aqueous acid solutions, where Ho is Hammett’s acidity function and Ho + lg{[H+]} represents the activity function lg(γSγH+/γSH+) for the nitroaniline reference bases to build Ho. ϕ is an empirical parameter that is determined by the slope of the linear correlation of lg([SH+]/[S]) − lg{[H+]} vs. Ho + lg{[H+]}.
Arguments in the lg functions should be of dimension 1. Thus, concentrations should be divided by the respective unit (unless they are eliminated as in the ratio of two concentrations), i.e., the reduced quantity should be used, indicated by curly brackets.
Note 1: These equations avoid using (or defining) an acidity function for each family of bases, including those for which such a definition is not possible. In many cases, ϕ (or ϕ − 1) values for base families defining an acidity function are very similar. Broadly, the value of ϕ is related to the degree of solvation of SH+.
Note 2: These equations are obsolete, and the Cox–Yates equation, with the equivalent parameter m* (= 1 − ϕ), is now preferred.
See also Cox–Yates equation.
rev[3]
β, βnuc, βlg
Parameter in a Brønsted relation expressing the sensitivity of the rate of deprotonation to basicity.
Note: βnuc and βlg are used to correlate nucleophilic reactivity and leaving-group ability, respectively.
β-elimination
1,2-elimination
Transformation of the general type
where the central atoms A and B are commonly, but not necessarily, carbon.
See also elimination.
cage
Aggregate of molecules, generally solvent molecules in the condensed phase, that surrounds fragments formed by thermal or photochemical dissociation.
Note: Because the cage hinders the separation of the fragments by diffusion, they may preferentially react with one another (“cage effect”) although not necessarily to re-form the precursor species.
Example:
See also geminate recombination.
[3]
cage compound
Polycyclic compound capable of encapsulating another compound.
Example (adamantane, where the central cavity is large enough to encapsulate He, Ne, or Na+):
Note: A compound whose cage is occupied is called an inclusion complex.
rev[3]
canonical form
resonance form
rev[3]
captodative effect
Combined action of an electron-withdrawing (“captor”) substituent and an electron-releasing (“dative”) substituent, both attached to a radical center, on the stability of a carbon-centered radical.
Example:
rev[3]
carbanion
Generic name for an anion containing an even number of electrons and having an unshared pair of electrons on a carbon atom which satisfies the octet rule and bears a negative formal charge in its Lewis structure or in at least one of its resonance forms.
Examples:
See also radical ion.
rev[3]
carbene
Generic name for the species H2C: (“methylidene”) and substitution derivatives thereof, containing an electrically neutral bivalent carbon atom with two nonbonding electrons.
Note 1: The nonbonding electrons may have antiparallel spins (singlet state) or parallel spins (triplet state).
Note 2: Use of the alternative name “methylene” as a generic term is not recommended.
See [39].
See also diradical.
[3]
carbenium ion
Generic name for a carbocation whose electronic structure can be adequately described by two-electron-two-center bonds.
Note 1: The name implies a hydronated carbene or a substitution derivative thereof.
Note 2: The term is a replacement for the previously used term carbonium ion, which now specifies a carbocation with penta- or higher-coordinate carbons. The names provide a useful distinction between tricoordinate and pentacoordinate carbons.
Note 3: To avoid ambiguity, the name should be avoided as the root for the nomenclature of carbocations. For example, the term “ethylcarbenium ion” might refer to either CH3CH2+ (ethyl cation, ethylium) or CH3CH2CH2+ (propyl cation, propylium).
rev[3]
carbenoid
Carbene-like chemical species but with properties and reactivity differing from the free carbene, arising from additional substituents bonded to the carbene carbon.
Example: R1R2C(Cl)M (M = metal)
rev[3]
carbocation
Positive ion containing an even number of electrons and with a significant portion of the excess positive charge located on one or more carbon atoms.
Note 1: This is a general term embracing carbenium ions, all types of carbonium ions, vinyl cations, etc.
Note 2: Carbocations may be named by adding the word “cation” to the name of the corresponding radical [97, 98].
Note 3: Such names do not imply structure (e.g., whether three-coordinated or five-coordinated carbon atoms are present) [98].
See also bridged carbocation, radical ion.
See [39].
[3]
carbonium ion
Carbocation that contains at least one carbon atom with a coordination number of five or greater.
Carbocation whose structure cannot adequately be described by only two-electron two-center bonds.
Example: methanium (CH5+).
Note 1: In most of the earlier literature, this term was used for all types of carbocations, including those that are now defined as a (tricoordinate) carbenium ion.
Note 2: To avoid ambiguity, the term should be avoided as the root for the nomenclature of carbocations. For example, the name “ethylcarbonium ion” might refer to either CH3CH2+ (ethyl cation) or CH3CH2CH2+ (propyl cation).
rev[3]
carbyne
methylidyne
Generic name for the species HC·: and substitution derivatives thereof, such as EtOCO–C·: (2-ethoxy-2-oxoethylidyne), containing an electrically neutral univalent carbon atom with three non-bonding electrons.
Note: Use of the alternative name “methylidyne” as a generic term is not recommended.
[3]
Catalán solvent parameters
Quantitative measure of solvent polarity, based on the solvent’s hydrogen-bond-donor ability, hydrogen-bond-acceptor ability, polarizability, and dipolarity.
See also solvent parameter.
catalyst
Substance that increases the rate of a chemical reaction (owing to a change of mechanism to one having a lower Gibbs energy of activation) without changing the overall standard Gibbs energy change (or position of equilibrium).
Note 1: The catalyst is both a reactant and product of the reaction, so that there is no net change in the amount of that substance.
Note 2: At the molecular level, the catalyst is used and regenerated during each set of microscopic chemical events leading from a molecular entity of reactant to a molecular entity of product.
Note 3: The requirement that there be no net change in the amount of catalyst is sometimes relaxed, as in the base catalysis of the bromination of ketones, where base is consumed, but this is properly called pseudo-catalysis.
Note 4: Catalysis can be classified as homogeneous, in which only one phase is involved, and heterogeneous, in which the reaction occurs at or near an interface between phases.
Note 5: Catalysis brought about by one of the products of a reaction is called autocatalysis.
Note 6: The terms catalyst and catalysis should not be used when the added substance reduces the rate of reaction (see inhibition).
Note 7: The above definition is adequate for isothermal-isobaric reactions, but under other experimental conditions, the state function that is lowered by the catalyst is not the Gibbs activation energy but the quantity corresponding to those conditions (e.g., the Helmholtz energy under isothermal-isochoric conditions).
See [12].
See also autocatalytic reaction, bifunctional catalysis, catalytic coefficient, electron-transfer catalysis, general acid catalysis, general base catalysis, intramolecular catalysis, micellar catalysis, Michaelis–Menten kinetics, phase-transfer catalysis, pseudo-catalysis, rate of reaction, specific catalysis.
rev[3]
catalytic antibody
abzyme
monoclonal antibody with enzymatic activity
Note 1: A catalytic antibody acts by binding its antigen and catalyzing a chemical reaction that converts the antigen into desired products. Despite the existence of natural catalytic antibodies, most of them were specifically designed to catalyze desired chemical reactions.
Note 2: Catalytic antibodies are produced through immunization against a transition-state analogue for the reaction of interest. The resulting antibodies bind strongly and specifically the transition-state analogue, so that they become catalysts for the desired reaction.
Note 3: The concept of catalytic antibodies and the strategy for obtaining them were advanced by W. P. Jencks [102]. The first catalytic antibodies were finally produced in 1986 [103, 104].
catalytic coefficient
If the rate of reaction v is expressible in the form
where A, B, … are reactants and Ci represents one of a set of catalysts, then the proportionality factor ki is the catalytic coefficient of the particular catalyst Ci.
Note: Normally, the partial order of reaction (ni) with respect to a catalyst will be unity, so that ki is an (a + b +⋯+ 1)th-order rate coefficient.
[3]
catalytic cycle
Sequence of reaction steps in the form of a loop. One step is binding of a reactant to the active catalyst (sometimes formed from a precatalyst), and another step is the release of product and regeneration of the catalyst.
Example: A + B → C, catalyzed by X formed from precatalyst X′.
cation-π interaction
Noncovalent attractive force between a positive ion (metal cation, protonated Brønsted base, etc.) and a π electron system.
See [105].
chain reaction
Reaction in which one or more reactive reactive intermediates (frequently radicals) are continuously regenerated through a repetitive cycle of elementary steps (“propagation steps”).
Example: Chlorination of methane by a radical mechanism, where Cl· is continually regenerated in the chain-propagation steps:
Note: In chain polymerization reactions, reactive intermediates of the same types, generated in successive steps or cycles of steps, differ in molecular mass, as in
See [106].
See also chain transfer, initiation, termination.
[3]
chain transfer
Chemical reaction during a chain polymerization in which the active center is transferred from the growing macromolecule to another molecule or to another site on the same molecule, often by abstraction of an atom by the radical end of the growing macromolecule.
Note 1: The growth of the polymer chain is thereby terminated but a new radical, capable of chain propagation and polymerization, is simultaneously created. For the example of alkene polymerization cited for a chain reaction, the reaction
represents a chain transfer, with the radical Cl3C⋅ inducing further polymerization.
Note 2: Chain transfer also occurs in other chain reactions, such as cationic or anionic polymerization, in which case the abstraction is by the reactive cationic or anionic end of the growing chain.
See [106].
See also telomerization.
[3]
charge density
See electron density.
See [11].
[3]
charge-transfer (CT) complex
Ground-state adduct that exhibits an electronic absorption corresponding to light-induced transfer of electronic charge from one region of the adduct to another.
See [8].
rev[3]
chelation
Formation or presence of bonds (or other attractive interactions) between a single central atom (or ion) and two or more separate binding sites within the same ligand.
Note 1: A molecular entity in which there is chelation (and the corresponding chemical species) is called a chelate, while the species that binds to the central atom is called a chelant.
Note 2: The terms bidentate, tridentate, … multidentate are used to indicate the number of potential binding sites of the ligand, at least two of which must be used by the ligand in forming a chelate. For example, the bidentate ethylenediamine forms a chelate with Cu+2 in which both nitrogen atoms of ethylenediamine are bonded to copper.
Note 3: The use of the term is often restricted to metallic central atoms or ions.
Note 4: The phrase “separate binding sites” is intended to exclude cases such as [PtCl3(CH2=CH2)]–, ferrocene, and (benzene)tricarbonylchromium, in which ethene, the cyclopentadienyl group, and benzene, respectively, are considered to present single binding sites to the respective metal atom.
See also ambident, cryptand.
See also [28].
[3]
cheletropic reaction
Cycloaddition across the terminal atoms of a π system with formation of two new σ bonds to a single atom of a monocentric reagent. There is formal loss of one π bond in the substrate and an increase in the coordination number of the relevant atom of the reagent.
Example: addition of sulfur dioxide to butadiene:
Note: The reverse of this type of reaction is designated “cheletropic elimination”.
See [107].
[3]
chelotropic reaction
Alternative (and etymologically more correct) name for cheletropic reaction.
See [50].
[3]
chemical flux
ϕ
Unidirectional rate of reaction, applicable to the progress of component reaction steps in a complex system or to the progress of reactions in a system at dynamic equilibrium (in which there are no observable concentration changes with time), excluding the reverse reaction and other reaction steps.
Note 1: Chemical flux is a derivative with respect to time, and has the dimensions of amount of substance per volume transformed per time. For example, for the mechanism
Note 2: The sum of all the chemical fluxes leading to C is designated the “total chemical flux into C” (symbol
Note 3: The net rate of appearance of C is then given by
Note 4: In this system, ϕ1 (or
Note 5: Even when there is no net reaction, chemical flux can often be measured by NMR methods.
See [1].
See also order of reaction, rate-limiting step, steady state.
rev[3]
chemical reaction
Process that results in the interconversion of chemical species.
Note 1: This definition includes experimentally observable interconversions of conformers and degenerate rearrangements.
Note 2: Chemical reactions may be elementary reactions or stepwise reactions.
Note 3: Detectable chemical reactions normally involve sets of molecular entities, as indicated by this definition, but it is often conceptually convenient to use the term also for changes involving single molecular entities (i.e., “microscopic chemical events”), whose reactions can now be observed experimentally.
See also identity reaction.
rev[3]
chemical relaxation
Passage of a perturbed system toward or into chemical equilibrium.
Note 1: A chemical reaction at equilibrium can be disturbed from equilibrium by a sudden change of some external parameter such as temperature, pressure, or electric-field strength.
Note 2: In many cases, and in particular when the displacement from equilibrium is slight, the progress of the system towards equilibrium can be expressed as a first-order process
where (ceq)1 and (ceq)2 are the equilibrium amount concentrations of one of the chemical species before and after the change in the external parameter, and c(t) is its amount concentration at time t. The time parameter τ, called relaxation time, is related to the rate coefficients of the chemical reaction involved. Such measurements are commonly used to follow the kinetics of very fast reactions.
Note 3: Relaxation, or the passage toward equilibrium, is more general than chemical relaxation, and includes relaxation of nuclear spins.
See relaxation.
rev[3]
chemical shift (NMR)
δ
Variation of the resonance frequency of a nucleus in nuclear magnetic resonance (NMR) spectrometry as a consequence of its environment.
Note 1: The chemical shift of a nucleus X, δX, expressed as its frequency, νX, relative to that of a standard, νref, and defined as
For 1H and 13C NMR, the reference signal is usually that of tetramethylsilane (SiMe4).
Note 2: Chemical shift is usually reported in “parts per million” or ppm, where the numerator has unit Hz, and the denominator has unit MHz, like the spectrometer’s operating frequency.
Note 3: For historical reasons that predate Fourier-transform NMR, if a resonance signal occurs at higher frequency than a reference signal, it is said to be downfield, and if resonance occurs at lower frequency, the signal is upfield. Resonances downfield from SiMe4 have positive δ-values, and resonances upfield from SiMe4 have negative δ-values. These terms have been superseded, and deshielded and shielded are preferred for downfield and upfield, respectively.
See [110].
See also shielding.
rev[3]
chemical species
Ensemble of chemically identical molecular entities that can explore the same set of molecular energy levels on the time scale of an experiment. The term is applied equally to a set of chemically identical atomic or molecular structural units in a solid array.
Note 1: For example, conformational isomers may be interconverted sufficiently slowly to be detectable by separate NMR spectra and hence to be considered to be separate chemical species on a time scale governed by the radiofrequency of the spectrometer used. On the other hand, in a slow chemical reaction, the same mixture of conformers may behave as a single chemical species, i.e., there is virtually complete equilibrium population of the total set of molecular energy levels belonging to the two conformers.
Note 2: Except where the context requires otherwise, the term is taken to refer to a set of molecular entities containing isotopes in their natural abundance.
Note 3: The definition given is intended to embrace not only cases such as graphite and sodium chloride but also a surface oxide, where the basic structural units may not be capable of isolated existence.
Note 4: In common chemical usage, and in this Glossary, generic and specific chemical names (such as radical or hydroxide ion) or chemical formulae refer either to a chemical species or to a molecular entity.
[3]
chemically induced dynamic nuclear polarization (CIDNP)
Non-Boltzmann nuclear spin-state distribution produced in thermal or photochemical reactions, usually from colligation and diffusion or disproportionation of radical pairs, and detected by NMR spectroscopy as enhanced absorption or emission signals.
rev[3]
chemiexcitation
Generation, by a chemical reaction, of an electronically excited molecular entity from reactants in their ground electronic states.
See [8].
[3]
chemoselectivity
Feature of a chemical reagent that reacts preferentially with one of two or more different functional groups.
Note 1: A reagent has a high chemoselectivity if reaction occurs with only a limited number of different functional groups. For example, sodium tetrahydridoborate (NaBH4) is a more chemoselective reducing agent than is lithium tetrahydridoaluminate (LiAlH4). The concept has not been defined in more quantitative terms.
Note 2: The term is also applied to reacting molecules or intermediates that exhibit selectivity towards chemically different reagents.
Note 3: Usage of the term chemospecificity for 100 % chemoselectivity is discouraged.
See [113].
See also regioselectivity, stereoselectivity, stereospecificity.
[3]
chemospecificity
obsolete
See chemoselectivity.
[3]
chirality
Property of a structure that is not superimposable on its mirror image.
[3]
chirality center
chiral center (superseded)
Atom with attached groups such that the arrangement is not superimposable on its mirror image.
Note: Often this is a tetrahedral atom with four different groups attached, such as CHBrClF, or C2 of CH3CHBrCH2CH3, or the sulfur of CH3S(=O)Ph, where the lone pair is considered as a fourth group.
See [10].
See also stereogenic center.
[3]
chiral feature
Structural characteristic rendering a molecule chiral.
Examples: four different substituents on a carbon atom (chirality center), conformational helix, and chiral axis (as in allenes XCH=C=CHX).
chiral recognition
Attraction between molecules through noncovalent interactions that exhibit complementarity only between partners with specific chirality.
See also molecular recognition.
chromophore
Part (atom or group of atoms) of a molecular entity in which the electronic transition responsible for a given spectral band is approximately localized.
Note 1: The term arose originally to refer to the groupings that are responsible for a dye’s color.
Note 2: The electronic transition can often be assigned as involving n, π, π*, σ, and/or σ* orbitals whose energy difference falls within the range of the visible or UV spectrum.
Note 3: The term has been extended to vibrational transitions in the infrared.
[3]
CIDNP
Acronym for chemically induced dynamic nuclear polarization.
[3]
cine-substitution
Substitution reaction (generally aromatic) in which the entering group takes up a position adjacent to that occupied by the leaving group.
Example:
See also tele-substitution.
[3]
classical carbocation
Carbocation whose electronic structure can be adequately described by two-electron-two-center bonds, i.e., synonymous with carbenium ion.
clathrate
See host, inclusion compound.
[3]
coarctate
Feature of a concerted transformation in which the primary changes in bonding occur within a cyclic array of atoms but in which two nonbonding atomic orbitals on an atom interchange roles with two bonding orbitals.
Example: epoxidation with dimethyldioxirane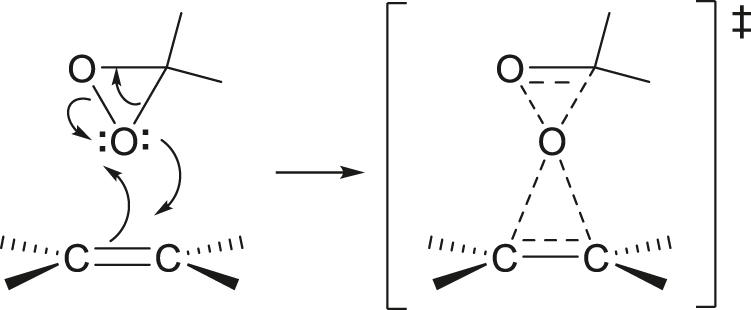
Note: Because the atomic orbitals that interchange roles are orthogonal, such a reaction does not proceed through a fully conjugated transition state and is thus not a pericyclic reaction. It is therefore not governed by the rules that express orbital symmetry restrictions applicable to pericyclic reactions.
See [116].
See also pseudopericyclic.
colligation
Formation of a covalent bond by combination or recombination of two radicals.
Note: This is the reverse of unimolecular homolysis.
Example:
[3]
collision complex
Ensemble formed by two reaction partners, where the distance between them is equal to the sum of the van der Waals radii of neighboring atoms.
See also encounter complex.
[3]
common-ion effect (on rates)
Reduction in the rate of certain reactions of a substrate RX in solution [by a path that involves a pre-equilibrium with formation of R+ (or R–) ions as reaction intermediates] caused by the addition to the reaction mixture of an electrolyte solute containing the “common ion” X– (or X+).
Example: the rate of solvolysis of chlorodiphenylmethane in acetone–water is reduced by the addition of salts of the common ion Cl–, which causes a decrease in the steady-state concentration of the diphenylmethyl cation:
Note: This retardation due to a common ion should be distinguished from the acceleration due to a salt effect of all ions.
rev[3]
compensation effect
Observation that a plot of T∆rS vs. ∆rH (frequently
Note: Frequently, such
See [117], [118], [119], [120].
See also isoequilibrium relationship, isokinetic relationship.
rev[3]
complex
Molecular entity formed by loose association involving two or more component molecular entities (ionic or uncharged) or the corresponding chemical species. The attraction between the components is often due to hydrogen-bonding or van der Waals attraction and is normally weaker than a covalent bond.
Note 1: The term has also been used with a variety of meanings in different contexts: it is therefore best avoided when a more explicit alternative is applicable, such as adduct when the association is a consequence of bond formation.
Note 2: In inorganic chemistry, the term “coordination entity” is recommended instead of “complex” [28, 121].
See also activated complex, adduct, charge transfer complex, electron-donor-acceptor complex, encounter complex, hydrogen bond, inclusion complex, σ-adduct, π-adduct, transition state, van der Waals forces.
[3]
composite reaction
Chemical reaction for which the expression for the rate of disappearance of a reactant (or rate of appearance of a product) involves rate constants of more than a single elementary reaction.
Examples: “opposing reactions” (where rate constants of two opposed chemical reactions are involved), “parallel reactions” (for which the rate of disappearance of any reactant is governed by the rate constants relating to several simultaneous reactions that form different products from a single set of reactants), and stepwise reactions.
[3]
comproportionation
Any chemical reaction of the type A′ + A″ → 2 A
Example:
Note: Other stoichiometries are possible, depending on the oxidation numbers of the species.
Reverse of disproportionation. The term “symproportionation” is also used.
See [122].
rev[3]
concerted
Feature of a process in which two or more primitive changes occur within the same elementary reaction. Such changes will normally be “energetically coupled”.
Note 1: The term “energetically coupled” means that the simultaneous progress of the primitive changes involves a transition state of lower energy than that for their successive occurrence.
Note 2: In a concerted process, the primitive changes may be synchronous or asynchronous.
See also bifunctional catalysis, potential-energy surface.
[3]
condensation
Reaction (usually stepwise) in which two or more reactants (or remote reactive sites within the same molecular entity) yield a product with accompanying formation of water or some other small molecule, e.g., ammonia, ethanol, acetic acid, hydrogen sulfide.
Note 1: The mechanism of many condensation reactions has been shown to comprise consecutive addition and elimination reactions, as in the base-catalyzed formation of (E)-but-2-enal (crotonaldehyde) from acetaldehyde, via dehydration of 3-hydroxybutanal (aldol). The overall reaction in this example is known as the aldol condensation.
Note 2: The term is sometimes also applied to cases where the formation of water or other simple molecule does not occur, as in “benzoin condensation”.
[3]
condensed formula
Linear representation of the structure of a molecular entity in which bonds are omitted.
Example: methyl 3-methylbutyl ether (isoamyl methyl ether, (CH3)2CHCH2CH2OCH3, sometimes condensed further to (CH3)2CH[CH2]2OCH3)
Note: This term is sometimes also called a line formula, because it can be written on a single line, but the line formula explicitly shows all bonds.
configuration (electronic)
Distribution of the electrons of an atom or a molecular entity over a set of one-electron wavefunctions called orbitals, according to the Pauli principle.
Note: From one configuration several states with different multiplicities may result. For example, the ground electronic configuration of the oxygen molecule (O2) is
resulting in
[3]
configuration (molecular)
Arrangement in space of the atoms of a molecular entity that distinguishes it from any other molecular entity having the same molecular formula and connectivity and that is not due to conformational differences (rotation about single bonds).
See [10].
[3]
conformations
Different spatial arrangements of a molecular entity that can be interconverted by rotation about one or more formally single bonds.
Note 1: Different conformations are often not considered to be stereoisomeric, because interconversion is rapid at the temperature under consideration.
Note 2: Different or equivalent spatial arrangements of ligands about a central atom, such as those interconverted by pyramidal inversion (of amines) or Berry pseudorotation (as of PF5) and other “polytopal rearrangements”, are sometimes considered conformations, but they are properly described as configurations.
See [10].
rev[3]
conformational isomers
conformers
conformer
Conformation of a molecular entity that corresponds to a minimum on the potential-energy surface of that molecular entity.
Note: The distinction between conformers and isomers is the height of the barrier for interconversion. Isomers are stable on macroscopic timescales because the barrier for interconversion is high, whereas a conformer cannot persist on a macroscopic timescale because the interconversion between conformations is achieved rapidly.
See [10].
conjugate acid
Brønsted acid BH+ formed on protonation (hydronation) of the base B.
Note 1: B is called the conjugate base of the acid BH+.
Note 2: The conjugate acid always carries one unit of positive charge more than the base, but the absolute charges of the species are immaterial to the definition. For example: the Brønsted acid HCl and its conjugate base Cl– constitute a conjugate acid–base pair and so do NH4+ and its conjugate base NH3.
rev[3]
conjugated system, conjugation
Molecular entity whose structure may be represented as a system of alternating multiple and single (or σ) bonds: e.g.,
Note: In such systems, conjugation is the interaction of one p-orbital with another p-orbital (or d-orbital) across an intervening σ bond, including the analogous interaction involving a p-orbital containing an unshared electron pair, e.g.,
or the interaction across a double bond whose π system does not interact with the p orbitals of the conjugated system, as in
See also cross-conjugation, delocalization, resonance, through-conjugation.
[3]
connectivity
Description of which atoms are bonded to which other atoms.
Note: Connectivity is often displayed in a line formula or other structure showing which atoms are bonded to which other atoms, but with minimal or no indication of bond multiplicity.
Example: The connectivity of propyne is specified by CH3CCH.
rev[3]
conrotatory
Stereochemical feature of an electrocyclic reaction in which the substituents at the interacting termini of the conjugated system rotate in the same sense (both clockwise or both counterclockwise).
See also disrotatory.
rev[3]
conservation of orbital symmetry
An approach to understanding pericyclic reactions that focuses on a symmetry element (e.g., a reflection plane) that is retained along a reaction pathway. If each of the singly or doubly occupied orbitals of the reactant(s) is of the same symmetry as a similarly occupied orbital of the product(s), that pathway is “allowed” by orbital symmetry conservation. If instead a singly or doubly occupied orbital of the reactant(s) is of the same symmetry as an unoccupied orbital of the product(s), and an unoccupied orbital of the reactant(s) is of the same symmetry as a singly or doubly occupied orbital of the product(s), that pathway is “forbidden” by orbital symmetry conservation.
Note 1: This principle permits the qualitative construction of correlation diagrams to show how molecular orbitals transform and how their energies change during chemical reactions.
Note 2: Considerations of orbital symmetry are frequently grossly simplified in that, for example, the π and π* orbitals of a carbonyl group in an asymmetric molecule are treated as having the same topology (pattern of local nodal planes) as those of a symmetric molecule (e.g., CH2=O), despite the absence of formal symmetry elements.
See also orbital symmetry.
constitutional isomers
Species (or molecular entities) with the same atomic composition (molecular formula) but with different connectivity.
Note: The term structural isomers is discouraged because all isomers differ in structure and because isomers may be constitutional, configurational, or conformational.
See [10].
contributing structure
resonance form
rev[3]
coordinate covalent bond
dative bond
coordination
Formation of a covalent bond, the two shared electrons of which come from only one of the two partners linked by the bond, as in the reaction of a Lewis acid and a Lewis base to form a Lewis adduct; alternatively, the bonding formed in this way.
Note: In the former sense, it is the reverse of unimolecular heterolysis.
See also dative bond, π-adduct.
rev[3]
coordination number
Number of other atoms directly linked to a specified atom in a chemical species regardless of the number of electrons in the bonds linking them [28, Rule IR-10.2.5]. For example, the coordination number of carbon in methane or of phosphorus in triphenylphosphane oxide (triphenylphosphine oxide) is four whereas the coordination number of phosphorus is five in phosphorus pentafluoride.
Note: The term is used in a different sense in the crystallographic description of ionic crystals.
rev[3]
coronate
See crown ether.
rev[3]
correlation analysis
Use of empirical correlations relating one body of experimental data to another, with the objective of finding quantitative relationships among the factors underlying the phenomena involved. Correlation analysis in organic chemistry often uses linear Gibbs-energy relations (formerly linear free-energy relation, LFER) for rates or equilibria of reactions, but the term also embraces similar analysis of physical (most commonly spectroscopic) properties and of biological activity.
See [124], [125], [126], [127], [128].
See also linear free-energy relation, quantitative structure–activity relationship (QSAR).
[3]
coupling constant
spin-spin coupling constant
J
(unit: Hz)
Quantitative measure of nuclear spin–spin coupling in nuclear magnetic resonance spectroscopy.
Note: Spin–spin coupling constants have been correlated with atomic hybridization and with molecular conformations.
rev[3]
covalent bond
Stabilizing interaction associated with the sharing of electron pairs between two atomic centers of a molecular entity, leading to a characteristic internuclear distance.
See also agostic, coordination, hydrogen bond, multi-center bond.
See [7].
rev[3]
Cox–Yates equation
Generalization of the Bunnett–Olsen equation of the form
where [H+] is the amount concentration of acid and X is the activity-coefficient ratio
lg(γsγH+/γSH+) for an arbitrary reference base, pKSH+ is the thermodynamic dissociation constant of SH+, and m* is an empirical parameter derived from linear regression of the left-hand side vs. X. Arguments in the lg functions should be unitless. Thus, the reduced quantities should be used: {[H+]} is [H+] divided by its unit.
Note: The function X is called excess acidity because it gives a measure of the difference between the acidity of a solution and that of an ideal solution of the same concentration. In practice, X = –(Ho + lg{[H+]}) and m* = 1 − ϕ, where Ho is the Hammett acidity function and ϕ is the slope in the Bunnett–Olsen equation.
See Bunnett–Olsen equation.
rev[3]
critical micellization concentration (cmc)
critical micelle concentration
Relatively small range of concentrations separating the limit below which virtually no micelles are detected and the limit above which virtually all additional surfactant molecules form micelles.
Note 1: Many physical properties of surfactant solutions, such as conductivity or light scattering, show an abrupt change at a particular concentration of the surfactant, which can be taken as the cmc.
Note 2: As values obtained using different properties are not quite identical, the method by which the cmc is determined should be clearly stated.
See [135].
See also inverted micelle.
rev[3]
cross-conjugation
Phenomenon of three conjugated groups, two pairs of which exhibit conjugation but without through-conjugation of all three, as in 2-phenylallyl, benzoate anion, divinyl ether, or m-xylylene [1,3-C6H4(CH2)2].
See [64].
rev[3]
crown ether
Molecular entity comprising a monocyclic ligand assembly that contains three or more binding sites held together by covalent bonds and capable of binding a guest in a central (or nearly central) position. The adducts formed are sometimes known as “coronates”. The best known members of this group are macrocyclic polyethers, such as 18-crown-6, containing several repeating units –CR2–CR2O– (where R is most commonly H).
See also host.
rev[3]
cryptand
Molecular entity comprising a cyclic or polycyclic assembly of binding sites, which contains three or more binding sites held together by covalent bonds and which defines a molecular cavity in such a way as to bind (and thus “hide” in the cavity) another molecular entity, the guest (a cation, an anion, or a neutral species), more strongly than do the separate parts of the assembly (at the same total concentration of binding sites).
Note 1: The adduct thus formed is called a “cryptate”. The term is usually restricted to bicyclic or oligocyclic molecular entities.
Example:
Note 2: Corresponding monocyclic ligand assemblies (crown ether) are sometimes included in this group, if they can be considered to define a cavity in which a guest can hide. The terms “podand” and “spherand” are used for certain specific ligand assemblies. Coplanar cyclic polydentate ligands, such as porphyrins, are not normally regarded as cryptands.
See [138].
See also host. See also [139].
[3]
curly arrows
Symbols for depicting the flow of electrons in a chemical reaction or to generate additional resonance forms.
Note 1: The tail of the curly arrow shows where an electron pair originates, and the head of the curly arrow shows where the electron pair goes.
Note 2: Single-headed curly arrows are used to depict the flow of unpaired electrons.
Examples: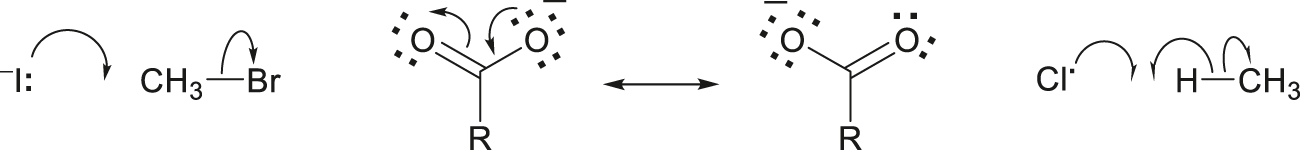
See electron pushing.
Curtin–Hammett principle
Statement that in a chemical reaction that yields one product (X) from one isomer (A′) and a different product (Y) from another isomer (A″) (and provided these two isomers are rapidly interconvertible relative to the rate of product formation, whereas the products do not undergo interconversion) the product composition is not directly related to the relative concentrations of the isomers in the substrate; it is controlled only by the difference in standard Gibbs energies (
Note 1: The product composition is given by [Y]/[X] = (kYk1)/(k−1kX) or KckY/kX, where Kc = [A″]/[A′] is the equilibrium constant, and where kY and kX are the respective rate constants of their reactions; these parameters are usually unknown.
Note 2: The energy diagram below represents the transformation of rapidly interconverting isomers A′ and A″ into products X and Y.

Note 3: A related concept is the Winstein-Holness equation for the overall rate of product formation in a system of rapidly interconverting isomers A′ and A″. The overall rate constant k is given by
where x′ and x″ are the respective mole fractions of the isomers at equilibrium [14]. However, because x′ and x″ are simple functions of Kc, the apparent dependence of the overall rate on the relative concentrations of A′ and A″ disappears. The overall rate constant is the sum kX + KckY, which is a function of the same quantities that determine the product ratio.
See [140]. See also [14, 141, 142].
rev[3]
cybotactic region
That part of a solution in the vicinity of a solute molecule in which the ordering of the solvent molecules is modified by the presence of the solute molecule. The term solvent “cosphere” of the solute has also been used.
[3]
cyclization
Formation of a ring compound from a chain by formation of a new bond.
See also annulation.
[3]
cycloaddition
Reaction in which two or more unsaturated molecules (or parts of the same molecule) combine with the formation of a cyclic adduct in which there is a net reduction of the bond multiplicity.
Note 1: The following two systems of notation have been used for the more detailed specification of cycloadditions, of which the second, more recent system (described under (2)) is preferred:
An (i+j+…) cycloaddition is a reaction in which two or more molecules (or parts of the same molecule), respectively, provide units of i, j,… linearly connected atoms: these units become joined at their respective termini by new σ bonds so as to form a cycle containing (i+j+…) atoms. In this notation, (a) a Diels–Alder reaction is a (4+2) cycloaddition, (b) the initial reaction of ozone with an alkene is a (3+2) cycloaddition, and (c) the reaction of norbornadiene below is a (2+2+2) cycloaddition. (Parentheses are used to indicate the numbers of atoms, but brackets are also used.)
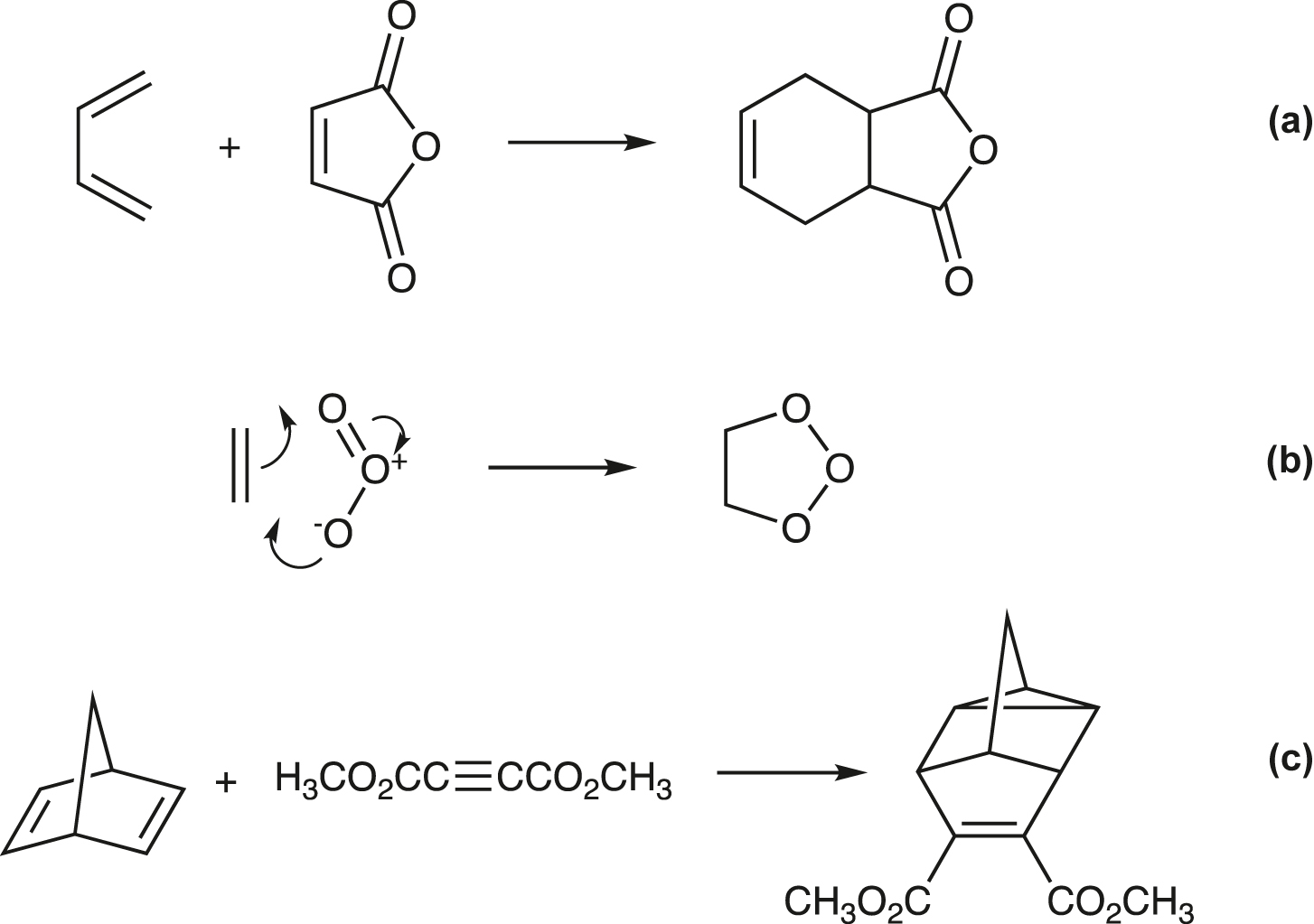
The symbolism [i+j+…] for a cycloaddition identifies the numbers i, j,… of electrons in the interacting units that participate in the transformation of reactants to products. In this notation the reaction (a) and (b) of the preceding paragraph would both be described as [2+4] cycloadditions, and (c) as a [2+2+2] cycloaddition. The symbol a or s (a: antarafacial, s: suprafacial) is often added (usually as a subscript) after the number to designate the stereochemistry of addition to each fragment. A subscript specifying the orbitals, viz., σ, π (with their usual significance), or n (for an orbital associated with a single atom), may be added as a subscript before the number. Thus, the normal Diels–Alder reaction is a [4s+2s] or [π4s + π2s] cycloaddition, whereas the reaction
 would be a [14a+2s] or [π14a + π2s] cycloaddition, leading to the stereoisomer shown, with hydrogens anti. (Brackets are used to indicate the numbers of electrons, and they are also used instead of parentheses to denote the numbers of atoms.)
would be a [14a+2s] or [π14a + π2s] cycloaddition, leading to the stereoisomer shown, with hydrogens anti. (Brackets are used to indicate the numbers of electrons, and they are also used instead of parentheses to denote the numbers of atoms.)
Note 2: Cycloadditions may be pericyclic reactions or (non-concerted) stepwise reactions. The term “dipolar cycloaddition” (Huisgen reaction) is used for cycloadditions of 1,3-dipolar compounds.
See also cheletropic reaction, ene reaction, pericyclic reaction.
rev[3]
cycloelimination
Reverse of cycloaddition. The term is preferred to the synonyms “cycloreversion”, “retro-addition”, and “retrocycloaddition”.
[3]
cycloreversion
obsolete
See cycloelimination.
[3]
dative bond
obsolescent
Coordination bond formed between two chemical species, one of which serves as a donor and the other as an acceptor of the electron pair that is shared in the bond.
Examples: the N–B bond in H3N+–B–H3 and the S–O bond in (CH3)2S+–O–.
Note 1: A distinctive feature of dative bonds is that their minimum-energy rupture in the gas phase or in inert solvent follows the heterolytic bond-cleavage path.
Note 2: The term is obsolescent because the distinction between dative bonds and ordinary covalent bonds is not useful, in that the precursors of the bond are irrelevant: H3N+–B–H3 is the same molecule, with the same bonds, regardless of whether the precursors are considered to have been H3N + BH3 or H3N+ + –BH3.
See [7].
See coordination.
rev[3]
degenerate chemical reaction
See identity reaction.
[3]
degenerate rearrangement
Chemical reaction in which the product is indistinguishable (in the absence of isotopic labelling) from the reactant.
Note 1: The term includes both “degenerate intramolecular rearrangements” and reactions that involve intermolecular transfer of atoms or groups (“degenerate intermolecular reactions”): both are degenerate isomerizations.
Note 2: The occurrence of degenerate rearrangements may be detectable by isotopic labelling or by dynamic NMR techniques. For example: the [3,3]sigmatropic rearrangement of hexa-1,5-diene (Cope rearrangement),
Note 3: Synonymous but less preferable terms are “automerization”, “permutational isomerism”, “isodynamic transformation”, and “topomerization”.
See [147].
See also fluxional, molecular rearrangement, narcissistic reaction, valence isomer.
[3]
delocalization
Quantum-mechanical concept most usually applied in organic chemistry to describe the redistribution of π electrons in a conjugated system, where each link has a fractional double-bond character or a non-integer bond order, rather than π electrons that are localized in double or triple bonds.
Note 1: There is a corresponding “delocalization energy”, identifiable with the stabilization of the system relative to a hypothetical alternative in which formal (localized) single and double bonds are present. Some degree of delocalization is always present and can be estimated by quantum mechanical calculations. The effects are particularly evident in aromatic systems and in symmetrical molecular entities in which a lone pair of electrons or a vacant p-orbital is conjugated with a double bond (e.g., carboxylate ions, nitro compounds, enamines, and the allyl cation).
Note 2: Delocalization in such species may be represented by partial bonds or by resonance (here symbolized by a two-headed arrow) between resonance forms.
These examples also illustrate the concomitant delocalization of charge in ionic conjugated systems. Analogously, delocalization of the spin of an unpaired electron occurs in conjugated radicals.
Note 3: Delocalization is not limited to π electrons. Hyperconjugation is the delocalization of electrons of σ bonds.
See [7].
[3]
dendrimer
Large molecule constructed from a central core with repetitive branching and multiple functional groups at the periphery.
See [148].
Example: (with 48 CH2OH at the periphery)
Note: The name comes from the Greek δενδρον (dendron), which translates to “tree”. Synonymous terms include arborols and cascade molecules.
deshielding
See shielding.
[3]
detachment
Reverse of attachment.
[3]
detailed balancing
Principle that when equilibrium is reached in a reaction system (containing an arbitrary number of components and reaction paths), as many atoms, in their respective molecular entities, will pass forward in a given finite time interval as will pass backward along each individual path.
Note 1: It then follows that the reaction path in the reverse direction must in every detail be the reverse of the reaction path in the forward direction (provided that the system is at equilibrium).
Note 2: The principle of detailed balancing is a consequence for macroscopic systems of the principle of microscopic reversibility.
[3]
diamagnetism
Property of substances having a negative magnetic susceptibility (χ), whereby they are repelled out of a magnetic field.
See also paramagnetism.
[3]
diastereoisomerism
Stereoisomerism other than enantiomerism.
See diastereoisomers [10].
[3]
diastereomeric excess
diastereoisomeric excess
x 1 − x2, where x1 and x2 (with x1 + x2 = 1) are the mole fractions of two diastereoisomers in a mixture, or the fractional yields of two diastereoisomers formed in a reaction.
Note: Frequently this term is abbreviated to d.e.
See stereoselectivity, diastereoisomers.
See [10].
diastereomeric ratio
x 1/x2, where x1 and x2 are the mole fractions of two diastereoisomers in a mixture formed in a reaction.
Note: Frequently this term is abbreviated to d.r.
See stereoselectivity, diastereoisomers.
See [10].
diastereoisomers
diastereomers
Stereoisomers not related as mirror images of each other.
Note: Diastereoisomers are characterized by differences in physical properties and by differences in chemical behavior toward chiral as well as achiral reagents.
See [10].
diastereoselectivity
Preferential formation in a chemical reaction of one diastereoisomer over another.
Note: This can be expressed quantitatively by the diastereoisomeric excess or by the diastereomeric ratio, which is preferable because it is more closely related to a Gibbs-energy difference.
See [10].
See selectivity.
dielectric constant
obsolete
See [11].
See permittivity (relative).
rev[3]
dienophile
Ene or yne component of a Diels–Alder reaction, including compounds with hetero-double bonds and hetero-triple bonds.
See cycloaddition.
rev[3]
diffusion-controlled rate
See encounter-controlled rate, microscopic diffusion control. Contrast mixing control.
[3]
dimerization
Transformation of a molecular entity A to give a molecular entity A2.
Examples:
See also association.
[3]
Dimroth–Reichardt ET parameter
Quantitative measure of solvent polarity, based on the wavelength, λmax, of the longest-wavelength intramolecular charge-transfer absorption band of the solvatochromic betaine dye 2,6-diphenyl-4-(2,4,6-triphenylpyridin-1-ium-1-yl)phenolate.
See solvent parameter.
rev[3]
dipolar aprotic solvent
See dipolar non-HBD solvent.
rev[3]
dipolar non-HBD solvent
Non-hydrogen-bond donating solvent
Solvent with a comparatively high relative permittivity (“dielectric constant”), greater than ca. 15, and composed of molecules that have a sizable permanent dipole moment and that, although it may contain hydrogen atoms, cannot donate suitably labile hydrogen atoms to form strong solvent–solute hydrogen bonds.
Examples: dimethyl sulfoxide, acetonitrile, acetone, as contrasted with methanol and N-methylformamide.
Note 1: The term “dipolar” refers to solvents whose molecules have a permanent dipole moment, in contrast to solvents whose molecules have no permanent dipole moment and should be termed “apolar” or “nonpolar”.
Note 2: Non-HBD solvents are often called aprotic, but this term is misleading because a proton can be removed by a sufficiently strong base. The aprotic nature of a solvent molecule means that its hydrogens are in only covalent C–H bonds and not in polar O–Hδ+ or N–Hδ+ bonds that can serve as hydrogen-bond donors. Use of “aprotic” is therefore discouraged, unless the context makes the term unambiguous.
Note 3: It is recommended to classify solvents according to their capability to donate or not donate, as well as to accept or not accept, hydrogen bonds to or from the solute, as follows:
Hydrogen-bond donating solvents (short: HBD solvents), formerly protic solvents
Non-hydrogen-bond donating solvents (short: non-HBD solvents), formerly aprotic solvents
Hydrogen-bond accepting solvents (short: HBA solvents)
Non-hydrogen-bond accepting solvents (short: non-HBA solvents)
dipole–dipole excitation transfer
Förster resonance-energy transfer (FRET)
See [8].
dipole–dipole interaction
Intermolecular or intramolecular interaction between molecules or groups having a permanent electric dipole moment. The strength of the interaction depends on the distance and relative orientation of the dipoles.
Note: A dipole/dipole interaction is a simplification of the electrostatic interactions between molecules that originate from asymmetries in the electron densities. Such interactions can be described more correctly by the use of higher-order multipole moments.
See also van der Waals forces.
rev[3]
dipole-induced dipole forces
See van der Waals forces.
[3]
diradical
biradical
Even-electron molecular entity with two (possibly delocalized) radical centers that act nearly independently of each other, e.g.,
Note 1: Species in which the two radical centers interact significantly are often referred to as “diradicaloids”. If the two radical centers are located on the same atom, the species is more properly referred to by its generic name: carbene, nitrene, etc.
Note 2: The lowest-energy triplet state of a diradical lies below or at most only a little above its lowest singlet state (usually judged relative to kBT, the product of the Boltzmann constant kB and the absolute temperature T). If the two radical centers interact significantly, the singlet state may be more stable. The states of those diradicals whose radical centers interact particularly weakly are most easily understood in terms of a pair of local doublets.
Note 3: Theoretical descriptions of low-energy states of diradicals display two unsaturated valences: the dominant valence-bond structures have two dots, the low-energy molecular orbital configurations have only two electrons in two approximately nonbonding molecular orbitals, and two of the natural orbitals have occupancies close to one.
See [8, 158], [159], [160], [161], [162], [163].
See also carbene, nitrene.
[3]
dispersion forces
See London forces, van der Waals forces.
[3]
disproportionation
Any chemical reaction of the type
Examples:
Note 1: The reverse of disproportionation is called comproportionation.
Note 2: A special case of disproportionation (or “dismutation”) is “radical disproportionation”, exemplified by
Note 3: A somewhat more restricted usage of the term prevails in inorganic chemistry, where A, A′, and A″ are of different oxidation states.
[3]
disrotatory
Stereochemical feature of an electrocyclic reaction in which the substituents at the interacting termini of the conjugated system rotate in opposite senses (one clockwise and the other counterclockwise).
See also conrotatory.
rev[3]
dissociation
Separation of a molecular entity into two or more molecular entities (or any similar separation within a polyatomic molecular entity).
Example:
Note 1: Although the separation of the constituents of an ion pair into free ions is a dissociation, the ionization that produces the ion pair is not a dissociation because the ion pair is a single molecular entity.
Note 2: The reverse of dissociation is association.
rev[3]
distonic ion
Radical ion in which charge and radical sites are separated.
Example: ·CH2CH2OCH2+
rev[3]
distortion interaction model
activation strain model
Method for analyzing activation energy as the sum of the energies to distort the reactants into the geometries they have in transition states plus the energy of interaction between the two distorted reactants.
See [166].
distribution ratio
partition ratio
Ratio of concentrations of a solute in a mixture of two immiscible phases at equilibrium.
See also Hansch constant.
See [167].
di-π-methane rearrangement
Photochemical reaction of a molecular entity with two π-systems separated by a saturated carbon, to form an ene-substituted cyclopropane.
Pattern:
donicity
also called donor number, DN, which is a misnomer
Quantitative experimental measure of the Lewis basicity of a molecule B, expressed as the negative enthalpy of the formation of the 1:1 complex B·SbCl5.
Note: donicity is often used as a measure of a solvent’s basicity.
See also acceptor parameter, Lewis basicity.
downfield
superseded but still widely used in NMR to mean deshielded.
See chemical shift, shielding.
rev[3]
driving force
Negative of the Gibbs energy change (∆rG°) on going from the reactants to the products of a chemical reaction under standard conditions. Also called affinity.
Qualitative term that relates the favorable thermodynamics of a reaction to a specific feature of molecular structure, such as the conversion of weaker bonds into stronger (CH3–H + Br–Br → CH3–Br + H–Br), neutralization of an acid (Claisen condensation of 2 CH3COOEt + EtO– to CH3COCH–COOEt + EtOH), or increase in entropy (cycloelimination of cyclohexene to butadiene + ethylene).
Note 1: This term is a misnomer, because favorable thermodynamics is due to energy, not force.
Note 2: This term has also been used in connection with photoinduced electron transfer reactions, to indicate the negative of the estimated standard Gibbs energy change for the outer sphere electron transfer (∆ETG°) [8].
rev[3]
dual substituent-parameter equation
Any equation that expresses substituent effects in terms of two parameters.
Note: In practice, the term is used specifically for an equation for modeling the effects of meta- and para-substituents X on chemical reactivity, spectroscopic properties, etc. of a probe site in benzene or other aromatic system.
where PX is the magnitude of the property for substituent X, expressed relative to the property for X = H; σI and σR are inductive (or polar) and resonance substituent constants, respectively, there being various scales for σR; ρI and ρR are the corresponding regression coefficients.
See also extended Hammett equation, Yukawa–Tsuno equation.
[3]
dynamic NMR
NMR spectroscopy of samples undergoing chemical reactions.
Note: Customarily, this does not apply to samples where the composition of the sample, and thus its NMR spectrum, changes with time, but rather to samples at equilibrium, without any net reaction. The occurrence of chemical reactions is manifested by features of the NMR line-shape or by magnetization transfer.
See chemical flux.
dyotropic rearrangement
Process in which two σ bonds simultaneously migrate intramolecularly.
Example
rev[3]
educt
deprecated: usage strongly discouraged
starting material, reactant
Note: This term should be avoided and replaced by reactant, because it means “something that comes out”, not “something that goes in”.
rev[3]
effective charge
Z eff
Net positive charge experienced by an electron in a polyelectronic atom, which is less than the full nuclear charge because of shielding by the other electrons.
rev[3]
effective molarity
effective concentration
Ratio of the first-order rate constant or equilibrium constant of an intramolecular reaction involving two functional groups within the same molecular entity to the second-order rate constant or equilibrium constant of an analogous intermolecular elementary reaction.
Note: This ratio has unit of concentration, mol dm−3 or mol L−1, sometimes denoted by M.
See [177].
See also intramolecular catalysis.
[3]
eighteen-electron rule
Electron-counting rule that the number of nonbonding electrons at a metal plus the number of electrons in the metal–ligand bonds should be 18.
Note: The 18-electron rule in transition-metal chemistry is an analogue of the Lewis octet rule.
[3]
electrocyclic reaction
electrocyclization
Molecular rearrangement that involves the formation of a σ bond between the termini of a fully conjugated linear π-electron system (or a linear fragment of a π-electron system) and a decrease by one in the number of π bonds, or the reverse of that process.
Examples:
Note: The stereochemistry of such a process is termed “conrotatory” if the substituents at the interacting termini of the conjugated system both rotate in the same sense, as in or “disrotatory” if one terminus rotates in a clockwise and the other in a counterclockwise sense, as in
or “disrotatory” if one terminus rotates in a clockwise and the other in a counterclockwise sense, as in
See also pericyclic reaction.
[3]
electrofuge
Leaving group that does not carry away the bonding electron pair.
Examples: In the nitration of benzene by NO2+, H+ is the electrofuge, and in an SN1 reaction, the carbocation is the electrofuge.
Note 1: Electrofugality characterizes the relative rates of atoms or groups to depart without the bonding electron pair. Electrofugality depends on the nature of the reference reaction and is not the reverse of electrophilicity [178].
Note 2: For electrofuges in SN1 reactions, see [179].
See also electrophile, nucleofuge.
rev[3]
electromeric effect
obsolete
rev[3]
electron acceptor
Molecular entity to which an electron may be transferred.
Examples: 1,4-dinitrobenzene,1,1’-dimethyl-4,4’-bipyridinium dication, and benzophenone.
Note 1: A group that accepts electron density from another group is not called an electron acceptor but an electron-withdrawing group.
Note 2: A Lewis acid is not called an electron acceptor but an electron-pair acceptor.
rev[3]
electron affinity
Energy released when an additional electron (without excess energy) attaches itself to a molecular entity (often electrically neutral but not necessarily).
Note 1: Equivalent to the minimum energy required to detach an electron from a singly charged negative ion.
Note 2: Measurement of electron affinities is possible only in the gas phase, but there are indirect methods for evaluating them from solution data, such as polarographic half-wave potentials or charge-transfer spectra.
rev[3]
electron capture
Transfer of an electron to a molecular entity, resulting in a molecular entity of (algebraically) increased negative charge.
electron-deficient bond
Bond between adjacent atoms that is formed by fewer than two electrons.
Example:
Note: The B–H–B bonds are also called “two-electron three-center bonds”.
[3]
electron density
The electron density at a point with coordinates x, y, z in an atom or molecular entity is the product of the probability P(x,y,z) (unit: m−3) of finding an electron at that point with the volume element dx dy dz (unit: m3).
Note: For many purposes (e.g., X-ray scattering and forces on atoms), the system behaves as if the electrons were spread out into a continuous distribution, which is a manifestation of the wave–particle duality.
See also atomic charge, charge density.
rev[3]
electron donor
Molecular entity that can transfer an electron to another molecular entity or to the corresponding chemical species.
Note 1: After the electron transfer, the two entities may separate or remain associated.
Note 2: A group that donates electron density to another group is an electron-donating group, regardless of whether the donation is of σ or π electrons.
Note 3: A Lewis base is not called an electron donor, but an electron-pair donor.
See also electron acceptor.
rev[3]
electron-donor-acceptor complex
obsolete
charge-transfer complex.
See also adduct, coordination.
electron-pair acceptor
Lewis acid.
[3]
electron-pair donor
Lewis base.
[3]
electron pushing
Method using curly arrows for showing the formal movement of an electron pair (from a lone pair or a σ or π bond) or of an unpaired electron, in order to generate additional resonance forms or to denote a chemical reaction.
Note 1: The electron movement may be intramolecular or intermolecular.
Note 2: When a single electron is transferred, a single-headed curly arrow or “fish-hook” is used, but the electron movement in the opposite direction is redundant and sometimes omitted.
Examples:
electron transfer
Transfer of an electron from one molecular entity to another or between two localized sites in the same molecular entity.
See also inner sphere (electron transfer), Marcus equation, outer sphere (electron transfer).
[3]
electron-transfer catalysis
Process describing a sequence of reactions such as shown in equations (1)–(3), leading from A to B, via species A·– and B·– that have an extra electron:
Note 1: An analogous sequence involving radical cations (A·+, B·+) also occurs.
Note 2: The most notable example of electron-transfer catalysis is the SRN1 (or T+DN+AN) reaction of haloarenes with nucleophiles.
Note 3: The term has its origin in an analogy to acid–base catalysis, with the electron instead of the proton. However, there is a difference between the two catalytic mechanisms, as the electron is not a true catalyst but rather behaves as the initiator of a chain reaction. “Electron-transfer induced chain reaction” is a more appropriate term for the mechanism described by equations (1)–(3).
[3]
electronation
obsolete
See reduction.
rev[3]
electronegativity
Measure of the power of an atom to attract electrons to itself.
Note 1: The concept has been quantified by a number of authors. The first, due to Pauling, is based on bond-dissociation energies, Ed (unit: eV), and is anchored by assigning the electronegativity of hydrogen as χr,H = 2.1. For atoms A and B
where χr denotes the Pauling relative electronegativity; it is relative in the senses that the values are of dimension 1 and that only electronegativity differences are defined on this empirical scale. See [11], section 2.5.
Note 2: Alternatively, the electronegativity of an element, according to the Mulliken scale, is the average of its atomic ionization energy and electron affinity. Other scales have been developed by Allred and Rochow, by Sanderson, and by Allen.
Note 3: Absolute electronegativity is property derived from density functional theory [7].
See [75, 184], [185], [186], [187], [188], [189], [190].
rev[3]
electronic effects (of substituents)
Changes exerted by a substituent on a molecular property or molecular reactivity, often distinguished as inductive (through-bond polarization), through-space electrostatics (field effect), or resonance, but excluding steric.
Note 1: The obsolete terms mesomeric and electromeric are discouraged.
Note 2: Quantitative scales of substituent effects are available.
See also polar effect.
rev[3]
electrophile (n.), electrophilic (adj.)
Reagent that forms a bond to its reaction partner (the nucleophile) by accepting both bonding electrons from that partner.
Note 1: Electrophilic reagents are Lewis acids.
Note 2: “Electrophilic catalysis” is catalysis by Lewis acids.
Note 3: The term “electrophilic” is also used to designate the apparent polar character of certain radicals, as inferred from their higher relative reactivities with reaction sites of higher electron density.
See also electrophilicity.
[3]
electrophilic substitution
Heterolytic reaction in which the entering group adds to a nucleophile and in which the leaving group, or electrofuge, relinquishes both electrons to its reaction partner, whereupon it becomes another potential electrophile.
Example (azo coupling):
Note: It is arbitrary to emphasize the electrophile and ignore the feature that this is also a nucleophilic substitution, but the distinction depends on the electrophilic nature of the reactant that is considered to react with the substrate.
See also substitution.
electrophilicity
Relative reactivity of an electrophile toward a common nucleophile.
Note: The concept is related to Lewis acidity. However, whereas Lewis acidity is measured by relative equilibrium constants toward a common Lewis base, electrophilicity is measured by relative rate constants for reactions of different electrophilic reagents towards a common nucleophilic substrate.
See also nucleophilicity.
rev[3]
element effect
Ratio of the rate constants of two reactions that differ only in the identity of the element in the leaving group.
Example: kBr/kCl for the reaction of N3– (azide) with CH3Br or CH3Cl.
rev[3]
elementary reaction
Reaction for which no reaction intermediates have been detected or need to be postulated in order to describe the chemical reaction on a molecular scale. An elementary reaction is assumed to occur in a single step and to pass through only one transition state or none.
See [12].
See also composite reaction, stepwise reaction.
rev[3]
elimination
Reverse of an addition reaction.
Note 1: In an elimination, two groups (called eliminands) are lost, most often from two different centers [1,2-elimination (β-elimination) or 1,3-elimination, etc.] with concomitant formation of an unsaturation (double bond or triple bond) in the molecule, or formation of a ring.
Note 2: If the groups are lost from a single carbon or nitrogen center (1,1-elimination, α-elimination), the resulting product is a carbene or nitrene, respectively.
rev[3]
empirical formula
List of the elements in a chemical species, with integer subscripts indicating the simplest possible ratios of all elements.
Note 1: In organic chemistry, C and H are listed first, then the other elements in alphabetical order.
Note 2: This differs from the molecular formula, in which the subscripts indicate how many of each element is included and which is an integer multiple of the empirical formula. For example, the empirical formula of glucose is CH2O while its molecular formula is C6H12O6.
Note 3: The empirical formula is the information provided by combustion analysis, which has been largely superseded by mass spectrometry, which provides the molecular formula.
enantiomer
One of a pair of stereoisomeric molecular entities that are non-superimposable mirror images of each other.
See [10].
See also stereoisomers.
[3]
enantiomeric excess
Absolute value of the difference between the mole fractions of two enantiomers:
where x+ + x− = 1.
Note: Enantiomeric excess can be evaluated experimentally from the observed specific optical rotatory power [α]obs, relative to [α]max, the (maximum) specific optical rotatory power of a pure enantiomer:
and also by chiral chromatography, NMR, and MS methods.
See [10].
rev[3]
enantiomeric ratio
mole fraction of one enantiomer in a mixture divided by the mole fraction of the other.
where x+ + x– = 1
See [10].
rev[3]
enantioselectivity
See stereoselectivity.
[3]
encounter complex
Complex of molecular entities produced at an encounter-controlled rate, and which occurs as an intermediate in a reaction.
Note 1: When the complex is formed from two molecular entities, it is called an “encounter pair”. A distinction between encounter pairs and (larger) encounter complexes may be relevant for mechanisms involving pre-association.
Note 2: The separation between the entities is small compared to the diameter of a solvent molecule.
See also [8].
rev[3]
encounter-controlled rate
Rate of reaction corresponding to the rate at which the reacting molecular entities encounter each other. This is also known as the “diffusion-controlled rate”, as rates of encounter are themselves controlled by diffusion rates (which in turn depend on the viscosity of the medium and the dimensions of the reacting molecular entities).
Note: At 25 °C in most solvents, including water, a bimolecular reaction that proceeds at an encounter-controlled rate has a second-order rate constant of 109 to 1010 dm3 mol−1 s−1.
See also microscopic diffusion control.
[3]
ene reaction
Addition of a compound with a double bond and an allylic hydrogen (the “ene”) to a compound with a multiple bond (the “enophile”) with transfer of the allylic hydrogen and a concomitant reorganization of the bonding.
Example, with propene as the ene and ethene as the enophile.
Note: The reverse is a “retro-ene” reaction.
[3]
energy of activation
E a or EA
Arrhenius energy of activation
activation energy
(derived SI unit: kJ mol−1)
Operationally defined quantity expressing the dependence of a rate constant on temperature according to
as derived from the Arrhenius equation, k(T) = A exp(−Ea/RT), where A is the pre-exponential factor and R the gas constant. As the argument of the ln function, k should be divided by its unit, i.e., by [k].
Note 1: According to collision theory, the pre-exponential factor A is the frequency of collisions with the correct orientation for reaction and Ea (or E0) is the threshold energy that collisions must have for the reaction to occur.
Note 2: The term Arrhenius activation energy is to be used only for the empirical quantity as defined above. There are other empirical equations with different activation energies, see [11].
See [12].
See also enthalpy of activation.
rev[3]
energy profile
See Gibbs energy diagram, potential-energy profile.
[3]
enforced concerted mechanism
Situation where a putative intermediate possesses a lifetime shorter than a bond vibration, so that the steps become concerted.
rev[3]
enthalpy of activation
standard enthalpy of activation
(derived SI unit: kJ mol−1)
Standard enthalpy difference between the transition state and the ground state of the reactants at the same temperature and pressure. It is related experimentally to the temperature dependence of the rate coefficient k according to equation (1) for first-order rate constants:
and to equation (2) for second order rate coefficients
This quantity can be obtained, along with the entropy of activation ∆‡S°, from the slope and intercept of the linear least-squares fit of rate coefficients k to the equation
for a first-order rate coefficient
and
for a second-order rate coefficient where kB is Boltzmann constant, h is Planck constant, and kB/h = 2.08366… × 1010 K−1 s−1.
Note 1: An advantage of the least-squares fit is that it can also give error estimates for
Note 2: It is also given by
where Ea is the energy of activation, provided that the rate coefficients for reactions other than first-order are expressed in temperature-independent concentration unit (e.g., mol kg−1, measured at a fixed temperature and pressure). The argument in a logarithmic function should be of dimension 1. Thus, k is divided by its unit, i.e., by [k], as in the alternative form, in terms of reduced variables.
See also entropy of activation, Gibbs energy of activation.
rev[3]
entropy of activation
standard entropy of activation
(derived SI unit: J mol−1 K−1)
Standard entropy difference between the transition state and the ground state of the reactants, at the same temperature and pressure. It is related to the Gibbs energy of activation and enthalpy of activation by the equation
provided that rate coefficients for reactions other than first-order reactions are expressed in temperature-independent concentration unit (e.g., mol dm−3, measured at fixed temperature and pressure). The numerical value of S depends on the standard state (and therefore on the concentration unit selected).
Note 1: It can also be obtained from the intercept of the linear least-squares fit of rate coefficients k to the equation
where kB/h = 2.08366… × 1010 K−1 s−1. k should be divided by its unit, i.e., by [k] = s−1 for first-order, and by [k] = (s−1 mol−1 dm3) for second-order rate coefficients and T should be divided by its unit [T] = K, as in the second equation, in terms of reduced variables.
Note 2: The information represented by the entropy of activation may alternatively be conveyed by the pre-exponential factor A, which reflects the fraction of collisions with the correct orientation for reaction (see energy of activation).
rev[3]
epimer
Diastereoisomer that has the opposite configuration at only one of two or more tetrahedral stereogenic centers in the respective molecular entity.
See [10].
[3]
epimerization
Interconversion of epimers by reversal of the configuration at one of the stereogenic centers.
See [10].
[3]
equilibrium, chemical
Situation in which reversible processes (processes that may be made to proceed in either the forward or reverse direction by the infinitesimal change of one variable) have reached a point where the rates in both directions are identical, so that the amount of each species no longer changes.
Note 1: In this situation, the Gibbs energy, G, is a minimum. Also, the sum of the chemical potentials of the reactants equals that of the products, so that
where the thermodynamic equilibrium constant, K (of dimension 1), is the product of product activities divided by the product of reactant activities.
Note 2: In dilute solutions, the numerical values of the thermodynamic activities may be approximated by the respective concentrations.
rev[3]
equilibrium control
See thermodynamic control.
[3]
E T-value
See Dimroth–Reichardt ET parameter, Z-value.
[3]
excess acidity
See Bunnett–Olsen equations, Cox–Yates equation.
[3]
excimer (“excited dimer”)
Complex formed by the interaction of an excited molecular entity with another identical molecular entity in its ground state.
Note: The complex is not stable in the ground state.
See [8].
See also exciplex.
[3]
exciplex
Electronically excited complex of definite stoichiometry that is non-bonding in the ground state.
Note: An exciplex is a complex formed by the noncovalent interaction of an excited molecular entity with the ground state of a different molecular entity, but an excimer, formed from two identical components, is often also considered to be an exciplex.
See [8].
See also excimer.
[3]
excited state
Condition of a system with energy higher than that of the ground state. This term is most commonly used to characterize a molecular entity in one of its electronically excited states, but may also refer to vibrational and/or rotational excitation in the electronic ground state.
See [8].
[3]
EXSY
NMR exchange spectroscopy
Two-dimensional NMR technique producing cross peaks corresponding to site-to site chemical exchange. The cross-peak amplitudes carry information about exchange rates.
See [198].
extended Hammett equation
Multiparameter extension of the Hammett equation for the description of substituent effects.
Note 1: The major extensions using two parameters (dual substituent-parameter equations) were devised for the separation of inductive and steric effects (Taft equation) or of inductive (or field) and resonance effects.
Note 2: Other parameters may be added (polarizability, hydrophobicity, etc.) when additional substituent effects are operative.
See [191].
See also Yukawa–Tsuno equation.
[3]
external return
external ion-pair recombination
Recombination of free ions formed in an SN1 reaction (as distinguished from ion-pair collapse).
See ion-pair recombination.
rev[3]
extrusion
Transformation in which an atom or group Y connected to two other atoms or groups X and Z is lost from a molecule, leading to a product in which X is bonded to Z, i.e.,
Example:
Note 1: When Y is a metal, this process is called a reductive elimination.
Note 2: The reverse of an extrusion is called an insertion.
See also cheletropic reaction.
[3]
field effect
Experimentally observable substituent effect (on reaction rates, etc.) of intramolecular electrostatic interaction between the center of interest and a monopole or dipole, by direct electric-field action through space rather than through bonds.
Note 1: The magnitude of the field effect depends on the monopole charge or dipole moment, on the orientation of the dipole, on the distance between the center of interest and the monopole or dipole, and on the effective dielectric constant that reflects how the intervening bonds are polarized.
Note 2: Although a theoretical distinction may be made between the field effect and the inductive effect as models for the Coulomb interaction between a given site and a remote monopole or dipole within the same entity, the experimental distinction between the two effects has proved difficult because the field effect and the inductive effect are ordinarily influenced in the same direction by structural changes.
Note 3: The substituent acts through the electric field that it generates, and it is an oversimplification to reduce that interaction to that of a monopole or dipole.
See also electronic effect, inductive effect, polar effect.
rev[3]
flash photolysis
Spectroscopic or kinetic technique in which an ultraviolet, visible, or infrared radiation pulse is used to produce transient species.
Note 1: Commonly, an intense pulse of short duration is used to produce a sufficient concentration of transient species, suitable for spectroscopic observation. The most common observation is of the absorption of the transient species (transient absorption spectroscopy).
Note 2: If only photophysical processes are involved, a more appropriate term would be “pulsed photoactivation”. The term “flash photolysis” would be correct only if chemical bonds are broken (the Greek “lysis” means dissolution or decomposition and in general lysis is used to indicate breaking). However, historically, the name has been used to describe the technique of pulsed excitation, independently of the process that follows the excitation.
See [8].
flash vacuum pyrolysis (FVP)
Thermal reaction of a molecule by exposure to a short thermal shock at high temperature, usually in the gas phase.
Example:
See [201], [202], [203], [204].
rev[3]
fluxionality
Property of a chemical species that undergoes rapid degenerate rearrangements (generally detectable by methods that allow the observation of the behavior of individual nuclei in a rearranged chemical species, e.g., NMR, X-ray).
Example: tricyclo[3.3.2.02,8]deca-3,6,9-triene (bullvalene), which has 1 209 600 (= 10!/3) interconvertible arrangements of the ten CH groups.
Note 1: Fluxionality differs from resonance, where no rearrangement of nuclear positions occurs.
Note 2: The term is also used to designate positional change among ligands of complex compounds and organometallics. In these cases, the change is not necessarily degenerate.
See also valence tautomerization.
rev[3]
force-field calculations
See molecular mechanics calculation.
GB
formal charge
Quantity (omitted if zero) attached to each atom in a Lewis structure according to
Examples: CH2=N+=N–, H3O+, (CH3)2C=N-O–, etc.
Note: This formalism assumes that electrons in bonds are shared equally, regardless of electronegativity.
Förster resonance-energy transfer (FRET)
dipole–dipole excitation transfer
Nonradiative mechanism for transfer of electronic excitation energy from one molecular entity to another, distant one. It arises from the interaction between the transition dipole moments of the two entities.
See [8].
fractionation factor, isotopic
Ratio [x1(A)/x2(A)]/[x1(B)/x2(B)], where x is the mole fraction of the isotope designated by the subscript, when the two isotopes are equilibrated between two different chemical species A and B (or between specific sites A and B in the same chemical species).
Note 1: The term is most commonly met in connection with deuterium solvent isotope effects, where the fractionation factor, symbolized by ϕ, expresses the ratio
for the exchangeable hydrogen atoms in the chemical species (or sites) concerned.
Note 2: The concept is also applicable to transition states.
See [1].
[3]
fragmentation
Heterolytic or homolytic cleavage of a molecule into more than two fragments, according to the general reaction (where a, b, c, d, and X are atoms or groups of atoms)
Examples:
See [205].
Breakdown of a radical or radical ion into a closed-shell molecule or ion and a smaller radical
Examples:
[3]
Franck–Condon Principle
Approximation that an electronic transition is most likely to occur without change in nuclear positions.
Note 1: The resulting state is called a Franck–Condon state, and the transition involved is called a vertical transition.
Note 2: As a consequence, the intensity of a vibronic transition is proportional to the square of the overlap integral between the vibrational wavefunctions of the two states that are involved in the transition.
See [8].
free energy
The thermodynamic function Gibbs energy (symbol G) or Helmholtz energy (symbol A) specifically defined as
and
where H is enthalpy, U is internal energy, and S is entropy. The possibility of spontaneous motion for a statistical distribution of an assembly of atoms (at absolute temperature T above 0 K) is governed by free energy and not by potential energy.
The IUPAC recommendation is to use the specific terms Gibbs energy or Helmholtz energy whenever possible. However, it is useful to retain the generic term “free energy” for use in contexts where the distinction between (on the one hand) either Gibbs energy or Helmholtz energy and (on the other hand) potential energy is more important than the distinction between conditions either of constant pressure or of constant volume; e.g., in computational modelling to distinguish between results of simulations performed for ensembles under conditions of either constant P or constant V at finite T and calculations based purely on potential energy.
Whereas motion of a single molecular entity is determined by the force acting upon it, which is obtained as the negative gradient of the potential energy, motion for an assembly of many molecular entities is determined by the mean force acting upon the statistical distribution, which is obtained as the negative gradient of the potential of mean force.
free radical
See radical.
[3]
frontier orbitals
Highest-energy Occupied Molecular Orbital (HOMO) and Lowest-energy Unoccupied Molecular Orbital (LUMO) of a molecular entity.
These terms should be limited to doubly occupied orbitals, and not to a singly occupied molecular orbital (sometimes designated as a SOMO), because HOMO and LUMO are ambiguous for molecular orbitals that are half filled and thus only partly occupied or unoccupied.
Examination of the mixing of frontier molecular orbitals of reacting molecular entities affords an approach to the interpretation of reaction behavior; this constitutes a simplified perturbation theory of chemical behavior.
In some cases, a subjacent orbital (Next-to-Highest Occupied Molecular Orbital (NHOMO) or a Second Lowest Unoccupied Molecular Orbital (SLUMO)) may affect reactivity.
See also SOMO, subjacent orbital.
rev[3]
frustrated Lewis acid–base pair
Acid and base for which adduct formation is prevented by steric hindrance.
fullerene
Molecular entity composed entirely of carbon, in the form of a hollow sphere, ellipsoid, or tube.
Note: Spherical fullerenes are also called buckyballs, and cylindrical ones are called carbon nanotubes or buckytubes.
functional group
Atom or group of atoms within molecular entities that are responsible for the characteristic chemical reactions of those molecular entities. The same functional group will undergo the same or similar chemical reaction(s) regardless of the size of the molecule it is a part of. However, its relative reactivity can be modified by nearby substituents.
rev[3]
gas-phase acidity
Standard reaction Gibbs energy (∆rG°) change for the gas-phase reaction.
Note 1: The symbol often found in the literature is ∆acidG or GA.
Note 2: The corresponding enthalpy ∆rH° is often symbolized by ∆acidH and called “enthalpy of acidity” or deprotonation enthalpy, abbreviated DPE.
rev[3]
gas-phase basicity
Negative of the standard reaction Gibbs energy (∆rG°) change for the gas-phase reaction
Note: An acronym commonly used in the literature is GB. The corresponding enthalpy ∆rH° is called proton affinity, PA, even though affinity properly refers to Gibbs energy. Moreover, such acronyms are not accepted by IUPAC.
rev[3]
Gaussian orbital
Function centered on an atom of the form ϕ(r)
See [7].
geminate pair
Pair of molecular entities in close proximity within a solvent cage and resulting from reaction (e.g., bond scission, electron transfer, group transfer) of a precursor.
Note: Because of the proximity, the pair constitutes only a single kinetic entity.
See also ion pair, radical pair.
rev[3]
geminate recombination
Recombination reaction of a geminate pair.
Example:
rev[3]
general acid catalysis
Catalysis of a chemical reaction by Brønsted acids (which may include the solvated hydrogen ion), where the rate of the catalyzed part of the reaction is given by ΣHAkHA[HA] multiplied by some function of substrate concentrations.
Note 1: General acid catalysis can be experimentally distinguished from specific catalysis by hydrogen cations (hydrons) if the rate of reaction increases with buffer concentration at constant pH and ionic strength.
Note 2: The acid catalysts HA are unchanged by the overall reaction. This requirement is sometimes relaxed, but the phenomenon is then properly called pseudo-catalysis.
See also catalysis, catalytic coefficient, intramolecular catalysis, pseudo-catalysis, specific catalysis.
rev[3]
general base catalysis
Catalysis of a chemical reaction by Brønsted bases (which may include the lyate ion), where the rate of the catalyzed part of the reaction is given by ΣBkB[B] multiplied by some function of substrate concentrations.
See also general acid catalysis.
[3]
Gibbs energy diagram
Diagram showing the relative standard Gibbs energies of reactants, transition states, reaction intermediates, and products, in the same sequence as they occur in a chemical reaction.
Note 1: The abscissa expresses the sequence of reactants, products, reaction intermediates, and transition states and is often an undefined “reaction coordinate” or only vaguely defined as a measure of progress along a reaction path. In some adaptations, the abscissas are however explicitly defined as bond orders, Brønsted exponents, etc.
Note 2: These points are often connected by a smooth curve (a “Gibbs energy profile”, commonly referred to as a “free-energy profile”, a terminology that is discouraged), but experimental observation can provide information on relative standard Gibbs energies only at the maxima and minima and not at the configurations between them.
Note 3: It should be noted that the use of standard Gibbs energies implies a common standard state for all chemical species (usually 1 M for reactions in solution). Contrary to statements in many textbooks, the highest point on a Gibbs energy diagram does not automatically correspond to the transition state of the rate-limiting step. For example, in a stepwise reaction consisting of two elementary reaction steps
C + D → E 2
one of the two transition states must (in general) have a higher standard Gibbs energy than the other. Under experimental conditions where all species have the standard-state concentration, the rate-limiting step is that whose transition state is of highest standard Gibbs energy. However, under (more usual) experimental conditions where all species do not have the standard-state concentration, the value of the concentration of D determines which reaction step is rate-limiting. However, if the particular concentrations of interest, which may vary, are chosen as the standard state, then the rate-limiting step is indeed the one of highest Gibbs energy.
See also potential-energy profile, potential-energy surface, reaction coordinate.
rev[3]
Gibbs energy of activation
standard free energy of activation
(derived SI unit: kJ mol−1)
Standard Gibbs energy difference between the transition state of an elementary reaction and the ground state of the reactants for that step. It is calculated from the rate constant k via the absolute rate equation:
where kB is Boltzmann’s constant, [k] represents the unit of k, and h is Planck’s constant. The values of the rate constants, and hence the Gibbs energies of activation, depend on the choice of concentration unit (or of the thermodynamic standard state).
Note 1: For a complex stepwise reaction, composed of many elementary reactions, ∆‡G°, the observed Gibbs energy of activation (activation free energy), can be calculated as RT [ln(kB/J K−1)/(h/J s) − ln(k′/[k′]/T/K)], where k′ is the observed rate constant, k′ should be divided by its unit, and T/K is the absolute temperature, of dimension 1, as the argument of a logarithmic function should be of dimension 1, as expressed by the reduced variables in the second form of the equation.
Note 2: Both
See also enthalpy of activation, entropy of activation.
rev[3]
graphene
Allotrope of carbon, whose structure is a one-atom-thick planar sheet of sp2-bonded carbon atoms in a honeycomb (hexagonal) crystal lattice.
ground state
State of the lowest Gibbs energy of a system [1].
Note: In photochemistry and quantum chemistry, the lowest-energy state of a chemical entity (ground electronic state) is usually meant.
See [8].
See also excited state.
rev[3]
group
See functional group, substituent.
rev[3]
Grunwald–Winstein equation
Linear Gibbs-energy relation (Linear free-energy relation)
expressing the dependence of the rate of solvolysis of a substrate on the ionizing power of the solvent, where the rate constant k0 applies to the reference solvent (80:20 ethanol–water by volume) and kS to the solvent S.
Note 1: The parameter m is characteristic of the substrate and is assigned the value unity for tert-butyl chloride (2-chloro-2-methylpropane). The value Y is intended to be a quantitative measure of the ionizing power of the solvent S.
Note 2: The equation was later extended [215] to the form
where N is the nucleophilicity of the solvent and l is a susceptibility parameter
Note 3: The equation has also been applied to reactions other than solvolysis.
Note 4: For the definition of other Y-scales, see [216], [217], [218], [219].
See also Dimroth–Reichardt ET parameter, polarity.
rev[3]
guest
Organic or inorganic ion or molecule that occupies a cavity, cleft, or pocket within the molecular structure of a host molecular entity and forms a complex with it or that is trapped in a cavity within the crystal structure of a host, but with no covalent bond being formed.
See also crown ether, cryptand, inclusion compound.
[3]
half-life
t 1/2
(SI unit: s)
Time required for the concentration of a particular reacting chemical species to fall to one-half of its initial value.
Note 1: Half-life is independent of initial concentration only for a first-order process.
Note 2: For first-order reactions, t1/2 = τ ln2, where τ is the lifetime.
See also lifetime.
rev[3]
halochromism
Color change that occurs on addition of acid or base to a solution of a compound as a result of chemical reaction or on addition of a salt as a result of changing the solvent polarity.
[3]
halogen bond
Association between a Lewis-acidic halogen atom in a molecular entity and a Lewis-basic region in another, or the same, molecular entity, which acts as an electron-pair donor, such that the balance of forces of attraction and repulsion results in net stabilization.
Note 1: Typical halogen-bond donors are I2, Br2, ICN, and IC≡CH.
Note 2: This is analogous to a hydrogen bond, in which the H is the acidic atom.
Note 3: The interaction provides a stabilization of a few kJ mol−1.
See Lewis acid, Lewis base
Hammett equation
Hammett relation
Equation of the form
or
expressing the influence of meta and para substituents X on the reactivity of the functional group Y in the benzene derivatives m- and p-XC6H4Y, where {kX} and {KX} are the numerical values of the rate or equilibrium constant, respectively, for the reactions of m- and p-XC6H4Y, σX is the substituent constant characteristic of m- or p-X, and ρ is the reaction constant characteristic of the given reaction of Y. The values lg {k0} and lg {K0} are intercepts for graphical plots of lg {kX} or lg {KX}, respectively, against σX for all substituents X (including X = H, if present).
Note 1: It is very common in the literature to find either of these equations written without the curly brackets denoting a reduced rate or equilibrium constant, i.e., k or K divided by its unit, [k] or [K], respectively.
Note 2: The Hammett equation is most frequently used to obtain the value of ρ which, as the slope of a graph or least-squares fit, is independent of the choice of unit for kX or KX. If the equation is used to estimate the unknown value of a rate or equilibrium constant for an X-substituted compound based upon the known value of σX, the ordinate must be multiplied by [kX] or [KX] in order to obtain the value with its appropriate unit.
Note 3: The equation is often encountered in a form with kH or KH incorporated into the logarithm on the left-hand side, where kH or KH corresponds to the reaction of parent C6H5Y, with X = H;
or
This form satisfies the requirement that arguments of the lg function must be of dimension 1, but it would suggest a one-parameter linear least-squares fit, whereas lg k0, lg K0, lg k0/[k0], and lg K0/[K0], in the other forms correctly represent the intercept in a two-parameter linear least-squares fit of lg k, lg K, lg k/[k], or lg K/[K] vs. σX.
See also extended Hammett equation, Taft equation, Yukawa–Tsuno equation, σ-constant, ρ-value.
rev[3]
Hammond postulate
Hammond-Leffler principle
Hypothesis that when a transition state leading to a high-energy reaction intermediate (or product) has nearly the same energy as that intermediate (or product), the two are interconverted with only a small reorganization of molecular structure.
Note 1: Essentially the same idea is sometimes referred to as “Leffler’s assumption”, namely, that the transition state bears a greater resemblance to the less stable species (reactant or reaction intermediate/product). Many textbooks and physical organic chemists, however, express the idea in Leffler’s form (couched in terms of Gibbs energies) but attribute it to Hammond (whose original conjecture concerns structure).
Note 2: As a corollary, it follows that a factor stabilizing a reaction intermediate will also stabilize the transition state leading to that intermediate.
Note 3: If a factor stabilizes a reaction intermediate (or reactant or product), then the position of the transition state along the minimum-energy reaction path (MERP) for that elementary step moves away from that intermediate, as shown in the energy diagram (a) below left, where the transition state moves from ‡ to ‡′ when the species to the right is stabilized. This behavior is often called a Hammond effect and is simply a consequence of adding a linear perturbation to the parabola.
Note 4: If a structure lying off the MERP is stabilized, then the position of the transition state moves toward that structure, as shown in the energy diagram (b) above right, where the transition state moves from ‡ to ‡′. This behavior is often called anti-Hammond and arises because the transition state is a maximum along the MERP but a minimum perpendicular to it.
See [227], [228], [229], [230].
See Leffler’s relation, More O’Ferrall–Jencks diagram, parallel effect, perpendicular effect.
rev[3]
Hansch constant
Measure of the contribution of a substituent to the partition ratio of a solute, defined as
where PX is the partition ratio for the compound with substituent X and PH is the partition constant for the parent.
rev[3]
hapticity
Topological description for the number of contiguous atoms of a ligand that are bonded to a central metal atom.
Note: The hapticity is indicated as a superscript following the Greek letter η.
Example: (C5H5)2Fe (ferrocene) = bis(η5-cyclopentadienyl)iron, where η5 can be read as eta-five or pentahapto.
See [28].
hard acid, base
Lewis acid with an acceptor center or Lewis base with a donor center (e.g., an oxygen atom) of low polarizability.
Note 1: A high polarizability characterizes a soft acid or base.
Note 2: Whereas the definition above is qualitative, a theoretically consistent definition of hardness η, in eV, is as half the second derivative of the calculated energy E with respect to N, the number of electrons, at constant potential ν due to the nuclei.
Note 3: Other things being equal, complexes of hard acids with hard bases or of soft acids with soft bases have an added stabilization (sometimes called the HSAB rule). For limitations of the HSAB rule, see [35, 233].
rev[3]
HBA solvent
Hydrogen-Bond Acceptor solvent
Strongly or weakly basic solvent capable of acting as a hydrogen-bond acceptor (HBA) and forming strong intermolecular solute-solvent hydrogen bonds.
Note: A solvent that is not capable of acting as a hydrogen-bond acceptor is called a non-HBA solvent.
See [16].
HBD solvent
Hydrogen-Bond Donating solvent, also dipolar HBD solvent and protic solvent
Solvent with a sizable permanent dipole moment that bears suitably acidic hydrogen atoms to form strong intermolecular solvent–solute hydrogen bonds.
Note: A solvent that is not capable of acting as hydrogen-bond donor is called a non-HBD solvent (formerly aprotic solvent).
heat capacity of activation
(derived SI unit: J mol−1 K−1)
Temperature coefficient of ∆‡H (enthalpy of activation) or ∆‡S (entropy of activation) at constant pressure according to the equations:
See [237].
rev[3]
Henderson–Hasselbalch equation
Equation of the form
relating the pH of a buffer solution to the ratio [HA]/[A–] and the dissociation constant Ka of the acid HA.
See [238].
rev[3]
heterobimetallic complex
Metal complex having two different metal atoms or ions.
rev[3]
heteroleptic
Characteristic of a transition metal or Main Group compound having more than one type of ligand.
See also homoleptic.
[3]
heterolysis, heterolytic bond fission
Cleavage of a covalent bond so that both bonding electrons remain with one of the two fragments between which the bond is broken, e.g.,
See also heterolytic bond-dissociation energy, homolysis.
[3]
heterolytic bond-dissociation energy
Energy required to break a given bond of a specific compound by heterolysis.
Note: For the dissociation of a neutral molecule AB in the gas phase into A+ and B–, the heterolytic bond-dissociation energy D(A+B–) is the sum of the homolytic bond-dissociation energy, D(A–B), and the adiabatic ionization energy of the radical A· minus the electron affinity of the radical B·.
[3]
high-throughput screening
Automated method to quickly assay large libraries of chemical species for the affinity of small organic molecules toward a target of interest.
Note: Currently more than 105 different compounds can be tested per day.
See [239].
highest occupied molecular orbital (HOMO)
Doubly filled molecular orbital of highest energy.
Note: Examination of the HOMO can predict whether an electrocyclic reaction is conrotatory or disrotatory.
See also frontier orbitals.
Hildebrand solubility parameter
δ[Derived unit: Pa1/2 = (kg m−1 s−2)1/2]
Ability of a solvent to dissolve a non-electrolyte, defined as the square root of the solvent’s cohesive energy density (also called cohesive pressure, equal to the energy of vaporization divided by the solvent’s molar volume and corresponding to the energy necessary to create a cavity in the solvent).
See [240].
rev[3]
Hofmann rule
Observation that when two or more alkenes can be produced in a β-elimination reaction, the alkene having the smallest number of alkyl groups attached to the double-bond carbon atoms is the predominant product.
Note: This orientation is observed in elimination reactions of quaternary ammonium salts and tertiary sulfonium (sulfanium) salts, and in certain other cases where there is steric hindrance.
See [241].
See also Saytzeff rule.
rev[3]
HOMO
Acronym for Highest Occupied Molecular Orbital.
See also frontier orbitals.
Prefix (in lower case) used to indicate a higher homologue of a compound, as homocysteine for HSCH2CH2CH(NH2)COOH, the homologue of cysteine, HSCH2CH(NH2)COOH.
rev[3]
homoaromatic
Showing features of aromaticity despite a formal discontinuity in the overlap of a cyclic array of p orbitals resulting from the presence of an sp3-hybridized atom at one or several positions within the cycle, in contrast to an aromatic molecule, where there is a continuous overlap of p orbitals over a cyclic array.
Note 1: Homoaromaticity arises because p-orbital overlap can bridge an sp3 center.
Note 2: Pronounced homoaromaticity is not normally associated with neutral molecules, but mainly with ionic species, e.g., the “homotropylium” cation, C8H9+,
Note 3: In bis-, tris-, (etc.) homoaromatic species, two, three, (etc.) single sp3 centers separately interrupt the π-electron system.
[3]
homodesmotic reaction
Subclass of isodesmic reactions in which reactants and products contain not only the same number of carbon atoms in each state of hybridization but also the same number of each CHn group joined to n hydrogen atoms.
Examples:
Note: The definition may be extended to molecules with heteroatoms.
See [7].
rev[3]
homoleptic
Characteristic of a transition-metal or Main Group compound having only one type of ligand, e.g., Ta(CH3)5
See also heteroleptic.
[3]
homolysis
Cleavage of a bond so that each of the molecular fragments between which the bond is broken retains one of the bonding electrons.
Note 1: A unimolecular reaction involving homolysis of a bond not forming part of a cyclic structure in a molecular entity containing an even number of (paired) electrons results in the formation of two radicals:
Note 2: Homolysis is the reverse of colligation.
See also bond dissociation energy, heterolysis.
[3]
host
Molecular entity that forms complexes (adducts) with organic or inorganic guests, or a chemical species that can accommodate guests within cavities of its crystal structure.
Examples include cryptands and crown ethers (where there are ion/dipole attractions between heteroatoms and cations), hydrogen-bonded molecules that form clathrates (e.g., hydroquinone or water), and host molecules of inclusion compounds (e.g., urea or thiourea), where intermolecular forces and hydrophobic interactions bind the guest to the host molecule.
rev[3]
Hückel molecular orbital (HMO) theory
Simplest molecular orbital theory of π-conjugated molecular systems. It uses the following approximations: π-electron approximation; LCAO representation of the π molecular orbitals; neglect of electron-electron and nuclear-nuclear repulsions. The diagonal elements of the effective Hamiltonian (Coulomb integrals) and the off-diagonal elements for directly bonded atoms (resonance integrals) are taken as empirical parameters, all overlap integrals being neglected.
See [7].
rev[3]
Hückel (4n + 2) rule
Principle that monocyclic planar (or almost planar) systems of trigonally (or sometimes digonally) hybridized atoms that contain (4n + 2) π electrons (where n is an integer, generally 0 to 5) exhibit aromatic character.
Note 1: This rule is derived from Hückel MO theory calculations on planar monocyclic conjugated hydrocarbons (CH)m where integer m is at least 3, according to which (4n + 2) electrons are contained in a closed-shell system.
Examples:
Note 2: Planar systems containing 4n π electrons (such as cyclobutadiene and cyclopentadienyl cation) are antiaromatic.
Note 3: Cyclooctatetraene, with 8 π electrons, is nonplanar and therefore neither aromatic nor antiaromatic but nonaromatic.
See [7].
See also conjugation, Möbius aromaticity.
rev[3]
Huisgen reaction
Cycloaddition of 1,3 dipolar compounds.
hybrid orbital
Atomic orbital constructed as a linear combination of atomic orbitals on an atom.
Note 1: Hybrid orbitals are often used to describe the bonding in molecules containing tetrahedral (sp3), trigonal (sp2), and digonal (sp) atoms, whose σ bonds are constructed using 1:3, 1:2, or 1:1 combinations, respectively, of s and p atomic orbitals.
Note 2: Construction of hybrid orbitals can also include d orbitals, as on an octahedral atom, with d2sp3 hybridization.
Note 3: Integer ratios are not necessary, and the general hybrid orbital made from s and p orbitals can be designated as spλ.
hydration
Addition of water or of the elements of water (i.e., H and OH) to a molecular entity or to a chemical species.
Example: hydration of ethene:
Note: In contrast to aquation, hydration, as in the incorporation of waters of crystallization into a protein or in the formation of a layer of water on a nonpolar surface, does not necessarily require bond formation.
See [47].
See also aquation.
rev[3]
hydrogen bond
An attractive interaction between a hydrogen atom from a molecule or a molecular fragment X–H in which X is more electronegative than H, and an atom or a group of atoms in the same or a different molecule, in which there is evidence of bond formation.
Note 1: Both electronegative atoms are usually (but not necessarily) from the first row of the Periodic Table, i.e., N, O, or F.
Note 2: A hydrogen bond is largely an electrostatic interaction, heightened by the small size of hydrogen, which permits proximity of the interacting dipoles or charges.
Note 3: Hydrogen bonds may be intermolecular or intramolecular.
Note 4: With few exceptions, usually involving hydrogen fluoride and fluoride and other ions, the associated energies are less than 20 to 25 kJ mol−1.
Note 5: Hydrogen bonds are important for many chemical structures, giving rise to the attraction between H2O molecules in water and ice, between the strands of DNA, and between aminoacid residues in proteins.
See electronegativity.
rev[3]
hydrolysis
Solvolysis by water, generally involving the rupture of one or more bonds in the reactant and involvement of water as nucleophile or base.
Example:
rev[3]
hydron
General name for the ion H+ either in natural abundance or where it is not desired to distinguish between the isotopes, as opposed to proton for 1H+, deuteron for 2H+ and triton for 3H+.
See [244].
[3]
hydronation
Attachment of the ion H+ either in natural abundance or where it is not desired to distinguish between the isotopes.
hydrophilicity
Capacity of a molecular entity or of a substituent to undergo stabilizing interactions with polar solvents, in particular with water and aqueous mixtures, to an extent greater than with a nonpolar solvent.
rev[3]
hydrophobic interaction
Tendency of lipophilic hydrocarbon-like groups in solutes to form intermolecular aggregates in an aqueous medium.
Note: The name arises from the attribution of the phenomenon to the apparent repulsion between water and hydrocarbons. However, the phenomenon is more properly attributed to the effect of the hydrocarbon-like groups to avoid disrupting the favorable water–water interactions.
[3]
hyperconjugation
Delocalization of electrons between σ bonds and a π network.
Note 1: The concept of hyperconjugation is often applied to carbenium ions and radicals, where the interaction is between σ bonds and an unfilled or partially filled p or π orbital. Resonance illustrating this for the tert-butyl cation is:
Note 2: A distinction is made between positive hyperconjugation, as above, and negative hyperconjugation, where the interaction is between a filled σ or π orbital and adjacent antibonding σ* orbitals, as for example in the fluoroethyl anion (2-fluoroethan-1-ide).
Note 3: Historically, conjugation involves only π bonds, and hyperconjugation is considered unusual in involving σ bonds.
See [245], [246], [247], [248].
See also σ, π, delocalization.
rev[3]
hypercoordinated
Feature of a main-group atom with a coordination number greater than four.
Example: the pentacoordinate carbon in the carbonium ion CH5+, where three C–H bonds may be regarded as two-electron bonds and the two electrons in the remaining CH2 fragment are delocalized over three atoms. Likewise, both hydrogens in the CH2 fragment are hypercoordinated.
See also agostic.
rev[3]
hypervalency
Ability of an atom in a molecular entity to expand its valence shell beyond the limits of the Lewis octet rule.
Examples: PF5, SO3, iodine(III) compounds, and pentacoordinate carbocations (carbonium ions).
Note: Hypervalent compounds are more common for the second- and subsequent-row elements in groups 15 to 18 of the periodic table. A proper description of the hypervalent bonding implies a transfer of electrons from the central (hypervalent) atom to the nonbonding molecular orbitals of the attached ligands, which are usually more electronegative.
See [7].
See also electronegativity, valence.
rev[3]
hypsochromic shift
Shift of a spectral band to higher frequency (shorter wavelength) upon substitution or change in medium.
Note: This is informally referred to as a blue shift and is opposite to a bathochromic shift (“red shift”), but these historical terms are discouraged because they apply only to visible transitions.
See [8].
rev[3]
identity reaction
Chemical reaction whose products are chemically identical with the reactants
Examples:
bimolecular exchange reaction of CH3I with I–
proton transfer between NH4+ and NH3
electron transfer between manganate(VI) MnO42– and permanganate MnO4–.
See also degenerate rearrangement.
rev[3]
imbalance
Feature that reaction parameters characterizing different bond-forming or bond-breaking processes in the same reaction change to different extents as the transition state is approached (along some arbitrarily defined reaction path).
Note: Imbalance is common in reactions such as elimination, addition, and other complex reactions that involve proton (hydron) transfer.
Example: the nitroalkane anomaly, where the Brønsted β exponent for hydron removal is smaller than the Brønsted α for the nitroalkane as acid because of imbalance between the extent of bond breaking and the extent of resonance delocalization in the transition state.
See [249].
See also Brønsted relation, synchronization (principle of nonperfect synchronization), synchronous.
rev[3]
inclusion compound
inclusion complex
Complex in which one component (the host) forms a cavity or, in the case of a crystal, a crystal lattice containing spaces in the shape of long tunnels or channels in which molecular entities of a second chemical species (the guest) are located.
Note: There is no covalent bonding between guest and host, the attraction being generally due to van der Waals forces. If the spaces in the host lattice are enclosed on all sides so that the guest species is “trapped” as in a cage, such compounds are known as clathrates or cage compounds.
See [39].
[3]
induction period
Initial slow phase of a chemical reaction whose rate later accelerates.
Note: Induction periods are often observed with radical reactions, but they may also occur in other reactions, such as those where a steady-state concentration of the reactants is not established immediately.
See [12].
[3]
inductive effect
Experimentally observable effect (on rates of reaction, etc.) of a substituent through transmission of charge through a chain of atoms by electrostatics.
Note: Although a theoretical distinction may be made between the field effect and the inductive effect as models for the Coulomb interaction between a given site within a molecular entity and a remote monopole or dipole within the same entity, the experimental distinction between the two effects has proved difficult (except for molecules of peculiar geometry, which may exhibit “reversed field effects“) because the inductive effect and the field effect are ordinarily influenced in the same direction by structural changes.
See also field effect, polar effect.
rev[3]
inert
Stable and unreactive under specified conditions.
[3]
inhibition
Decrease in the rate of reaction brought about by the addition of a substance (inhibitor), by virtue of its effect on the concentration of a reactant, catalyst, or reaction intermediate.
For example, molecular oxygen or 1,4-benzoquinone can act as an inhibitor in many chain reactions involving radicals as intermediates by virtue of its ability to act as a scavenger toward those radicals.
Note: If the rate of a reaction in the absence of inhibitor is vo and that in the presence of a certain amount of inhibitor is v, the degree of inhibition (i) is given by
See also mechanism-based inhibition.
[3]
initiation
Reaction or process generating radicals (or some other reactive reaction intermediates) which then induce a chain reaction or catalytic cycle.
Example: In the chlorination of alkanes by a radical mechanism, the initiation step may be the dissociation of molecular chlorine.
rev[3]
inner-sphere electron transfer
Feature of an electron transfer between two metal centers that in the transition state share a ligand or atom in their coordination shells.
Note: The definition has been extended to any situation in which the interaction between the electron-donor and electron-acceptor centers in the transition state is significant (greater than 20 kJ mol−1).
See [8].
See also outer-sphere electron transfer.
[3]
insertion
Chemical reaction or transformation of the general type
in which the connecting atom or group Y replaces the bond joining the parts X and Z of the reactant XZ.
Example: carbene insertion reaction
Note: The reverse of an insertion is called an extrusion.
[3]
intermediate
reactive intermediate
Molecular entity in a stepwise chemical reaction with a lifetime appreciably longer than a molecular vibration (corresponding to a local potential energy minimum of depth greater than RT and thus distinguished from a transition state) that is formed (directly or indirectly) from the reactants and reacts further to give (directly or indirectly) the products of a chemical reaction.
See also elementary reaction, reaction step, stepwise reaction.
[3]
intermolecular
Descriptive of any process that involves a transfer (of atoms, groups, electrons, etc.) or interactions between two or more molecular entities.
Relating to a comparison between different molecular entities.
See also intramolecular.
[3]
internal return
See ion-pair recombination.
rev[3]
intramolecular
Descriptive of any process that involves a transfer (of atoms, groups, electrons, etc.) or interactions between different parts of the same molecular entity.
Relating to a comparison between different groups within the same molecular entity.
See also intermolecular.
[3]
intramolecular catalysis
Acceleration of a chemical transformation at one site of a molecular entity through the involvement of another functional (“catalytic”) group in the same molecular entity, without that group appearing to have undergone change in the reaction product.
Note 1: The use of the term should be restricted to cases for which analogous intermolecular catalysis by a chemical species bearing that catalytic group is observable.
Note 2: Intramolecular catalysis can be detected and expressed in quantitative form by a comparison of the reaction rate with that of a comparable model compound in which the catalytic group is absent, or by measurement of the effective molarity of the catalytic group.
See also catalyst, effective molarity, neighboring group participation.
[3]
intrinsic barrier Δ ‡ G 0
Gibbs energy of activation in the limiting case where ΔrG° = 0, i.e., when the effect of thermodynamic driving force is eliminated, as in an identity reaction, X* + AX → X*A + X, where A may be an atom, ion, or group of atoms or ions, and where the equilibrium constant K is equal to 1.
Note: According to the Marcus equation, originally developed for outer-sphere electron transfer reactions, the intrinsic barrier is related to λ, the reorganization energy of the reaction, by the equation
For a non-identity reaction, Y + AX → YA + X, the intrinsic barrier Δ‡G0(Y,X) is estimated as ½[Δ‡G(X,X) + Δ‡G(Y,Y)], where the latter two terms are the Gibbs energies of activation of the identity reactions X* + AX → X*A + X and Y* + AY → Y*A + Y, respectively.
See [251], [252], [253], [254].
rev[3]
intrinsic reaction coordinate (IRC)
Minimum-energy path leading from the saddle point (corresponding to the transition structure) on the potential-energy surface for an elementary reaction, obtained by tracing the steepest descent in mass-weighted coordinates in both directions.
Note: The IRC is mathematically well defined, in contrast to the (generally) vague reaction coordinate. Strictly, the IRC is a specific case of a minimum-energy reaction path, and its numerical value at any point along this path is usually taken to be zero at the saddle point, positive in the direction of the products, and negative in the direction of the reactants.
rev[3]
inverted micelle
reverse micelle
Association colloid formed reversibly from surfactants in non-polar solvents, leading to aggregates in which the polar groups of the surfactants are concentrated in the interior and the lipophilic groups extend toward and into the non-polar solvent.
Note: Such an association is often of the type
and a critical micelle concentration is consequently not observed.
[3]
ion pair
Pair of oppositely charged ions held together by Coulomb attraction without formation of a covalent bond.
Note 1: Experimentally, an ion pair behaves as one unit in determining conductivity, kinetic behavior, osmotic properties, etc.
Note 2: Following Bjerrum, oppositely charged ions with their centers closer together than a distance
are considered to constitute an ion pair. Here Z+ and Z– are the charge numbers of the ions, e is the elementary charge, ε0 is the vacuum permittivity, εr is the relative permittivity (“dielectric constant”) of the medium, kB is Boltzmann’s constant, and T is the absolute temperature. This is the distance at which the Coulomb energy equals the thermal energy, and e2/(4πε0kB) is approximately equal to 1.671 × 10−5 m K−1.
Note 3: An ion pair, the constituent ions of which are in direct contact (and not separated by intervening solvent or by other neutral molecule) is designated as a “tight ion pair” (or “intimate” or “contact ion pair”). A tight ion pair of R+ and X– is symbolically represented as R+X–.
Note 4: By contrast, an ion pair whose constituent ions are separated by one or several solvent or other neutral molecules is described as a “loose ion pair”, symbolically represented as R+||X–. The components of a loose ion pair can readily interchange with other free or loosely paired ions in the solution. This interchange may be detectable (e.g., by isotopic labelling) and thus afford an experimental distinction between tight and loose ion pairs.
Note 5: A further conceptual distinction has sometimes been made between two types of loose ion pairs. In “solvent-shared ion pairs” for which the ionic constituents of the pair are separated by only a single solvent molecule, whereas in “solvent-separated ion pairs” more than one solvent molecule intervenes. However, the term “solvent-separated ion pair” must be used and interpreted with care since it has also widely been used as a less specific term for “loose” ion pair.
See [256].
See also common-ion effect, dissociation, ion-pair recombination, special salt effect.
[3]
ion-pair recombination (formerly ion-pair return)
Recombination of a pair of ions R+ and X– formed from ionization of RX.
Note: Ion-pair recombination can be distinguished as external or internal, depending on whether the ion pair did or did not undergo dissociation to free ions.
See ion pair.
rev[3]
ionic liquid (ionic solvent, molten salt)
Liquid that consists exclusively or almost exclusively of equivalent amounts of oppositely charged ions.
Note 1: In practice, the ions are monocations and monoanions.
Note 2: Ionic liquids that are liquid at or around room temperature are called room-temperature ionic liquids (RTILs).
Note 3: The term ionic liquid has been often restricted to those water-free liquids that have melting points (or glass-transition temperatures) below 100 °C, following a definition given by Walden [257], who prepared the first ionic liquid, ethylammonium nitrate, CH3CH2NH3+ NO3−, mp. 13–14 °C, for conductivity measurements.
Note 4: The terminology for ionic liquids is not yet settled, as stated by Welton [258]. Room-temperature ionic liquid, non-aqueous ionic liquid, molten salt, liquid organic salt, and fused salt are often synonymous.
ionic strength
I
(In concentration basis, Ic, SI unit: mol m−3, more commonly mol dm−3 or mol L−1, in molality basis, Im, SI unit: mol kg−1)
In concentration basis: Ic = 0.5 ΣiciZi2, in which ci is the concentration of a fully dissociated electrolyte in solution and Zi the charge number of ionic species i.
In molality basis: Im = 0.5 ΣimiZi2, in which mi is the molality of a fully dissociated electrolyte in solution and Zi the charge number of ionic species i.
rev[3]
ionization
Generation of one or more ions.
Note 1: This may occur, e.g., by loss or gain of an electron from a neutral molecular entity, by the unimolecular heterolysis of that entity into two or more ions, or by a heterolytic substitution reaction involving neutral molecules, such as
M → M+• + e– (photoionization, electron ionization in mass spectrometry)
SF6 + e– → SF6–• (electron capture)
Ph3CCl → Ph3C+ Cl– (ion pair, which may dissociate to free ions)
CH3COOH + H2O → H3O+ + CH3CO2– (dissociation)
Note 2: In mass spectrometry ions may be generated by several methods, including electron ionization, photoionization, laser desorption, chemical ionization, and electrospray ionization.
Note 3: Loss of an electron from a singly, doubly, etc. charged cation is called second, third, etc. ionization.
See also dissociation, ionization energy.
rev[3]
ionization energy
E i
(derived SI unit: J)
Minimum energy required to remove an electron from an isolated molecular entity (in its vibrational ground state) in the gaseous phase.
Note 1: If the resulting molecular entity is in its vibrational ground state, the energy is the “adiabatic ionization energy”.
Note 2: If the molecular entity produced possesses the vibrational energy determined by the Franck–Condon principle (according to which the electron ejection takes place without an accompanying change in molecular geometry), the energy is the “vertical ionization energy”.
Note 3: The name ionization energy is preferred to the somewhat misleading earlier name “ionization potential”.
See also ionization.
[3]
ionizing power
Tendency of a particular solvent to promote ionization of a solute.
Note: The term has been used in both kinetic and thermodynamic contexts.
See also Dimroth–Reichardt ETparameter, Grunwald–Winstein equation, Z-value.
[3]
ipso attack
Attachment of an entering group to a position in an aromatic compound already carrying a substituent group other than hydrogen.
Note: The entering group may displace that substituent group or may itself be expelled or migrate to a different position in a subsequent step. The term “ipso-substitution” is not used, since it is synonymous with substitution.
Example:
where E+ is an electrophile and Z is a substituent other than hydrogen.
See [262].
See also cine-substitution, tele-substitution.
[3]
isentropic
See isoentropic.
isodesmic
Property of a reaction (actual or hypothetical) in which the types of bonds that are made in forming the products are the same as those that are broken in the reactants.
Examples:
Note 1: Such processes have advantages for theoretical treatment.
Note 2: The Hammett equation as applied to equilibria, as in (1), succeeds because it deals with isodesmic processes.
Note 3: For the use of isodesmic processes in quantum chemistry, see [263].
See [7].
See also homodesmotic.
rev[3]
isoelectronic
Having the same number of valence electrons and the same structure, i.e., number and connectivity of atoms, but differing in some or all of the elements involved.
Examples:
CO, N2, and NO+
CH2=C=O and CH2=N+=N–
[3]
isoentropic
Isentropic
Property of a reaction series in which the individual reactions have the same standard entropy or entropy of activation.
[3]
isoequilibrium relationship
Feature of a series of related substrates or of a single substrate under a series of reaction conditions whereby the enthalpies and entropies of reaction can be correlated by the equation
Note: The parameter β is called the isoequilibrium temperature.
See also compensation effect, isokinetic relationship.
[3]
isokinetic relationship
Feature of a series of related substrates or of a single substrate under a series of reaction conditions whereby the enthalpies of activation and entropies of activation can be correlated by the equation
Note 1: The parameter β is called the isokinetic temperature. At this temperature, all members of the series react at the same rate.
Note 2: Isokinetic relationships as established by direct correlation of Δ‡H with Δ‡S are often spurious, and the calculated value of β is meaningless, because errors in Δ‡H lead to compensating errors in Δ‡S. Satisfactory methods of establishing such relationships have been devised.
See also compensation effect, isoequilibrium relationship, isoselective relationship.
[3]
isolobal
Feature of two molecular fragments for which the number, symmetry properties, approximate energy, shape of the frontier orbitals, and number of electrons in them are similar.
Example:
See also isoelectronic.
rev[3]
isomer
One of several species (or molecular entities) all of which have the same atomic composition (molecular formula) but differ in their connectivity or stereochemistry and hence have different physical and/or chemical properties.
Note: Conformational isomers that interconvert by rapid rotation about single bonds and configurations that interconvert by rapid pyramidal inversion are often not considered as separate isomers.
rev[3]
isomerization
Chemical reaction in which the product is an isomer of the reactant.
See also molecular rearrangement.
rev[3]
isosbestic point
Wavelength (or frequency) at which two or more components in a mixture have the same molar absorption coefficients.
Note 1: Isosbestic points are commonly met when electronic spectra are taken (a) on a solution in which a chemical reaction is in progress (in which case the two absorbing components concerned are a reactant and a product, A → B) or (b) on a solution in which the two absorbing components are in equilibrium and their relative proportions are controlled by the concentration of some other component, typically the concentration of hydrogen ions, as in an acid–base indicator equilibrium.
Note 2: The effect may also appear (c) in the spectra of a set of solutions of two or more non-interacting components having the same total concentration.
Note 3: In all these examples, A (and/or B) may be either a single chemical species or a mixture of chemical species present in invariant proportion.
Note 4: If absorption spectra of the types considered above intersect not at one or more isosbestic points but over a progressively changing range of wavelengths, this is prima facie evidence (a) for the formation of a reaction intermediate in substantial concentration (A → C → B) or (b) for the involvement of a third absorbing species in the equilibrium, e.g.,
or (c) for some interaction of A and B, e.g.,
Note 5: Isobestic is a misspelling and is discouraged.
rev[3]
isoselective relationship
Relationship analogous to the isokinetic relationship, but applied to selectivity data of reactions.
Note: At the isoselective temperature, the selectivities of the series of reactions following the relationship are identical, within experimental error.
See [266].
See also isoequilibrium relationship, isokinetic relationship, selectivity.
[3]
isotope effect
Relative difference between, or the ratio of, the rate coefficients or equilibrium constants of two reactions that differ only in the isotopic composition of one or more of their otherwise chemically identical components.
Note: The ratio kl/kh of rate constants (or Kl/Kh of equilibrium constants) for “light” and “heavy” reactions is most often used in studies of chemical reaction mechanisms. However, the opposite ratios are frequently used in environmental chemistry and geochemistry, i.e., kh/kl or Kh/Kl. Furthermore the relative difference kh/kl − 1 or Kh/Kl − 1 (often expressed as the percentage deviation of the ratio from unity) is commonly used to quantify isotopic fractionation.
See isotope effect (equilibrium), kinetic isotope effect.
rev[3]
isotope effect, equilibrium
isotope effect, thermodynamic
Effect of isotopic substitution on an equilibrium constant
Note 1: For example, the effect of isotopic substitution in reactant A that participates in the equilibrium:
is the ratio Kl/Kh of the equilibrium constant for the reaction in which A contains the light isotope to that in which it contains the heavy isotope. The ratio can also be expressed as the equilibrium constant for the isotopic exchange reaction:
in which reactants such as B that are not isotopically substituted do not appear.
Note 2: The potential-energy surfaces of isotopic molecules are identical to a high degree of approximation, so thermodynamic isotope effects can arise only from the effect of isotopic mass on the nuclear motions of the reactants and products, and can be expressed quantitatively in terms of nuclear partition functions:
Note 3: Although the nuclear partition function is a product of the translational, rotational, and vibrational partition functions, the isotope effect is usually determined almost entirely by the last named, specifically by vibrational modes involving motion of isotopically different atoms. In the case of light atoms (i.e., protium vs. deuterium or tritium) at moderate temperatures, the isotope effect is dominated by zero-point energy differences.
See [267].
See also fractionation factor.
rev[3]
isotope effect, heavy-atom
Isotope effect due to isotopes other than those of hydrogen.
[3]
isotope effect, intramolecular
Kinetic isotope effect observed when a single substrate, in which the isotopic atoms occupy equivalent reactive positions, reacts to produce a non-statistical distribution of isotopologue products.
Example:
The intramolecular isotope effect kH/kD can be measured from the D content of product (PhCH2Br + PhCHDBr), which is experimentally much easier than measuring the intermolecular isotope effect kH/kD from the separate rates of reaction of PhCH3 and PhCD3.
rev[3]
isotope effect, inverse
Kinetic isotope effect in which kl/kh < 1, i.e., the heavier substrate reacts more rapidly than the lighter one, as opposed to the more usual “normal” isotope effect, in which kl/kh > 1.
Note: The isotope effect will be “normal” when the vibrational frequency differences between the isotopic transition states are smaller than in the reactants. Conversely, an inverse isotope effect can be taken as evidence for an increase in the force constants on passing from the reactant to the transition state.
[3]
isotope effect, kinetic
Effect of isotopic substitution on a rate constant.
For example in the reaction
the effect of isotopic substitution in reactant A is expressed as the ratio of rate constants kl/kh, where the subscripts l and h represent reactions in which the molecules A contain the light and heavy isotopes, respectively.
Note 1: Within the framework of transition-state theory, where the reaction is rewritten as
k l/kh can be regarded as if it were the equilibrium constant for an isotope exchange reaction between the transition state [TS]‡ and the isotopically substituted reactant A and calculated from their vibrational frequencies as in the case of a thermodynamic isotope effect (see thermodynamic (equilibrium) isotope effect).
Note 2: Isotope effects like the above, involving a direct or indirect comparison of the rates of reaction of isotopologues, are called “intermolecular”, in contrast to intramolecular isotope effects (see intramolecular isotope effect), in which a single substrate reacts to produce a non-statistical distribution of isotopologue product molecules.
[3]
isotope effect, primary
Kinetic isotope effect attributable to isotopic substitution of an atom to which a bond is made or broken in the rate-limiting step or in a pre-equilibrium step of a reaction.
Note: The corresponding isotope effect on the equilibrium constant of a reaction in which one or more bonds to isotopic atoms are broken is called a primary equilibrium isotope effect.
See also secondary isotope effect.
[3]
isotope effect, secondary
Kinetic isotope effect that is attributable to isotopic substitution of an atom to which bonds are neither made nor broken in the rate-limiting step or in a pre-equilibrium step of a specified reaction.
Note 1: The corresponding isotope effect on the equilibrium constant of such a reaction is called a secondary equilibrium isotope effect.
Note 2: Secondary isotope effects can be classified as α, β, etc., where the label denotes the position of isotopic substitution relative to the reaction center.
Note 3: Although secondary isotope effects have been discussed in terms of conventional electronic effects, e.g., induction, hyperconjugation, and hybridization, such an effect is not electronic but vibrational in origin.
See [269].
See also steric isotope effect.
rev[3]
isotope effect, solvent
Kinetic or equilibrium isotope effect resulting from change in the isotopic composition of the solvent.
See also kinetic isotope effect, equilibrium isotope effect.
[3]
isotope effect, steric
Secondary isotope effect attributed to the different vibrational amplitudes of isotopologues.
Note 1: For example, both the mean and mean-square amplitudes of vibrations associated with C–H bonds are greater than those of C–D bonds. The greater effective bulk of molecules containing the former may be manifested by a steric effect on a rate or equilibrium constant.
Note 2: Ultimately the steric isotope effect arises from changes in vibrational frequencies and zero-point energies, as normally do other isotope effects.
rev[3]
isotope effect, thermodynamic
See isotope effect, equilibrium.
[3]
isotope exchange
Chemical reaction in which the reactant and product chemical species are chemically identical but have different isotopic composition.
Note: In such a reaction, the isotope distribution tends towards equilibrium (as expressed by fractionation factors) as a result of transfers of isotopically different atoms or groups. For example,
[3]
isotopic perturbation, method of
Measurement of the NMR shift difference due to the isotope effect on a fast (degenerate) equilibrium between two species that are equivalent except for isotopic substitution.
Note: This method can distinguish a rapidly equilibrating mixture with time-averaged symmetry from a single structure with higher symmetry.
See [270], [271], [272], [273].
[3]
isotopic scrambling
Achievement of, or the process of achieving, a redistribution of isotopes within a specified set of atoms in a chemical species or group of chemical species.
Examples
(where * denotes the position of an isotopically different atom.)
See also fractionation factor.
[3]
isotopologues
Molecular entities that differ only in isotopic composition (number of isotopic substitutions), e.g., CH4, CH3D, CH2D2,
Note: These are isotopic homologues. It is a misnomer to call them isotopomers because they are not isomers with the same atoms.
rev[3]
isotopomers
Isomers having the same number of each isotopic atom but differing in their positions. The term is a contraction of “isotopic isomer”.
Note: Isotopomers can be either constitutional isomers (e.g., CH2DCH=O and CH3CD=O) or isotopic stereoisomers (e.g., (R)- and (S)-CH3CHDOH or (Z)- and (E)-CH3CH=CHD).
See [10].
[3]
Kamlet–Taft solvent parameters
Quantitative measure of solvent polarity, based on the solvent’s hydrogen-bond donor, hydrogen-bond acceptor, dipolarity/polarizability, and cohesive-pressure properties.
See [274], [275], [276], [277], [278], [279].
See solvent parameter.
rev[3]
Kaptein–Closs rules
Rules used to predict the sign of chemically induced dynamic nuclear polarization (CIDNP) effects.
Kekulé structure
Representation of a molecular entity (usually aromatic) with fixed alternating single and double bonds, in which interactions between multiple bonds are ignored.
Example: For benzene, the Kekulé structures are
Note: The distinction among Lewis structure, Kekulé structure, and line formula is now not generally observed, nor is the restriction to aromatic molecular entities.
See also non-Kekulé structure.
rev[3]
keto-enol tautomerization
Interconversion of ketone and enol tautomers by hydron migration, accelerated by acid or base catalysis.
Example:
See also tautomerization.
kinetic ambiguity
deprecated
See kinetic equivalence.
kinetic control (of product composition)
Conditions (including reaction times) that lead to reaction products in a proportion governed by the relative rates of the parallel (forward) reactions by which those products are formed, rather than by the equilibrium constants.
See also thermodynamic control.
[3]
kinetic electrolyte effect
kinetic ionic-strength effect
General effect of an added electrolyte (i.e., other than, or in addition to, that due to its possible involvement as a reactant or catalyst) on the rate constant of a reaction in solution.
Note 1: At low concentrations (when only long-range coulombic forces need to be considered), the effect on a given reaction is determined only by the ionic strength of the solution and not by the chemical identity of the ions. This concentration range is roughly the same as the region of validity of the Debye–Hückel limiting law for activity coefficients. At higher concentrations, the effect of an added electrolyte depends also on the chemical identity of the ions. Such specific actions can sometimes be interpreted as the incursion of a reaction path involving an ion of the electrolyte as the reactant or catalyst, in which case the action is not properly to be regarded just as a kinetic electrolyte effect. At higher concentrations, the effect of an added electrolyte does not necessarily involve a new reaction path, but merely the breakdown of the Debye–Hückel law, whereby ionic activity coefficients vary with the ion.
Note 2: Kinetic electrolyte effects are also called kinetic salt effects.
Note 3: A kinetic electrolyte effect ascribable solely to the influence of the ionic strength on activity coefficients of ionic reactants and transition states is called a primary kinetic electrolyte effect. A kinetic electrolyte effect arising from the influence of the ionic strength of the solution upon the pre-equilibrium concentration of an ionic species that is involved in a subsequent rate-limiting step of a reaction is called a secondary kinetic electrolyte effect. A common case is the secondary electrolyte effect on the concentration of hydrogen ion (acting as catalyst) produced from the ionization of a weak acid in a buffer solution. To eliminate the complication of kinetic electrolyte effects in buffer solutions, it is advisable to maintain constant the ionic strength.
See [283].
See also common-ion effect, order of reaction.
[3]
kinetic equivalence
Property of two reaction schemes that imply the same rate law.
Example: Schemes (i) and (ii) for the formation of C from A under conditions that B does not accumulate as a reaction intermediate:
Both equations for d[C]/dt are of the form where r and s are constants (sometimes called rate coefficients). The equations are identical in their dependence on concentrations and do not distinguish whether HO– catalyzes the formation of B and its reversion to A or is involved only in its further transformation to C. The two schemes are therefore kinetically equivalent.
where r and s are constants (sometimes called rate coefficients). The equations are identical in their dependence on concentrations and do not distinguish whether HO– catalyzes the formation of B and its reversion to A or is involved only in its further transformation to C. The two schemes are therefore kinetically equivalent.
[3]
Koppel–Palm solvent parameters
Quantitative measure of solvent polarity, based on the solvent’s permittivity, refractive index, basicity or nucleophilicity, and acidity or electrophilicity.
See [284].
See solvent parameter.
rev[3]
Kosower Z value
See Z-value.
[3]
labile
Property of a chemical species that is relatively unstable and transient or reactive.
Note: This term must not be used without explanation of the intended meaning.
See also inert, persistent, reactivity, unreactive.
[3]
Laurence solvent parameters
Quantitative measures of solvent polarity, based on the solvent’s dispersion/induction interactions, electrostatic interactions between permanent multipoles, solute Lewis base/solvent Lewis acid interactions, and solute HBD/solvent HBA interactions.
See [285].
least nuclear motion, principle of
hypothesis of least motion
Hypothesis that, for given reactants, the reactions involving the smallest change in nuclear positions will have the lowest energy of activation.
Note: The basis for this hypothesis is that the energy of a structural deformation leading toward reaction is proportional to the sum of the squares of the changes in nuclear positions, which holds only for small deformations and is therefore not always valid.
rev[3]
leaving group
Species (charged or uncharged) that carries away the bonding electron pair when it becomes detached from another fragment in the residual part of the substrate.
Example 1: In the heterolytic solvolysis of (bromomethyl)benzene (benzyl bromide) in acetic acid
the leaving group is Br–.
Example 2: In the reaction
the leaving group is NMe3.
Note: The historical term “leaving group” is ambiguous, because in the heterolysis of R–X, both R+ and X− are fragments that leave from each other. For that reason, X– (as well as Br– and NMe3 in examples 1 and 2) can unambiguously be called nucleofuges, whereas R+ is an electrofuge.
See also electrofuge, entering group, nucleofuge.
rev[3]
Leffler’s relation
Leffler’s assumption
In a series of elementary reactions, the changes in Gibbs activation energies are often found to be proportional to the changes in Gibbs energies for the overall reaction.
This relation was interpreted in terms of the simple assumption that a small change in any transition-state property P‡ is a linear combination of changes in reactant- and product-state properties, PR and PP.
Within the limits of this assumption, the parameter α is an approximate measure of the fractional displacement of the transition state along the minimum-energy reaction path from reactants to products.
See [109].
Note: There are many exceptions to the validity of Leffler’s assumption that α is a measure of the position of the transition state.
See [287].
See Gibbs energy of activation, Hammond postulate.
rev[3]
left-to-right convention
Arrangement of the structural formulae of the reactants so that the bonds to be made or broken form a linear array in which the electron pushing proceeds from left to right.
See [288].
[3]
levelling effect
Tendency of a solvent to make all Brønsted acids whose acidity exceeds a certain value appear equally acidic.
Note 1: This phenomenon is due to the complete transfer of a hydron to a Brønsted-basic solvent from a dissolved acid stronger than the conjugate acid of the solvent. The only acid present to any significant extent in all such solutions is the lyonium ion.
Note 2: For example, the solvent water has a levelling effect on the acidities of HClO4, HCl, and HI. Aqueous solutions of these acids at the same (moderately low) concentrations have the same acidities.
Note 3: A corresponding levelling effect applies to strong bases in protogenic solvents.
[3]
Lewis acid
Molecular entity (and the corresponding chemical species) that is an electron–pair acceptor and therefore able to react with a Lewis base to form a Lewis adduct by sharing the electron pair furnished by the Lewis base.
Example:
Me3B (Lewis acid) + :NH3 (Lewis base) → Me3B––N+H3 (Lewis adduct)
See also coordination.
[3]
Lewis acidity
Thermodynamic tendency of a substrate to act as a Lewis acid.
Note 1: This property is defined quantitatively by the equilibrium constant or Gibbs energy for Lewis adduct formation of a series of Lewis acids with a common reference Lewis base.
Note 2: An alternative measure of Lewis acidity in the gas phase is the enthalpy of Lewis adduct formation for that Lewis acid with a common reference Lewis base.
See also electrophilicity, Gibbs energy diagram, Lewis basicity.
rev[3]
Lewis adduct
Adduct formed between a Lewis acid and a Lewis base.
[3]
Lewis base
Molecular entity (and the corresponding chemical species) able to provide a pair of electrons and thus capable of coordination to a Lewis acid, thereby producing a Lewis adduct.
[3]
Lewis basicity
Thermodynamic tendency of a substance to act as a Lewis base.
Note 1: This property is defined quantitatively by the equilibrium constant or Gibbs energy of Lewis adduct formation for that Lewis base with a common reference Lewis acid.
Note 2: An alternative measure of Lewis basicity in the gas phase is the enthalpy of Lewis adduct formation for that Lewis base with a common reference Lewis acid.
See also donicity, Lewis acidity, nucleophilicity, proton affinity.
rev[3]
Lewis structure
electron-dot structure, Lewis formula
Representation of molecular structure in which (a) nonbonded valence electrons are shown as dots placed adjacent to the atoms with which they are associated and in which (b) a pair of bonding valence electrons in a covalent bond is shown as two dots between the bonded atoms and in which (c) formal charges (e.g., +, –, 2+) are attached to atoms to indicate the difference between the nuclear charge (atomic number) and the total number of electrons associated with that atom, on the formal basis that bonding electrons are shared equally between atoms they join.
Examples:
Note 1: A double bond is represented by two pairs of dots, and a triple bond by three pairs, as in the last example above.
Note 2: Bonding pairs of electrons are usually denoted by lines, representing covalent bonds, as in line formulas, rather than as a pair of dots, and the lone pairs are sometimes omitted.
Examples:
Note 3: The distinction among Lewis structure, Kekulé structure, and line formula is now not generally observed.
rev[3]
lifetime
mean lifetime
τ
Time needed for the concentration of a chemical species that decays in a first-order process to decrease to 1/e of its original value. i.e., c(t = τ) = c(t = 0)/e.
Note 1: Statistically, it represents the mean life expectancy of the species.
Note 2: Mathematically: τ = 1/k = 1/(Σiki), where ki is the first-order rate constant for the i-th decay process of the species.
Note 3: Lifetime is sometimes applied to processes that are not first-order. However, in such cases, the lifetime depends on the initial concentration of the entity or of a quencher and, therefore, only an initial lifetime can be defined. In this case, it should be called decay time.
See [8].
See also chemical relaxation, half-life, rate of reaction.
[3]
ligand
Atom or group bound to a central atom in a polyatomic molecular entity (if it is possible to indicate such a central atom).
See [28].
Note 1: The term is generally used in connection with metallic central atoms.
Note 2: In biochemistry, a part of a polyatomic molecular entity may be considered central, and atoms, groups, or molecules bound to that part are considered ligands.
See [289], p. 335.
[3]
line formula
Two-dimensional representation of molecular entities in which atoms are shown joined by lines representing single bonds (or multiple lines for multiple bonds), without any indication or implication concerning the spatial direction of bonds.
Examples: methanol, ethene
See also condensed formula, Kekulé formula, Lewis formula, skeletal formula.
rev[3]
linear free-energy relation
linear Gibbs-energy relation
[3]
linear Gibbs-energy relation
linear free-energy relation (LFER)
Linear correlation between the logarithm of a rate constant or equilibrium constant for a series of reactions and the logarithm of the rate constant or equilibrium constant for a related series of reactions.
Typical examples of such relations are the Brønsted relation and the Hammett equation (see also σ-value).
Note: The name arises because the logarithm of the value of an equilibrium constant (at constant temperature and pressure) is proportional to a standard Gibbs energy (free energy) change, and the logarithm of the value of a rate constant is a linear function of the Gibbs energy (free energy) of activation.
See Gibbs energy diagram.
[3]
linear solvation-energy relationship (LSER)
Application of solvent parameters in the form of a single- or multi-parameter equation expressing the solvent effect on a given property: e.g., rate of reaction, equilibrium constant, spectroscopic shift, etc.
Note: The solvent effect may be estimated as a linear combination of elementary effects on a given property P, relative to the property P0 in the reference solvent:
where A, B, DP, etc. are acidity, basicity, dipolarity, etc. parameters, and a, b, p… are the sensitivity of the property to each effect.
See Catalán solvent parameters, Dimroth–Reichardt ETparameter, Kamlet–Taft solvent parameters, Koppel–Palm solvent parameters, Laurence solvent parameters, Z-value.
rev[3]
line-shape analysis
Method for determination of rate constants for dynamic chemical exchange from the shapes of spectroscopic signals, most often used in nuclear magnetic resonance (NMR) spectroscopy.
[3]
Lineweaver–Burk plot
See Michaelis–Menten kinetics.
[3]
lipophilic
Feature of molecular entities (or parts of molecular entities) that have a tendency to dissolve in fat-like solvents (e.g., hydrocarbons).
See also hydrophilic, hydrophobic interaction.
rev[3]
London forces
dispersion forces
Attractive forces between molecules due to their mutual polarizability.
Note: London forces are the principal components of the forces between nonpolar molecules.
See [290].
See also van der Waals forces.
[3]
lone (electron) pair (nonbonding electron pair)
Two spin-paired electrons localized in the valence shell on a single atom.
Note: In structural formulas, lone pairs should be designated as two dots.
See also Lewis structure.
rev[3]
LUMO
Acronym for Lowest Unoccupied Molecular Orbital
See frontier orbitals.
rev[3]
lyate ion
Anion produced by hydron (proton, deuteron, triton) removal from a solvent molecule.
Example: the hydroxide ion is the lyate ion of water.
[3]
lyonium ion
Cation produced by hydronation (protonation, deuteronation, or tritonation) of a solvent molecule.
Example: CH3OH2+ is the lyonium ion of methanol.
See also onium ion.
[3]
macroscopic diffusion control
See mixing control.
[3]
magnetic equivalence
Property of nuclei that have the same resonance frequency in nuclear magnetic resonance (NMR) spectroscopy and also identical spin–spin interactions with each nucleus of a neighboring group.
Note 1: The spin–spin interaction between magnetically equivalent nuclei is not manifested in the spectrum, and has no effect on the multiplicity of the respective NMR signals.
Note 2: Magnetically equivalent nuclei are necessarily chemically equivalent, but the reverse is not necessarily true.
[3]
magnetization transfer
NMR method for determining kinetics of chemical exchange by perturbing the magnetization of nuclei in a particular site or sites and following the rate at which magnetic equilibrium is restored.
Note: The most common perturbations are saturation and inversion, and the corresponding techniques are often called “saturation transfer” and “selective inversion-recovery”.
See also saturation transfer.
[3]
Marcus equation
General expression that correlates the Gibbs energy of activation (
where λ is the reorganization energy and Δ‡G0 is the intrinsic barrier, with λ = 4Δ‡G0.
Note: Originally developed for outer-sphere electron transfer reactions, the Marcus equation applies to many atom- and group-transfer reactions. The Marcus equation captures earlier ideas that reaction thermodynamics can influence reaction barriers: e.g., the Brønsted relation (1926), the Bell–Evans–Polanyi principle (1936–38), Leffler’s relation (1953), and the Hammond postulate (1955) [227]. It also implies that changes in intrinsic barriers may dominate over changes of reaction Gibbs energies and thus account for the fact that reaction rates may not be controlled by the relative thermodynamic stabilities of the products.
See [291], [292], [293], [294], [295], [296].
rev[3]
Markovnikov (Markownikoff) rule
Statement of the common mechanistic observation that in electrophilic addition reactions the more electropositive atom (or part) of a polar molecule becomes attached to the carbon bearing more hydrogens.
Note 1: This rule was originally formulated by Markownikoff (Markovnikov) as “In the addition of hydrogen halides to [unsaturated] hydrocarbons, the halogen atom becomes attached to the carbon bearing the lesser number of hydrogen atoms”.
Note 2: This rule can be rationalized as the addition of the more electropositive atom (or part) of the polar molecule to the end of the multiple bond that would result in the more stable carbenium ion (regardless of whether the carbenium ion is a stable intermediate or a transient structure along the minimum-energy reaction path).
Note 3: Addition in the opposite sense, as in radical addition reactions, is commonly called anti-Markovnikov addition.
See [297].
[3]
mass action, law of
Statement that the velocity of a reaction depends on the active mass, i.e., the concentrations of the reactants.
Example: for an association reaction (1) and its reverse (2)
the forward velocity is v1 = k1[A][B], with k1 being the rate constant for the association reaction. For the dissociation reaction 2, the velocity is v2 = k2 [AB]. This is valid only for elementary reactions. Furthermore, the law of mass action states that when a reversible chemical reaction reaches equilibrium at a given temperature, the forward rate is the same as the backward rate. Therefore, the concentrations of the chemicals involved bear a constant relation to each other, described by the equilibrium constant, i.e., for
in equilibrium, v1 = k1 [A][B] = v2 = k2 [AB] and one form of the equilibrium constant for the above chemical reaction is the ratio
Note: First recognized in 1864 as the kinetic law of mass action by Guldberg and Waage, who first introduced the concept of dynamic equilibrium, but incorrectly assumed that the rates could be deduced from the stoichiometric equation [298]. Only after the work of Horstmann [299] and van’t Hoff [300] a mathematical derivation of the reaction rates considering the order of the reaction involved was correctly made.
rev[3]
matrix isolation
Technique for preparation of a reactive or unstable species by dilution in an inert solid matrix (argon, nitrogen, etc.), usually condensed on a window or in an optical cell at low temperature, to preserve its structure for identification by spectroscopic or other means.
See [304].
[3]
Mayr–Patz equation
Rate constants for the reactions of sp2-hybridized electrophiles with nucleophiles can be expressed by the correlation
where E is the nucleophile-independent electrophilicity parameter, N is the electrophile-independent nucleophilicity parameter, and sN is the electrophile-independent nucleophile-specific susceptibility parameter.
Note 1: This equation is equivalent to the conventional linear Gibbs energy relationship lg k = Nu + sNE, where Nu = sNN. The use of N is preferred, because it provides an approximate ranking of relative reactivities of nucleophiles.
Note 2: The correlation should not be applied to reactions with bulky electrophiles, where steric effects cannot be neglected. Because of the way of parametrization, the correlation is applicable only if one or both reaction centers are carbon.
Note 3: As the E parameters of the reference electrophiles are defined as solvent-independent, all solvent effects are shifted into the parameters N and sN.
Note 4: The equation transforms into the Ritchie equation for sN = 1.
Note 5: Applications to SN2-type reactions are possible if an electrophile-specific susceptibility parameter is introduced.
See [305], [306], [307], [308].
See also Ritchie equation, Swain–Scott equation.
The Mayr scale is available at http://www.cup.lmu.de/oc/mayr/DBintro.htm
mechanism
Detailed description of the process leading from the reactants to the products of a reaction, including a characterization as complete as possible of the composition, structure, energy, and other properties of reaction intermediates, products, and transition states.
Note 1: An acceptable mechanism of a specified reaction (and there may be a number of such alternative mechanisms not excluded by the evidence) must be consistent with the reaction stoichiometry, the rate law, and with all other available experimental data, such as the stereochemical course of the reaction.
Note 2: Inferences concerning the electronic motions that dynamically interconvert successive species along the reaction path (as represented by curved arrows, for example) are often included in the description of a mechanism.
rev[3]
mechanism-based inhibition
suicide inhibition
Irreversible inhibition of an enzyme by formation of covalent bond(s) between the enzyme and the inhibitor, which is a substrate analogue that is converted by the enzyme into a species that reacts with the enzyme.
rev[3]
medium
Phase (and composition of the phase) in which chemical species and their reactions are studied.
[3]
Meisenheimer adduct
Jackson-Meisenheimer adduct
Lewis adduct formed in nucleophilic aromatic substitution from a nucleophile (Lewis base) and an aromatic or heteroaromatic compound,
Note 1: In cases where the substrate lacks electron-withdrawing groups, and depending also on the nucleophile, the Meisenheimer adduct is not a minimum on the potential-energy surface but a transition state.
See [309], [310], [311], [312].
Note 2: Analogous cationic adducts, such as
which are intermediates in electrophilic aromatic substitution reactions, are instead called Wheland intermediates or σ-adducts (or the discouraged term σ-complexes).
rev[3]
melting point (corrected/uncorrected)
Temperature at which liquid and solid phases coexist in equilibrium, as measured with a thermometer whose reading was corrected (or not) for the emergent stem that is in ambient air.
Note: In current usage, the qualification often means that the thermometer was/(was not) calibrated or that its accuracy was/(was not verified). This usage is inappropriate and should be abandoned.
[3]
mesolytic cleavage
Cleavage of a bond in a radical ion whereby a radical and an ion are formed. The term reflects the mechanistic duality of the process, which can be viewed as homolytic or heterolytic depending on how the electrons are assigned to the fragments.
[3]
mesomerism, mesomeric structure
obsolete
Resonance, resonance form
rev[3]
mesophase
Phase of a liquid crystalline compound between the crystalline and the isotropic liquid phase.
See [317].
[3]
metastable (chemical species)
deprecated
Transient (chemical species).
[3]
metathesis
Process formally involving the redistribution of fragments between similar chemical species so that the bonds to those fragments in the products are identical (or closely similar) to those in the reactants.
Example:
Note: The term has its origin in inorganic chemistry, with a different meaning, but that older usage is not applicable in physical organic chemistry.
[3]
micellar catalysis
Acceleration of a chemical reaction in solution by the addition of a surfactant at a concentration higher than its critical micelle concentration so that the reaction can proceed in the environment of surfactant aggregates (micelles).
Note 1: Rate enhancements may be due to a higher concentration of the reactants in that environment, or to a more favorable orientation and solvation of the species, or to enhanced rate constants in the micellar pseudophase of the surfactant aggregate.
Note 2: Micelle formation can also lead to a decreased reaction rate.
See [318].
See also catalyst.
[3]
micelle
Aggregate of 1- to 1000-nm diameter formed by surfactants in solution, which exists in equilibrium with the molecules or ions from which it is formed.
See [135].
See also inverted micelle.
rev[3]
Michaelis–Menten kinetics
Appearance of saturation behavior in the dependence of the initial rate of reaction v0 on the initial concentration [S]0 of a substrate when it is present in large excess over the concentration of an enzyme or other catalyst (or reagent) E, following the equation,
where v0 is the observed initial rate, Vmax is its limiting value at substrate saturation (i.e., when [S]0 >> KM, so that all the enzyme is bound to the substrate), and KM is the substrate concentration at which v0 = Vmax/2.
Note 1: Empirical definition, applying to any reaction that follows an equation of this general form.
Note 2: Often, only initial rates are measured, at low conversion, so that [S]0 can be considered as time-independent but varied from run to run.
Note 3: The parameters Vmax and KM (the Michaelis constant) can be evaluated from slope and intercept of a linear plot of 1/v0 against 1/[S]0 (Lineweaver–Burk plot) or from slope and intercept of a linear plot of [S]0/v0 against [S]0 (Eadie-Hofstee plot), but a nonlinear fit, which is readily performed with modern software, is preferable.
Note 4: This equation is also applicable to the condition where E is present in large excess, in which case [E] appears in the equation instead of [S]0.
Note 5: The term has been used to describe reactions that proceed according to the scheme
in which case KM = (k−1 + kcat)/k1 (Briggs-Haldane conditions). It has more usually been applied only to the special case in which k−1 >> kcat and KM = k−1/k1; in this case KM is a true dissociation constant (Michaelis–Menten conditions).
[3]
microscopic diffusion control
encounter control
Observable consequence of the limitation that the rate of a bimolecular chemical reaction in a homogeneous medium cannot exceed the rate of encounter of the reacting molecular entities.
Note: The maximum rate constant is usually in the range 109 to 1010 dm3 mol−1 s−1 in common solvents at room temperature.
See also mixing control.
rev[3]
microscopic reversibility, principle of
In a reversible reaction the mechanism in one direction is exactly the reverse of the mechanism in the other direction.
See also chemical reaction, detailed balancing.
[3]
migration
Transfer (usually intramolecular) of an atom or group during the course of a molecular rearrangement.
Example (of a hydrogen migration):
rev[3]
migratory aptitude
Tendency of a group to participate in a molecular rearrangement, relative to that of another group, often in the same molecule.
Example: In the Baeyer-Villiger rearrangement of PhCOCH3, via intermediate PhC(OH)(OOCOAr)CH3, the major product is CH3COOPh, by phenyl migration, rather than PhCOOCH3.
Note: In nucleophilic rearrangements (migration to an electron-deficient center) the migratory aptitude of a group is loosely related to its capacity to stabilize a partial positive charge, but exceptions are known, and the position of hydrogen in the series is often unpredictable.
rev[3]
migratory insertion
Reaction that involves the migration of a group to another position on a metal center, with insertion of that group into the bond between the metal and the group that is in that other position.
Examples:
CO insertion
Ziegler–Natta reaction
Note: On repetition of the reaction the substituent alternates between the two positions.
rev[3]
minimum structural change, principle of
Principle that a chemical reaction is expected to occur with a minimum of bond changes or with a minimum redistribution of electrons (although more complex reaction cascades are also possible).
Example:
where the transfer of Cl is accompanied by migration of only one carbon, rather than a more extensive molecular rearrangement involving migration of three methyls.
See [286].
See also least nuclear motion, principle of.
rev[3]
minimum-energy reaction path (MERP)
Path of steepest-descent from a saddle point on a potential-energy surface in each direction towards adjacent energy minima; equivalent to the energetically easiest route from reactants to products.
See intrinsic reaction coordinate.
See [7].
mixing control
Experimental limitation of the rate of reaction in solution by the rate of mixing of solutions of the two reactants.
Note 1: Mixing control can occur even when the reaction rate constant is several orders of magnitude less than that for an encounter-controlled reaction.
Note 2: Analogous (and more important) effects of the limitation of reaction rates by the rate of mixing are encountered in heterogeneous (solid/liquid, solid/gas, liquid/gas) systems.
See [321].
See also microscopic diffusion control, stopped flow.
[3]
Möbius aromaticity
Feature of a monocyclic array of π orbitals in which there is a single out-of-phase overlap (or, more generally, an odd number of out-of-phase overlaps), whereby the pattern of aromatic character is opposite to Hückel systems; with 4n π electrons it is stabilized (aromatic), whereas with 4n + 2, it is destabilized (antiaromatic).
Note 1: The name is derived from the topological analogy of such an arrangement of orbitals to a Möbius strip.
Note 2: The concept has been applied to transition states of pericyclic reactions.
Note 3: In the electronically excited state 4n + 2 Möbius π-electron systems are stabilized, and 4n systems are destabilized.
Note 4: A few examples of ground-state Möbius π systems are known [322, 323].
See also aromatic, Hückel (4n + 2) rule, molecular orbitals.
rev[3]
molecular entity
Any constitutionally or isotopically distinct atom, molecule, ion, ion pair, radical, radical ion, complex, conformer, etc., identifiable as a separately distinguishable entity.
Note 1: Molecular entity is used in this glossary as a general term for singular entities, irrespective of their nature, while chemical species stands for sets or ensembles of molecular entities. Note that the name of a substance may refer to the molecular entity or to the chemical species, e.g., methane, may mean either a single molecule of CH4 (molecular entity) or an ensemble of such species, specified or not (chemical species), participating in a reaction.
Note 2: The degree of precision necessary to describe a molecular entity depends on the context. For example, “hydrogen molecule” is an adequate definition of a certain molecular entity for some purposes, whereas for others, it may be necessary to distinguish the electronic state and/or vibrational state and/or nuclear spin, etc. of the molecule.
[3]
molecular formula
List of the elements in a chemical species or molecular entity, with subscripts indicating how many atoms of each element are included.
Note: In organic chemistry, C and H are listed first, then the other elements in alphabetical order.
See [28].
See also empirical formula.
molecular mechanics (MM)
empirical force-field calculation
Computational method intended to give estimates of structures and energies for molecules.
Note: Even though such calculations can be made with either classical or quantum mechanics (or both), the term molecular mechanics is widely understood as a classical mechanics method that does not explicitly describe the electronic structure of the molecular entities. It is based on the assumption of preferred bond lengths and angles, deviations from which lead to strain, and the existence of torsional interactions and attractive and repulsive van der Waals and Coulomb forces between non-bonded atoms, all of which are parametrized to fit experimental properties such as energies or structures. In contrast, in the quantum mechanical implementation no such assumptions/parameters are needed.
rev[3]
molecular metal
Non-metallic material whose properties, such as conductivity, resemble those of metals, usually following oxidative doping.
Example: polyacetylene following oxidative doping with iodine.
[3]
molecular orbital
One-electron wavefunction describing a single electron moving in the field provided by the nuclei and all other electrons of a molecular entity of more than one atom.
Note 1: Molecular orbitals describing valence electrons are often delocalized over several atoms of a molecule. They are conveniently expressed as linear combinations of atomic orbitals. They can be described as two-center, multi-center, etc. in terms of the number of nuclei (or “centers”) encompassed.
Note 2: For molecules with a plane of symmetry, a molecular orbital can be classed as sigma (σ) or pi (π), depending on whether the orbital is symmetric or antisymmetric with respect to reflection in that plane.
See atomic orbital, orbital.
See [7].
rev[3]
molecular rearrangement
Reaction of a molecular entity that involves a change of connectivity.
Note 1: The simplest type of rearrangement is an intramolecular reaction in which the product is isomeric with the reactant (intramolecular isomerization). An example is the first step of the Claisen rearrangement.
Note 2: The definition of molecular rearrangements includes reactions in which there is a migration of an atom or bond (unexpected on the basis of the principle of minimum structural change), as in the reaction
where the rearrangement step can formally be represented as the “1,2-shift” of hydride between adjacent carbon atoms in a carbocation
Note 3: Such migrations also occur in radicals, e.g.,
Note 4: The definition also includes reactions in which an entering group takes up a different position from the leaving group, with accompanying bond migration, such as in the “allylic rearrangement”:
Note 5: A distinction can be made between intramolecular rearrangements (or “true” molecular rearrangements) and intermolecular rearrangements (or apparent rearrangements). In the former case, the atoms and groups that are common to a reactant and a product never separate into independent fragments during the rearrangement stage, whereas in an intermolecular rearrangement, a migrating group becomes completely free from the parent molecule and is re-attached to a different position in a subsequent step, as in the Orton reaction:
See [327].
rev[3]
molecular recognition
Attraction between specific molecules through noncovalent interactions that often exhibit electrostatic and stereochemical complementarity between the partners.
Note: The partners are usually designated as host and guest, where the host recognizes and binds the guest with high selectivity over other molecules of similar size and shape.
molecularity
Number of reactant molecular entities that are involved in the “microscopic chemical event” constituting an elementary reaction.
Note 1: For reactions in solution, this number is always taken to exclude molecular entities that form part of the medium and which are involved solely by virtue of their solvation of solutes.
Note 2: A reaction with a molecularity of one is called unimolecular, one with a molecularity of two, “bimolecular”, and of three, “termolecular”.
See also chemical reaction, order of reaction.
[3]
molecule
An electrically neutral entity consisting of more than one atom.
Note: Rigorously, a molecule must correspond to a depression on the potential-energy surface that is deep enough to confine at least one vibrational state.
See also molecular entity.
[3]
More O’Ferrall–Jencks diagram
Conceptual visualization of the potential-energy surface for a reacting system, as a function of two coordinates, usually bond lengths or bond orders.
Note 1: The diagram is useful for analyzing structural effects on transition-state geometry and energy.
Note 2: According to the Hammond postulate, stabilization of the products relative to the reactants (an effect parallel to the minimum-energy reaction path, MERP) shifts the transition state away from the product geometry, whereas destabilization of the products shifts the transition state towards the product geometry. As first noted by Thornton, stabilization of a structure located off the assumed MERP in a direction perpendicular to it shifts the transition state toward that more stabilized geometry (a perpendicular effect); destabilization shifts the transition state in the opposite direction.
Figure. More O’Ferrall–Jencks diagram for β-elimination reaction, with reactants, products, and possible intermediates at the four corners.
Case 1: In a concerted elimination, the transition state (‡1) is a saddle point near the center of the diagram, and the assumed MERP follows close to the diagonal.
Case 2: If the carbanion intermediate is stabilized, the transition state shifts toward that intermediate, to a transition state (‡2) in which the C–H bond is more extensively broken and the C–X bond is more intact. Conversely, if the carbocation intermediate is stabilized, the transition state shifts towards the top-left corner of the Figure, with less C–H bond breaking and more C–X bond making
Case 3: If leaving group X– is stabilized, energy decreases along the dotted vertical arrow. It follows that in the resulting transition state (‡3), the C–H bond is more intact but there is little change in the C–X bond, the shift of the transition state is the resultant of a parallel (Hammond) component away from the top-right corner and a perpendicular (anti-Hammond) component towards the top-left corner, yielding a transition state (‡3) with less C–H bond breaking but approximately no change in the extent of C–X bond making.
See [229, 328], [329], [330], [331].
See also anti-Hammond effect, Hammond Postulate.
rev[3]
Morse potential
V(r)
(unit: J)
Empirical function relating the potential energy of a molecule to the interatomic distance r accounting for the anharmonicity of bond stretching:
where De is the bond-dissociation energy, re is the equilibrium bond length, and a is a parameter characteristic of a given molecule.
See [7].
mu
µ or µ
Symbol µ used to designate (as a prefix) a ligand that bridges two or more atoms.
Note: If there are more than two atoms being bridged, μ carries a subscript to denote the number of atoms bridged.
Symbol µ used to designate dipole moment as well as many other terms in physics and physical chemistry.
multi-center bond
Bond in which an electron pair is shared among three or more atomic centers.
Note 1: This may be needed when the representation of a molecular entity solely by localized two-electron two-center bonds is unsatisfactory, or when there are not enough electrons to allow one electron pair shared between two adjacent atoms.
Note 2: This is restricted to σ bonds and does not apply to species with delocalized π electrons.
Examples include the three-center bonds in diborane B2H6 and in bridged carbocations.
rev[3]
multident
multidentate
See ambident.
[3]
nanomaterial
Substance whose particles are in the size range of 1 to 100 nm.
Note: This may have chemical properties different from those of the corresponding bulk material.
narcissistic reaction
Chemical reaction that can be described as the automerization or enantiomerization of a reactant into its mirror image (regardless of whether the reactant is chiral).
Examples are cited under degenerate rearrangement and fluxional.
See [332].
rev[3]
neighboring-group participation
Direct interaction of the reaction center (usually, but not necessarily, an incipient carbenium-ion center) with a lone pair of electrons of an atom or with the electrons of a σ or π bond contained within the parent molecule but not conjugated with the reaction center.
Example:
Note 1: A distinction is sometimes made between n, σ, and π participation.
Note 2: The neighboring group serves as a nucleophile, as in SN2 reactions, except that the nucleophile is intramolecular, so that this step is unimolecular.
Note 3: A rate increase due to neighboring-group participation is known as anchimeric assistance.
Note 4: Synartetic acceleration is the name given to the special case of participation by electrons on a substituent attached to a β-carbon, relative to the leaving group attached to the α-carbon, as in the example above. This term is deprecated.
See also intramolecular catalysis, multi-center bond.
rev[3]
NHOMO
Next-to-highest occupied molecular orbital.
See subjacent orbital.
rev[3]
NIH shift
Intramolecular hydrogen migration that can be observed in enzymatic and chemical hydroxylations of aromatic rings, as evidenced by appropriate deuterium labelling, as in
Note 1: In enzymatic reactions, the NIH shift is thought to derive from the rearrangement of arene oxide intermediates, but other pathways have been suggested.
Note 2: NIH stands for National Institutes of Health, where the shift was discovered.
See [333].
[3]
nitrene
Generic name for the species HN and substitution derivatives thereof, containing an electrically neutral univalent nitrogen atom with four nonbonding electrons.
Note 1: Two nonbonding electrons may have antiparallel spins (singlet state) or parallel spins (triplet state).
Note 2: The name is the strict analogue to carbene and, as a generic name, it is preferred to a number of alternative proposed (imene, imine radical, aminylene, azene, azylene, azacarbene, imin, and imidogen).
[3]
no-bond resonance
double-bond-no-bond resonance
Inclusion of one or more contributing structures that lack the σ bond of another contributing structure.
Example:
See hyperconjugation.
rev[3]
nonclassical carbocation
Carbocation that has delocalized (bridged) bonding σ electrons.
rev[3]
non-Kekulé structure
Compound with unpaired electrons for which no Lewis structures are possible with all bonding electrons paired in single or double bonds.
Examples:

Note: For the second example (“trimethylenemethane”), the isomer shown (valence tautomer methylidenecyclopropane) is a Kekulé structure.
See [336].
nucleofuge
Leaving group that carries away the bonding electron pair in a nucleophilic substitution reaction.
Example: In the hydrolysis of a chloroalkane, Cl– is the nucleofuge.
Note 1: Nucleofugality, commonly called leaving-group ability, characterizes the relative rates of atoms or groups to depart with the bonding electron pair from a reference substrate. Nucleofugality depends on the nature of the reference reaction and is not the reverse of nucleophilicity.
Note 2: Protofugality is a special type of nucleofugality, characterizing the relative rates of proton transfer from a series of Brønsted acids H–X to a common Brønsted base.
See [194].
See also electrofuge, nucleophile.
rev[3]
nucleophile (n.), nucleophilic (adj.)
Reactant that forms a bond to its reaction partner (the electrophile) by donating both of its bonding electrons.
Note 1: A “nucleophilic substitution reaction” is a heterolytic reaction in which the reagent supplying the entering group acts as a nucleophile.
Example:
Note 2: Nucleophilic reagents are Lewis bases.
Note 3: The term “nucleophilic” is also used to designate the apparent polar character of certain radicals, as inferred from their higher relative reactivity with reaction sites of lower electron density.
See also nucleophilicity, order of reaction.
[3]
nucleophilic catalysis
Catalysis by a Lewis base, involving conversion of a substrate with low electrophilicity into an intermediate with higher electrophilicity.
Example 1: hydrolysis of acetic anhydride in aqueous solution, catalyzed by 4-(dimethylamino)pyridine
Example 2: SN2 reaction of CH3CH2Cl with HO–, catalyzed by I–:
See also nucleophilicity.
rev[3]
nucleophilic substitution
Heterolytic reaction in which an entering group adds to the electrophilic part of the substrate and in which the leaving group, or nucleofuge, retains both electrons of the bond that is broken, whereupon it becomes another potential nucleophile.
Example:
Note 1: It is arbitrary to emphasize the nucleophile and ignore the feature that this is also an electrophilic substitution, but the distinction depends on the nucleophilic nature of the reactant that is considered to react with the substrate.
Note 2: Nucleophilic substitution reactions are designated as SN1 or SN2, depending on whether they are unimolecular or bimolecular, respectively. Mechanistically, these correspond to two-step and one-step processes, respectively. SN1 reactions follow first-order kinetics but SN2 reactions do not always follow second-order kinetics.
See order of reaction, rate coefficient.
nucleophilicity
Relative reactivity of a nucleophile toward a common electrophile.
Note 1: The concept is related to Lewis basicity. However, whereas the Lewis basicity of a base B: is measured by its equilibrium constant for adduct formation with a common acid A, the nucleophilicity of a Lewis base B: is measured by the rate coefficient for reaction with a common substrate A–Z, often involving formation of a bond to carbon.
Note 2: Protophilicity is a special case of nucleophilicity, describing the relative rates of reactions of a series of Lewis bases B: with a common Brønsted acid H–Z. The term “protophilicity” is preferred over the alternative term “kinetic basicity” because basicity refers to equilibrium constants, whereas “philicity” (like “fugality”) refers to rate constants.
See also Brønsted basicity, electrophilicity, Lewis basicity, Mayr–Patz equation, Ritchie equation, Swain–Scott equation.
rev[3]
n-σ* delocalization
n-σ* no-bond resonance
Delocalization of a lone pair (n) into an antibonding σ-orbital (σ*).
See also anomeric effect, hyperconjugation, resonance.
[3]
octanol–water partition ratio
K OW
Equilibrium concentration of a substance in octan-1-ol divided by its equilibrium concentration in water.
Note: This is a measure of the lipophilicity of a substance. It is used in pharmacological studies and in the assessment of environmental fate and transport of organic chemicals.
See also partition ratio.
octet rule
Electron-counting rule that the number of lone-pair electrons on a first-row atom plus the number of electron pairs in that atom’s bonds should be 8.
onium ion
Cation derived by addition of a hydron to a mononuclear parent hydride of the nitrogen, chalcogen, and halogen family, e.g., H4N+ ammonium ion.
Derivative formed by substitution of the above parent ions by univalent groups, e.g., (CH3)2SH+ dimethylsulfonium (dimethylsulfanium), (CH3CH2)4N+ tetraethylammonium.
See [97].
See also carbenium ion, carbonium ion.
rev[3]
optical yield
Ratio of the optical purity of the product to that of the chiral precursor or reactant.
Note 1: This should not be confused with enantiomeric excess.
Note 2: The optical yield is not related to the chemical yield of the reaction.
See [10].
See stereoselectivity.
[3]
orbital (atomic or molecular)
Wavefunction depending on the spatial coordinates of only one electron.
Note: An orbital is often illustrated by sketching contours, often very approximate, on which the wavefunction has a constant value or by indicating schematically the envelope of the region of space in which there is an arbitrarily fixed high probability (say 95 %) of finding the electron occupying that region, and affixing also the algebraic sign (+ or –) of the wavefunction in each part of that region, or suggesting the sign by shading.
Examples:
See atomic orbital, molecular orbital.
See [7].
rev[3]
orbital steering
Concept expressing the principle that the energetically favorable stereochemistry of approach of two reacting species is governed by the most favorable overlap of their appropriate orbitals.
rev[3]
orbital symmetry
Behavior of an atomic orbital or molecular orbital under molecular symmetry operations, such that under reflection in a symmetry plane or rotation by 180° around a symmetry axis the phase of the orbital is either unchanged (symmetric) or changes sign (antisymmetric), whereby positive and negative lobes are interchanged.
Examples:
The orbital of an idealized single bond is σ, with cylindrical symmetry.
A p-orbital or π-bond orbital has π symmetry, i.e., it is antisymmetric with respect to reflection in a plane passing through the atomic centers with which it is associated.
The HOMO of 1,3-butadiene (illustrated below by its component atomic orbitals) is antisymmetric with respect to 180° rotation about an axis through the C2–C3 bond and perpendicular to the molecular plane.

See also conservation of orbital symmetry, sigma, pi.
rev[3]
order of reaction
Exponent α, independent of concentration and time, in the differential rate equation (rate law) relating the macroscopic (observed, empirical, or phenomenological) rate of reaction v to cA the concentration of one of the chemical species present, as defined by
The argument in the ln function should be of dimension 1. Thus, reduced quantities should be used, i.e., the quantity divided by its unit, {v} = v/(mol dm−3 s−1) and
Note 1: A rate equation can often be expressed in the form v = k [A]α[B]β…, describing the dependence of the rate of reaction on the concentrations [A], [B],…, where exponents α, β, … are independent of concentration and time and k is independent of [A], [B], … In this case, the reaction is said to be of order α with respect to A, of order β with respect to B,…, and of (total or overall) order n = α + β + … The exponents α, β,… sometimes called “partial orders of reaction”, can be positive or negative, integral, or rational nonintegral numbers.
Note 2: For an elementary reaction, a partial order of reaction is the same as the stoichiometric number. The overall order is then the same as the molecularity. For stepwise reactions there is no general connection between stoichiometric numbers and partial orders. Such reactions may have more complex rate laws, so that an apparent order of reaction may vary with the concentrations of the chemical species involved and with the progress of the reaction: in such cases, it is not useful to speak of orders of reaction, although apparent orders of reaction may be deducible from initial rates.
Note 3: In a stepwise reaction, orders of reaction may in principle be assigned to the elementary steps.
Note 4: For chemical rate processes occurring in systems for which concentration changes are not measurable, as in the case of a dynamic equilibrium
then the reaction is of order α with respect to A… and of total (or overall) order (=α + β +…).
Note 5: If the overall rate of reaction is given by
but [B] remains constant in any particular sample (but can vary from sample to sample), then the order of the reaction in A will be α, and the rate of disappearance of A can be expressed in the form
The proportionality factor kobs is called the “observed rate coefficient” and is related to the rate constant k by
Note 6: For the frequent case where α = 1, kobs is often referred to as a “pseudo-first-order rate coefficient”.
See [12].
See also kinetic equivalence, rate coefficient, rate constant.
rev[3]
organocatalysis
Catalysis by small organic molecules, as distinguished from catalysis by (transition) metals or enzymes.
Note 1: Frequently used organocatalysts are secondary amines (covalent catalysis by generation of enamines or of iminium ions as reactive intermediates) and thioureas (hydrogen-bonding catalysis).
Note 2: Organocatalysts are often employed for enantioselectivity.
Note 3: The mechanisms employed by organocatalysts are examples of general acid catalysis, general base catalysis, nucleophilic catalysis, specific acid catalysis, specific base catalysis.
outer-sphere electron transfer
Feature of an electron transfer between redox centers not sharing a common atom or group.
Example:
Note 1: In the transition state, the interaction between the relevant electronic orbitals of the two centers is weak (below 20 kJ mol−1), and the electron(s) must tunnel through space.
Note 2: If instead the donor and the acceptor exhibit a strong electronic coupling, often through a ligand that bridges both, the reaction is described as inner-sphere electron transfer.
Note 3: These two terms derive from studies of metal complexes, and for organic reactions, the terms “nonbonded” and “bonded” electron transfer are often used.
rev[3]
oxidation
Removal of one or more electrons from a molecular entity.
Increase in the oxidation number of an atom within a substrate, see [344].
Gain of oxygen and/or loss of hydrogen by an organic substrate.
Note 1: All oxidations meet criterion (2) and many meet criterion (3), but this is not always easy to demonstrate. Alternatively, an oxidation can be described as the transformation of an organic substrate by removal of one or more electrons from the substrate, often accompanied by gain or loss of water, hydrons, and/or hydroxide, or by nucleophilic substitution, or by molecular rearrangement.
Note 2: This formal definition allows the original idea of oxidation (combination with oxygen), together with its extension to removal of hydrogen, as well as processes closely akin to this type of transformation (and generally regarded in organic chemistry to be oxidations and to be effected by “oxidizing agents”) to be descriptively related to definition (1). For example, the oxidation of methane to chloromethane may be considered as
followed by
rev[3]
oxidation number
oxidation state
Number assigned to a carbon atom in a covalent organic compound according to
where NX, NO, NN, NH, and NM are the numbers of bonds to halogen, oxygen (or sulfur), nitrogen, hydrogen, and a metal, respectively.
Examples:
Note 1: This assignment is based on the convention that each attached atom more electronegative than carbon contributes +1, while each atom less electronegative (including H) contributes −1, and an attached carbon contributes zero.
Note 2: Oxidation numbers are not significant in themselves, but changes in oxidation number are useful for recognizing whether a reaction is an oxidation or a reduction or neither.
Note 3: A different system is used for transition-metal species [345].
See also electronegativity, oxidation.
rev[3]
oxidative addition
Insertion of the metal of a metal complex into a covalent bond involving formally an overall two-electron loss on one metal or a one-electron loss on each of two metals, i.e.,
rev[3]
oxidative coupling
Coupling of two molecular entities through an oxidative process, usually catalyzed by a transition-metal compound.
Example (where the oxidant can be O2 or Cu(II) or others):
rev[3]
parallel effect
Change of the position of the transition state upon stabilization or destabilization of a structure (or structures) along the assumed minimum-energy reaction path.
See Hammond postulate, More O’Ferrall–Jencks diagram.
parallel reaction
See composite reaction.
[3]
paramagnetism
Property of substances having a magnetic susceptibility greater than 0, whereby they are drawn into a magnetic field.
See also diamagnetism.
[3]
partial rate factor
p f Z, mfZ
Rate constant for substitution at one specific site in an aromatic compound divided by the rate constant for substitution at one position in benzene.
Note 1: The partial rate factor pfZ for para-substitution in a monosubstituted benzene C6H5Z is related to the rate constants k(C6H5Z) and k(C6H6) for the total reactions (i.e., at all positions) of C6H5Z and benzene, respectively, and fpara (the fraction of para-substitution in the total product formed from C6H5Z, usually expressed as a percentage) by the relation
Similarly for meta-substitution:
The symbols fpZ, fmZ, foZ are also in use.
Note 2: The term applies also to the ipso position, and it can be extended to other substituted substrates undergoing parallel reactions at different sites with the same reagent according to the same rate law.
See also selectivity.
rev[3]
partition ratio
partition constant, distribution ratio
P
Concentration of a substance in one phase divided by its concentration in another phase, at equilibrium.
Example, for an aqueous/organic system the partition ratio (or distribution ratio D) is given by
Note 1: The most common way of applying P in correlation analysis or quantitative structure–activity relationships is as lg P.
Note 2: The parameter P is extensively used as an indicator of the capacity of a molecular entity to cross biological membranes by passive diffusion.
Note 3: The term partition coefficient is in common usage in toxicology but is not recommended for use in chemistry and should not be used as a synonym for partition constant, partition ratio, or distribution ratio.
See also Hansch constant, octanol–water partition ratio.
pericyclic reaction
Chemical reaction in which concerted reorganization of bonding takes place throughout a cyclic array of continuously bonded atoms.
Note 1: It may be viewed as a reaction proceeding through a fully conjugated cyclic transition state.
Note 2: The term embraces a variety of processes, including cycloaddition, cheletropic reaction, electrocyclic reaction, sigmatropic rearrangement, etc. (provided they are concerted).
See also pseudopericyclic.
[3]
permittivity, relative
dielectric constant (obsolete)
ε r
Measure of the reduction of the magnitude of the potential energy of interaction between two charges on going from vacuum to a condensed medium, expressed as the ratio of the former to the latter.
Note: The term dielectric constant is obsolete. Moreover, the dielectric constant is not a constant because it depends on frequency.
See [11].
perpendicular effect
Change of the position of the transition state upon stabilization or destabilization of a structure (or structures) that lies off the assumed minimum-energy reaction path.
See anti-Hammond effect, Hammond postulate, More O’Ferrall - Jencks diagram.
rev[3]
persistence
Characteristic of a molecular entity that has an appreciable lifetime (minutes or nanoseconds or other, depending on context).
Note 1: Dilute solution or inert solvent may be required for persistence.
Note 2: Persistence is a kinetic or reactivity property, whereas, in contrast, stability (being stable) is a thermodynamic property.
See [348].
See also transient.
rev[3]
pH-rate profile
Plot of observed rate coefficient, or more usually the decadic logarithm of its numerical value, against solution pH, other variables being kept constant.
[3]
phase-transfer catalysis
Enhancement of the rate of a reaction between chemical species located in different phases (immiscible liquids or solid and liquid) by addition of a small quantity of an agent (called the phase-transfer catalyst) that extracts one of the reactants, most commonly an anion, into the other phase so that reaction can proceed.
Note 1: These catalysts are often onium ions (e.g., tetraalkylammonium ions) or complexes of inorganic cations (e.g., as crown ether complexes).
Note 2: The catalyst cation is not consumed in the reaction although an anion exchange does occur.
Example:
rev[3]
phenonium ion
See bridged carbocation.
[3]
photochromism
Reversible transformation of a molecular entity between two forms, A and B, having different absorption spectra, induced in one or both directions by absorption of electromagnetic radiation.
Note 1: The thermodynamically stable form A is transformed by irradiation into form B. The back reaction can occur thermally (photochromism of type T) or photochemically (photochromism of type P).
Note 2: The spectral change is typically, but not necessarily, of visible color.
Note 3: An important parameter is the number of cycles that a photochromic system can undergo.
See [8].
photolysis
Bond cleavage induced by ultraviolet, visible, or infrared radiation.
Example:
Note: The term is used incorrectly to describe irradiation of a sample without any bond cleavage, although in the term “flash photolysis” this usage is accepted.
See [8].
[3]
photostationary state
Steady state reached by a chemical system undergoing photochemical reaction, such that the rates of formation and disappearance are equal for each of the transient molecular entities formed.
See [8].
pi-adduct
See π-adduct.
pi-bond
See σ, π.
polar aprotic solvent
See dipolar non-HBD solvent.
[3]
polar effect
All the interactions whereby a substituent on a reactant molecule RY modifies the electrostatic forces operating at the reaction center Y, relative to the reference standard RoY.
Note 1: These forces may be governed by charge separations arising from differences in the electronegativity of atoms (leading to the presence of dipoles), by the presence of monopoles, or by electron delocalization.
Note 2: It is distinguished from a steric effect.
Note 3: Sometimes, however, the term polar effect is taken to refer to the influence, other than steric, that non-conjugated substituents exert on reaction rates or equilibria, thus excluding effects of electron delocalization between a substituent and the molecular framework to which it is attached.
See also electronic effects (of substituents), field effect, inductive effect.
[3]
polar solvent
Liquid composed of molecules with a significant dipole moment, capable of dissolving ions or other molecules with significant dipole moments.
See polarity.
rev[3]
polarity (of a bond)
Characteristic of a bond between atoms of different electronegativity, such that the electrons in that bond are not shared equally.
polarity (of a solvent)
Overall solvation capability (solvation power) of a solvent toward solutes, which depends on the action of all possible intermolecular interactions between solute ions or molecules and solvent molecules, excluding interactions leading to definite chemical alterations of the ions or molecules of the solute.
Note: Quantitative measures of solvent polarity include relative permittivity (dielectric constant) and various spectroscopic parameters.
See [16].
See solvent parameter.
rev[3]
polarizability
electric polarizability
α
(derived SI unit: C m2 V−1)
Induced dipole moment, μinduced, divided by applied electric field strength E
Note 1: The polarizability represents the ease of distortion of the electron cloud of a molecular entity by an electric field (such as that due to the proximity of a charged species).
Note 2: Polarizability is more often expressed as polarizability volume, with unit cm3, where ε0 is the permittivity of vacuum:
Note 3: In general, polarizability is a tensor that depends on direction, for example, depending on whether the electric field is along a bond or perpendicular to it, and the induced dipole may not even be along the direction of the electric field. However, in ordinary usage, the term refers to the mean polarizability, the average over three rectilinear axes of the molecule.
rev[3]
polydent
polydentate
See ambident.
[3]
potential-energy profile
Curve describing the variation of the potential energy of a system of atoms as a function of a single coordinate.
Note 1: For an elementary reaction, the relevant coordinate is the reaction coordinate, which is a measure of progress along the minimum-energy reaction path (MERP) from a saddle point on a potential-energy surface in each direction toward adjacent energy minima. For a stepwise reaction it is the succession of reaction coordinates for the successive individual reaction steps. For a reaction involving a bifurcation, each branch requires a different reaction coordinate and has its own profile.
Note 2: A profile constructed as a function of an arbitrary internal coordinate (for example, a bond distance) is not guaranteed to pass through a saddle point on the corresponding potential-energy surface; in order to do so, it must be smooth, continuous, and follow the path of lowest energy connecting the reactant and product energy minima.
Examples: (one-step reaction and two-step reaction)
See [349].
See also Gibbs energy diagram, potential-energy surface.
rev[3]
potential-energy surface (PES)
Surface describing the variation within the Born–Oppenheimer approximation of the potential energy of a system of atoms as a function of a set of internal coordinates.
Note 1: A minimum on a PES is characterized by positive curvature in all directions and corresponds to a structure that is stable with respect to small displacements away from its equilibrium geometry; for this structure (a reactant, intermediate, or product), all vibrational frequencies are real. A saddle point is characterized by positive curvature in all directions except for one with negative curvature and corresponds to a transition structure, for which one vibrational frequency is imaginary. A local maximum is characterized by negative curvatures in two (or more) directions and has two (or more) imaginary frequencies; it is sometimes called a second-order (or higher-order) saddle point.
Note 2: It is usual to select only two coordinates in order to represent the surface, with potential energy as the third dimension, or alternatively as a two-dimensional contour map. For example, a PES for a simple reacting triatomic system A–B + C → A + B–C could be constructed using the A···B and B···C distances as two internal coordinates; the third independent coordinate could be the ABC angle or the A···C distance, and its value could either be kept fixed or else be relaxed to minimize the energy at each point on the (A···B, B···C) surface.
Note 3: The path of steepest-descent from a saddle point in each direction towards adjacent energy minima defines a minimum-energy reaction path (MERP) that is equivalent to the energetically easiest route from reactants to products. The change in potential energy along this path across the PES defines a potential-energy profile for the elementary reaction. Progress along this path is measured by the value of the reaction coordinate.
Note 4: In general there is neither a unique set of internal coordinates nor a unique choice of two coordinates with which to construct a PES of reduced dimensionality. Consequently, there is no guarantee of a smooth and continuous PES containing a saddle point connecting reactant and product minima.
See bifurcation, minimum-energy reaction path, potential-energy profile, reaction coordinate, transition structure.
rev[3]
potential of mean force (PMF)
Free energy as a function of a set of coordinates, the negative gradient of which gives the average force acting on that configuration averaged over all other coordinates and momenta within a statistical distribution. If the averaging is performed within a canonical ensemble (constant volume, temperature, and number of particles), the PMF is equivalent to the Helmholtz energy, but if it is performed within an isobaric–isothermal ensemble (constant pressure, temperature, and number of particles), the PMF is equivalent to the Gibbs energy.
Note 1: Commonly, the PMF acting upon a selected geometric variable and averaged over the coordinates and momenta of all other geometric variables is evaluated for a succession of constrained values of the selected variable, thereby generating (generically) a free-energy profile with respect to the selected reaction coordinate (e.g., a bond distance or angle or a combination of internal coordinates); specifically, this is either a Helmholtz-energy or a Gibbs-energy profile, depending upon the choice of ensemble for the statistical averaging within a computational simulation.
Note 2: Selection of two geometric variables as reaction coordinates allows a free-energy surface to be computed as a two-dimensional PMF.
Note 3: Molecular simulations often yield Helmholtz energies, not Gibbs energies, but for condensed phases the difference is usually neglected.
See free energy, reaction coordinate.
pre-association
Step on the reaction path of some stepwise reactions in which the molecular entity C forms an encounter pair or encounter complex with A prior to the reaction of A to form product.
Example:
Note 1: In this mechanism, the chemical species C may but does not necessarily assist the formation of B from A, which may itself be a bimolecular reaction with some other reagent.
Specific example (aminolysis of phenyl acetate):
Note 2: Pre-association is important when B is too short-lived to permit B and C to come together by diffusion. In the specific example, T± would dissociate faster than general acid HA can diffuse to it. Experimentally, the Brønsted α is > 0, which is inconsistent with rate-limiting diffusion and hydron transfer.
See [352].
See also Brønsted relation, microscopic diffusion control, spectator mechanism.
rev[3]
precursor complex
See encounter complex.
[3]
pre-equilibrium
prior equilibrium
Rapid reversible step preceding the rate-limiting step in a stepwise reaction.
Example:
See also kinetic equivalence, steady state.
[3]
pre-exponential factor
See energy of activation, entropy of activation.
[3]
principle of nonperfect synchronization
Consideration applicable to reactions in which there is a lack of synchronization between bond formation or bond rupture and other changes that affect the stability of products and reactants, such as resonance, solvation, electrostatic, hydrogen bonding, and polarizability effects.
Note: The principle states that a product-stabilizing factor whose development lags behind bond changes at the transition state, or a reactant-stabilizing factor whose loss is ahead of bond changes at the transition state, increases the intrinsic barrier and decreases the rate constant of a reaction. For a product-stabilizing factor whose development is ahead of bond changes, or a reactant-stabilizing factor whose loss lags behind bond changes, the opposite relations hold. The reverse effects are observable for factors that destabilize a reactant or product.
See [108].
See also imbalance, synchronous.
prior equilibrium
See pre-equilibrium.
[3]
product-determining step
Step of a stepwise reaction in which the product distribution is determined.
Note: The product-determining step may be identical to, or may occur later than, the rate-determining step in the reaction.
[3]
product-development control
Case of kinetic control in which the selectivity of a reaction parallels the relative (thermodynamic) stabilities of the products.
Note: Product-development control arises because whatever effect stabilizes or destabilizes a product is already operative at the transition state. Therefore, it is usually associated with a transition state occurring late on the minimum-energy reaction path.
See also steric-approach control, thermodynamic control.
[3]
promotion
See pseudo-catalysis.
[3]
propagation
See chain reaction.
[3]
propargylic substitution
See allylic substitution reaction.
protic
See protogenic.
[3]
protic solvent
Solvent that is capable of acting as a hydrogen-bond donor.
See HBD solvent.
protofugality
Special case of nucleofugality, describing the relative rates of transfer of a proton (more generally: hydron) from a series of Brønsted acids H–X to a common Brønsted base.
Note: This term has the advantage over the commonly used “kinetic acidity” that philicity and fugality are associated with kinetics, while acidity and basicity are associated with thermodynamics.
See [194].
See also Brønsted acidity, nucleofugality, protophilicity.
protogenic (solvent)
HBD (hydrogen bond donor) solvent.
Capable of acting as a proton (hydron) donor.
Note 1: Such a solvent may be a strong or weak Brønsted acid.
Note 2: The term is preferred to the synonym protic or to the more ambiguous expression acidic.
See protophilic solvent.
[3]
protolysis
proton (hydron)-transfer reaction.
Note: Because of its misleading similarity to hydrolysis, photolysis, etc., this use is discouraged.
See also autoprotolysis.
rev[3]
proton affinity
Negative of the enthalpy change in the gas phase reaction between a proton (more appropriately hydron) and the chemical species concerned, usually an electrically neutral or anionic species, to give the conjugate acid of that species.
Note 1: For an anion A–, the proton affinity is the negative of the enthalpy of the heterolytic dissociation (in the gas phase) of the Brønsted acid HA.
Note 2: Proton affinity is often, but unofficially, abbreviated as PA.
Note 3: Affinity properly refers to Gibbs energy.
See also gas phase acidity, gas phase basicity.
rev[3]
protonation
Attachment of the ion 1H+ of relative isotopic mass approximately equal to 1.
See also hydronation.
proton–transfer reaction
Chemical reaction, the main feature of which is the intermolecular or intramolecular transfer of a proton (hydron) from one binding site to another.
Example:
Note: In the detailed description of proton-transfer reactions, especially of rapid proton transfers between electronegative atoms, it should always be specified whether the term is used to refer to the overall process (including the more-or-less encounter-controlled formation of a hydrogen-bonded complex and the separation of the products) or just to the proton-transfer event (including solvent rearrangement) by itself.
See also autoprotolysis, microscopic diffusion control, tautomerism.
[3]
protophilic (solvent)
See HBA (hydrogen bond acceptor) solvent.
rev[3]
protophilicity
Special case of nucleophilicity, describing the relative rates of reactions of a series of Lewis bases with a common Brønsted acid. This term has the advantage over the commonly used “kinetic basicity” that philicity and fugality are associated with kinetics, while acidity and basicity are associated with thermodynamics.
See [194].
See also Brønsted basicity, nucleophilicity, protofugality.
prototropic rearrangement (prototropy)
See tautomerization.
[3]
pseudo-catalysis
Increase in the rate of a reaction by an acid or base present in nearly constant concentration throughout a reaction in solution (owing to buffering or to the use of a large excess), even though that acid or base is consumed during the process, so that the acid or base is not a catalyst and the phenomenon strictly cannot be called catalysis according to the established meaning of these terms in chemical kinetics.
Note 1: Although the mechanism of such a process is often closely related to that of a catalyzed reaction, it is recommended that the term pseudo-catalysis be used in these and analogous cases. For example, if a Brønsted acid accelerates the hydrolysis of an ester to a carboxylic acid and an alcohol, this is properly called acid catalysis, whereas the acceleration, by the same acid, of the hydrolysis of an amide should be described as pseudo-catalysis because the acid pseudo-catalyst is stoichiometrically consumed during the reaction through formation of an ammonium ion.
Note 2: The terms general-acid pseudo-catalysis and general-base pseudo-catalysis may be used as the analogues of general acid catalysis and general base catalysis.
Note 3: The terms acid- and base-promoted, acid- and base-accelerated, and acid- and base-induced are sometimes used for reactions that are pseudo-catalyzed by acids or bases. However, the term promotion also has a different meaning in other chemical contexts.
rev[3]
pseudo-first-order rate coefficient
See order of reaction, rate coefficient.
[3]
pseudopericyclic
Feature of a concerted transformation in which the primary changes in bonding occur within a cyclic array of atoms but in which one (or more) nonbonding and bonding atomic orbitals interchange roles.
Examples: enol-to-enol tautomerism of 4-hydroxypent-3-en-2-one (where the electron pairs in the O–H bond and in the lone pair on the other O are in σ orbitals whereas the other three electron pairs are in π orbitals) and hydroboration (where the B uses an sp2 bonding orbital and a vacant p orbital)

Note: Because the atomic orbitals that interchange roles are orthogonal, such a reaction does not proceed through a fully conjugated transition state and is thus not a pericyclic reaction. It is therefore not governed by the rules that express orbital symmetry restrictions applicable to pericyclic reactions.
See also coarctate.
rev[3]
push-pull conjugation
Feature of an extended conjugated π system bearing an electron donor group at one end and an electron acceptor group at the other end.
See [356].
See also cross-conjugation.
pyrolysis
Thermolysis, usually associated with exposure to a high temperature.
See also flash vacuum pyrolysis.
[3]
π-adduct
pi-adduct
Adduct formed by electron-pair donation from a π orbital into a σ* orbital, or from a σ orbital into a π* orbital, or from a π orbital into a π* orbital.
Example:
Note: Such an adduct has commonly been known as a π complex, but, as the bonding is not necessarily weak, it is better to avoid the term complex, in accordance with the recommendations in this Glossary.
See also coordination.
[3]
π-bond
pi bond
Interaction between two atoms whose p orbitals overlap sideways.
Note: The designation as π is because the p orbitals are antisymmetric with respect to a defining plane containing the two atoms.
See sigma, pi.
rev[3]
π-complex
See π-adduct.
[3]
π-electron acceptor
Substituent capable of electron withdrawal by resonance (e.g., NO2).
See electronic effect, polar effect, π-electron donor, σ-constant.
rev[3]
π-electron donor
Substituent capable of electron donation by resonance (e.g., OCH3).
See electronic effect, polar effect, π-electron acceptor, σ-constant.
rev[3]
quantitative structure–activity relationship (QSAR)
Regression model to correlate biological activity or chemical reactivity with predictor parameters based on measured or calculated features of molecular structure.
See also correlation analysis.
rev[3]
quantitative structure–property relationship (QSPR)
Regression model to correlate chemical properties such as boiling point or chromatographic retention time with predictor parameters based on measured or calculated features of molecular structure.
See [359].
See also correlation analysis.
quantum yield
Number of defined events that occur per photon absorbed by the system.
Note 1: The integral quantum yield Φ is the number of events divided by the number of photons absorbed in a specified wavelength range.
Note 2: For a photochemical reaction, Φ(λ) is the amount of reactant consumed or product formed divided by the number of photons absorbed at wavelength λ.
Note 3: The differential quantum yield for a homogeneous system is
where |dx/dt| is the rate of change of a quantity x that measures the progress of a reaction, qp,λ is the spectral photon flux (mol or its non-SI equivalent einstein) incident per unit time at wavelength λ, and A(λ) is the decadic absorbance at the excitation wavelength λ.
Note 4: When the quantity x is an amount concentration, it is convenient to use in the denominator the rate (in moles) of photons absorbed per volume.
rev[3]
radical
free radical (obsolete)
Molecular entity possessing an unpaired electron.
Examples: ·CH3, ·SnR3, and Cl·
Note 1: In these formulae, the dot, symbolizing the unpaired electron, should be placed so as to indicate the atom of highest spin density, if possible.
Note 2: Paramagnetic metal ions are not normally regarded as radicals. However, in the isolobal analogy, the similarity between certain paramagnetic metal ions and radicals becomes apparent.
Note 3: Depending on the core atom that possesses the highest spin density, the radicals can be described as carbon-, oxygen-, nitrogen-, or metal-centered radicals.
Note 4: If the unpaired electron occupies an orbital having considerable s or more or less pure p character, the respective radicals are termed σ or π radicals.
Note 5: The term radical has also been used to designate a substituent group within a molecular entity, as opposed to “free radical”, which is now simply called radical. The bound entities may be called groups or substituents, but should no longer be called radicals.
See also diradical.
rev[3]
radical combination
Formation of a covalent bond by reaction of one radical with another.
See colligation.
rev[3]
radical ion
Radical that carries a net electric charge.
Note 1: A positively charged radical is called a radical cation (e.g., the benzene radical cation C6H6•+); a negatively charged radical is called a radical anion (e.g., the benzene radical anion C6H6•– or the benzophenone radical anion Ph2C–O•–).
Note 2: Unless the positions of unpaired spin and charge can be associated with specific atoms, superscript dot and charge designations should be placed in the order ⋅+ or ⋅–, as suggested by the name radical ion. However, the usage in mass spectrometry is to place the charge symbol before the dot [7].
Note 3: In the first edition of this Glossary, it was recommended to place the charge designation directly above the dot. This format is now discouraged because of the difficulty of extending it to ions bearing more than one charge and/or more than one unpaired electron.
[3]
radical pair
geminate pair
Two radicals in close proximity in solution, within a solvent cage.
Note 1: The two radicals may be formed simultaneously by some unimolecular process, e.g., peroxide decomposition or photolysis, or they may have come together by diffusion.
Note 2: While the radicals are together, correlation of the unpaired electron spins of the two species cannot be ignored: this correlation is responsible for the CIDNP phenomenon.
See also geminate recombination.
[3]
radiolysis
Cleavage of one or several bonds resulting from exposure to high-energy radiation.
Note: The term is also often used loosely to specify the method of irradiation (e.g., pulse radiolysis) used in any radiochemical reaction, not necessarily one involving bond cleavage.
[3]
rate coefficient
Empirical constant k in the equation for the rate of a reaction that is expressible by an equation of the form
Note 1: It is recommended that the term rate constant be confined to reactions that are believed to be elementary reactions.
Note 2: When a rate coefficient relates to a reaction occurring by a composite mechanism, it may vary not only with temperature and pressure but also with the concentration of reactants. For example, in the case of a unimolecular gas reaction the rate at sufficiently high pressures is given by
whereas at low pressures the rate expression is
Similarly, for a second-order reaction, the rate is given by
But under conditions where [B] remains constant at [B]0, as when B is a catalyst or is present in large excess,
where the rate coefficient k = k2[B]0, which varies with [B]0.
Such a rate coefficient k, which varies with the concentration [B], is called a first-order rate coefficient for the reaction, or a pseudo first-order rate constant even though it is not a rate constant.
Note: Rate constant and rate coefficient are often used as synonyms, see [12], section 2.12, p.63.
See [12].
See order of reaction.
rev[3]
rate constant
k
Term generally used for the rate coefficient of a reaction that is believed to be an elementary reaction. See [12].
Note 1: In contrast to a rate coefficient, a rate constant should be independent of concentrations, but in general both rate constant and rate coefficient vary with temperature.
Note 2: Rate constant and rate coefficient are often used as synonymous, see [11], section 2.12, p.63.
See order of reaction.
rev[3]
rate-controlling step
See rate-determining states, rate-limiting step.
rev[3]
rate law
empirical differential rate equation
Expression for the rate of reaction in terms of concentrations of chemical species and constant parameters (normally rate coefficients and partial orders of reaction) only.
For examples of rate laws, see the equations under kinetic equivalence, and under steady state.
[3]
rate-limiting step
rate-controlling step
rate-determining step
Step in a multistep reaction that is the last step in the sequence whose rate constant appears in the rate equation.
See [12].
Example: Two-step reaction of A with B to give intermediate C, which then reacts further with D to give products:
If [C] reaches a steady state, then the observed rate is given by
Case 1:
If k2[D] >> k−1,then the observed rate simplifies to
Because k2 disappears from the rate equation and k1 is the last rate constant to remain, step (1) is said to be rate-limiting.
Case 2:
If k2[D] << k−1, then the observed rate is given by
where K, equal to k1/k−1, is the equilibrium constant for the pre-equilibrium (Step 1). Because k2 remains in the rate equation, Step 2 is said to be rate-limiting. Notice that in this case, where Step 2 involves another reactant D, which step is rate-limiting can depend on [D]: Step 1 at high [D] and Step 2 at low [D].
Specific example: Acid-catalyzed bromination of a methyl ketone
where
According to the steady-state approximation,
or
If k4 >> k−3, k−3 can be ignored in the parentheses, so that kobs simplifies to k1k2k3k4/{k2k3k4 + k−1k3k4 + k−1k−2k4}. Then, if k3 >> k−2, kobs simplifies further to k1k2k3k4/{k2k3k4 + k−1k3k4}, where k3k4 cancels and kobs becomes k1k2/{k−1 + k2}, so that k2 is the last rate constant remaining in kobs. The second step is rate-limiting, and the rate is independent of k3 or of [Br2].
Note 1: Although the expressions rate-controlling, rate-determining, and rate-limiting are often regarded as synonymous, rate-limiting is to be preferred, because in Case 2, all three rate constants enter into the rate equation, so that all three are rate-controlling and rate-determining, but the first step is not rate-limiting.
Note 2: If the concentration of any intermediate builds up to an appreciable extent, then the steady-state approximation no longer holds, and the reaction should be analyzed as though that intermediate is the reactant.
Note 3: It should be noted that a catalytic cycle does not have a rate-determining step. Instead, under steady-state conditions, all steps proceed at the same rate because the concentrations of all intermediates adjust so as to offset the differences in the corresponding rate constants [360].
See also Gibbs energy diagram, microscopic diffusion control, mixing control.
rev[3]
rate of reaction
v
(unit: mol dm−3 s−1 or mol L−1 s−1)
For the general chemical reaction
occurring under constant-volume conditions, without an appreciable build-up of reaction intermediates, the rate of reaction v is defined as
where symbols inside square brackets denote concentrations (conventionally expressed in unit mol dm−3). The symbols R and r are also used instead of v. It is recommended that the unit of time be the second.
Example:
Note: For a stepwise reaction, this definition of rate of reaction will apply only if there is no accumulation of intermediate or formation of side products. It is therefore recommended that the term rate of reaction be used only in cases where it is experimentally established that these conditions apply. More generally, it is recommended that, instead, the terms rate of disappearance or rate of consumption of A (i.e., –d[A]/dt) or rate of appearance of P (i.e., d[P]/dt) be used, depending on the particular chemical species that is actually observed. In some cases, reference to the chemical flux observed may be more appropriate.
See [12].
See also chemical relaxation, lifetime, order of reaction.
rev[3]
reaction coordinate
Parameter that changes during the conversion of one (or more) reactant molecular entities into one (or more) product molecular entities and whose value can be taken as a measure of the progress along a minimum-energy reaction path.
Note 1: The term “reaction coordinate” is often used to refer to a geometric variable itself (typically a bond distance or bond angle, or a combination of distances and/or angles) as well as (or instead of) the value of that variable. Although strictly incorrect, this usage is very commonly encountered.
Note 2: In cases where the location of the transition structure is unknown, an internal coordinate of the system (e.g., a geometric variable or a bond order, or an energy gap between reactant-like and product-like valence-bond structures) is often selected as a reaction coordinate. A potential-energy profile obtained by energy minimization over other coordinates for a succession of fixed values of an arbitrary reaction coordinate is not guaranteed to pass through the transition structure unless it is a continuous function of that reaction coordinate. Similarly, a free-energy profile obtained as a potential of mean force with respect to an arbitrary reaction coordinate is not guaranteed to pass through the lowest-energy transition state.
Note 3: “Reaction coordinate” is sometimes used as an undefined label for the horizontal axis of a potential-energy profile or a Gibbs energy diagram.
See also Gibbs energy diagram, potential-energy profile, potential-energy surface.
rev[3]
reaction path
Synonym for mechanism.
Trajectory on the potential-energy surface.
reaction step
Elementary reaction constituting one of the stages of a stepwise reaction in which a reaction intermediate (or, for the first step, the reactants) is converted into the next reaction intermediate (or, for the last step, the products) in the sequence of intermediates between reactants and products.
[3]
reactive intermediate
intermediate
reactivity (n.), reactive (adj.)
Kinetic property of a chemical species by which (for whatever reason) it has a different rate constant for a specified elementary reaction than some other (reference) species.
Note 1: The term has meaning only by reference to some explicitly stated or implicitly assumed set of conditions. It is not to be used for reactions or reaction patterns of compounds in general.
Note 2: Term also used more loosely as a phenomenological description not restricted to elementary reactions. When applied in this sense, the property under consideration may reflect not only rate constants but also equilibrium constants.
See also stable, unreactive, unstable.
[3]
reactivity index
Numerical quantity derived from quantum-mechanical model calculations or from a linear Gibbs-energy relationship (linear free-energy relationship) that permits the prediction or correlation of relative reactivities of different molecular sites.
Note: Many indices are in use, based on a variety of theories and relating to various types of reaction. The more successful applications have been to the substitution reactions of conjugated systems, where relative reactivities are determined largely by changes of π-electron energy and π-electron density.
rev[3]
reactivity-selectivity principle (RSP)
Idea that the more reactive a reagent is, the less selective it is.
Note: There are many examples in which the RSP is followed, but there are also many counterexamples. Although the RSP is in accordance with intuitive feeling, it is now clear that selectivity can decrease, increase, or remain constant as reactivity increases, so that the RSP is unreliable as a guide to reactivity.
See [126, 346, 362], [363], [364], [365].
rev[3]
rearrangement
See degenerate rearrangement, molecular rearrangement, sigmatropic rearrangement.
[3]
reduction
Transfer of one or more electrons to a molecular entity, usually inorganic.
Decrease in the oxidation number of any atom within any substrate [344].
Loss of oxygen or halogen and/or gain of hydrogen of an organic substrate.
See oxidation.
rev[3]
reductive elimination
Reverse of oxidative addition.
[3]
regioselectivity (n.), regioselective (adj.)
Property of a reaction in which one position of bond making or breaking occurs preferentially over all other possible positions.
Note 1: The resulting regioisomers are constitutional isomers.
Note 2: Reactions are termed completely (100 %) regioselective if the discrimination is complete, or partially (x %) if the product of reaction at one site predominates over the product of reaction at other sites. The discrimination may also be referred to semi-quantitatively as high or low regioselectivity.
Note 3: Historically the term was restricted to addition reactions of unsymmetrical reagents to unsymmetrical alkenes.
Note 4: In the past, the term regiospecificity was proposed for 100 % regioselectivity. This terminology is not recommended, owing to inconsistency with the terms stereoselectivity and stereospecificity.
See also chemoselectivity.
rev[3]
Reichardt ET parameter
See Dimroth–Reichardt ET(30) parameter.
relaxation
Passage of a system that has been perturbed from equilibrium, by radiation excitation or otherwise, toward or into thermal equilibrium with its environment.
See [8].
See also chemical relaxation.
rev[3]
reorganization energy
Gibbs energy required to distort the reactants (and their associated solvent molecules) from their relaxed nuclear configurations to the relaxed nuclear configurations of the products (and their associated solvent molecules).
Note 1: This approach was originally formulated for one-electron transfer reactions, A + D → A–• + D+•, in the framework of the Marcus equation, assuming weak coupling between the reactants [368].
Note 2: Reorganization energy is not the same as distortion energy, which is the energy required to distort the reactants to the nuclear configuration of the transition state.
Note 3: This approach has been extended to enzyme-catalyzed reactions [369].
Note 4: Marcus theory has been shown to be valid for some complex reactions (cycloaddition, SN2), even though the weak-coupling assumption is clearly not valid. In these cases, the reorganization energy (in terms of activation strain) is counteracted by stabilizing interactions (electrostatic and orbital) [166].
See also distortion interaction model, intrinsic barrier, Marcus equation.
rev[3]
resonance
Representation of the electronic structure of a molecular entity in terms of contributing Lewis structures.
Note 1: Resonance means that the wavefunction is represented by mixing the wavefunctions of the contributing Lewis structures.
Note 2: The contributing Lewis structures are represented as connected by a double-headed arrow (↔), rather than by the double arrow
Note 3: This concept is the basis of the quantum-mechanical valence-bond methods. The resulting stabilization is linked to the quantum-mechanical concept of resonance energy. The term resonance is also used to refer to the delocalization phenomenon itself.
Note 4: This term has a completely different meaning in physics.
See also [371].
rev[3]
resonance effect
Experimentally observable influence (on reactivity, etc.) of a substituent through electron delocalization to or from the substituent.
See also inductive effect.
rev[3]
resonance energy
Difference in potential energy between the actual molecular entity and the contributing Lewis structure of lowest potential energy.
Note: The resonance energy cannot be measured experimentally, but only estimated, as contributing Lewis structures are not observable molecular entities.
See resonance.
rev[3]
resonance form
resonance structure, contributing structure, canonical form
One of at least two Lewis structures, with fixed single, double, and triple bonds, that is a contributing structure to the valence-bond wavefunction of a molecule that cannot be described by a single Lewis structure.
Note 1: Although the valence-bond wavefunction is a linear combination of the wavefunctions of the individual resonance forms, the coefficients and the relative contributions of the various resonance forms are usually kept qualitative. For example, the major resonance forms for the conjugate base of acetone are CH2=C(CH3)–O– and H2C––C(CH3)=O, with the former contributing more.
Note 2: Resonance forms are connected by a double-headed arrow (↔). This must not be confused with the double arrow connecting species in equilibrium
See also delocalization, Kekulé structure, resonance.
resonance hybrid
Molecular entity whose electronic structure is represented as the superposition of two or more resonance forms (or Lewis structures) with different formal arrangements of electrons but identical arrangements of nuclei.
Note 1: Whereas each contributing resonance form represents a localized arrangement of electrons which, considered by itself, would imply different bond lengths (say) for formal single and double bonds, resonance between two or more contributors requires each to have the same geometry, namely that of the resulting hybrid, which represents a delocalized arrangement of electrons. A particular bond in the hybrid may have a length that is, loosely, an “average” of formal single-bond and double-bond values implied by its contributing individual resonance forms, but the hybrid does not oscillate among these as if they were in equilibrium.
Note 2: The resonance forms are connected by a double-headed arrow (↔), rather than by the double arrow
retrocycloaddition
deprecated
Cycloelimination.
[3]
Ritchie equation
Linear Gibbs-energy relation (linear free-energy relation)
applied to the reactions between nucleophiles and certain large and relatively stable organic cations, e.g., arenediazonium, triarylmethyl, and aryltropylium cations, in various solvents, where {kN} is the (reduced) second-order rate constant for reaction of a given cation with a given nucleophilic system (i.e., given nucleophile in a given solvent). {k0} is the (reduced) first-order rate constant for the same cation with water in water, and N+ is a parameter characteristic of the nucleophilic system and independent of the electrophilic reaction partner.
The discrepancy between second-order and first-order rate constants must be reconciled by writing the equation with arguments of the logarithms of dimension 1, i.e., by using reduced rate constants (as denoted by the curly brackets in the defining equation):
Note 1: A surprising feature of the equation is the absence of a coefficient of N+ characteristic of the substrate (cf. the s in the Swain–Scott equation), even though values of N+ vary over 13 decadic log (lg) units. The equation thus involves a gigantic breakdown of the reactivity-selectivity principle.
Note 2: The Ritchie equation is a special case of the more general Mayr–Patz equation.
See [193, 372, 373]. See also [125, 374].
rev[3]
ρ-value
rho-value
Quantitative measure of the susceptibility of the rate constant or equilibrium constant of an organic reaction to the influence of substituent groups, usually on an aromatic ring.
Note 1: Defined by Hammett to describe the effects of substituents at the meta- and para-positions on rate or equilibrium of a reaction on the side chain of a substituted benzene, the empirical equation has the general form
in which σX is a constant characteristic of the substituent X and of its position in the reactant molecule.
Note 2: More generally (not only for aromatic series), ρ-values (modified with appropriate subscripts and superscripts) are used to designate the susceptibility to substituent effects of reactions of families of organic compounds, as given by the modified set of σ-constants in an empirical correlation.
Note 3: Reactions with a positive ρ-value are accelerated (or the equilibrium constants are increased) by substituents with positive σ-constants. Because the sign of σ was defined so that substituents with a positive σ increase the acidity of benzoic acid, such substituents are generally described as attracting electrons away from the aromatic ring. It follows that reactions with a positive ρ-value involve a transition state (or reaction product) with an increased electron density at the reactive site of the substrate.
See also Hammett equation, σ-constant, Taft equation.
rev[3]
ρσ-equation
rho-sigma equation
See Hammett equation, ρ-value, σ-constant, Taft equation.
[3]
salt effect
See kinetic electrolyte effect.
[3]
saturation transfer
See magnetization transfer.
rev[3]
Saytzeff rule
Preferential removal of a hydrogen from the β carbon that has the fewest hydrogens in dehydrohalogenation of secondary and tertiary haloalkanes.
Note 1: The rule was originally formulated by A. Saytzeff (Zaitsev) to generalize the orientation in β-elimination reactions of haloalkanes. It has been extended and modified, as follows: When two or more olefins can be produced in an elimination reaction, the thermodynamically most stable alkene will predominate.
Note 2: Exceptions to the Saytzeff rule are exemplified by the Hofmann rule.
See [375].
See also Markovnikov rule.
[3]
scavenger
Substance that reacts with (or otherwise removes) a trace component (as in the scavenging of trace metal ions) or traps a reactive intermediate.
See also inhibition.
[3]
selectivity
Discrimination shown by a reagent in competitive attack on two or more substrates or on two or more positions or diastereotopic or enantiotopic faces of the same substrate.
Note 1: Selectivity is quantitatively expressed by the ratio of rate constants of the competing reactions or by the decadic logarithm of such a ratio.
Note 2: In the context of aromatic substitution (usually electrophilic, for monosubstituted benzene derivatives), the selectivity factor Sf (expressing discrimination between p- and m-positions in PhZ) is defined as
where the partial rate factors pfZ and mfZ express the reactivity of para and meta positions in the aromatic compound PhZ relative to that of a single position in benzene.
See [347].
See also isoselective relationship, partial rate factor, regioselectivity, stereoselectivity.
rev[3]
self-assembly
Process whereby a system of single-molecule components spontaneously forms an organized structure, owing to molecular recognition.
shielding
Extent to which the effective magnetic field is reduced for a nucleus in a molecule immersed in an external magnetic field, relative to that experienced by a bare nucleus in that field.
Note 1: The reduction is due to the circulation of the electrons around the observed and the neighboring nuclei. The external field induces a magnetic moment that is oriented in the opposite direction to the external field, so that the local field at the central nucleus is weakened, although it may be strengthened at other nuclei (deshielding).
Note 2: This phenomenon is the origin of the structural dependence of the resonance frequencies of the nuclei.
See also chemical shift.
[3]
shift reagent
Paramagnetic substance that induces an additional change of the NMR resonance frequency of a nucleus near any site in a molecule to which the substance binds.
See chemical shift
sigma, pi
See σ, π
rev[3]
sigmatropic rearrangement
Molecular rearrangement that involves both the creation of a new σ bond between atoms previously not directly linked and the breaking of an existing σ bond.
Note 1: There is normally a concurrent relocation of π bonds in the molecule concerned, but neither the number of π bonds nor the number of σ bonds changes.
Note 2: The transition state of such a reaction may be visualized as an association of two fragments connected at their termini by two partial σ bonds, one being broken and the other being formed as, for example, the two allyl fragments in (a’). Considering only atoms within the (real or hypothetical) cyclic array undergoing reorganization, if the numbers of these in the two fragments are designated i and j, then the rearrangement is said to be a sigmatropic change of order [i,j] (conventionally i ≤ j). Thus rearrangement (a) is of order [3,3], whereas rearrangement (b) is a [1,5]sigmatropic shift of hydrogen. (By convention the square brackets […] here refer to numbers of atoms, in contrast with current usage in the context of cycloaddition.)
The descriptors a and s (antarafacial and suprafacial) may also be annexed to the numbers i and j; (b) is then described as a [1s,5s] sigmatropic rearrangement, since it is suprafacial with respect to both the hydrogen atom and the pentadienyl system:
See also cycloaddition, tautomerization
rev[3]
silylene
Generic name for H2Si: and substitution derivatives thereof, containing an electrically neutral bivalent silicon atom with two non-bonding electrons. (The definition is analogous to that given for carbene.)
The silanediyl group (H2Si<), analogous to the methylene group (H2C<).
[3]
single-electron transfer mechanism (SET)
Reaction mechanism characterized by the transfer of a single electron between two species, occurring in one of the steps of a multistep reaction.
[3]
single-step reaction
one-step reaction
Reaction that proceeds through a single transition state (or no transition state).
[3]
skeletal formula
bond-line formula
Two-dimensional representation of a molecular entity in which bonds are indicated as lines between vertices representing octet carbon atoms with attached hydrogens omitted and in which other atoms are represented by their chemical symbols.
Examples: ethanol, cyclohexene, 4-pyridone
See line formula.
Slater-type orbital (STO)
Function centered on an atom for which the radial dependence has the form
Note 1: n is the effective principal quantum number and ζ is the orbital exponent (screening constant) derived from empirical considerations.
Note 2: The angular dependence is usually introduced by multiplying the radial function by a spherical harmonic Ylm(θ,ϕ).
Note 3: Owing to difficulties in computing the integrals of STOs analytically for molecules with more than two atoms they are often replaced by linear combinations of Gaussian orbitals.
See [7].
rev[3]
solvation
Stabilizing interaction between a solute (or solute moiety) and the solvent.
Note: Such interactions generally involve electrostatic forces and van der Waals forces, as well as chemically more specific effects such as hydrogen bond formation.
See also cybotactic region.
[3]
solvatochromic relationship
Linear Gibbs-energy relationship (linear free-energy relationship) based on solvatochromism.
See also Dimroth–Reichardt ET parameter, Kamlet–Taft solvent parameters.
[3]
solvatochromism
Pronounced change in position and sometimes intensity of an electronic absorption or emission band, accompanying a change in the polarity of the medium.
Note: Negative (positive) solvatochromism corresponds to a hypsochromic shift (bathochromic shift) with increasing solvent polarity.
See also Dimroth–Reichardt ETparameter, Z-value.
[3]
solvatomers
Isomers that differ in their solvation environment.
Note 1: Because the solvation environment fluctuates rapidly, solvatomers interconvert rapidly.
Note 2: Species that differ in the type of solvent molecules should not be called solvatomers because they are not isomers.
solvent parameter
Quantity that expresses the capability of a solvent for interaction with solutes, based on experimentally determined physicochemical quantities, in particular: relative permittivity, refractive index, rate constants, Gibbs energies and enthalpies of reaction, and ultraviolet-visible, infrared, and NMR spectra.
Note 1: Solvent parameters are used in correlation analysis of solvent effects, either in single-parameter or in multiple-parameter equations.
Note 2: Solvent parameters include those representing a bulk property, such as relative permittivity (dielectric constant) as well as those that describe a more localized solute/solvent interaction, such as hydrogen-bonding acceptance or donation and Lewis acid/base adduct formation.
See also acceptor number, Catalán solvent parameters, Dimroth–Reichardt ETparameter, Grunwald–Winstein equation, Kamlet–Taft solvent parameters, Laurence solvent parameters, Koppel–Palm solvent parameters, linear solvation energy relationship, Z-value.
rev[3]
solvolysis
Reaction with a solvent.
Note 1: Such a reaction generally involves the rupture of one or more bonds in the solute. More specifically, the term is used for substitution, elimination, and fragmentation reactions in which a solvent species serves as nucleophile or base.
Note 2: A solvolysis can also be classified as a hydrolysis, alcoholysis, or ammonolysis, etc., if the solvent is water, alcohol, or ammonia, etc.
Note 3: Often a solvolysis is a nucleophilic substitution (usually SN1, accompanied by E1 elimination), where the nucleophile is a solvent molecule.
rev[3]
SOMO
Singly Occupied Molecular Orbital (such as the half-filled HOMO of a radical).
See also frontier orbitals.
[3]
special salt effect
Steep increase of the rate of certain solvolysis reactions observed at low concentrations of some non-common-ion salts.
Note: The effect is attributed to trapping of an intimate ion pair that would revert to reactant in the absence of the salt.
See also kinetic electrolyte effect.
rev[3]
specific catalysis
Acceleration of a reaction by a unique catalyst, rather than by a family of related substances.
Note: The term is most commonly used in connection with specific hydrogen-ion or hydroxide-ion (lyonium ion or lyate ion) catalysis.
See also general acid catalysis, general base catalysis, pseudo-catalysis.
[3]
spectator mechanism
Pre-association mechanism in which one of the molecular entities, C, is already present in an encounter pair with A during formation of B from A, but does not assist the formation of B, e.g.,
Note: The formation of B from A may itself be a bimolecular reaction with some other reagent. Because C does not assist the formation of B, it is described as being present as a spectator.
See also microscopic diffusion control.
[3]
spin adduct
See spin trapping.
[3]
spin counting
See spin trapping.
[3]
spin density
Unpaired electron density at a position of interest, usually at carbon, in a radical or a triplet state.
Note: Spin density is often measured experimentally by electron paramagnetic resonance/electron spin resonance (EPR/ ESR) spectroscopy through hyperfine splitting of the signal by neighboring magnetic nuclei.
See also radical center.
[3]
spin label
Stable paramagnetic group (typically an aminoxy radical, R2NO•) that is attached to a part of a molecular entity whose chemical environment may be revealed by its electron spin resonance (ESR) spectrum.
Note: When a paramagnetic molecular entity is used without covalent attachment to the molecular entity of interest, it is frequently referred to as a spin probe.
[3]
spin trapping
Formation of a more persistent radical from interaction of a transient radical with a diamagnetic reagent.
Note 1: The product radical accumulates to a concentration where detection and, frequently, identification are possible by EPR/ESR spectroscopy.
Note 2: The key reaction is usually one of attachment; the diamagnetic reagent is said to be a spin trap, and the persistent product radical is then the spin adduct. The procedure is referred to as spin trapping, and is used for monitoring reactions involving the intermediacy of reactive radicals at concentrations too low for direct observation. Typical spin traps are C-nitroso compounds and nitrones, to which reactive radicals will rapidly add to form aminoxy radicals.
Example:
Note 3: A quantitative development in which essentially all reactive radicals generated in a particular system are intercepted has been referred to as spin counting.
Note 4: Spin trapping has also been adapted to the interception of radicals generated in both gaseous and solid phases. In these cases, the spin adduct is in practice transferred to a liquid solution for EPR/ESR observation.
[3]
stable
Having a lower standard Gibbs energy, compared to a reference chemical species.
Note 1: Quantitatively, in terms of Gibbs energy, a chemical species A is more stable than its isomer B if ∆rG° is positive for the (real or hypothetical) reaction A → B.
Note 2: For the two reactions
if ∆rG1° > ∆rG2°, then P is more stable relative to its product Y than is Q relative to Z.
Note 3: Both in qualitative and quantitative usage, the term stable is therefore always used in reference to some explicitly stated or implicitly assumed standard.
Note 4: The term should not be used as a synonym for unreactive or less reactive because this confuses thermodynamics and kinetics. A relatively more stable chemical species may be more reactive than some reference species towards a given reaction partner.
See also inert, unstable.
[3]
stationary state
(in quantum mechanics): Wavefunction whose probability density |Ψ|2 remains constant and whose observable properties do not evolve with time.
(in kinetics): See steady state.
[3]
steady state
or stationary state
Approximation that the kinetic analysis of a complex reaction involving unstable intermediates in low concentration can be simplified by setting the rate of change of each such intermediate equal to zero, so that the rate equation can be expressed as a function of the concentrations of chemical species present in macroscopic amounts.
For example, if X is an unstable intermediate in the reaction sequence:
Because [X] is negligibly small, d[X]/dt, the rate of change of [X], can be set equal to zero. The steady state approximation then permits solving the following equation
to obtain the steady-state [X]:
whereupon the rate of reaction is expressed:
Note: The steady-state approximation does not imply that [X] is even approximately constant, only that its absolute rate of change is very much smaller than that of [A] and [D].
Regime in a stirred flow reactor such that all concentrations are independent of time.
See [12].
[3]
stepwise reaction
Chemical reaction with at least one reaction intermediate and involving at least two consecutive elementary reactions.
See also composite reaction, reaction step.
[3]
stereoelectronic
Pertaining to the dependence of the properties (especially energy or reactivity) of a molecular entity or of a transition state on the relative disposition of electron pairs owing to the nuclear geometry.
Note: Stereoelectronic effects are ascribed to the differing overlaps of atomic orbitals in different conformations.
See [10].
[3]
stereogenic center
Atom within a molecule bearing groups such that interchanging any two of them leads to a stereoisomer of the original molecule.
stereoisomers
Isomers that have the same bonds (connectivity) but differ in the arrangement of their atoms and cannot be interconverted by rapid rotation around single bonds.
See [10].
stereoselectivity
stereoselective
Preferential formation in a chemical reaction of one stereoisomer over another.
Note 1: When the stereoisomers are enantiomers, the phenomenon is called enantioselectivity and is quantitatively expressed by the enantiomeric excess or enantiomeric ratio; when they are diastereoisomers, it is called diastereoselectivity and is quantitatively expressed by the diastereomeric excess or diastereomeric ratio.
Note 2: Reactions are termed 100 % stereoselective if the preference is complete, or partially (x %) stereoselective if one product predominates. The preference may also be referred to semiquantitatively as high or low stereoselectivity
[3]
stereospecificity
stereospecific
Property of those chemical reactions in which different stereoisomeric reactants are converted into different stereoisomeric products.
Example: electrocyclization of trans,cis,trans-octa-2,4,6-triene produces cis-5,6-dimethylcyclohexa-1,3-diene, whereas cis,cis,trans-octa-2,4,6-triene produces racemic trans-5,6-dimethylcyclohexa-1,3-diene.
Note 1: A stereospecific process is necessarily stereoselective but not all stereoselective processes are stereospecific. Stereospecificity may be total (100 %) or partial.
Note 2: The term is also applied to situations where reaction can be performed with only one stereoisomer. For example the exclusive formation of racemic trans-1,2-dibromocyclohexane upon bromination of cyclohexene is a stereospecific process, even though the analogous reaction with (E)-cyclohexene has not been performed.
Note 3: Stereospecificity does NOT mean very high stereoselectivity. This usage is unnecessary and is strongly discouraged.
For the term stereospecific polymerization, see [379].
rev[3]
steric-approach control
Situation in which the stereoselectivity of a reaction under kinetic control is governed by steric hindrance to attack of the reagent, which is directed to the less hindered face of the molecule.
Note: Partial bond making at the transition state must be strong enough for steric control to take place, but the transition state should not be so close to products that the steric demand of the reagent at the transition state is the same as the steric demand of the group as present in the product.
An example is LiAlH4 reduction of 3,3,5-trimethylcyclohexan-1-one, where steric hindrance by an axial methyl directs hydride addition to the equatorial position, even though the more stable product has the H axial and the OH equatorial.
See also product-development control.
rev[3]
steric effect
Consequences for molecular geometry, thermochemical properties, spectral features, solvation, or reaction rates resulting from the fact that atoms repel each other at close distance. The repulsion is due to the quantum-mechanical Pauli exclusion principle. Substitution of hydrogen atoms by groups with a larger van der Waals radius may lead to situations where atoms or groups of atoms repel each other, thereby affecting distances and angles.
Note 1: It is in principle difficult to separate the steric effect from other electronic effects.
Note 2: For the purpose of correlation analysis or linear Gibbs-energy relations (linear free-energy relations) various scales of steric parameters have been proposed, notably A values, Taft’s ES, and Charton’s v scales.
Note 3: A steric effect on a rate process may result in a rate increase (steric acceleration) or a decrease (steric retardation) depending on whether the transition state or the reactant state is more affected by the steric effect.
Note 4: Bulky groups may also attract each other if at a suitable distance.
See Taft equation, van der Waals forces.
rev[3]
steric hindrance
Steric effect whereby the crowding of substituents around a reaction center retards the attack of a reagent.
rev[3]
stopped flow
Technique for following the kinetics of reactions in solution (usually in the millisecond time range) in which two, or more, reactant solutions are rapidly mixed by being forced through a mixing chamber. The flow of the mixed solution along a uniform tube is then suddenly arrested. At a fixed position along the tube the solution is monitored as a function of time following the stoppage of the flow by a method with a rapid response (e.g., optical absorption spectroscopy).
See mixing control.
rev[3]
strain
Feature of a molecular entity or transition structure for which the energy is increased because of unfavorable non-bonded (steric) interactions, bond lengths, bond angles, or dihedral angles (torsional strain), relative to a standard.
Note 1: The strain energy is quantitatively defined as the standard enthalpy of a structure relative to that of a strainless structure (real or hypothetical) made up from the same atoms with the same types of bonding.
For example, the enthalpy of formation of cyclopropane is +53.6 kJ mol−1, whereas the hypothetical enthalpy of formation based on three “normal” methylene groups, from acyclic models, is −62 kJ mol−1. On this basis cyclopropane is destabilized by ca. 115 kJ mol−1 of strain energy.
See molecular mechanics.
[3]
structural isomers
discouraged term for constitutional isomers.
subjacent orbital
Next-to-Highest Occupied Molecular Orbital (NHOMO, also called HOMO–1).
Note: Subjacent and superjacent orbitals sometimes play an important role in the interpretation of molecular interactions according to the frontier orbital approach.
See [381].
[3]
substituent
Any atom or group of bonded atoms that can be considered to have replaced a hydrogen atom (or two hydrogen atoms in the special case of bivalent groups) in a parent molecular entity (real or hypothetical).
[3]
substitution
Chemical reaction, elementary or stepwise, of the form A–B + C → A–C + B, in which one atom or group in a molecular entity is replaced by another atom or group.
Examples:
Note: A substitution reaction can be distinguished as an electrophilic substitution or a nucleophilic substitution, depending on the nature of the reactant that is considered to react with the substrate.
rev[3]
substrate
Chemical species, the reaction of which with some other chemical reagent is under observation (e.g., a compound that is transformed under the influence of a catalyst).
Note: The term should be used with care. Either the context or a specific statement should always make it clear which chemical species in a reaction is regarded as the substrate.
See also transformation.
[3]
successor complex
Chemical species formed by the transfer of an electron or of a hydrogen (atom or ion) from a donor D to an acceptor A after these species have diffused together to form the precursor or encounter complex:
rev[3]
suicide inhibition
See mechanism-based inhibition.
[3]
superacid
Medium having a high acidity, generally greater than that of 100 % sulfuric acid. The common superacids are made by dissolving a powerful Lewis acid (e.g., SbF5) in a suitable Brønsted acid, such as HF or HSO3F.
Note 1: An equimolar mixture of HSO3F and SbF5 is known by the trade name Magic Acid.
Note 2: An uncharged gas-phase substance having an endothermicity (enthalpy) of deprotonation (dehydronation) lower than that of H2SO4 is also called a superacid. Nevertheless, such a superacid is much less acidic in the gas phase than similar cationic acids.
See acidity, superbase.
rev[3]
superbase
Compound having a very high basicity.
Examples include amide bases such as LDA, potassium tert-butoxide + organolithium, some phosphazenes.
See superacid.
See [385].
rev[3]
superjacent orbital
Second Lowest Unoccupied Molecular Orbital (SLUMO).
Note: Subjacent and superjacent orbitals sometimes play an important role in the interpretation of molecular interactions according to the frontier orbital approach.
See [381].
suprafacial
See antarafacial.
[3]
supramolecular
Description of a system of two or more molecular entities that are held together and organized by means of intermolecular (noncovalent) binding interactions.
See [386].
[3]
Swain–Lupton equation
Dual-parameter approach to the correlation analysis of substituent effects, which involves a field constant (F) and a resonance constant (R).
Note 1: The original treatment was modified later.
Note 2: The procedure has often been applied, but also often criticized.
See [388], [389], [390], [391], [392].
[3]
Swain–Scott equation
Linear Gibbs-energy relation (linear free-energy relation) of the form
applied to the variation of reactivity of a given electrophilic substrate towards a series of nucleophilic reagents, where k0 is a rate constant for reaction with water, k is the corresponding rate constant for reaction with any other nucleophilic reagent, n is a measure of the nucleophilicity of the reagent (n = 0.0 for water), and s is a measure of the sensitivity of the substrate to the nucleophilicity of the reagent (s = 1.0 for CH3Br).
See [192].
See also Mayr–Patz equation, Ritchie equation.
[3]
symproportionation
comproportionation
[3]
syn
See anti.
See [10].
[3]
synartetic acceleration
See neighboring group participation.
[3]
synchronization
See principle of nonperfect synchronization.
rev[3]
synchronous
Feature of a concerted process in which all the changes (generally bond rupture and bond formation) have progressed to the same extent at the transition state.
Note 1: A synchronous reaction is distinguished from (1) a concerted reaction, which takes place in a single kinetic step without being synchronous, (2) a reaction where some of the changes in bonding take place earlier, followed by the rest, and (3) a two-step reaction, which takes place in two kinetically distinct steps, via a stable intermediate.
Note 2: The progress of the bonding changes (or other primitive changes) is not defined quantitatively in terms of a single parameter applicable to different bonds. The concept therefore does not admit an exact definition except in the case of concerted processes involving changes in two identical bonds. If the bonds are not identical, the process should simply be described as concerted.
See [393], [394], [395]. For an index of synchronicity see [396].
See also imbalance.
rev[3]
σ, π
Symmetry designations that distinguish molecular orbitals as being symmetric (σ) or antisymmetric (π) with respect to a defining plane containing at least one atom.
Note 1: In practice, the terms are used both in this rigorous sense (for orbitals encompassing the entire molecule) and for localized two-center orbitals or bonds. In the case of localized two-center bonds, a π bond has a nodal plane that includes the internuclear bond axis, whereas a σ bond has no such nodal plane. (A δ bond in organometallic or inorganic chemical species has two nodes). Radicals are classified by analogy into σ and π radicals.
Note 2: Such two-center orbitals may take part in molecular orbitals of σ or π symmetry. For example, the methyl group in propene contains three C–H bonds, each of which is of local σ symmetry (i.e., without a nodal plane including the internuclear axis), but these three σ bonds can in turn be combined to form a set of group orbitals one of which has π symmetry with respect to the principal molecular plane and can accordingly interact with the two-center orbital of π symmetry (π bond) of the double-bonded carbon atoms, to form a molecular orbital of π symmetry. Such an interaction between the CH3 group and the double bond is an example of hyperconjugation. This cannot rigorously be described as σ-π conjugation because σ and π here refer to different defining planes, and interaction between orbitals of different symmetries (with respect to the same defining plane) is forbidden.
Note 3: Conjugation between a π system and a lone pair of (for example) an ether oxygen involves a lone pair of π symmetry with respect to the defining plane. It is incorrect to consider this as an interaction between the π system and one of two identical sp3-hybrid lone pairs, which are neither σ nor π.
Note 4: The two lone pairs on a carbonyl oxygen are properly classified as σ or π with respect to the plane perpendicular to the molecular plane (dotted line perpendicular to the plane of the page), rather than as two sp2-hybridized lone pairs. This distinction readily accounts for the facts that there are two different lone-pair ionization energies and two different n-π* excited states.
See also [397].
σ-adduct
Product formed by the attachment of an electrophilic or nucleophilic entering group or of a radical to a ring carbon of an aromatic species so that a new σ bond is formed and the original conjugation is disrupted.
Note 1: This has generally been called a σ complex or a Wheland complex from electrophilic addition, but adduct is more appropriate.
Note 2: The term may also be used for analogous adducts to π systems.
See also Meisenheimer adduct.
rev[3]
σ-constant
Substituent constant for meta- and para-substituents in benzene derivatives as defined by Hammett on the basis of the ionization constant of a substituted benzoic acid, i.e., lg (KaX/KaH), where KaX is the ionization constant (acid-dissociation constant) of a m- or p-substituted benzoic acid and KaH is that of benzoic acid itself.
Note 1: A large positive σ-value implies high electron-withdrawing power by an inductive and/or resonance effect, relative to H; a large negative σ-value implies high electron-releasing power relative to H.
Note 2: The term is also used as a collective description for related electronic substituent constants based on other standard reaction series, of which, σ+, σ– and σo are typical; also for constants which represent dissected electronic effects, such as σI and σR. For example, σ– (sigma-minus) constants are defined on the basis of the ionization constants of para-substituted phenols, where such substituents as nitro show enhanced electron-withdrawing power.
See [124], [125], [126, 191, 226, 398].
See also Hammett equation, ρ-value, Taft equation.
[3]
Taft equation
Linear free-energy relation (linear Gibbs-energy relation) involving the polar substituent constant σ* and the steric substituent constant Es, as derived from reactivities of aliphatic esters
The argument of the lg function should be of dimension 1. Thus, the reduced rate constants should be used, i.e., the rate coefficient divided by its unit: {k} = k/[k] and {k0} = k0/[k0].
Note: The standard reaction (k0) is the hydrolysis of methyl acetate, whereby Es is evaluated from the rate of acid-catalyzed hydrolysis, relative to that of methyl acetate, and σ* is evaluated from the ratio of the rates of base- and acid-catalyzed hydrolysis.
See also Hammett equation, ρ-value, σ-constant.
rev[3]
tautomerization
Rapid isomerization of the general form
where the isomers (called tautomers) are readily interconvertible.
Note 1: The atoms of the groups X, Y, and Z are typically any of C, H, N, O, or S, and G is a group that becomes an electrofuge or nucleofuge during isomerization.
Note 2: The commonest case, when the electrofuge is H+, is also known as a prototropic rearrangement.
Examples:
Note 3: The group Y may itself be a three-atom (or five-atom) chain extending the conjugation, as in
Note 4: Ring-chain tautomerization is the case where addition across a double bond leads to ring formation, as in
Note 5: Valence tautomerization is the case of rapid isomerization involving the formation and rupture of single and/or double bonds, without migration of atoms: for example
See also ambident, fluxional, sigmatropic rearrangement, valence tautomerization.
rev[3]
tautomers
Constitutional isomers that can interconvert more or less rapidly, often by migration of a hydron.
See tautomerization.
tele-substitution
Substitution reaction in which the entering group takes up a position more than one atom away from the atom to which the leaving group was attached.
Example
See also cine-substitution.
rev[3]
termination
Step(s) in a chain reaction in which reactive intermediates are destroyed or rendered inactive, thus ending the chain.
[3]
tetrahedral intermediate
Reaction intermediate in which the bond arrangement around an initially double-bonded carbon atom (typically a carbonyl carbon) has been transformed from tricoordinate to tetracoordinate (with a coordination number of 4).
Example:
rev[3]
thermodynamic control (of product composition)
equilibrium control
Conditions (including reaction times) that lead to reaction products in a proportion specified by the equilibrium constant for their interconversion.
See also kinetic control.
rev[3]
thermolysis
Uncatalyzed cleavage of one or more covalent bonds resulting from exposure of a molecular entity to a raised temperature, or a process of which such cleavage is an essential part.
See also pyrolysis.
[3]
through-conjugation
Phenomenon whereby electrons can be delocalized from any of three (or more) groups to any other.
Example: p-XC6H4Y, where an electron pair can be delocalized from electron-donating X not only to ring carbons but also to electron-withdrawing Y.
Note 1: This may be contrasted with cross-conjugation.
Note 2: In Hammett-type correlations (linear Gibbs-energy relationships), this situation can lead to exalted substituent constants σ+ or σ–, as in solvolysis of p-CH3OC6H4C(CH3)2Cl or acidity of p-nitrophenol, respectively.
TICT
See twisted intramolecular charge transfer.
torquoselectivity
Preference for inward or outward rotation of substituents in conrotatory or disrotatory electrocyclic ring-opening and ring-closing reactions, often owing to a preference for electron donors (especially including large groups) to rotate outward and acceptors to rotate inward.

rev[3]
transferability
Assumption that a chemical property associated with an atom or group of atoms in a molecule will have a similar (but not identical) value in other circumstances.
Examples: equilibrium bond length, bond force constant, NMR chemical shift.
[3]
transformation
Conversion of a substrate into a particular product, irrespective of the specific reagents or mechanisms involved.
Example: transformation of aniline (C6H5NH2) into N-phenylacetamide (C6H5NHCOCH3), which may be effected with acetyl chloride or acetic anhydride or ketene.
Note: A transformation is distinct from a reaction, the full description of which would state or imply all the reactants and all the products.
See [406].
[3]
transient (chemical) species
Short-lived reaction intermediate.
Note 1: Transiency can be defined only in relation to a time scale fixed by the experimental conditions and by the limitations of the technique employed in the detection of the intermediate. The term is a relative one.
Note 2: Transient species are sometimes also said to be metastable. However, this latter term should be avoided because it relates a thermodynamic term to a kinetic property, although most transients are also thermodynamically unstable with respect to reactants and products.
See also persistent.
[3]
transition dipole moment
Vector quantity describing the oscillating electronic moment induced by an electromagnetic wave, given by an integral involving the electric dipole moment operator m and the ground- and excited-state wavefunctions:
Note: The magnitude of this quantity describes the allowedness of an electronic transition. It can often be separated into an electronic factor and a nuclear-overlap factor known as the Franck–Condon factor.
See [8].
transition state
State of a molecular system from which there are equal probabilities of evolving toward states of lower energy, generally considered as reactants and products of an elementary reaction.
Note 1: The transition state corresponds to the maximum along the minimum-Gibbs-energy path connecting reactants and products.
Note 2: The transition state can be considered to be a chemical species of transient existence.
Note 3: The term transition-state structure refers to a structure inferred from kinetic and stereochemical investigations; it does not necessarily coincide with a transition state or with a transition structure, which corresponds to a saddle point on a potential-energy surface, although it may represent an average structure.
Note 4: The assembly of atoms at the transition state has also been called an activated complex, although it is not a complex according to the definition in this Glossary.
Note 5: There are also reactions, such as the gas-phase colligation of simple radicals or the reactions of some reactive intermediates in solution, that do not require activation and do not involve a transition state.
Note 6: Ultrafast spectroscopy permits observation of transition states in some special cases.
See also Gibbs energy of activation, potential-energy profile, reaction coordinate, transition structure.
rev[3]
transition-state analogue
Species designed to mimic the geometry and electron density of the transition state of a reaction, usually enzymatic.
Note: A transition-state analogue is usually not a substrate for the enzyme, but rather an inhibitor.
rev[3]
transition structure
Molecular entity corresponding to a saddle point on a potential-energy surface, with one negative force constant and its associated imaginary frequency.
Note 1: Whereas the transition state is not a specific molecular structure, but a set of structures between reactants and products, a transition structure is one member of that set with a specific geometry and energy.
Note 2: Although the saddle point coincides with the potential-energy maximum along a minimum-energy reaction path, it does not necessarily coincide with the maximum of Gibbs energy for an ensemble of chemical species.
Note 3: The term transition-state structure is not a synonym for transition structure.
See also activated complex, transition state.
rev[3]
transition vector
Normal mode of vibration of a transition structure corresponding to the single imaginary frequency and tangent to the intrinsic reaction coordinate at the saddle point. Sometimes called the reaction-coordinate vibrational mode.
Note 1: Infinitesimal motion along the transition vector in the two opposite senses determines the initial direction leading either toward reactants or toward products.
Note 2: The term “transition coordinate” was used in the 1994 Glossary of Terms in Physical Organic Chemistry in the sense defined here for transition vector, but apparently this usage was unique in the literature at that time and has not been generally adopted since.
See reaction coordinate, transition state.
transport control
See encounter-controlled rate, microscopic diffusion control.
[3]
trapping
Interception of a reactive molecule or reaction intermediate so that it is removed from the system or converted into a more stable form for study or identification.
See also scavenger.
[3]
triplet state
State having a total electron spin quantum number of 1.
See [8].
tunnelling
Quantum-mechanical phenomenon by which a particle or a set of particles penetrates a barrier on its potential-energy surface without having the energy required to surmount that barrier.
Note 1: In consequence of Heisenberg’s Uncertainty Principle, a molecular entity has a nonzero probability of adopting a geometry that is classically forbidden because it corresponds to a potential energy greater than the total energy.
Note 2: Tunnelling is often considered to be a correction to an (over)simplified version of transition-state theory.
Note 3: Because the rate of tunnelling increases with decreasing mass, it is significant in the context of isotope effects, especially of hydrogen isotopes.
rev[3]
twisted intramolecular charge transfer (TICT)
Feature of an excited electronic state formed by intramolecular electron transfer from an electron donor (D) to an electron acceptor (A), where interaction between electron and hole is restricted because D+ and A– are perpendicular to each other.
See [412].
umpolung
Process by which the nucleophilic or electrophilic property of a functional group is reversed.
Note: Umpolung is often achieved by temporary exchange of heteroatoms N or O by others, such as P, S, or Se, as in the conversion of electrophilic RCH=O to RCH(SR')2 and then with base to nucleophilic RC(SR')2–. Also, the transformation of a haloalkane RX into a Grignard reagent RMgCl is an umpolung.
See [413].
rev[3]
unimolecular
Feature of a reaction in which only one molecular entity is involved.
See molecularity.
rev[3]
unreactive
Failing to react with a specified chemical species under specified conditions.
Note: The term should not be used in place of stable, which refers to a thermodynamic property, as a relatively more stable species may nevertheless be more reactive than some reference species towards a given reaction partner.
[3]
unstable
Opposite of stable, i.e., the chemical species concerned has a higher molar Gibbs energy than some assumed standard.
Note: The term should not be used in place of reactive or transient, although more reactive or transient species are frequently also more unstable.
rev[3]
upfield
superseded but still widely used to mean shielded.
See chemical shift.
[3]
valence
Maximum number of single bonds that can be commonly formed by an atom or ion of the element under consideration.
Note: Often there is a most common maximum for a given element, and atoms in compounds where this number is exceeded, such as pentacoordinate carbocations (“carbonium ions”) and iodine(III) compounds, are called hypervalent.
rev[3]
valence isomer
Constitutional isomer related to another by pericyclic reaction.
Examples: Dewar benzene, prismane, and benzvalene are valence isomers of benzene.
Note: Valence isomers are separable, as distinguished from valence tautomers, which interconvert rapidly. See valence tautomerization.
rev[3]
valence tautomerization
Rapid isomerization involving the formation and rupture of single and/or double bonds, without migration of atoms.
Example:
See tautomerization.
rev[3]
van der Waals forces
Attractive or repulsive forces between molecular entities (or between groups within the same molecular entity) other than those due to bond formation or to the electrostatic interaction of ions or of ionic groups with one another or with neutral molecules.
Note 1: The term includes dipole–dipole, dipole-induced dipole, and London (instantaneous induced dipole-induced dipole) forces, as well as quadrupolar forces.
Note 2: The term is sometimes used loosely for the totality of nonspecific attractive or repulsive intermolecular forces.
Note 3: In the context of molecular mechanics, van der Waals forces correspond only to the London dispersion forces plus the Pauli repulsion forces. Interactions due to the average charge distribution (and not to fluctuations around the average or to induced dipoles), including dipole–dipole, ion–ion and ion–dipole forces, are called Coulombic forces.
See also dipole-dipole interaction, dipole-induced dipole forces, London forces.
rev[3]
volume of activation
∆‡V
Quantity derived from the pressure dependence of the rate constant of a reaction, defined by the equation
provided that rate constants of all reactions (except first-order reactions) are expressed in pressure-independent concentration unit, such as mol dm−3 at a fixed temperature and pressure. The argument in the lg function should be of dimension 1. Thus, the rate constant should be divided by its unit, [k].
Note: The volume of activation is interpreted as the difference between the partial molar volume ‡V of the transition state and the sums of the partial molar volumes of the reactants at the same temperature and pressure, i.e.,
where ρ is the order in the reactant R and VR its partial molar volume.
See [12].
See order of reaction.
rev[3]
water/octanol partition coefficient
partition ratio
See octanol–water partition ratio
wavefunction
A mathematical expression whose form resembles the wave equations of physics, supposed to contain all the information associated with a particular atomic or molecular system. In particular, a solution of the Schrödinger wave equation, Hψ = Eψ, as an eigenfunction ψ of the hamiltonian operator H, which involves the electronic and/or nuclear coordinates.
Note 1: The wavefunction contains all the information describing an atomic or molecular system that is consistent with the Heisenberg Uncertainty Principle.
Note 2: When a wavefunction is operated on by certain quantum-mechanical operators, a theoretical evaluation of physical and chemical observables for that system (the most important one being energy) can be carried out.
Wheland intermediate
See Meisenheimer adduct, σ-adduct.
[3]
Woodward–Hoffmann rules
See orbital symmetry.
ylide
Chemical species that can be produced by loss of a hydron from an atom directly attached to the central atom of an onium ion.
Example:
See [39].
[3]
Yukawa–Tsuno equation
Multiparameter extension of the Hammett equation to quantify the role of enhanced resonance effects on the reactivity of para–substituted benzene derivatives,
where the parameter r expresses the enhancement and where σ° is a substituent constant based on reactivities of phenylacetic acids and similar substrates, where resonance interaction is weak or absent. The argument in the lg function should be of dimension 1. Thus, reduced rate constants should be used {k} = k/[k] and {ko} = ko/[ko].
See also dual substituent-parameter equation, ρ-value, σ-constant, through-conjugation.
rev[3]
Zaitsev rule
See Saytzeff rule.
[3]
zero-point energy
Extent, in consequence of Heisenberg’s Uncertainty Principle, by which a particle or a set of particles has an energy greater than that of the minimum on the potential-energy surface.
Note 1: Because of zero-point energy, a molecular entity has a nonzero probability of adopting a geometry whose energy is greater than that of the energy minimum.
Note 2: A molecular entity with zero-point energy may even adopt a geometry with a potential energy greater than its total energy, a possibility that permits tunnelling.
Note 3: Because the magnitude of zero-point energy increases with decreasing mass, it is significant in the context of isotope effects, especially of hydrogen isotopes.
Zucker–Hammett hypothesis
Assumption that if lg {k1} (= k1/[k1], reduced pseudo-first-order rate constant of an acid-catalyzed reaction) is linear in Ho (Hammett acidity function), then water is not involved in the transition state of the rate-controlling step, whereas if lg {k1} is linear in lg {[H+]}, then water is involved. The argument in the lg function should be of dimension 1. Thus, reduced concentration, {[H+]}, should be used, i.e., concentration of protons divided by its unit.
Note: This has been shown to be an overinterpretation.
See also Bunnett–Olsen equation, Cox–Yates equation.
rev[3]
Z-value
Quantitative measure of solvent polarity based on the UV-vis spectrum of 1-ethyl-4-(methoxycarbonyl)pyridinium iodide.
See [143].
See solvent parameter.
rev[3]
zwitterion
Highly dipolar, net uncharged (neutral) molecule having full electrical charges of opposite sign, which may be delocalized within parts of the molecule but for which no uncharged canonical resonance structure can be written.
Examples: glycine (H3N+–CH2–CO2−), betaine (Me3N+–CH2–CO2−).
Note 1: Sometimes also referred to as inner salts or ampholytes.
Note 2: Mesoionic compounds, such as sydnones, in which both positive and negative charge are delocalized, are sometimes considered as zwitterions, but species with a localized nonzero formal charge, such as a nitrone, CH3CH=N+(–O–)CH3, are not.
See [418].
rev[3]
Membership of sponsoring body
The Subcommittee on Structural and Mechanistic Chemistry is a subcommittee of the IUPAC Division III (Organic and Biomolecular Chemistry). Its present membership is
Ian Williams (Chair), Manabu Abe, Igor Alabugin, Moisés Canle, Jin-Pei Cheng, Maria Cristiano, Jason Harper, Eduardo Humeres, Miroslav Ludwig, Anat Milo, Henrik Ottosson, Charles Perrin, Hans-Ullrich Siehl, Raghavan B. Sunoj, Pietro Tundo, Einar Uggerud.
The present membership of Division III is
President: Amélia P. Rauter; Past President: Nikolay Nifantiev; Vice-President: Einar Uggerud; Secretary: Slawomir Jarosz; Titular Members: Jason Harper, Ari Koskinen, Cristina Nativi, Paolo Scrimin, Zbigniew Witczak, Makoto Yamashita; Associate Members: Axel Griesbeck, Mohamed Hegazy, John F. Honek, Priyani Paranagama, Ian H. Williams, Zhen Xi; National Representatives: Claudia Bonfio, Morgan Donnard, Eun Joo Kang, Palangpon Kongsaeree, Mindy Levine, Emilia Naydenova, Romano Orrù, Vinod K. Singh, Shih-Sheng Sun, Zuriati Zakaria.
Article note:
Sponsoring body: IUPAC Subcommittee on Structural and Mechanistic Chemistry: see more details on page 526.
This work was started under the project 2009-002-1-300: Update of IUPAC Glossary of Physical Organic Chemistry with membership of Charles L. Perrin (Task group Chair), Israel Agranat, Alessandro Bagno,† Silvia E. Braslavsky, Pedro Alexandrino Fernandes, Jean-François Gal, Guy C. Lloyd-Jones, Herbert Mayr, Joseph R. Murdoch, Norma Sbarbati Nudelman, Leo Radom, Zvi Rappoport,† Marie-Françoise Ruasse,† Hans-Ullrich Siehl, Yoshito Takeuchi, Thomas T. Tidwell, Einar Uggerud, and Ian H. Williams. These Recommendations were prepared for final publication by Charles L. Perrin, Silvia E. Braslavsky and Ian H. Williams.
(† Deceased)
References
[1] V. Gold. Pure Appl. Chem.55, 1281 (1983) (IUPAC Recommendations 1982), https://doi.org/10.1351/pac198355081281.Search in Google Scholar
[2] P. Müller. Pure Appl. Chem.66, 1077 (1994) (IUPAC Recommendations 1994), https://doi.org/10.1351/pac199466051077.Search in Google Scholar
[3] https://www.qmul.ac.uk/sbcs/iupac/gtpoc.Search in Google Scholar
[4] https://doi.org/10.1351/goldbook.Search in Google Scholar
[5] A. Lavoisier. Traité élémentaire de chimie, v–xxxii, Chez Cuchet (1789).Search in Google Scholar
[6] K. K. Murray, R. K. Boyd, M. N. Eberlin, G. J. Langley, L. Li, Y. Naito. Pure Appl. Chem.85, 1515 (2013) (IUPAC Recommendations 2013), https://doi.org/10.1351/PAC-REC-06-04-06.Search in Google Scholar
[7] V. I. Minkin. Pure Appl. Chem.71, 1919 (1999) (IUPAC Recommendations 1999), https://doi.org/10.1351/pac199971101919.Search in Google Scholar
[8] S. E. Braslavsky. Pure Appl. Chem.79, 293 (2007) (IUPAC Recommendations 2006), https://doi.org/10.1351/pac200779030293.Search in Google Scholar
[9] S. E. Braslavsky, A. M. Braun, A. E. Cassano, A. V. Emeline, M. I. Litter, L. Palmisano, V. N. Parmon, N. Serpone. Pure Appl. Chem.83, 931 (2011) (IUPAC Recommendations 2011), https://doi.org/10.1351/PAC-REC-09-09-36.Search in Google Scholar
[10] G. P. Moss. Pure Appl. Chem.68, 2193 (1996) (IUPAC Recommendations 1996), https://doi.org/10.1351/pac199668122193.Search in Google Scholar
[11] E. R. Cohen, T. Cvitas, J. G. Frey, B. Holmström, K. Kuchitsu, R. Marquardt, I. Mills, F. Pavese, M. Quack, J. Stohner, H. L. Strauss, M. Takami, A. J. Thor. Quantities, Units and Symbols in Physical Chemistry, IUPAC Green Book, 3rd ed. (2007).10.1039/9781847557889Search in Google Scholar
[12] K. J. Laidler. Pure Appl. Chem.68, 149 (1996) (IUPAC Recommendations 1996), https://doi.org/10.1351/pac199668010149.Search in Google Scholar
[13] E. V. Anslyn, D. A. Dougherty. Modern Physical Organic Chemistry. University Science Books, Sausalito (2006).Search in Google Scholar
[14] S. Winstein, N. J. Holness. J. Am. Chem. Soc.77, 5562 (1955), https://doi.org/10.1021/ja01626a037.Search in Google Scholar
[15] V. Gutmann. Coord. Chem. Rev.18, 225 (1976), https://doi.org/10.1016/s0010-8545(00)82045-7.Search in Google Scholar
[16] C. Reichardt, T. Welton. Solvents and Solvent Effects in Organic Chemistry, Wiley, Weinheim, 4th ed. (2011).Search in Google Scholar
[17] G. A. Olah, G. K. Surya Prakash, Á. Molnár, J. Sommer. Superacid Chemistry, John Wiley & Sons, Hoboken, NJ, 2nd ed. (2009).10.1002/9780470421604Search in Google Scholar
[18] C. H. Rochester. Acidity Functions, Academic Press, NY (1970).Search in Google Scholar
[19] R. A. Cox, K. Yates. Can. J. Chem.61, 2225 (1983), https://doi.org/10.1139/v83-388.Search in Google Scholar
[20] L. P. Hammett. Physical Organic Chemistry, McGraw Hill, New York (1940).Search in Google Scholar
[21] L. P. Hammett. Physical Organic Chemistry: Reaction Rates, Equilibria and Mechanisms; McGraw-Hill Series in Advanced Chemistry, McGraw-Hill, New York (1970).Search in Google Scholar
[22] S. W. Benson, N. Cohen. Chem. Rev.93, 2419 (1993).10.1021/cr00023a005Search in Google Scholar
[23] S. W. Benson. Thermochemical Kinetics, John Wiley & Sons, New York, London, Sydney, Toronto, 2nd ed. (1976).Search in Google Scholar
[24] K. A. Dill. J. Biol. Chem.272, 701 (1997), https://doi.org/10.1074/jbc.272.2.701.Search in Google Scholar PubMed
[25] M. Brookhart, M. L. H. Green. J. Organomet. Chem.250, 395 (1983), https://doi.org/10.1016/0022-328x(83)85065-7.Search in Google Scholar
[26] X.-L. Luo, R. H. Crabtree. J. Am. Chem. Soc.111, 2527 (1989), https://doi.org/10.1021/ja00189a026.Search in Google Scholar
[27] W. Scherer, G. S. McGrady. Angew. Chem. Int. Ed.43, 1782 (2004), https://doi.org/10.1002/anie.200200548.Search in Google Scholar PubMed
[28] N. G. Connelly, T. Damhus, R. M. Hartshorn, A. T. Hutton. Nomenclature of Inorganic Chemistry (IUPAC Recommendations 2005; Red Book), Royal Society of Chemistry, Cambridge, UK (2015). See also R. M. Hartshorn, K.-H. Hellwich, A. Yerin, T. Damhus, A. T. Hutton. Pure Appl. Chem. 87, 1039.Search in Google Scholar
[29] C. A. Coulson, G. S. Rushbrooke. Proc. Cambridge Phil. Soc.36, 193 (1940), https://doi.org/10.1017/s0305004100017163.Search in Google Scholar
[30] A. D. McLachlan. Mol. Phys.2, 271 (1959), https://doi.org/10.1080/00268975900100261.Search in Google Scholar
[31] M. J. S. Dewar, R. C. Dougherty. The PMO Theory of Organic Chemistry, Plenum Press, New York (1975).10.1007/978-1-4613-4404-9Search in Google Scholar
[32] R. Gompper. Angew. Chem. Int. Ed.3, 525 (1964), https://doi.org/10.1002/anie.196405601.Search in Google Scholar
[33] R. Gompper, H.-U. Wagner. Angew. Chemie Int. Ed.15, 321 (1976), https://doi.org/10.1002/anie.197603211.Search in Google Scholar
[34] P. A. S. Smith, G. L. DeWall. J. Am. Chem. Soc.99, 5751 (1977), https://doi.org/10.1021/ja00459a036.Search in Google Scholar
[35] H. Mayr, M. Breugst, A. R. Ofial. Angew. Chem. Int. Ed.50, 6470 (2011), https://doi.org/10.1002/anie.201007100.Search in Google Scholar PubMed
[36] V. I. Minkin, L. P. Olekhnovich, A. Yu. Zhdanov. Molecular Design of Tautomeric Compounds, Springer, Netherlands (1988).10.1007/978-94-009-1429-2Search in Google Scholar
[37] R. L. Danheiser, S. K. Gere, H. Sard. J. Am. Chem. Soc.104, 7670 (1982), https://doi.org/10.1021/ja00390a054.Search in Google Scholar
[38] M. E. Jung. Tetrahedron32, 3 (1976), https://doi.org/10.1016/0040-4020(76)80016-6.Search in Google Scholar
[39] G. P. Moss, P. A. S. Smith, D. Tavernier. Pure Appl. Chem.67, 1307 (1995), https://doi.org/10.1351/pac199567081307.Search in Google Scholar
[40] P. v. R. Schleyer. J. Am. Chem. Soc.107, 6393 (1985), https://doi.org/10.1021/ja00308a041.Search in Google Scholar
[41] E. Juaristi. Tetrahedron48, 5019 (1992), https://doi.org/10.1016/s0040-4020(01)90118-8.Search in Google Scholar
[42] K. B. Wiberg, W. F. Bailey, K. M. Lambert, Z. D. Stempel. J. Org. Chem.83, 5242 (2018), https://doi.org/10.1021/acs.joc.8b00707.Search in Google Scholar PubMed
[43] C. M. Filloux. Angew. Chem. Int. Ed. 54, 8880 (2015), https://doi.org/10.1002/anie.201411185.Search in Google Scholar PubMed
[44] J. Rigaudy, S. P. Klesney. Nomenclature of Organic Chemistry, Pergamon Press, Oxford (1979).Search in Google Scholar
[45] H. A. Favre, W. H. Powell. Nomenclature of Organic Chemistry: IUPAC Recommendations and Preferred Names, Royal Society of Chemistry, Cambridge (2013).Search in Google Scholar
[46] W. Klyne, V. Prelog. Experientia16, 521 (1960), https://doi.org/10.1007/bf02158433.Search in Google Scholar
[47] S. Ahrland. Pure Appl. Chem.51, 201 (1979), https://doi.org/10.1351/pac197951102019.Search in Google Scholar
[48] J. Kruszewski, T. M. Krygowski. Tetrahedron Lett.13, 3839 (1972), https://doi.org/10.1016/s0040-4039(01)94175-9.Search in Google Scholar
[49] P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao, N. J. R. van Eikema Hommes. J. Am. Chem. Soc.118, 6317 (1996), https://doi.org/10.1021/ja960582d.Search in Google Scholar PubMed
[50] M. J. S. Dewar. Angew. Chem. Int. Ed.10, 761 (1971), https://doi.org/10.1002/anie.197107611.Search in Google Scholar
[51] H. E. Zimmerman. Acc. Chem. Res.4, 272 (1971), https://doi.org/10.1021/ar50044a002.Search in Google Scholar
[52] C. L. Perrin. Chem. Brit.8, 163 (1972).Search in Google Scholar
[53] Chem. Rev. 101, 1115 (2001); all articles.10.1021/cr0103221Search in Google Scholar PubMed
[54] R. G. Bergman. Acc. Chem. Res.6, 25 (1973), https://doi.org/10.1021/ar50061a004.Search in Google Scholar
[55] H. Maskill. The Physical Basis of Organic Chemistry, Oxford University Press, Oxford (1985).Search in Google Scholar
[56] A. V. Bandura, S. N. Lvov. J. Phys. Chem. Ref. Data35, 15 (2006), https://doi.org/10.1063/1.1928231.Search in Google Scholar
[57] http://www.iapws.org/relguide/Ionization.html.Search in Google Scholar
[58] S. Hoz, E. Buncel. Isr. J. Chem.26, 313 (1985), https://doi.org/10.1002/ijch.198500113.Search in Google Scholar
[59] E. Juaristi, G. dos Passos Gomes, A. O. Terent’ev, R. Notario, I. V. Alabugin. J. Am. Chem. Soc.139, 10799 (2017), https://doi.org/10.1021/jacs.7b05367.Search in Google Scholar PubMed
[60] I.-K. Um, M.-Y. Kim, H.-J. Cho, J. M. Dust, E. Buncel. Can. J. Chem.93, 1109 (2015), https://doi.org/10.1139/cjc-2015-0073.Search in Google Scholar
[61] J. E. Baldwin. J. Chem. Soc. Chem. Commun.18, 734 (1976), https://doi.org/10.1039/c39760000734.Search in Google Scholar
[62] K. Gilmore, I. V. Alabugin. Chem. Rev.111, 6513 (2011), https://doi.org/10.1021/cr200164y.Search in Google Scholar PubMed
[63] C. Laurence, J.-F. Gal. Lewis Basicity and Affinity Scales: Data and Measurement, Wiley, Chichester (2010).10.1002/9780470681909Search in Google Scholar
[64] M. J. S. Dewar. Molecular Orbital Theory of Organic Chemistry, McGraw-Hill, New York (1969).Search in Google Scholar
[65] W. P. Jencks. Chem. Rev.85, 511 (1985), https://doi.org/10.1021/cr00070a001.Search in Google Scholar
[66] R. P. Bell. Proc. R. Soc. Lond. Ser. A154, 414 (1936).10.1098/rspa.1936.0060Search in Google Scholar
[67] M. G. Evans, M. Polanyi. J. Chem. Soc. Faraday Trans.32, 1340 (1936), https://doi.org/10.1039/tf9363201333.Search in Google Scholar
[68] B. A. Cunningham, G. L. Schmir. J. Am. Chem. Soc.89, 917 (1967), https://doi.org/10.1021/ja00980a032.Search in Google Scholar
[69] M. Shibasaki, M. Kanai, S. Matsunaga, N. Kumagai. Acc. Chem. Res.42, 1117 (2009), https://doi.org/10.1021/ar9000108.Search in Google Scholar PubMed
[70] R. Noyori, M. Yamakawa, S. Hashiguchi. J. Org. Chem.66, 7931 (2001), https://doi.org/10.1021/jo010721w.Search in Google Scholar PubMed
[71] G. Della Sala, A. Russo, A. Lattanzi. Current Org. Chem.15, 2147 (2011), https://doi.org/10.2174/138527211796150741.Search in Google Scholar
[72] J. Aleman, A. Parra, H. Jiang, K. A. Jorgensen. Chem. Eur. J.17, 6890 (2011), https://doi.org/10.1002/chem.201003694.Search in Google Scholar PubMed
[73] D. H. Ess, S. E. Wheeler, R. G. Iafe, L. Xu, N. Celebi-Olcum, K. N. Houk. Angew. Chem. Int. Ed.47, 7592 (2008), https://doi.org/10.1002/anie.200800918.Search in Google Scholar PubMed PubMed Central
[74] S. R. Hare, D. J. Tantillo. Pure Appl. Chem.89, 679 (2017), https://doi.org/10.1515/pac-2017-0104.Search in Google Scholar
[75] L. Pauling. The Nature of the Chemical Bond, Cornell University Press, Ithaca, New York, 3rd ed. (1960).Search in Google Scholar
[76] S. J. Blanksby, G. B. Ellison. Acc. Chem. Res.36, 255 (2003), https://doi.org/10.1021/ar020230d.Search in Google Scholar PubMed
[77] H. S. Johnston, C. Parr. J. Am. Chem. Soc.85, 2544 (1963), https://doi.org/10.1021/ja00900a002.Search in Google Scholar
[78] P. Gütlich, H. A. Goodwin, D. N. Hendrickson. Angew. Chem. Int. Ed.33, 425 (1994), https://doi.org/10.1002/anie.199404251.Search in Google Scholar
[79] G. Parkin. Acc. Chem. Res.25, 455 (1992), https://doi.org/10.1021/ar00022a004.Search in Google Scholar
[80] G. Parkin. Chem. Rev.93, 887 (1993), https://doi.org/10.1021/cr00019a003.Search in Google Scholar
[81] J. M. Schulman, R. L. Disch. J. Am. Chem. Soc.113, 11153 (1993), https://doi.org/10.1021/ja00077a012.Search in Google Scholar
[82] J. Bredt. Justus Liebig Ann. Chem.437, 1 (1924), https://doi.org/10.1002/jlac.19244370102.Search in Google Scholar
[83] F. S. Fawcett. Chem. Rev.47, 219 (1950), https://doi.org/10.1021/cr60147a003.Search in Google Scholar PubMed
[84] J. R. Wiseman. J. Am. Chem. Soc.89, 5966 (1967), https://doi.org/10.1021/ja00999a050.Search in Google Scholar
[85] R. Keese, R. P. Krebs. Angew. Chem. Int. Ed. Engl.11, 518 (1972), https://doi.org/10.1002/anie.197205181.Search in Google Scholar
[86] W. F. Maier, P. v. R. Schleyer. J. Am. Chem. Soc.103, 1891 (1981), https://doi.org/10.1021/ja00398a003.Search in Google Scholar
[87] J. R. Wiseman, J. A. Chong. J. Am. Chem. Soc.91, 7775 (1969), https://doi.org/10.1021/ja50001a061.Search in Google Scholar
[88] G. A. Olah, G. K. Surya Prakash, M. Saunders. Acc. Chem. Res.16, 440 (1983), https://doi.org/10.1021/ar00096a003.Search in Google Scholar
[89] F. Scholz, D. Himmel, F. W. Heinemann, P. v. R. Schleyer, K. Meyer, I. Krossing. Science341, 62 (2013), https://doi.org/10.1126/science.1238849.Search in Google Scholar PubMed
[90] J. F. Bunnett, F. P. Olsen. Can. J. Chem.44, 1899 (1966), https://doi.org/10.1139/v66-286.Search in Google Scholar
[91] G. A. Olah, P. v. R. Schleyer. Cage Hydrocarbons, Wiley, New York (1990).Search in Google Scholar
[92] D. Moran, H. L. Woodcock, Z. Chen, H. F. Schaefer, P. v. R. Schleyer. J. Am. Chem. Soc.125, 11443 (2003), https://doi.org/10.1021/ja0345470.Search in Google Scholar PubMed
[93] F. G. Bordwell, T. Y. Lynch. J. Am. Chem. Soc.111, 7558 (1989), https://doi.org/10.1021/ja00201a043.Search in Google Scholar
[94] R. Sustmann, H.-G. Korth. Adv. Phys. Org. Chem.26, 131 (1990), https://doi.org/10.1016/s0065-3160(08)60045-3.Search in Google Scholar
[95] H. G. Viehe, Z. Janousek, R. Mereny, L. Stella. Acc. Chem. Res.18, 148 (1985), https://doi.org/10.1021/ar00113a004.Search in Google Scholar
[96] E. Buncel, J. M. Dust. Carbanion Chemistry-Structures and Mechanism, Oxford University Press, New York (2003).Search in Google Scholar
[97] R. Panico, W. H. Powell, J. C. Richer. A Guide to IUPAC Nomenclature of Organic Compounds (IUPAC Recommendations 1993), Blackwell Scientific Publications, Oxford (1993).Search in Google Scholar
[98] G. A. Olah, P. v. R. Schleyer. Carbonium Ions 3: Major Types (continued) (Reactive Intermediates in Organic Chemistry), Wiley, New York (1972).Search in Google Scholar
[99] G. A. Olah, G. K. S. Prakash, R. E. Williams, L. D. Field, K. Wade. Hypercarbon Chemistry, Wiley-Interscience, New York (1987).Search in Google Scholar
[100] J. Catalán, C. Díaz, F. García Blanco. J. Org. Chem.17, 5846 (2001), https://doi.org/10.1021/jo010415i.Search in Google Scholar PubMed
[101] J. Catalán. J. Phys. Chem. B17, 5951 (2009).10.1021/jp8095727Search in Google Scholar PubMed
[102] W. P. Jencks. Catalysis in Chemistry and Enzymology, pp. 288, McGraw-Hill, New York (1969).Search in Google Scholar
[103] A. Tramontano, K. D. Janda, R. A. Lerner. Science234, 1566 (1986), https://doi.org/10.1126/science.3787261.Search in Google Scholar PubMed
[104] S. J. Pollack, J. W. Jacobs, P. G. Schultz. Science234, 1570 (1986), https://doi.org/10.1126/science.3787262.Search in Google Scholar PubMed
[105] J. C. Ma, D. A. Dougherty. Chem. Rev.97, 1303 (1997), https://doi.org/10.1021/cr9603744.Search in Google Scholar PubMed
[106] S. Penczek, G. Moad. Pure Appl. Chem.80, 2163 (2008), https://doi.org/10.1351/pac200880102163.Search in Google Scholar
[107] R. B. Woodward, R. Hoffmann. Angew. Chem. Int. Ed.8, 781 (1969), https://doi.org/10.1002/anie.196907811.Search in Google Scholar
[108] C. F. Bernasconi. Relaxation Kinetics, Academic Press, New York (1976).Search in Google Scholar
[109] J. E. Leffler, E. Grunwald. Rates and Equilibria of Organic Reactions, Wiley, New York (1963).Search in Google Scholar
[110] R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, P. Granger, R. E. Hoffman, K. W. Zilm. Pure Appl. Chem.80, 59 (2008), https://doi.org/10.1351/pac200880010059.Search in Google Scholar
[111] L. T. Muus, P. W. Atkins, K. A. McLauchlan, J. B. Pedersen (Eds.), Chemically Induced Magnetic Polarisation, D. Reidel, Dordrecht (1977).10.1007/978-94-010-1265-2Search in Google Scholar
[112] P. J. Hore, R. W. Broadhurst. Progr. NMR Spectr.25, 345 (1993), https://doi.org/10.1016/0079-6565(93)80002-b.Search in Google Scholar
[113] B. M. Trost. Acc. Chem. Res.13, 385 (1980), https://doi.org/10.1021/ar50155a001.Search in Google Scholar
[114] O. N. Witt. Berichte der deutschen chemischen Ges.9, 522 (1876), https://doi.org/10.1002/cber.187600901164.Search in Google Scholar
[115] H. Zollinger. Color Chemistry: Syntheses, Properties, and Applications of Organic Dyes and Pigments, Wiley, Weinheim, 3rd revised ed. (2003).Search in Google Scholar
[116] R. Herges. Angew. Chem. Int. Ed. 33, 255 (1994), https://doi.org/10.1002/anie.199402551.Search in Google Scholar
[117] K. A. Connor. Chemical Kinetics, VCH, New York (1991).Search in Google Scholar
[118] L. Liu, Q.-Xi. Guo. Chem. Rev.101, 673 (2001), https://doi.org/10.1021/cr990416z.Search in Google Scholar PubMed
[119] O. Exner. Coll. Czech. Chem. Commun.29, 1094 (1964), https://doi.org/10.1135/cccc19641094.Search in Google Scholar
[120] P. J. Barrie. Phys. Chem. Chem. Phys.14, 327 (2012), https://doi.org/10.1039/c1cp22667c.Search in Google Scholar PubMed
[121] G. J. Leigh (Ed.), Nomenclature of Inorganic Chemistry, Blackwell Scientific Publications, Oxford (1990).Search in Google Scholar
[122] J. Hartmanns, K. Klenke, J. O. Metzger. Chem. Ber.119, 488 (1986), https://doi.org/10.1002/cber.19861190212.Search in Google Scholar
[123] E. A. Halevi. Orbital Symmetry and Reaction Mechanism: The OCAMS View, Springer, Berlin, Heidelberg (1992).10.1007/978-3-642-83568-1Search in Google Scholar
[124] N. B. Chapman, J. Shorter (Eds.), Advances in Linear Free Energy Relationships, Plenum Press, London (1972).10.1007/978-1-4615-8660-9Search in Google Scholar
[125] N. B. Chapman, J. Shorter (Eds.), Correlation Analysis in Chemistry, Recent Advances (1978).10.1007/978-1-4615-8831-3Search in Google Scholar
[126] O. Exner. Correlation Analysis of Chemical Data, Plenum Press, New York (1988).Search in Google Scholar
[127] R. I. Zalewski, T. M. Krygowski, J. Shorter. Similarity Models in Organic Chemistry, Biochemistry and Related Fields, in Studies in Organic Chemistry, Elsevier, Amsterdam, Vol. 42 (1991).Search in Google Scholar
[128] A. Williams. Free Energy Relationships in Organic and Bio-organic Chemistry, The Royal Society of Chemistry, Cambridge (2003).10.1039/9781847550927Search in Google Scholar
[129] H. S. Gutowsky, D. W. McCall, C. P. Slichter. Phys. Rev.84, 589 (1951), https://doi.org/10.1103/physrev.84.589.2.Search in Google Scholar
[130] N. F. Ramsey, E. M. Purcell. Phys. Rev.85, 143 (1952), https://doi.org/10.1103/physrev.85.143.Search in Google Scholar
[131] P. S. Pregosin, H. Rueegger. Nuclear magnetic resonance spectroscopy, in Comprehensive Coordination Chemistry II, J. A. McCleverty, T. J. Meyer (Eds.), Pergamon, Oxford, p. 1, Vol. 2 (2004).10.1016/B0-08-043748-6/01061-6Search in Google Scholar
[132] R. A. Cox, K. Yates. J. Am. Chem. Soc.100, 3861 (1978), https://doi.org/10.1021/ja00480a033.Search in Google Scholar
[133] R. A. Cox, K. Yates. Can. J. Chem.59, 2116 (1981), https://doi.org/10.1139/v81-306.Search in Google Scholar
[134] V. Lucchini, G. Modena, G. Scorrano, R. A. Cox, K. Yates. J. Am. Chem. Soc.104, 1958 (1982), https://doi.org/10.1021/ja00371a026.Search in Google Scholar
[135] D. H. Everett. Pure Appl. Chem.31, 577 (1972), https://doi.org/10.1351/pac197231040577.Search in Google Scholar
[136] C. J. Pedersen. J. Am. Chem. Soc.89, 7017 (1967), https://doi.org/10.1021/ja01002a035.Search in Google Scholar
[137] D. J. Cram. Angew. Chem. Int. Ed.25, 1039 (1986), https://doi.org/10.1002/anie.198610393.Search in Google Scholar
[138] B. Dietrich, J. M. Lehn, J. P. Sauvage. Tetrahedron Lett.34, 2889 (1969), https://doi.org/10.1016/s0040-4039(01)88300-3.Search in Google Scholar
[139] E. Weber, F. Vögtle. Angew. Chem. Int. Ed.19, 1030 (1980), https://doi.org/10.1002/anie.198010301.Search in Google Scholar
[140] E. L. Eliel. Stereochemistry of Carbon Compounds, McGraw-Hill, New York (1962).Search in Google Scholar
[141] J. I. Seeman, H. V. Secor, H. Hartung, R. Galzerano. J. Am. Chem. Soc.102, 7741 (1980), https://doi.org/10.1021/ja00546a017.Search in Google Scholar
[142] J. I. Seeman. Chem. Rev.83, 83 (1983), https://doi.org/10.1021/cr00054a001.Search in Google Scholar
[143] E. M. Kosower. An Introduction to Physical Organic Chemistry, John Wiley, New York (1968).Search in Google Scholar
[144] G. W. Stewart, R. M. Morrow. Proc. Natl. Acad. Sci. U. S. A.13, 222 (1927).10.1073/pnas.13.4.222Search in Google Scholar PubMed PubMed Central
[145] R. Huisgen. Angew. Chem. Int. Ed.7, 321 (1968), https://doi.org/10.1002/anie.196803211.Search in Google Scholar
[146] R. Huisgen, R. Grashey, J. Sauer. Cycloaddition reactions of alkenes, in The Chemistry of the Alkenes, S. Patai (Ed.), John Wiley & Sons, New York, Vol. 1 (1964).Search in Google Scholar
[147] G. Binsch, E. L. Eliel, H. Kessler. Angew. Chem., Int. Ed.10, 570 (1971), https://doi.org/10.1002/anie.197105701.Search in Google Scholar
[148] A. Fradet, J. Chen, K.-H. Hellwich, K. Horie, J. Kahovec, W. Mormann, R. F. T. Stepto, J. Vohlídal, E. S. Wilks. Pure Appl. Chem.91, 523 (2018).Search in Google Scholar
[149] E. Buhleier, W. Wehner, F. Vögtle. Synthesis2, 155 (1978), https://doi.org/10.1055/s-1978-24702.Search in Google Scholar
[150] D. A. Tomalia, J. B. Christensen. Dendrimers, Dendrons and Dendritic Polymers, Cambridge University Press, Cambridge (2012).10.1017/CBO9781139048859Search in Google Scholar
[151] D. Astruc, E. Boisselier, C. Ornelas. Chem. Rev.110, 1857 (2010), https://doi.org/10.1021/cr900327d.Search in Google Scholar PubMed
[152] J. M. Frechet, D. A. Tomalia. Dendrimers and Other Dendritic Polymers, John Wiley & Sons, New York, NY (2002).10.1002/0470845821Search in Google Scholar
[153] K. Dimroth, C. Reichardt, T. Siepmann, F. Bohlmann. Liebigs Ann. Chem.661, 1 (1963), https://doi.org/10.1002/jlac.19636610102.Search in Google Scholar
[154] C. Reichardt, E. C. Harbusch-Görnert. Liebigs Ann. Chem.721 (1983).10.1002/jlac.198319830502Search in Google Scholar
[155] V. G. Machado, R. I. Stock, C. Reichardt. Chem. Rev.114, 10429 (2014), https://doi.org/10.1021/cr5001157.Search in Google Scholar PubMed
[156] A. J. Parker. Quart. Rev. Chem. Soc.16, 163 (1962), https://doi.org/10.1039/qr9621600163.Search in Google Scholar
[157] C. Reichardt, T. Welton. Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH Verlag GmbH & Co, Weinheim, 4th ed. (2011).10.1002/9783527632220Search in Google Scholar
[158] V. Bonacic-Koutecký, J. Koutecký, J. Michl. Angew. Chem. Int. Ed.26, 170 (1987).10.1002/anie.198701701Search in Google Scholar
[159] M. J. Lenington, P. G. Wenthold. J. Phys. Chem.114, 1334 (2010), https://doi.org/10.1021/jp905757p.Search in Google Scholar PubMed
[160] T. Ichino, S. M. Villano, A. J. Gianola, D. J. Goebbert, L. Velarde, A. Sanov, S. J. Blankby, X. Zhou, D. A. Hrovat, W. T. Borden, W. C. Lineberger. Angew. Chem. Int. Ed.48, 8509 (2009), https://doi.org/10.1002/anie.200904417.Search in Google Scholar PubMed
[161] W. T. Borden. Diradicals, Wiley, New York (1982).Search in Google Scholar
[162] W. T. Borden in ACS Symposium Series, 1209 (Foundations of Physical Organic Chemistry: Fifty Years of the James Flack Norris Award), Vol. 251 (2015).10.1021/bk-2015-1209.ch011Search in Google Scholar
[163] M. Abe. Chem. Rev.113, 7011 (2013), https://doi.org/10.1021/cr400056a.Search in Google Scholar PubMed
[164] B. F. Yates, W. J. Bouma, L. Radom. Tetrahedron42, 6225 (1986), https://doi.org/10.1016/s0040-4020(01)88084-4.Search in Google Scholar
[165] S. Hammerum. Mass Spectrometry Rev.7, 123 (1988), https://doi.org/10.1002/mas.1280070202.Search in Google Scholar
[166] F. M. Bickelhaupt, K. N. Houk. Angew. Chem. Int. Ed. 56, 10070 (2017), https://doi.org/10.1002/anie.201701486.Search in Google Scholar PubMed PubMed Central
[167] N. M. Rice, H. M. N. H. Irving, M. A. Leonard. Pure Appl. Chem.65, 2373 (1993), https://doi.org/10.1351/pac199365112373.Search in Google Scholar
[168] H. E. Zimmerman, D. Armesto. Chem. Rev.96, 3065 (1996), https://doi.org/10.1021/cr910109c.Search in Google Scholar PubMed
[169] V. Gutmann, A. Steininger, E. Wychera. Monatsh. Chem.97, 460 (1966), https://doi.org/10.1007/bf00905266.Search in Google Scholar
[170] V. Gutmann. The Donor-Acceptor Approach to Molecular Interactions, Plenum Press, New York (1978).10.1007/978-1-4615-8825-2Search in Google Scholar
[171] S. Ehrenson, R. T. C. Brownlee, R. W. Taft. Prog. Phys. Org. Chem.10, 1 (1973).10.1002/9780470171899.ch1Search in Google Scholar
[172] R. W. Taft, R. D. Topsom. Prog. Phys. Org. Chem.16, 1 (1987).10.1002/9780470171950Search in Google Scholar
[173] T. M. Krygowski, B. T. Stepien. Chem. Rev.105, 3482 (2005), https://doi.org/10.1021/cr030081s.Search in Google Scholar PubMed
[174] M. T. Reetz. Angew. Chem. Int. Ed.11, 129 (1972), https://doi.org/10.1002/anie.197201291.Search in Google Scholar
[175] M. T. Reetz. Angew. Chem. Int. Ed.11, 130 (1972), https://doi.org/10.1002/anie.197201301.Search in Google Scholar
[176] I. Fernández, F. P. Cossío, M. A. Sierra. Chem. Rev.109, 6687 (2009), https://doi.org/10.1021/cr900209c.Search in Google Scholar PubMed
[177] A. J. Kirby. Adv. Phys. Org. Chem.17, 183 (1980), https://doi.org/10.1016/s0065-3160(08)60129-x.Search in Google Scholar
[178] C. L. Perrin. J. Org. Chem.36, 420 (1971), https://doi.org/10.1021/jo00802a013.Search in Google Scholar
[179] N. Streidl, B. Denegri, O. Kronja, H. Mayr. Acc. Chem. Res.43, 1537 (2010), https://doi.org/10.1021/ar100091m.Search in Google Scholar PubMed
[180] P. Kebarle, S. Chowdury. Chem. Rev.87, 513 (1987), https://doi.org/10.1021/cr00079a003.Search in Google Scholar
[181] E. C. M. Chen, E. S. D. Chen. The Electron Capture Detector and the Study of Reactions with Thermal Electrons, Wiley, Hoboken, New Jersey (2004).10.1002/0471659894Search in Google Scholar
[182] L. Eberson. Electron Transfer Reactions in Organic Chemistry, Springer, Berlin (1987).10.1007/978-3-642-72544-9Search in Google Scholar
[183] A. Studer, D. P. Curran. Nat. Chem.6, 765 (2014), https://doi.org/10.1038/nchem.2031.Search in Google Scholar PubMed
[184] L. Pauling. J. Am. Chem. Soc.54, 3570 (1932), https://doi.org/10.1021/ja01348a011.Search in Google Scholar
[185] L. C. Allen. J. Am. Chem. Soc.111, 9003 (1989), https://doi.org/10.1021/ja00207a003.Search in Google Scholar
[186] R. J. Boyd, S. L. Boyd. J. Am. Chem. Soc.114, 1652 (1992), https://doi.org/10.1021/ja00031a018.Search in Google Scholar
[187] K. D. Sen, C. K. Jorgenson (Eds.), Structure and Bonding, 66: Electronegativity, Springer, Berlin (1987).10.1007/BFb0029833Search in Google Scholar
[188] M. Rahm, T. Zeng, R. Hoffmann. J. Am. Chem. Soc.141, 342 (2019), https://doi.org/10.1021/jacs.8b10246.Search in Google Scholar PubMed
[189] R. T. Sanderson. J. Am. Chem. Soc.105, 2259 (1983), https://doi.org/10.1021/ja00346a026.Search in Google Scholar
[190] R. T. Sanderson. J. Chem. Educ.65, 227 (1988), https://doi.org/10.1021/ed065p227.Search in Google Scholar
[191] C. Hansch, A. Leo, R. W. Taft. Chem. Rev.91, 165 (1991), https://doi.org/10.1021/cr00002a004.Search in Google Scholar
[192] C. G. Swain, C. B. Scott. J. Am. Chem. Soc.75, 141 (1953), https://doi.org/10.1021/ja01097a041.Search in Google Scholar
[193] C. D. Ritchie. Can. J. Chem.64, 2239 (1986), https://doi.org/10.1139/v86-370.Search in Google Scholar
[194] H. Mayr, A. R. Ofial. Acc. Chem. Res.49, 952 (2016), https://doi.org/10.1021/acs.accounts.6b00071.Search in Google Scholar PubMed
[195] A. Williams. Chem. Soc. Rev.23, 93 (1994), https://doi.org/10.1039/cs9942300093.Search in Google Scholar
[196] W. P. Jencks. Ann. Rev. Biochem.66, 1 (1997), https://doi.org/10.1146/annurev.biochem.66.1.1.Search in Google Scholar
[197] J. P. Richard. Adv. Carbocation Chem.1, 121 (1989).Search in Google Scholar
[198] C. L. Perrin, T. J. Dwyer. Chem. Rev.90, 935 (1990), https://doi.org/10.1021/cr00104a002.Search in Google Scholar
[199] L. M. Stock. J. Chem. Educ.49, 400 (1971).10.1021/ed049p400Search in Google Scholar
[200] O. Exner. J. Phys. Org. Chem.12, 265 (1999), https://doi.org/10.1002/(sici)1099-1395(199904)12:4<265::aid-poc124>3.0.co;2-o.10.1002/(SICI)1099-1395(199904)12:4<265::AID-POC124>3.0.CO;2-OSearch in Google Scholar
[201] W. S. Trahanovsky, M.-G. Park. J. Am. Chem. Soc.95, 5412 (1973), https://doi.org/10.1021/ja00797a051.Search in Google Scholar
[202] W. S. Trahanovsky, M.-G. Park. J. Org. Chem.39, 1448 (1974), https://doi.org/10.1021/jo00926a037.Search in Google Scholar
[203] C. Wentrup. Aust. J. Chem.67, 1150 (2014), https://doi.org/10.1071/ch14096.Search in Google Scholar
[204] C. Wentrup. Chem. Rev.117, 4562 (2017), https://doi.org/10.1021/acs.chemrev.6b00738.Search in Google Scholar
[205] C. A. Grob. Angew. Chem. Int. Ed. Engl.8, 535 (1969), https://doi.org/10.1002/anie.196905351.Search in Google Scholar
[206] K. Fukui, Y. Tejiro, S. Haruo. J. Chem. Phys.20, 722 (1952), https://doi.org/10.1063/1.1700523.Search in Google Scholar
[207] I. Fleming. Frontier Orbitals and Organic Chemical Reactions, John Wiley & Sons, New York (1976).Search in Google Scholar
[208] D. W. Stephan. J. Am. Chem. Soc.137, 10018 (2015), https://doi.org/10.1021/jacs.5b06794.Search in Google Scholar PubMed
[209] D. W. Stephan, G. Erker. Angew. Chem. Int. Ed. 54, 6400 (2015), https://doi.org/10.1002/anie.201409800.Search in Google Scholar PubMed
[210] H. W. Kroto, J. R. Heath, S. C. O’Brien, R. F. Curl, R. E. Smalley. Nature318, 162 (1985), https://doi.org/10.1038/318162a0.Search in Google Scholar
[211] J. A. Bartmess, J. E. Scott, R. T. McIverJr. J. Am. Chem. Soc.101, 6046 (1979), https://doi.org/10.1021/ja00514a030.Search in Google Scholar
[212] S. G. Lias, J. E. Bartmess, J. F. Liebman, J. L. Holmes, R. D. Levin, W. G. Mallard. J. Phys. Chem. Ref. Data17 (1988), National Institute of Standards and Technology (NIST, Gaithersburg, Maryland, USA) data base (http://webbook.nist.gov/).Search in Google Scholar
[213] E. P. L. Hunter, S. G. Lias. J. Phys. Chem. Ref. Data27, 413 (1998), National Institute of Standards and Technology (NIST, Gaithersburg, Maryland, USA) database (http://webbook.nist.gov/chemistry/guide/#ion).Search in Google Scholar
[214] J.-F. Gal, P.-C. Maria, E. D. Raczynska. J. Mass Spectrom.36, 699 (2001), https://doi.org/10.1002/jms.202.Search in Google Scholar PubMed
[215] S. Winstein, E. Grunwald, H. W. Jones. J. Am. Chem. Soc.73, 2700 (1951), https://doi.org/10.1021/ja01150a078.Search in Google Scholar
[216] T. W. Bentley, P. v. R. Schleyer. Adv. Phys. Org. Chem.14, 1 (1977), https://doi.org/10.1016/s0065-3160(08)60107-0.Search in Google Scholar
[217] T. W. Bentley, G. Llewellyn. Prog. Phys. Org. Chem.17, 121 (1990).10.1002/9780470171967.ch5Search in Google Scholar
[218] E. Grunwald, S. Winstein. J. Am. Chem. Soc.70, 846 (1948), https://doi.org/10.1021/ja01182a117.Search in Google Scholar
[219] A. H. Fainberg, S. Winstein. J. Phys. Chem.75, 1708 (1956).Search in Google Scholar
[220] J. Falbe, M. Regitz. Römpp Chemie Lexikon, Georg-Thieme Verlag, Stuttgart, 9th ed., Vol. 3, p. 1714 (1990).Search in Google Scholar
[221] C. Reichardt, S. Asharin-Fard, G. Schäfer. Chem. Ber.126, 143 (1993), https://doi.org/10.1002/cber.19931260122.Search in Google Scholar
[222] A. C. Legon. Phys. Chem. Chem. Phys.12, 7736 (2010), https://doi.org/10.1039/c002129f.Search in Google Scholar PubMed
[223] T. M. Beale, M. G. Chudzinski, M. G. Sarwar, M. S. Taylor. Chem. Soc. Rev.42, 1667 (2013), https://doi.org/10.1039/c2cs35213c.Search in Google Scholar PubMed
[224] G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati, K. Rissanen. Pure Appl. Chem.85, 1711 (2013), https://doi.org/10.1351/pac-rec-12-05-10.Search in Google Scholar
[225] A. V. Jentzsch. Pure Appl. Chem.87, 15 (2015), https://doi.org/10.1515/pac-2014-0807.Search in Google Scholar
[226] C. D. Johnson. The Hammett Equation, Cambridge University Press, Cambridge (1973).Search in Google Scholar
[227] G. S. Hammond. J. Am. Chem. Soc.77, 334 (1955), https://doi.org/10.1021/ja01607a027.Search in Google Scholar
[228] J. E. Leffler. Science117, 340 (1953), https://doi.org/10.1126/science.117.3039.340.Search in Google Scholar PubMed
[229] E. R. Thornton. J. Am. Chem. Soc.89, 2915 (1967), https://doi.org/10.1021/ja00988a020.Search in Google Scholar
[230] D. Farcasiu. J. Chem. Educ.52, 76 (1975), https://doi.org/10.1021/ed052p76.Search in Google Scholar
[231] C. Hansch, A. Leo. Substituent Constants for Correlation Analysis in Chemistry and Biology, Wiley, New York, NY (1979).Search in Google Scholar
[232] J. Sangster. Octanol–Water Partition Coefficients: Fundamentals and Physical Chemistry. Wiley Series in Solution Chemistry, John Wiley, Chichester, Vol. 2 (1997).Search in Google Scholar
[233] R. B. Martin. Inorg. Chim. Acta339, 27 (2002), https://doi.org/10.1016/s0020-1693(02)00922-2.Search in Google Scholar
[234] R. G. Pearson. J. Am. Chem. Soc.85, 3533 (1963), https://doi.org/10.1021/ja00905a001.Search in Google Scholar
[235] R. G. Pearson. Inorg. Chem.12, 712 (1973), https://doi.org/10.1021/ic50121a052.Search in Google Scholar
[236] P. W. Ayers, R. G. Parr, R. G. Pearson. J. Chem. Phys.124, 194107 (2006), https://doi.org/10.1063/1.2196882.Search in Google Scholar PubMed
[237] G. Kohnstam. Adv. Phys. Org. Chem.5, 121 (1967), https://doi.org/10.1016/s0065-3160(08)60310-x.Search in Google Scholar
[238] H. N. Po, N. M. Senozan. J. Chem. Educ.78, 1499 (2001), https://doi.org/10.1021/ed078p1499.Search in Google Scholar
[239] J. Inglese, D. S. Auld. Application of High Throughput Screening (HTS) Techniques: Applications in Chemical Biology, Wiley Encyclopedia of Chemical Biology, Wiley & Sons, Inc., Hoboken, NJ, Vol. 2, p. 260 (2009).10.1002/9780470048672.wecb223Search in Google Scholar
[240] J. H. Hildebrand, R. L. Scott. Solubility of Non-Electrolytes, Reinhold, New York, 3rd ed. (1964).Search in Google Scholar
[241] A. W. Hofmann. Philos. Trans. R. Soc. Lond.141, 357 (1851).10.1098/rstl.1851.0017Search in Google Scholar
[242] L. Pauling. Proc. Natl. Acad. Sci. U. S. A.14, 359 (1928).10.1073/pnas.14.4.359Search in Google Scholar PubMed PubMed Central
[243] E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci, D. J. Nesbitt. Pure Appl. Chem.83, 1619 (2011), https://doi.org/10.1351/PAC-REP-10-01-01.Search in Google Scholar
[244] J. F. Bunnett, R. A. Y. Jones. Pure Appl. Chem.60, 1115 (1988), https://doi.org/10.1351/pac198860071115.Search in Google Scholar
[245] L. Radom. Prog. Theor. Org. Chem.3, 1 (1982).Search in Google Scholar
[246] J. D. Roberts, R. L. Webb, E. A. McElhill. J. Am. Chem. Soc.72, 408 (1950), https://doi.org/10.1021/ja01157a111.Search in Google Scholar
[247] P. v. R. Schleyer, A. J. Kos. Tetrahedron39, 1141 (1983), https://doi.org/10.1016/s0040-4020(01)91877-0.Search in Google Scholar
[248] R. Gleiter, G. Haberhauer. Aromaticity and Other Conjugation Effects, Wiley-VCH, Weinheim (2012).Search in Google Scholar
[249] C. F. Bernasconi. Adv. Phys. Org. Chem.27, 119 (1992), https://doi.org/10.1016/s0065-3160(08)60065-9.Search in Google Scholar
[250] O. Exner, S. Boehm. J. Phys. Org. Chem.17, 124 (2004), https://doi.org/10.1002/poc.701.Search in Google Scholar
[251] R. A. Marcus. Disc. Faraday Soc.29, 21 (1960), https://doi.org/10.1039/df9602900021.Search in Google Scholar
[252] W. J. Albery, M. M. Kreevoy. Adv. Phys. Org. Chem.16, 87 (1978), https://doi.org/10.1016/s0065-3160(08)60087-8.Search in Google Scholar
[253] R. D. Cannon. Electron Transfer Reactions, Butterworths, London (1980).10.1016/B978-0-408-10646-7.50012-8Search in Google Scholar
[254] C. J. Schlesener, C. Amatore, J. K. Kochi. J. Phys. Chem.90, 3747 (1986), https://doi.org/10.1021/j100407a049.Search in Google Scholar
[255] K. Fukui. Acc. Chem Res.14, 363 (1981), https://doi.org/10.1021/ar00072a001.Search in Google Scholar
[256] M. Szwarc (Ed.), Ions and Ion Pairs in Organic Reactions, Wiley-Interscience, New York, Vol. 1 and 2 (1972–1974).Search in Google Scholar
[257] P. Walden. Bull. Acad. Imper. Sci. St. Petersbourg8, 405 (1914).Search in Google Scholar
[258] T. Welton. Biophys. Rev.10, 691 (2018), https://doi.org/10.1007/s12551-018-0419-2.Search in Google Scholar PubMed PubMed Central
[259] N. V. Plechkova, K. R. Seddon. Chem. Soc. Rev.37, 123 (2008), https://doi.org/10.1039/b006677j.Search in Google Scholar PubMed
[260] P. Wasserscheid, T. Welton (Eds.), Ionic Liquids in Synthesis, Wiley-VCH Verlag GmbH & Co, 2nd ed. (2008).10.1002/9783527621194Search in Google Scholar
[261] M. Freemantle. An Introduction to Ionic Liquids, Royal Society of Chemistry, Cambridge (2009).10.1039/9781839168604Search in Google Scholar
[262] C. L. Perrin, G. A. Skinner. J. Am. Chem. Soc.93, 3389 (1971), https://doi.org/10.1021/ja00743a015.Search in Google Scholar
[263] W. J. Hehre, R. Ditchfield, L. Radom, J. A. Pople. J. Am. Chem. Soc.92, 4796 (1970), https://doi.org/10.1021/ja00719a006.Search in Google Scholar
[264] O. Exner. Chemicke Listy67, 135 (1973).10.1016/S0003-2670(01)80241-5Search in Google Scholar
[265] J. E. Leffler. J. Org. Chem.20, 1202 (1955), https://doi.org/10.1021/jo01126a009.Search in Google Scholar
[266] B. Giese. Acc. Chem. Res.17, 438 (1984), https://doi.org/10.1021/ar00108a005.Search in Google Scholar
[267] M. Wolfsberg. Acc. Chem. Res.5, 225 (1972), https://doi.org/10.1021/ar50055a001.Search in Google Scholar
[268] L. Melander, W. H. Saunders. Reaction Rates of Isotopic Molecules, Wiley, New York (1980).Search in Google Scholar
[269] C. L. Perrin, A. Flach. Angew. Chem. Int. Ed.50, 7674 (2011), https://doi.org/10.1002/anie.201102125.Search in Google Scholar PubMed
[270] M. Saunders, L. Telkowski, M. R. Kates. J. Am. Chem. Soc.99, 8070 (1977), https://doi.org/10.1021/ja00466a060.Search in Google Scholar
[271] H. U. Siehl. Adv. Phys. Org. Chem.23, 63 (1987), https://doi.org/10.1016/s0065-3160(08)60203-8.Search in Google Scholar
[272] C. L. Perrin, K. D. Burke. J. Am. Chem. Soc.136, 4355 (2014), https://doi.org/10.1021/ja500174y.Search in Google Scholar PubMed
[273] P. E. Hansen. Molecules20, 2405 (2015), https://doi.org/10.3390/molecules20022405.Search in Google Scholar PubMed PubMed Central
[274] M. J. Kamlet, J. L. M. Abboud, R. W. Taft. Prog. Phys. Org. Chem.13, 485 (1981).10.1002/9780470171929.ch6Search in Google Scholar
[275] M. J. Kamlet, J. L. M. Abboud, R. W. Taft. J. Am. Chem. Soc.99, 6027 (1977), https://doi.org/10.1021/ja00460a031.Search in Google Scholar
[276] M. J. Kamlet, J. L. M. Abboud, R. W. Taft, M. H. Abraham. J. Org. Chem.48, 2877 (1983), https://doi.org/10.1021/jo00165a018.Search in Google Scholar
[277] J. L. M. Abboud, R. Notario. Pure Appl. Chem.71, 645 (1999), https://doi.org/10.1351/pac199971040645.Search in Google Scholar
[278] C. Laurence, P. Nicolet, M. T. Dalati, J. L. M. Abboud, R. Notario. J. Phys. Chem.23, 5807 (1994), https://doi.org/10.1021/j100074a003.Search in Google Scholar
[279] L. Crowhurst, R. Falcone, N. Llewellyn Lancaster, V. Llopis-Mestre, T. Welton. J. Org. Chem.71, 8847 (2006), https://doi.org/10.1021/jo0615302.Search in Google Scholar PubMed
[280] G. L. Closs. J. Am. Chem. Soc.91, 4552 (1969), https://doi.org/10.1021/ja01044a043.Search in Google Scholar
[281] R. Kaptein, L. J. Oosterhoff. Chem. Phys. Lett.4, 214 (1969), https://doi.org/10.1016/0009-2614(69)80105-3.Search in Google Scholar
[282] O. B. Morozova, K. L. Ivanov. ChemPhysChem20, 197 (2019) (Special Issue: BioNMR Spectroscopy), https://doi.org/10.1002/cphc.201800566.Search in Google Scholar PubMed
[283] B. Perlmutter-Hayman. Prog. Reaction Kinetics6, 240 (1971).Search in Google Scholar
[284] I. A. Koppel, V. A. Palm. The influence of the solvent on organic reactivity (Chapter 5). In: Advances in Linear Free-Energy Relationships, N. B. Chapman, J. Shorter (Eds.) (1972).10.1007/978-1-4615-8660-9_5Search in Google Scholar
[285] C. Laurence, J. Legros, A. Chantzis, A. Planchat, D. Jacquemin. J. Phys. Chem. B119, 3174 (2015), https://doi.org/10.1021/jp512372c.Search in Google Scholar PubMed
[286] J. Hine. Adv. Phys. Org. Chem.15, 1 (1977).Search in Google Scholar
[287] A. Pross. Theoretical and Physical Principles of Organic Reactivity, pp. 177–182, Wiley, New York (1995).Search in Google Scholar
[288] R. D. Guthrie. Pure Appl. Chem.61, 23 (1989), https://doi.org/10.1351/pac198961010023.Search in Google Scholar
[289] C. Liébecq (Ed.), Biochemical Nomenclature and Related Documents, Portland Press, London, 2nd ed. (1992).Search in Google Scholar
[290] J. P. Wagner, P. R. Schreiner. Angew. Chem. Int. Ed.54, 12274 (2015), https://doi.org/10.1002/anie.201503476.Search in Google Scholar PubMed
[291] R. A. Marcus. Ann. Rev. Phys. Chem.15, 155 (1964), https://doi.org/10.1146/annurev.pc.15.100164.001103.Search in Google Scholar
[292] A. O. Cohen, R. A. Marcus. J. Phys. Chem.72, 4249 (1968), https://doi.org/10.1021/j100858a052.Search in Google Scholar
[293] J. R. Murdoch. J. Am. Chem. Soc.94, 4410 (1972), https://doi.org/10.1021/ja00768a002.Search in Google Scholar
[294] W. J. Albery. Ann. Rev. Phys. Chem.31, 227 (1980), https://doi.org/10.1146/annurev.pc.31.100180.001303.Search in Google Scholar
[295] R. A. Marcus, N. Sutin. Biochim. Biophys. Acta811, 265 (1985), https://doi.org/10.1016/0304-4173(85)90014-x.Search in Google Scholar
[296] J. M. Mayer. Acc. Chem. Res.44, 36 (2011), https://doi.org/10.1021/ar100093z.Search in Google Scholar PubMed PubMed Central
[297] W. Markownikoff. Liebigs Ann. Chem.153, 228 (1870), https://doi.org/10.1002/jlac.18701530204.Search in Google Scholar
[298] C. M. Guldberg, P. Waage. Christiania Vid. Selks, Forhandl (Oslo)35, 111 (1864), C. M. Guldberg, P. Waage. Etudes sur les affinities chimiques, Brögger and Christie, Christiania, 1867; J. Prakt. Chem. 19, 69 (1879).10.1002/prac.18790190111Search in Google Scholar
[299] A. F. Horstmann. Ann. Chem.170, 192 (1873), https://doi.org/10.1002/jlac.18731700118, Ber. 4, 635 (1871).Search in Google Scholar
[300] J. H. van’t Hoff. Z. Physik Chem.1, 481 (1887), https://doi.org/10.1515/zpch-1887-0151.Search in Google Scholar
[301] A. B. Koudriavtsev, R. F. Jameson, W. Linert. The Law of Mass Action, Springer, Berlin, Heidelberg (2001).10.1007/978-3-642-56770-4Search in Google Scholar
[302] E. A. Guggenheim. J. Chem. Ed.33, 544 (1956), https://doi.org/10.1021/ed033p544.Search in Google Scholar
[303] K. J. Laidler. Arch. History Exact Sci.32, 43 (1985), https://doi.org/10.1007/bf00327865.Search in Google Scholar
[304] J. G. Calvert. Pure Appl. Chem.62, 2167 (1990), https://doi.org/10.1351/pac199062112167.Search in Google Scholar
[305] H. Mayr, M. Patz. Angew. Chem. Int. Ed.33, 938 (1994), https://doi.org/10.1002/anie.199409381.Search in Google Scholar
[306] H. Mayr, A. R. Ofial. Pure Appl. Chem.77, 1807 (2005), https://doi.org/10.1351/pac200577111807.Search in Google Scholar
[307] H. Mayr, A. R. Ofial. J. Phys. Org. Chem.21, 584 (2008), https://doi.org/10.1002/poc.1325.Search in Google Scholar
[308] H. Mayr. Tetrahedron71, 5095 (2015), https://doi.org/10.1016/j.tet.2015.05.055.Search in Google Scholar
[309] J. Meisenheimer. Liebigs Ann. Chem.323, 205 (1902), https://doi.org/10.1002/jlac.19023230205.Search in Google Scholar
[310] I. Fernández, G. Frenking, E. Uggerud. J. Org. Chem.75, 2971 (2010), https://doi.org/10.1021/jo100195w.Search in Google Scholar PubMed
[311] E. E. Kwan, Y. Zeng, H. A. Besser, E. N. Jacobsen. Nat. Chem.10, 917 (2018), https://doi.org/10.1038/s41557-018-0079-7.Search in Google Scholar PubMed PubMed Central
[312] S. Rohrbach, A. J. Smith, J. H. Pang, D. L. Poole, T. Tuttle, S. Chiba, J. A. Murphy. Angew. Chem. Int. Ed.58, 16368 (2019), https://doi.org/10.1002/anie.201902216.Search in Google Scholar PubMed PubMed Central
[313] C. L. Jackson, F. H. Gazzolo. J. Am Chem. Soc.23, 376 (1900).10.1021/ja02037a004Search in Google Scholar
[314] F. Terrier. Chem. Rev.84, 77 (1984).Search in Google Scholar
[315] P. Maslak. Top. Curr. Chem.168, 1 (1993).Search in Google Scholar
[316] E. Baciocchi, M. Bietti, O. Lanzalunga. Acc. Chem. Res.33, 243 (2000), https://doi.org/10.1021/ar980014y.Search in Google Scholar PubMed
[317] M. Baron, R. F. T. Stepto. Pure Appl. Chem.74, 493 (2001).10.1351/pac200274030493Search in Google Scholar
[318] L. S. Romsted, C. A. Bunton, J. Yao. Curr. Opin. Colloid Interface Sci.2, 622 (1997), https://doi.org/10.1016/s1359-0294(97)80055-6.Search in Google Scholar
[319] G. E. Briggs, J. B. S. Haldane. Biochem. J.19, 338 (1925), https://doi.org/10.1042/bj0190338.Search in Google Scholar PubMed PubMed Central
[320] J. Labuda. Pure Appl. Chem.90, 1121 (2018).10.1515/pac-2016-1120Search in Google Scholar
[321] P. Rys. Pure Appl. Chem.53, 209 (1981), https://doi.org/10.1351/pac198153010209.Search in Google Scholar
[322] D. M. Walba, R. M. Richards, R. Curtis Haltiwanger. J. Am. Chem. Soc.104, 3219 (1982), https://doi.org/10.1021/ja00375a051.Search in Google Scholar
[323] D. Ajami, O. Oeckler, A. Simon, R. Herges. Nature426, 819 (2003), https://doi.org/10.1038/nature02224.Search in Google Scholar PubMed
[324] E. Heilbronner. Tetrahedron Lett.1964, 1923 (1964), https://doi.org/10.1016/s0040-4039(01)89474-0.Search in Google Scholar
[325] H. E. Zimmerman. Acc. Chem. Res.4, 272 (1971), https://doi.org/10.1021/ar50044a002.Search in Google Scholar
[326] U. Burkert, N. L. Allinger. J. Comput. Chem.3, 40 (1982), https://doi.org/10.1002/jcc.540030108.Search in Google Scholar
[327] P. de Mayo (Ed.), Rearrangements in Ground and Excited States. Organic Chemistry Series, Vol. 42, Part 1, Academic Press, London (1980), Reedition (2013).Search in Google Scholar
[328] R. A. More O’Ferrall. J. Chem. Soc. B1970, 274 (1970).10.1039/J29700000274Search in Google Scholar
[329] W. P. Jencks. Chem. Rev.72, 705 (1972), https://doi.org/10.1021/cr60280a004.Search in Google Scholar
[330] W. P. Jencks. Acc. Chem. Res.13, 161 (1980), https://doi.org/10.1021/ar50150a001.Search in Google Scholar
[331] A. Winey, E. R. Thornton. J. Am. Chem. Soc.97, 3012 (1975), https://doi.org/10.1021/ja00844a030.Search in Google Scholar
[332] L. Salem. Acc. Chem. Res.4, 322 (1971), https://doi.org/10.1021/ar50045a005.Search in Google Scholar
[333] P. Hanzlik, K. Hogberg, C. M. Judson. Biochemistry23, 3048 (1984), https://doi.org/10.1021/bi00308a031.Search in Google Scholar PubMed
[334] C. Wentrup. Angew. Chem.57, 11508 (2018), https://doi.org/10.1002/anie.201804863.Search in Google Scholar PubMed
[335] P. D. Bartlett (Ed.), Nonclassical Ions, W. A. Benjamin, New York (1965).Search in Google Scholar
[336] P. Dowd. J. Am. Chem. Soc.88, 2587 (1966), https://doi.org/10.1021/ja00963a039.Search in Google Scholar
[337] J. Inczedy, T. Lengyel, A. M. Ure. Compendium of Analytical Nomenclature, Blackwell Science, Oxford, 3rd ed. (1998), Orange Book.Search in Google Scholar
[338] A. M. Wilkinson, A. D. McNaught. Compendium of Chemical Terminology (IUPAC Recommendations), Blackwell Science, Oxford (1997).Search in Google Scholar
[339] A. Berkessel, H. Groeger. Asymmetric Organocatalysis, Wiley-VCH, Weinheim(2005).10.1002/3527604677Search in Google Scholar
[340] B. List (Ed.), Organocatalysis. Special Issue of Chem. Rev. p. 5413, Vol. 107 (2007).10.1021/cr078412eSearch in Google Scholar
[341] P. I. Dalko (Ed.), Enantioselective Organocatalysis, Wiley-VCH, Weinheim (2007).10.1002/9783527610945Search in Google Scholar
[342] E. Vedejs, S. E. Denmark (Eds.), Lewis Base Catalysis in Organic Synthesis, Wiley‐VCH, Weinheim (2016).10.1002/9783527675142Search in Google Scholar
[343] J. S. Littler. Essays on Free-Radical Chemistry. Special Publication No. 24. Chemical Society, London (1970).Search in Google Scholar
[344] J. B. Hendrickson, D. J. Cram, G. S. Hammond. Organic Chemistry, McGraw-Hill, New York (1970).Search in Google Scholar
[345] P. Karen, P. McArdle, J. Takats. Pure Appl. Chem.86 (2014), https://doi.org/10.1515/pac-2013-0505, Pure Appl. Chem. 88, 831 (2016).Search in Google Scholar
[346] C. K. Ingold. Structure and Mechanism in Organic Chemistry, G. Bell and Sons, Ltd., London (1953).Search in Google Scholar
[347] L. M. Stock, H. C. Brown. Adv. Phys. Org. Chem.1, 35 (1963), https://doi.org/10.1016/s0065-3160(08)60277-4.Search in Google Scholar
[348] D. Griller, K. U. Ingold. Acc. Chem. Res.9, 13 (1976), https://doi.org/10.1021/ar50097a003.Search in Google Scholar
[349] I. Tuñón, I. H. Williams. Adv. Phys. Org. Chem.53, 29 (2019).10.1016/bs.apoc.2019.09.001Search in Google Scholar
[350] H. Eyring. J. Am. Chem. Soc.53, 2537 (1931), https://doi.org/10.1021/ja01358a014.Search in Google Scholar
[351] B. K. Carpenter. Potential Energy Surfaces and Reaction Dynamics. In: Reactive Intermediate Chemistry, R. A. Moss, M. S. Platz, M. Jones Jr (Eds.); Wiley, Hoboken, pp. 925–960 (2004).10.1002/0471721492.ch21Search in Google Scholar
[352] M. M. Cox, W. P. Jencks. J. Am. Chem. Soc.103, 572 (1981), https://doi.org/10.1021/ja00393a013.Search in Google Scholar
[353] S. G. Lias, J. F. Liebman, R. D. Levin. J. Phys. Chem. Ref. Data13, 695 (1984), https://doi.org/10.1063/1.555719.Search in Google Scholar
[354] J. A. Ross, R. P. Seiders, D. M. Lemal. J. Am. Chem. Soc.98, 4325 (1976), https://doi.org/10.1021/ja00430a060.Search in Google Scholar
[355] C. Zhou, D. M. Birney. J. Am. Chem. Soc.124, 5231 (2002), https://doi.org/10.1021/ja017559z.Search in Google Scholar PubMed
[356] H. Meier. Angew. Chem. Int. Ed.44, 2482 (2005), https://doi.org/10.1002/anie.200461146.Search in Google Scholar PubMed
[357] C. Hansch, A. Leo. In Exploring QSAR: Fundamentals and Applications in Chemistry and Biology, American Chemical Society, Washington, DC (1995).Search in Google Scholar
[358] C. Hansch, D. Hoekman, A. Leo, D. Weininger, C. D. Selassie. Chem. Rev.102, 783 (2002), https://doi.org/10.1021/cr0102009.Search in Google Scholar PubMed
[359] A. R. Katritzky, V. S. Lobanov, M. Karelson. Chem. Soc. Rev.279 (1995).10.1039/cs9952400279Search in Google Scholar
[360] B. Bosnich. Acc. Chem. Res.31, 667 (1998), https://doi.org/10.1021/ar970095i.Search in Google Scholar
[361] R. A. Marcus. J. Chem. Phys.45, 4493 (1966), https://doi.org/10.1063/1.1727528.Search in Google Scholar
[362] O. Exner. J. Chem. Soc. Perkin Trans.2, 973 (1993), https://doi.org/10.1039/p29930000973.Search in Google Scholar
[363] A. Argile, A. R. E. Carey, G. Fukata, M. Harcourt, R. A. More O’Ferrall, M. G. Murphy. Isr. J. Chem.26, 303 (1985), https://doi.org/10.1002/ijch.198500112.Search in Google Scholar
[364] E. Buncel, H. Wilson. J. Chem. Educ.64 (1987), https://doi.org/10.1021/ed064p475.Search in Google Scholar
[365] H. Mayr, A. R. Ofial. Angew. Chem. Int. Ed.45, 1844 (2006), https://doi.org/10.1002/anie.200503273.Search in Google Scholar PubMed
[366] D. L. Adams. J. Chem. Educ.69, 451 (1992), https://doi.org/10.1021/ed069p451.Search in Google Scholar
[367] A. Hassner. J. Org. Chem.33, 2684 (1968), https://doi.org/10.1021/jo01271a015.Search in Google Scholar
[368] M. N. Paddon-Row. Adv. Phys. Org. Chem.38, 1 (2003).10.1016/S0065-3160(03)38001-3Search in Google Scholar
[369] W. R. Cannon, S. J. Benkovic. J. Biol. Chem.273, 26257 (1988).10.1074/jbc.273.41.26257Search in Google Scholar PubMed
[370] P. W. Atkins. Quanta: A Handbook of Concepts, Clarendon Press, Oxford (1974).Search in Google Scholar
[371] S. Shaik, A. Shurki, D. Danovich, P. C. Hiberty. Chem. Rev.101, 1501 (2001), https://doi.org/10.1021/cr990363l.Search in Google Scholar PubMed
[372] C. D. Ritchie. Acc. Chem. Res.5, 348 (1972), https://doi.org/10.1021/ar50058a005.Search in Google Scholar
[373] C. D. Ritchie. Pure Appl. Chem.50, 1281 (1978), https://doi.org/10.1351/pac197850111281.Search in Google Scholar
[374] S. Minegishi, H. Mayr. J. Am. Chem. Soc.125, 286 (2003), https://doi.org/10.1021/ja021010y.Search in Google Scholar PubMed
[375] A. Saytzeff. Justus Liebigs Ann. Chem.179, 296 (1875), https://doi.org/10.1002/jlac.18751790304.Search in Google Scholar
[376] E. Buncel, S. Rajagopal. Acc. Chem. Res.23, 226 (1990), https://doi.org/10.1021/ar00175a004.Search in Google Scholar
[377] E. Buncel, R. A. Stairs. Solvent Effects in Chemistry, Wiley, Hoboken/NJ, USA, 2nd ed. (2016).10.1002/9781119044307Search in Google Scholar
[378] K. Mislow, J. S. Siegel. J. Am. Chem. Soc.106, 3319 (1984), https://doi.org/10.1021/ja00323a043.Search in Google Scholar
[379] R. C. Jones, J. Kahoves, R Stepto, E. S. Wilks, M. Hess, T. Kitayama, W. V. Metanowski. Pure Appl. Chem.84, 2167 (2012), https://doi.org/10.1351/PAC-REP-12-03-05.Search in Google Scholar
[380] M. Charton. Prog. Phys. Org. Chem.16, 287 (1987).10.1002/9780470171950.ch6Search in Google Scholar
[381] J. A. Berson. Acc. Chem. Res.5, 406 (1972), https://doi.org/10.1021/ar50060a003.Search in Google Scholar
[382] R. J. Gillespie. Acc. Chem. Res.1, 202 (1968), https://doi.org/10.1021/ar50007a002.Search in Google Scholar
[383] G. A. Olah, A. T. Ku, J. A. Olah. J. Org. Chem.35, 3904 (1970), https://doi.org/10.1021/jo00836a070.Search in Google Scholar
[384] I. A. Koppel, P. Burk, I. Koppel, I. Leito, T. Sonoda, M. Mishima. J. Am. Chem. Soc.122, 5114 (2000), https://doi.org/10.1021/ja0000753.Search in Google Scholar
[385] T. Ishikawa. Superbases for Organic Synthesis: Guanidines, Amidines, Phosphazenes and Related Organocatalysts, Wiley, Chichester (2009).10.1002/9780470740859Search in Google Scholar
[386] J.-M. Lehn. Science260, 1762 (1993), https://doi.org/10.1126/science.8511582.Search in Google Scholar PubMed
[387] C. G. Swain, E. C. Lupton. J. Am. Chem. Soc.90, 4328 (1968), https://doi.org/10.1021/ja01018a024.Search in Google Scholar
[388] W. F. Reynolds, R. D. Topsom. J. Org. Chem.49, 1989 (1984), https://doi.org/10.1021/jo00185a031.Search in Google Scholar
[389] A. J. Hoefnagel, W. Osterbeek, B. M. Wepster. J. Org. Chem.49, 1993 (1984), https://doi.org/10.1021/jo00185a032.Search in Google Scholar
[390] M. Charton. J. Org. Chem.49, 1997 (1984), https://doi.org/10.1021/jo00185a033.Search in Google Scholar
[391] C. G. Swain. J. Org. Chem.49, 2005 (1984), https://doi.org/10.1021/jo00185a035.Search in Google Scholar
[392] C. G. Swain, S. H. Unger, N. R. Rosenquist, M. S. Swain. J. Am. Chem. Soc.105, 492 (1983), https://doi.org/10.1021/ja00341a032.Search in Google Scholar
[393] M. J. S. Dewar. J. Am. Chem. Soc.106, 209 (1984), https://doi.org/10.1021/ja00313a042.Search in Google Scholar
[394] W. T. Borden, R. J. Loncharich, K. N. Houk. Ann. Rev. Phys. Chem.39, 213 (1988), https://doi.org/10.1146/annurev.pc.39.100188.001241.Search in Google Scholar
[395] P. Merino, M. A. Chiacchio, L. Legnani, I. Delso, T. Tejero. Org. Chem. Front.4, 1541 (2017), https://doi.org/10.1039/c7qo00233e.Search in Google Scholar
[396] A. Moyano. J. Org. Chem.54, 573 (1989), https://doi.org/10.1021/jo00264a014.Search in Google Scholar
[397] W. L. Jorgensen, L Salem. The Organic Chemist’s Book of Orbitals, Academic Press, New York (1973).Search in Google Scholar
[398] J. Shorter. Correlation Analysis in Organic Chemistry: An Introduction to Linear Free Energy Relationships, Oxford University Press, Oxford (1973).Search in Google Scholar
[399] R. W. Taft. J. Am. Chem. Soc.74, 3120 (1952), https://doi.org/10.1021/ja01132a049.Search in Google Scholar
[400] R. W. Taft. J. Am. Chem. Soc.75, 4231 (1953), https://doi.org/10.1021/ja01113a027.Search in Google Scholar
[401] M. Tisler. Synthesis-Stuttgart3, 123 (1973).10.1055/s-1973-22145Search in Google Scholar
[402] J. E. Torr, J. M. Large, P. N. Horton, M. B. Hursthouse, E. McDonald. Tetrahedron Lett.47, 31 (2006), https://doi.org/10.1016/j.tetlet.2005.10.146.Search in Google Scholar
[403] J. Suwiński, K. Świerczek. Tetrahedron57, 1639 (2001).10.1016/S0040-4020(00)01067-XSearch in Google Scholar
[404] C. W. Jefford, G. Bernardinelli, Y. Wang, D. C. Spellmeyer, A. Buda, K. N. Houk. J. Am. Chem. Soc.114, 1157 (1992), https://doi.org/10.1021/ja00030a005.Search in Google Scholar
[405] W. R. Dolbier, H. Koroniak, K. N. Houk, C. Sheu. Acc. Chem. Res.29, 471 (1996), https://doi.org/10.1021/ar9501986.Search in Google Scholar
[406] R. A. Y. Jones, J. F. Bunnett. Pure Appl. Chem.61, 725 (1989), https://doi.org/10.1351/pac198961040725.Search in Google Scholar
[407] J. C. Polanyi, A. H. Zewail. Acc. Chem. Res.28, 119 (1995), https://doi.org/10.1021/ar00051a005.Search in Google Scholar
[408] H. B. Schlegel. J. Comput. Chem.3, 214 (1982), https://doi.org/10.1002/jcc.540030212.Search in Google Scholar
[409] R. E. Stanton, J. W. McIver. J. Am. Chem. Soc.97, 3632 (1975), https://doi.org/10.1021/ja00846a012.Search in Google Scholar
[410] R. P. Bell. The Tunnel Effect in Chemistry, Chapman and Hall, London (1980).10.1007/978-1-4899-2891-7Search in Google Scholar
[411] D. Ley, D. Gerbig, P. R. Schreiner. Org. Biomol. Chem.10, 3781 (2012), https://doi.org/10.1039/c2ob07170c.Search in Google Scholar PubMed
[412] Z. R. Grabowski, K. Rotkiewicz, W. Rettig. Chem. Rev.103, 3899 (2003), https://doi.org/10.1021/cr940745l.Search in Google Scholar PubMed
[413] D. Seebach. Angew. Chem. Int. Ed.18, 239 (1979), https://doi.org/10.1002/anie.197902393.Search in Google Scholar
[414] J. N. Israelachvili. Intermolecular and Surface Forces, Academic Press, London, 3rd ed. (2011).10.1016/B978-0-12-391927-4.10001-5Search in Google Scholar
[415] Y. Yukawa, Y. Tsuno. Bull. Chem. Soc. Jpn.32, 971 (1959), https://doi.org/10.1246/bcsj.32.971.Search in Google Scholar
[416] Y. Tsuno, M. Fujio. Adv. Phys. Org. Chem.32, 267 (1999), https://doi.org/10.1016/s0065-3160(08)60009-x.Search in Google Scholar
[417] M. A. Paul, F. A. Long. Chem. Rev.57, 1 (1957), https://doi.org/10.1021/cr50013a001.Search in Google Scholar
[418] A. Schmidt. Adv. Heterocycl. Chem.85, 67 (2003), https://doi.org/10.1016/s0065-2725(03)85002-x.Search in Google Scholar
© 2022 IUPAC & De Gruyter. This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
This work is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
Articles in the same Issue
- Frontmatter
- IUPAC Recommendations
- Glossary of terms used in physical organic chemistry (IUPAC Recommendations 2021)
Articles in the same Issue
- Frontmatter
- IUPAC Recommendations
- Glossary of terms used in physical organic chemistry (IUPAC Recommendations 2021)