DFG-Research Unit 2795: “Synapses under Stress: Early events induced by metabolic failure at glutamatergic synapses”
-
Christine R. Rose
1990: Diploma in Biology, University of Konstanz. 1993: Dissertation (Dr. rer. nat.), University of Kaiserslautern. 1994–1997: Postdoc at the Dept Neurology, Yale School of Medicine, New Haven, CT, USA. 1998-1999: Postdoc at the Dept of Physiology, Saarland University, Homburg. 2000–2003: Postdoc at the Dept of Physiology, TU and LMU Munich. 2002: Professional dissertation (Habilitation) in Physiology, LMU Munich. 2003–2005: Heisenberg-Stipend of the DFG, Dept of Physiology LMU Munich. Since 2005: Full Professor and Head of the Institute of Neurobiology at the Heinrich Heine University Duesseldorf.

The brain is one of the major energy consumers of the human body. Most of the ATP is used by the Na+/K+-ATPase (NKA; Fig. 1) which sets the Na+ and K+ gradients across the plasma membrane and compensates for changes in intra- and extracellular Na+ and K+ concentrations occurring upon activation of voltage- and ligand-gated channels (Sweadner 1989). The inwardly-directed Na+ gradient generated by the NKA also provides the energy for a plethora of secondary active transporters, e. g. the sodium/calcium exchanger (NCX) and various transporters that terminate synaptic transmission via efficient re-uptake of neurotransmitters (Rose and Chatton 2016; Somjen 2002) (Fig. 1). Other ATP-consuming ion transporters, such as the plasma membrane Ca2+-ATPase, are vital for intracellular Ca2+ homeostasis (Harris et al. 2012). The vacuolar-type H+-ATPase (v-ATPase) creates the proton gradient necessary for neurotransmitter accumulation in presynaptic vesicles (Cotter et al. 2015) (Fig. 1).
Despite their considerable energy consumption, neurons themselves do not contain significant energy stores, but depend on close metabolic interactions with astrocytes (Allaman et al. 2011; Rose and Chatton 2016). Astrocytes furthermore shape excitatory synaptic transmission by controlling extracellular ion concentrations and by taking up glutamate (Rose et al. 2018; Verkhratsky and Nedergaard 2018) (Fig. 1); again processes directly or indirectly linked to activation of NKA and ATP consumption. Finally, astrocytes can release neuroactive substances, among them D-serine, feeding back onto surrounding neurons (Henneberger et al. 2010) and are involved in neurovascular coupling (Petzold and Murthy 2011). The term “tripartite” synapse highlights the strong functional interplay between neurons and astrocytes and emphasizes the fact that understanding synaptic function and dysfunction requires knowledge about pre- and postsynaptic neurons, as well as the surrounding glia (Allen and Eroglu 2017).
Conditions under which the brain’s energy demands exceed its availability, referred to as metabolic stress, give rise to rapid functional changes. An extreme form of metabolic stress is caused by brain ischemia, which can result in tissue damage and severe neurological deficits and which represents one of the leading causes of disability and death in our ageing population. The main mechanisms of delayed cell death, including the so-called “excitotoxic” action of the transmitter glutamate, are well described. In contrast, there are significant gaps in our understanding of the early changes in neuronal and glial function during reduced energy availability. As these synaptic changes are among the earliest and most ”upstream” events in the ischemic cascade, a better understanding of what causes metabolic stress in synapses during ischemia is translationally relevant.
The Research Unit (RU) “Synapses under stress: Early events induced by metabolic failure at glutamatergic synapses” will close this gap and will combine molecular biology, biochemistry, imaging, electrophysiology and optogenetic approaches together with mathematical simulations to unravel the dependence of synaptic function on cellular metabolism. The main scientific goals of the RU are to gain inclusive knowledge about the energy dependence of glutamatergic synapses of the mouse forebrain and to reveal the exact sequence of early events induced by transient moderate energy depletion. Analysing the main cellular compartments (pre- and postsynaptic elements, perisynaptic astrocyte processes) and the extracellular space will provide a novel integrated view of synaptic function and involved neuron-glia interactions, and enable the deciphering of existing feed-back and feed-forward loops between the different compartments. Investigating the reversibility of the induced effects will enable to gain better insight into the processes within the ischemic penumbra and to understand the factors driving cells towards irreversible demise after an ischemic stroke.
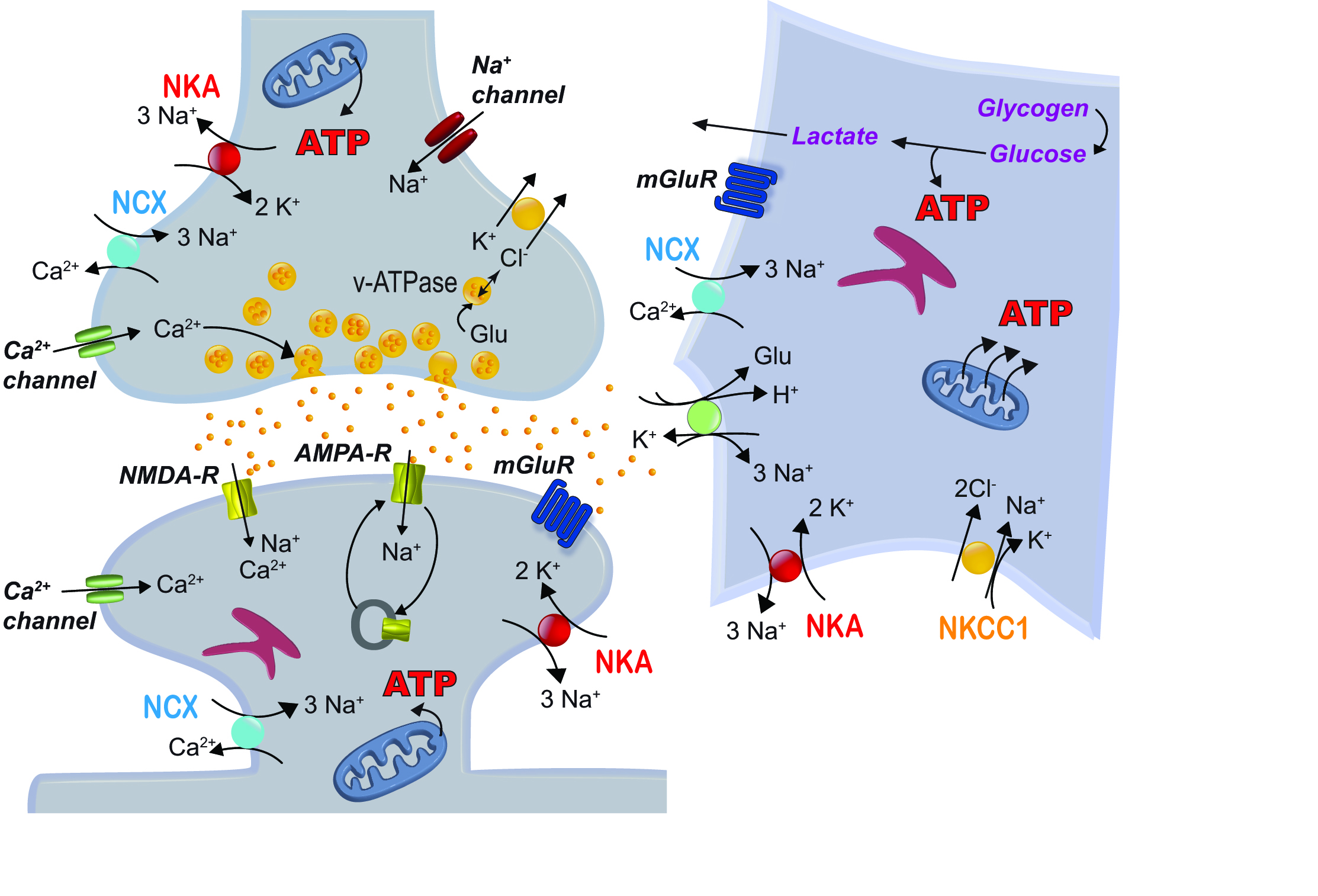
Ion fluxes and ATP-consuming processes at tripartite glutamatergic synapses (summarized from Harris et al., 2012 and Rose & Chatton, 2016).
The RU will start its work at the beginning of 2019. In its first funding period, it will receive about 2 million € over three years. The RU is led by Christine R. Rose at the Heinrich Heine University Düsseldorf together with her deputy Christoph Fahlke at the Forschungszentrum Jülich. Further members are Christian Henneberger (Rheinische Friedrich-Wilhelms University Bonn), Gabor Petzold (German Center for Neurodegenerative Diseases and University Bonn), Jürgen Klingauf (Westfälische-Wilhelms University Münster), Andreas Reiner (Ruhr University Bochum) and Nadine Erlenhardt (Heinrich Heine University Düsseldorf). Moreover, Michel J.A.M. van Putten and Stephan A. van Gils, two Mercator Fellows from the University of Twente (The Netherlands) are members of the consortium.
The following main questions will be addressed:
What are the immediate effects of acute metabolic stress on ion homeostasis?
What are the consequences of metabolic stress for astroglial function and neuron-glia interaction?
What are the consequences of metabolic stress on presynaptic function and glutamate release?
How do postsynaptic properties and ion signaling change in response to metabolic stress?
More specifically, the RU will analyse acute changes in ion concentrations, transmitter homeostasis, as well as the function and the subcellular distribution of ligand- and voltage-gated ion channels which control electrical and chemical signaling. The consortium will study the major cellular components of glutamatergic synapses, i. e. pre- and postsynaptic neuronal compartments as well as perisynaptic astrocytes. All groups will focus on glutamatergic synapses of the mouse cortex as a joint model system and use a common protocol for induction of transient chemical ischemia. Adaptive and pathological processes will be addressed at the molecular, cellular and systems’ level, employing experimental systems of increasing complexity that range from primary cell culture to acute and organotypic tissue slices to in vivo models of ischemic stroke. Experimental data generated will be used to develop a mathematical model. This should make it possible to simulate the processes taking place at the synapses “in silico”, i. e. on the computer, providing a novel, concerted view on early functional changes of synapses under stress.
Altogether, the research programme will lead to a thorough understanding of immediate responses of the tripartite synapse to transient energy shortage, of their functional consequences, as well as of the potential reversibility of the induced effects. This will generate a new, integrative understanding of basic pathomechanisms of metabolic failure, which is urgently needed to develop better therapeutic strategies to combat stroke-induced brain damage.
About the author

1990: Diploma in Biology, University of Konstanz. 1993: Dissertation (Dr. rer. nat.), University of Kaiserslautern. 1994–1997: Postdoc at the Dept Neurology, Yale School of Medicine, New Haven, CT, USA. 1998-1999: Postdoc at the Dept of Physiology, Saarland University, Homburg. 2000–2003: Postdoc at the Dept of Physiology, TU and LMU Munich. 2002: Professional dissertation (Habilitation) in Physiology, LMU Munich. 2003–2005: Heisenberg-Stipend of the DFG, Dept of Physiology LMU Munich. Since 2005: Full Professor and Head of the Institute of Neurobiology at the Heinrich Heine University Duesseldorf.
Literature
Allaman I, Belanger M, Magistretti PJ. 2011. Astrocyte-neuron metabolic relationships: for better and for worse. Trends Neurosci 34:76–87.10.1016/j.tins.2010.12.001Search in Google Scholar PubMed
Allen NJ, Eroglu C. 2017. Cell Biology of Astrocyte-Synapse Interactions. Neuron 96:697–708.10.1016/j.neuron.2017.09.056Search in Google Scholar PubMed PubMed Central
Cotter K, Stransky L, McGuire C, Forgac M. 2015. Recent Insights into the Structure, Regulation, and Function of the V-ATPases. Trends Biochem Sci 40:611–22.10.1016/j.tibs.2015.08.005Search in Google Scholar PubMed PubMed Central
Harris JJ, Jolivet R, Attwell D. 2012. Synaptic energy use and supply. Neuron 75:762–77.10.1016/j.neuron.2012.08.019Search in Google Scholar PubMed
Henneberger C, Papouin T, Oliet SH, Rusakov DA. 2010. Long-term potentiation depends on release of D-serine from astrocytes. Nature 463:232–6.10.1038/nature08673Search in Google Scholar PubMed PubMed Central
Petzold GC, Murthy VN. 2011. Role of astrocytes in neurovascular coupling. Neuron 71:782–97.10.1016/j.neuron.2011.08.009Search in Google Scholar PubMed
Rose CR, Chatton JY. 2016. Astrocyte sodium signaling and neuro-metabolic coupling in the brain. Neuroscience 323:121–34.10.1016/j.neuroscience.2015.03.002Search in Google Scholar PubMed
Rose CR, Ziemens D, Untiet V, Fahlke C. 2018. Molecular and cellular physiology of sodium-dependent glutamate transporters. Brain Res Bull 136:13–16.10.1016/j.brainresbull.2016.12.013Search in Google Scholar PubMed
Somjen GG. 2002. Ion regulation in the brain: implications for pathophysiology. Neuroscientist 8:254–67.10.1177/1073858402008003011Search in Google Scholar PubMed
Sweadner KJ. 1989. Isozymes of the Na+/K+-ATPase. Biochim Biophys Acta 988:185–220.10.1016/0304-4157(89)90019-1Search in Google Scholar PubMed
Verkhratsky A, Nedergaard M. 2018. Physiology of Astroglia. Physiol Rev 98:239–389.10.1007/978-981-13-9913-8_3Search in Google Scholar PubMed PubMed Central
Appendix
German Summary: DFG-Forschungsgruppe (FOR) 2795: Synapsen unter Stress: akute Veränderungen durch mangelnde Energiezufuhr an glutamatergen Synapsen
Das Gehirn des menschlichen Körpers hat einen im Vergleich zu anderen Organen sehr hohen Energieverbrauch. Wenn sein Bedarf die Menge an bereitstehender Energie überschreitet, führt dies zu raschen Veränderungen in seiner Funktion. Eine besonders dramatische Form des Energiemangels tritt bei einem ischämischen Insult auf. Dieser kann zu Gewebsuntergang und schwerwiegenden neurologischen Ausfällen führen und stellt eine der häufigsten Ursachen für Behinderungen und Tod in unserer alternden Gesellschaft dar. Die Gründe für den in Folge eines Schlaganfalls beobachteten verzögerten Zelltod sind recht gut verstanden, darunter die sog. exzitotoxische Wirkung des synaptischen Botenstoffs Glutamat. Die frühen Prozesse, die durch eine mangelnde Energieversorgung an Synapsen hervorgerufen werden, sind hingegen noch weitgehend unbekannt. Ein besseres Verständnis ihrer Ursachen und Auswirkungen ist jedoch geboten, da sie die ersten Ereignisse der ischämischen Kaskade darstellen.
Die Forschungsgruppe (FOR) 2795 „Synapsen unter Stress: akute Veränderungen durch mangelnde Energiezufuhr an glutamatergen Synapsen“ wird diese Fragen adressieren und die frühen zellulären Antworten nach Unterbrechung der Energieversorgung an Synapsen des Großhirns der Maus in den Blick nehmen. Dabei wird eine Kombination von Molekularbiologie, Biochemie, Elektrophysiologie, Imaging und Optogenetik zur Anwendung kommen, die durch mathematische Simulationen ergänzt werden. Die FOR 2795 wird sich insbesondere auf Änderungen in Ionenkonzentrationen, in der Transmitterhomöostase, sowie in der Funktion und subzellulären Verteilung von Ionenkanälen fokussieren. Wesentlich hierbei ist die Einbeziehung sowohl von neuronalen Kompartimenten (Prä- und Postsynapse) als auch von Gliazellen (Astrozyten) in die funktionelle Betrachtung, welche eine neue Sichtweise auf Schädigungsmechanismen eröffnen wird. Die einzelnen Teilprojekte der FOR werden eine gemeinsame Strategie für die Induktion von metabolischem Stress anwenden, jedoch unterschiedlich komplexe Systeme bearbeiten. Letztere reichen von Zellkulturen über Gewebeschnitte bis hin zu in vivo-Systemen und einer computergestützten Modellierung. Die FOR 2795 wird somit die molekularen und zellulären Prozesse an Synapsen, die in direkter Abhängigkeit vom zellulären Energiestatus stehen, entschlüsseln. Des Weiteren wird sie Mechanismen identifizieren, die für akute Störungen der synaptischen Funktion sowie für Zellschädigungen nach Zusammenbruch der Energieversorgung verantwortlich sind. Sie wird damit ein neues, ganzheitliches Verständnis grundlegender Pathomechanismen generieren, welches dringend für die Entwicklung besserer therapeutischer Strategien zur Behandlung von Schlaganfall-induzierten Hirnschädigungen benötigt wird.
© 2018 Walter de Gruyter GmbH, Berlin/Boston
Articles in the same Issue
- Titelseiten
- Editorial
- Wechsel des Chefredakteurs und Umstellung auf Englisch
- Review Article
- Fight or flee? Lessons from insects on aggression
- The first cortical circuits: Subplate neurons lead the way and shape cortical organization
- Neuromodulation of early sensory processing in the olfactory system
- Impact of Diet and the Gut Microbiome on Neurodegeneration and Regeneration in Neurological Disorders
- Small domain, large consequences: the axon initial segment as a key player in neuronal excitability
- Forschungsförderung
- DFG-Research Unit 2795: “Synapses under Stress: Early events induced by metabolic failure at glutamatergic synapses”
- DFG-Graduiertenkolleg 2185 “Situierte Kognition”
- DFG-Graduiertenkolleg 2154 „Materials for Brain: Dünnschichtbasierte Funktionsmaterialien für die minimal-invasive Therapie von Erkrankungen des Gehirns“
- Rezension
- Hana Roš, Matteo Farinella: Das Gehirn
- Nachrichten
- Die Otto-Loewi-Medaille der NWG
- Forschungspreise der Neurowissenschaftlichen Gesellschaft 2019
Articles in the same Issue
- Titelseiten
- Editorial
- Wechsel des Chefredakteurs und Umstellung auf Englisch
- Review Article
- Fight or flee? Lessons from insects on aggression
- The first cortical circuits: Subplate neurons lead the way and shape cortical organization
- Neuromodulation of early sensory processing in the olfactory system
- Impact of Diet and the Gut Microbiome on Neurodegeneration and Regeneration in Neurological Disorders
- Small domain, large consequences: the axon initial segment as a key player in neuronal excitability
- Forschungsförderung
- DFG-Research Unit 2795: “Synapses under Stress: Early events induced by metabolic failure at glutamatergic synapses”
- DFG-Graduiertenkolleg 2185 “Situierte Kognition”
- DFG-Graduiertenkolleg 2154 „Materials for Brain: Dünnschichtbasierte Funktionsmaterialien für die minimal-invasive Therapie von Erkrankungen des Gehirns“
- Rezension
- Hana Roš, Matteo Farinella: Das Gehirn
- Nachrichten
- Die Otto-Loewi-Medaille der NWG
- Forschungspreise der Neurowissenschaftlichen Gesellschaft 2019