The proteostasis burden of aneuploidy
-
Prince Saforo Amponsah

 and
Zuzana Storchová
and
Zuzana Storchová

Abstract
Aneuploidy refers to chromosome number abnormality that is not an exact multiple of the haploid chromosome set. Aneuploidy has largely negative consequences in cells and organisms, manifested as so-called aneuploidy-associated stresses. A major consequence of aneuploidy is proteotoxic stress due to abnormal protein expression from imbalanced chromosome numbers. Recent advances have improved our understanding of the nature of the proteostasis imbalance caused by aneuploidy and highlighted their relevance with respect to organellar homeostasis, dosage compensation, or mechanisms employed by cells to mitigate the detrimental stress. In this review, we highlight the recent findings and outline questions to be addressed in future research.
1 Introduction
Aneuploidy, defined as chromosome copy number aberrations that deviate from multiples of the haploid chromosome set, affects whole chromosomes or chromosome parts. Aneuploidy impacts the number and expression pattern of genes encoded on the affected chromosomes and is often associated with additional structural chromosome rearrangements and DNA damage, thereby contributing to genome instability. Aneuploidy is widespread in cancer and aging-related diseases (Naylor and van Deursen 2016). During cell division, nuclear chromosomes are segregated and evenly distributed into two daughter cells. However, errors in chromosome segregation during meiosis or mitosis can result in the loss (monosomy) or gain (polysomy) of one or more chromosome(s) in one daughter cell (Santaguida and Amon 2015b). In some instances, complete failure of cell division leads to genome doubling, which induces aneuploidy, as polyploid cells are inherently prone to chromosomal instability (Vittoria et al. 2023). Meiotic errors lead to aneuploid or polyploid embryos, mostly resulting in embryonal death (Nagaoka et al. 2012). In humans, the only chromosome gains compatible with survival are trisomy of chromosome 21 (Down’s syndrome), 13 (Patau’s syndrome), 18 (Edward’s syndrome), and gains of the sex chromosomes; only a loss of one sex chromosome can be tolerated. These syndromes are associated with a plethora of developmental defects. Aneuploidy arising from mitotic errors is rare in healthy somatic cells but widespread in cancer. Indeed, nearly 90 % of solid tumors are composed of cells with complex karyotypes, characterized by multiple numerical and structural copy number changes (Sdeor et al. 2024). Aneuploidy and chromosomal instability are the driving force of tumorigenesis and tightly linked to increased accumulation of genomic changes, elevated drug resistance and poor patient prognosis.
Despite its long recognition, significant advances in understanding the consequences of aneuploidy have been achieved only recently through the development of chromosomal engineering techniques, enabling comparisons between isogenic cells with euploid and defined aneuploid karyotypes (Chunduri and Storchová 2019; Lakhani et al. 2023). Studies of diverse laboratory-engineered models with one or more additional chromosomes, from yeast to human cells, revealed that genes on the extra chromosomes are transcribed and translated, leading to altered protein composition (Dephoure et al. 2014; Stingele et al. 2012). Similarly, monosomic cells, lacking one homologous chromosome, exhibited reduced expression of genes located on the missing chromosome (Beach et al. 2017; Chunduri et al. 2021). These gene expression changes, driven by altered gene dosage, impose significant stress on cellular homeostasis and induce secondary gene expression alterations. As a result, lab engineered aneuploid cells experience profound cellular stresses that adversely affect their proliferation (Chunduri and Storchová 2019; Zhu et al. 2018).
Proteotoxic stress, manifesting as protein misfolding and aggregation, is frequently observed in aneuploid cells with extra chromosomes, likely due to the presence of extra genes that overwhelm the protein homeostasis (proteostasis) network (Donnelly et al. 2014; Ohashi et al. 2015; Oromendia et al. 2012; Santaguida et al. 2015). Proteotoxic stress is also frequently observed in cancer cells (Deshaies 2014), yet little is known about its causes, possible links to aneuploidy, and its consequences for the cells. In the past few years, significant effort has been dedicated to understanding the burden that aneuploidy places on cellular proteostasis in a variety of model systems. In this review, we summarize the recent progress made in this field, with a focus on chromosome gain, which has been extensively studied. We also present findings on the diverse ways in which aneuploidy modulates cellular proteostasis, highlighting its impact on cellular organelles such as mitochondria and its implication for disease.
2 Gene expression changes related to the altered chromosomes
The development of several model systems in budding yeast, plants, murine and human cells, C. elegans and in Drosophila has accelerated the analysis of the consequences of aneuploidy (e.g., Huettel et al. 2008; Joy et al. 2021; Pavelka et al. 2010; Singla et al. 2020a; Stingele et al. 2012; Torres et al. 2007; Weaver et al. 2007). These model systems utilize genome engineering techniques including targeted addition or removal of individual chromosomes, introduction of structural chromosomal alterations, induced chromosome missegregation, or whole genome doubling (Lakhani et al. 2023). An advantage of these model systems is a defined chromosomal change in cells derived from parental near diploid cells lines, which serve as isogenic controls. Analysis of protein homeostasis in aneuploid cells arising from erroneous mitosis – e.g., by inducing missegregation using microtubule poisons or spindle assembly checkpoint inhibitors – revealed acute response to aneuploidy (Ohashi et al. 2015; Santaguida et al. 2015). These findings have been complemented by analysis of cells from patients with trisomy syndromes, and by computational analysis of the large databases of cancer cell lines and tumors (Bökenkamp et al. 2025; Hwang et al. 2021; Krivega et al. 2021; Schukken and Sheltzer 2022).
First analysis of transcriptome and proteome changes in aneuploid budding yeasts as well as in murine and human cells, demonstrated that the genes on the gained chromosome are fully transcribed and translated (Pavelka et al. 2010; Stingele et al. 2012; Torres et al. 2007). Interestingly, the abundance of the mRNA usually scales with the chromosome copy number changes, while the protein levels of up to 25 % of proteins encoded on the extra chromosome often resemble the abundance observed in euploids (Dephoure et al. 2014; Stingele et al. 2012). This dosage compensation affects mainly subunits of macromolecular complexes and suggests that mechanisms exist to, at least partially, compensate for chromosome copy number changes. Dosage compensation has also been observed in samples from patients with trisomy syndromes (Hwang et al. 2021; Liu et al. 2017), as well as in naturally occurring aneuploid budding yeasts (Muenzner et al. 2024). In monosomic cells, the mRNA and protein expression from the affected chromosome decrease, but here too buffering of the gene dosage was observed (Chunduri et al. 2021). Cancer cell lines also show significant gene dosage compensation (Schukken and Sheltzer 2022). This demonstrates that imbalanced protein abundance imposes a burden on aneuploid cells, but there are mechanisms that counteract altered protein expression to mitigate the effects (Figure 1). Buffering of the protein abundance may be an adaptive process, at least in naturally occurring yeast, however, further evidence is required to evaluate whether the buffering helps to improve proliferation of aneuploid cells.
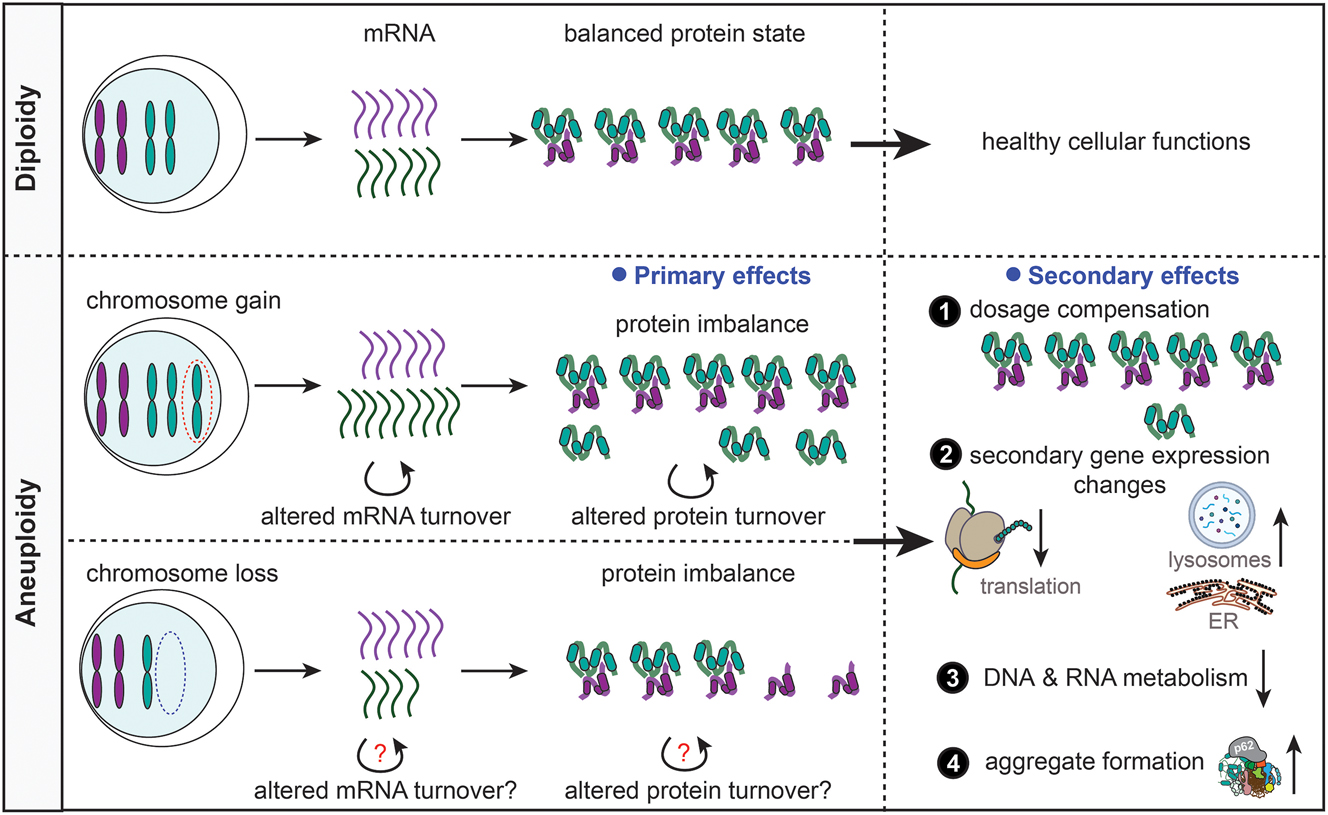
The effects of aneuploidy on protein homeostasis and gene expression. Aneuploidy (bottom), in contrast to diploidy (top), alters the mRNA and protein abundance in cells. The changes in molecular content place an enormous burden on the cells leading to dysregulation of various cellular pathways including decreased translation and DNA/RNA metabolism, increased lysosomal activity and ER stress, and elevated protein aggregation. Aneuploid cells suffer from proteotoxic stress because of the proteome imbalance. However, dosage compensation on both mRNA and protein levels partially alleviates the effects of the imbalance.
Apart from the primary gene expression changes that directly reflect the chromosome copy number alterations, there are also striking, conserved secondary gene expression changes in aneuploid cells (Durrbaum et al. 2014; Sheltzer et al. 2012) (Figure 1). Immediately after chromosome missegregation, cells show upregulated integrated stress response (ISR), altered autophagy, and downregulation of DNA replication factors (Ohashi et al. 2015; Santaguida et al. 2015). Additionally, acute aneuploidy induction causes altered ribosome and RNA metabolism, elevated oxidative stress, and activated metabolic responses (Durrbaum et al. 2014; Ippolito et al. 2024; Zhu et al. 2018). A chronic response to constitutive aneuploidy includes reduced expression of factors required for DNA and RNA metabolism, ribosomes, and G2 and M phases of the cell cycle. In contrast, pathways related to vesicle trafficking, endoplasmic reticulum (ER), endosomes, lysosomes, autophagy, and ISR are frequently upregulated (Durrbaum et al. 2014; Sheltzer 2013; Stingele et al. 2012). These findings suggest that the proteostasis network becomes disrupted by chromosome copy number changes, and this disruption affects multiple aspects of the network.
3 The proteostasis network and its modulation by aneuploidy
The proteostasis network refers to a set of mechanisms that safeguard protein quality in cells (Balch et al. 2008; Hipp et al. 2019; Klaips et al. 2018). This includes protein synthesis (i.e., translation) by ribosomes, protein folding and conformational stability maintenance by chaperones, and protein degradation by the ubiquitin-proteasome system (UPS) or autophagy. Aneuploidy alters all these processes (Figure 2).

The changes in proteostasis network in aneuploid cells. Aneuploidy impacts the proteostasis network, from protein translation to turnover. The changes do not only affect the quality of proteins, but also impair the machineries controlling the protein quality. Up and down arrows indicate increased and decreased activity or levels, respectively.
3.1 Translation and integrated stress response
Analysis of aneuploid cells showed an increased sensitivity to drugs that inhibit translation (Torres et al. 2007). Proteomic analysis of constitutively polysomic human cells revealed decreased levels of components of cytosolic ribosomes and translation machinery, likely in response to the stress (Stingele et al. 2012). In acutely aneuploid human cells, a decrease in global protein translation occurs due to ISR and unfolded protein response (UPR), aimed at buffering the protein imbalance (Ippolito et al. 2024; Ohashi et al. 2015). ISR is also activated in Down syndrome mouse models and patient cell lines (Sheltzer 2013; Zhu et al. 2019). Downregulation of ribosome biogenesis and translation appears to be a general consequence of aneuploidy. In monosomic RPE1-hTERT cell lines, altered regulation of genes involved in ribosome biogenesis and translation leads to decreased protein synthesis, as evidenced by reduced puromycin incorporation in cell culture (Chunduri et al. 2021). Hence, aneuploidy strongly affects translation, mostly through the activation of ISR, and thus aneuploid cancer cells might be particularly sensitive to ribosome-targeting drugs.
3.2 Protein folding and misfolding in response to aneuploidy
As explained above, protein levels largely scale with the gene dosage in aneuploid cells. In cells with extra chromosomes, the excess proteins compete for the limited number of chaperones for folding and maintenance of their native conformation. Consequently, many proteins become orphaned, randomly misfold, and form cytosolic aggregates. Defective protein folding is a prevalent phenotype in aneuploidy, rendering aneuploid cells more sensitive to drugs that interfere with protein folding (Donnelly et al. 2014; Oromendia et al. 2012; Tang et al. 2011; Torres et al. 2007). Interestingly, activation of the heat shock response rarely occurs in response to aneuploidy. Biochemical analysis of aneuploid budding yeast and human cells reveals no significant changes in the levels of many chaperones compared to their euploid counterparts (Donnelly et al. 2014; Oromendia et al. 2012). This suggests that chronic aneuploidy does not lead to chaperone overexpression, an intriguing aspect of chronic proteotoxic stress. Rather, the protein folding defects are related to limited chaperone capacity to deal with surplus proteins (Donnelly and Storchová 2014). Accordingly, constitutively polysomic human cells show remarkable deficiency in HSF1 transcription factor activity and HSP90-dependent protein folding, which could be rescued by transient overexpression of a constitutively active form of HSF1 (ca. HSF1). Notably, ca. HSF1 overexpression also recues sensitivity of parental disomic cells to the pharmacological HSP90 inhibitor 17-AAG (Donnelly et al. 2014). The vulnerability of aneuploid cells to chaperone inhibition, thus, enables its exploitation in cancer therapy.
Protein folding defects in cells with extra chromosomes cause accumulation of misfolded proteins and protein aggregates (Brennan et al. 2019; Stingele et al. 2012). In budding yeast, the aggregates are enriched for hydrophobic proteins, and largely consist of ribosomal proteins and subunits of macromolecular complexes (Brennan et al. 2019). Our recent analyses of polysomic human cells revealed that the aggregates are enriched, among others, for ribosomal and mitochondrial proteins, and that their accumulation can be alleviated upon transient overexpression of the chaperones HSP27 and alpha subunit of HSP90, as well as the wildtype transcription factor HSF1 and ca. HSF1 (Amponsah et al. 2024). Thus, the surplus proteins in aneuploid cells cause overall decreased protein folding activity and increased aggregate formation compared to isogenic euploids.
3.3 Ubiquitin proteasome system and protein turnover mitigate the effects of aneuploidy
Cells rely on protein degradation systems such as the UPS and autolysosome to deal with unneeded proteins and protein aggregates. In response to chromosome gain, the UPS and autolysosome are frequently upregulated (Durrbaum et al. 2014; Stingele et al. 2012). Intuitively, one can interpret the upregulation as an adaptive response to degrade misfolded proteins and protein aggregates, and thus prevent the toxic effects of their accumulation in the cells.
The 26S proteasome, consisting of 20S core and 19S regulatory particle, contributes to the degradation of damaged, misfolded, or unwanted proteins. Ubiquitin ligases add polyubiquitin chains to the amino acid residues of the proteins destined for proteasomal degradation. This enables the ATP-dependent recognition of the proteins by the 19S subunit before their delivery to the 20S subunit for proteolysis (Arkinson et al. 2025). Analysis of data for more than 900 cancer cell lines from the cancer cell line encyclopedia (CCLE) revealed increased levels of both the 19S and 20S proteasome subunits in highly aneuploid cells, which correlated with their proliferation capacity. Experimentally, highly aneuploid cells showed an increased vulnerability to UPS inhibition with bortezomib (Ippolito et al. 2024). In constitutive human aneuploids, increased UPS activity is also observed (Donnelly et al. 2014). Furthermore, naturally occurring aneuploid budding yeast strains undergo extensive global protein turnover in response to proteome imbalance through elevated expression and activity of the UPS. This likely enhances protein dosage compensation, as shown by recent high throughput multiomics analyses of hundreds of wild yeast isolates and lab strains (Muenzner et al. 2024). The importance of the UPS for survival and proliferation of aneuploid cells was further emphasized by a genetic screen aimed at identifying mutations that enable tolerance of aneuploidy in laboratory budding yeast strains. Strikingly, a loss of function of the gene encoding the de-ubiquitinases UBP6 and UBP3, increased proteasomal degradation, reduced protein aggregate burden, and improved proliferation of the aneuploid cells (Dodgson et al. 2016; Torres et al. 2010). Importantly, these cells showed increased sensitivity to the proteasome inhibitor MG132 (Ohashi et al. 2015; Torres et al. 2007). Together, these observations highlight the role of the UPS in enabling tolerance to aneuploidy-induced proteostasis crisis.
In contrast, both murine and human Down syndrome models show an impairment of proteasomal activity (Aivazidis et al. 2017; Di Domenico et al. 2013), attributed to an increased abundance of misfolded proteins, and decreased synthesis of ATP needed for proteasomal functioning (Rodriguez-Sureda et al. 2015; Venkatraman et al. 2004). Why there is such a difference in Down syndrome cells remains currently unclear.
3.4 Autophagy is an important player in response to aneuploidy
While the UPS is mainly dedicated to the degradation of ubiquitinated proteins, the autolysosome degrades a broader cargo, from protein aggregates to damaged organelles. How is the autophagy pathway modulated by aneuploidy? Murine aneuploids are sensitive to autophagy inhibition with chloroquine (Tang et al. 2011). In acutely aneuploid human cells, saturation of autolysosomal activity is frequently observed, as evidenced by increased accumulation of pathway components and reduced autophagic flux (Ohashi et al. 2015; Santaguida and Amon 2015a). To counteract this, cells activate and increase nuclear localization of the transcription factor TFEB, which upregulates expression of the autolysosomal machinery (Santaguida and Amon 2015a; Santaguida et al. 2015). Adapted, constitutively polysomic human cells exhibit active autophagy flux (Stingele et al. 2012), and upregulate TFEB-dependent transcription, at least partly via the cGAS-STING pathway in a type I interferon (IFN) innate immune response, induced by the leakage of double-stranded DNA from the nucleus and its accumulation in the cytoplasm (Krivega et al. 2021). While autophagy regulation in response to physiological signals is mainly mediated via the mTOR pathway (Rabanal-Ruiz et al. 2017), constitutive trisomies (Krivega et al. 2021) and aneuploid mouse embryos (Singla et al. 2020b) are less reliant on this pathway for autophagy activation. Similarly, in aneuploid human preimplantation embryos, a p53-dependent pathway drives an active autophagy flux (Regin et al. 2023). In summary, the early autophagic response to aneuploidy differs from the response to chronic aneuploidy and is controlled by different upstream effectors. It is possible that in cancer, autophagy plays a dual role in response to aneuploidy – as both a tumor suppressor and promoter. Presumably, autophagy may perform a tumor suppressive role in early stages of oncogenesis, and acts as a tumor promoter in advanced stages. However, this proposition warrants further investigation.
3.5 Impaired protein homeostasis spurs aggresome formation in aneuploids
Defects in the UPS and autophagy, induced genetically or by chemical treatment, worsen protein misfolding and aggregation (Brennan et al. 2019; Isono et al. 2005; Shaid et al. 2013). In aneuploid cells, however, it remains unclear whether proteins aggregate because of impaired degradation systems, or if protein aggregation occurs as an independent mechanism of proteostasis maintenance. In lab-engineered aneuploids, protein aggregation increases despite frequent upregulation of expression and activities of the UPS and autolysosome (Amponsah et al. 2024; Brennan et al. 2019; Oromendia et al. 2012; Stingele et al. 2012). In response to aneuploidy in budding yeast, accumulation of foci positive for HSP104, a disaggregase that localizes to protein aggregates, demonstrates increased protein aggregation (Oromendia et al. 2012). Aneuploid human cells also accumulate protein aggregates, which are frequently positive for ubiquitin and the autophagy receptor p62 (Amponsah et al. 2024; Ohashi et al. 2015; Stingele et al. 2012). Interestingly, in both lab yeast and human aneuploids, protein aggregation helps to reduce the levels of excess subunits of protein complexes encoded on the aneuploid chromosome, although this class of proteins is also often degraded by the UPS and autolysome (Brennan et al. 2019; Dephoure et al. 2014). Analysis of primary fibroblasts from patients with trisomy syndromes revealed an increased accumulation of protein aggregates due to dysregulated proteostasis (Nawa et al. 2019). Aneuploid Drosophila cells also accumulate p62- and ubiquitin-positive protein aggregates due to near-saturation of the UPS and autophagy (Dekanty et al. 2012; Joy et al. 2021). Thus, while protein aggregation in response to aneuploidy can occur when protein degradation is limiting, it may also serve as an independent mechanism to maintain proteostasis. Analysis of aggregated proteins in a wide variety of aneuploid models would provide better insights into how protein aggregation helps to mitigate the proteostasis imbalance.
For a long time, accumulation of protein aggregates in cells was deemed cytotoxic (Stefani 2004). For example, through their sequestration into aggregates, essential proteins are depleted from the global proteome, thereby affecting the cellular context in which they function (Hipp and Hartl 2024; Huiting and Bergink 2021). However, benefits for cytosolic protein aggregation are becoming more evident (Cohen et al. 2006; Fassler et al. 2021; Newby and Lindquist 2013). For instance, aggregation of misfolded proteins in budding yeast and mammalian cell culture enable their refolding or degradation to prevent toxicity (Kaganovich et al. 2008). Amyloid-like protein aggregates called P granules ensure normal development of C. elegans embryos, as their disassembly leads to cell division defects, embryonic arrest, and lethality (Skuodas et al. 2020). In wild yeast, accumulation of prions enables the inheritance of beneficial traits that confer survival advantages in fluctuating physical environments (Halfmann et al. 2012). Another example of beneficial protein aggregation was observed in lab budding yeast strains experiencing mitochondrial import stress. Here, to prevent their cytotoxicity, mitochondrial precursor proteins transiently aggregate into cytosolic granules called “MitoStores”, which are subsequently unfolded by chaperones for import when favorable conditions permit (Kramer et al. 2023). Similar aggregation of mitochondrial precursor proteins was observed in constitutively polysomic human cells (Amponsah et al. 2024). Thus, in response to aneuploidy, increase in protein aggregation likely helps to alleviate the effects of protein imbalance in a manner similar to, yet distinct from, protein degradation (Brennan et al. 2019).
3.6 Extracellular extrusion of protein aggregates and damaged organelles contributes to proteostasis maintenance
Packaging of cellular materials in membrane-bound structures occurs in many physiological and pathological processes such as immune response, inflammation, cell proliferation, tumorigenesis, and neurodegeneration (Goetzl et al. 2018; Marar et al. 2021; Takahashi et al. 2017). A noteworthy observation revealed the formation of extracellular vesicles, which aid in proteostasis maintenance in response to aneuploidy. Extracellular release of protein aggregates and mitochondrial components in mitovesicles occurs in mouse and human Down syndrome brains (D’Acunzo et al. 2021). In aneuploid Drosophila cells, damaged mitochondria were observed in extracellular vesicles (Joy et al. 2021). Thus, this mechanism of proteostasis maintenance is probably widespread in aneuploid cancer cells and may be driven by the proteotoxic stress.
4 Factors required for proteostasis maintenance in aneuploidy
Maintenance of proteostasis in cells is a multifaceted process requiring the contributions of many different groups of proteins. The impacts of specific proteins in the mitigation of aneuploidy-induced proteotoxic stress have consistently been reported, and in some cases, used as markers of proteostasis defects. Here, we summarize findings relating to three of these proteins, which have been the most extensively characterized to date.
4.1 Sequestosome 1 (SQSTM1/p62)
p62 is a multidomain protein involved in many cellular processes including oxidative stress response, nutrient sensing, and response to inflammation. It contains an N-terminal Phox-BEM1 (PB1) domain, which allows its oligomerization, and an intrinsically disordered region that facilitates its condensation. p62 also possesses binding sites for many proteins including autophagy receptors of the microtubule-associated protein 1 light chain 3 (MAP1LC3) and GABA type A receptor-associated protein (GABARAP) families. Additionally, it contains a C-terminal ubiquitin binding (UBA) domain that selectively recognizes and sequesters (poly)ubiquitinated cargo (i.e., misfolded proteins, protein aggregates, damaged organelles). p62 is a common constituent of cytoplasmic inclusions and is also found in protein aggregates in the extracellular environment (Berkamp et al. 2021). Together, these features place p62 at the nexus of the key pathways of proteostasis maintenance in cells, including the UPS, autophagy, protein aggregation, and extracellular extrusion. Furthermore, it functions as a key mediator in regulation of mitochondrial dynamics and turnover (Geisler et al. 2010; Yamada et al. 2018, 2019). To finetune its function p62 is post-translationally regulated by phosphorylation, oxidation, acetylation, SUMOylation, and ubiquitylation (Berkamp et al. 2021). Dysregulation of p62 manifests in pathological conditions, including neurodegenerative disorders, aging-related diseases, and cancer (Kumar et al. 2022).
In both acutely and constitutively aneuploid human cells, p62 accumulates in cytosolic deposits. While in acutely aneuploid cells this accumulation is possibly a consequence of autophagy saturation and thereby stabilization of p62 (Ohashi et al. 2015; Santaguida et al. 2015), in constitutively polysomic cells the expression of p62 increases on both transcriptome and proteome levels, likely triggered as a secondary response to cellular stresses (Krivega et al. 2021; Singla et al. 2020b; Stingele et al. 2012). Interestingly, both p62 expression and gene dependency correlate with aneuploidy score of cancer cells (Amponsah et al. 2024). Thus, it is conceivable that increased p62 expression contributes to the fitness of aneuploid cells through alleviation of proteotoxic stress.
4.2 Heat shock protein 104 (HSP104)
HSP104 is a budding yeast chaperone and member of the HSP100/Clp family of ATPases, which function as disaggregases to rescue denatured and aggregated proteins for refolding (Bosl et al. 2006). While other chaperones typically prevent protein aggregation through solubilization or facilitating degradation, HSP104 rather disassembles the protein aggregates (Glover and Lindquist 1998). Thus, its activity not only reduces proteotoxicity, but also restores protein functions that were lost due to their aggregation (Bosl et al. 2006; Wallace et al. 2015). Interestingly, HSP104 participates in both the formation and disruption of prion amyloids in budding yeast (Halfmann et al. 2012; Sweeny and Shorter 2016; Sweeny et al. 2015). HSP104 is also central to the cellular response to aneuploidy in yeast. Transcriptomic meta-analyses revealed upregulation and scaling of HSP104 levels with degree of aneuploidy in budding yeast (Sheltzer et al. 2012), and aneuploid engineered budding yeast strains show elevated HSP104 protein levels compared to isogenic euploid strains (Oromendia et al. 2012). Additionally, confocal imaging of HSP104-eGFP foci revealed an increase in protein aggregation in various aneuploid yeast strains (Oromendia et al. 2012). The accumulation of HSP104 in response to aneuploidy correlates with the defects in protein folding and increased protein aggregation, which is a conserved feature of aneuploid cells (Oromendia and Amon 2014). Due to their ability to reverse deleterious protein misfolding and aggregation, protein disaggregases, including HSP104, have attracted significant attention as potential therapeutic targets in neurodegeneration (March et al. 2019; Shorter 2017). It would be interesting to decipher the contribution of its human ortholog, CLPB, to aneuploidy tolerance in human cancers.
4.3 Suppressor of SIT4 deletion (SSD1)
SSD1 is a member of the RNase II family of nucleases and a conserved RNA-binding translational regulator (Jansen et al. 2009). In response to stress, SSD1 localizes to P-bodies and stress granules, thus suppressing mRNA translation (Kurischko et al. 2011). SSD1 also impacts mitochondrial biogenesis through binding and regulation of nuclear-encoded mitochondrial mRNAs, and alleviates protein misfolding and aggregation, possibly via mRNA binding and suppression of translation of aggregation-prone proteins. SSD1 was identified as a factor that alleviates the cellular response to aneuploidy by comparing naturally occurring aneuploid yeast, which tolerate aneuploidy well, and lab-engineered aneuploid strains, which show proliferation defects. This revealed that SSD1, which is functional in natural yeast, but defective in many laboratory strains, contributes to aneuploidy tolerance (Hose et al. 2020). Consequently, reduced SSD1 activity in natural yeasts recapitulated many of the aneuploidy features of lab yeast, such as proteotoxic stress, metabolic defects, and cell cycle impairment (Hose et al. 2020). The intriguing SSD1-associated phenotypes clearly show that the cell's genetic background can strongly affect its response to aneuploidy, and that regulation of mRNA translation may be a crucial step of alleviating the aneuploidy-associated proteotoxic stresses.
5 Aneuploidy impacts organellar homeostasis
Organellar homeostasis is intimately linked to cytosolic proteostasis. Cytosolic protein aggregates often exert their toxicity through interaction and interference with organellar membranes (Rinauro et al. 2024). In such circumstances, mitochondria are often the major victims (Bossy-Wetzel et al. 2008; Cenini et al. 2016; Ludtmann et al. 2018; Moreira et al. 2010; Vehvilainen et al. 2014; Yano et al. 2014). The consequences of aneuploidy for mitochondrial homeostasis in human cells are multifaceted (Figure 3). Firstly, aneuploidy-induced competition for a limited number of chaperones can make mitochondrial precursor proteins more susceptible to misfolding in the cytosol, leading to their sequestration into protein aggregates. This hinders their import and may cause their eventual degradation. Indeed, our recent analysis of p62-positive protein aggregates in constitutively polysomic human cells revealed enrichment of both chaperones and mitochondrial proteins (Amponsah et al. 2024). A similar analysis in acutely aneuploid human cells also revealed enrichment of mitochondrial proteins with p62 (Martin et al. 2024). Secondly, cytosolic protein aggregates can impede mitochondrial protein import through their direct interaction with mitochondrial translocases (Yano et al. 2014). Thirdly, in constitutive polysomic human cells, mitochondrial structural and functional defects are conspicuous. Among others, the mitochondrial network appears more perinuclearly clustered compared to an extend network is parental diploid cells, oxygen consumption capacity is decreased, and precursor protein import is affected (Amponsah et al. 2024; Yim et al. 2020). Interestingly, similar mitochondrial defects arise in many human and murine Down syndrome models (Al-Mehdi et al. 2012; Frieden et al. 2004; Izzo et al. 2017, 2018; Piccoli et al. 2013; Tan et al. 2023). While some of the mitochondrial phenotypes in Down syndrome may be a consequence of the aberrant expression of specific genes encoded on chromosome 21, including DYRK1A (Dowjat et al. 2007; Ortega et al. 2022) and SOD1 (Busciglio and Yankner 1995; Cowley et al. 2017), a general dysfunction in cellular proteostasis also likely contributes to the mitochondrial alterations.

Organellar homeostasis in response to aneuploidy. The functions of many cellular organelles are altered in response to aneuploidy, ultimately affecting cellular homeostasis and function.
In Drosophila, aneuploidy-induced proteostasis defects result in the accumulation of fragmented mitochondria, as compared to a filamentous array in wildtype cells. Here, the mitochondria showed aberrant Ca2+ homeostasis, decreased membrane potential, and elevated reactive oxygen species (ROS) production (Joy et al. 2021). High-resolution microscopy also revealed saturated mitophagy and the accumulation of defective mitochondria in the cytosol and extracellular vesicles (Joy et al. 2021). While it is evident that aneuploidy impacts mitochondrial function, it would be compelling to explore how this disruption might, for example, contribute to cancer progression.
Apart from its impact on mitochondria, aneuploidy also affects the ER. Strikingly, it causes an expansion of this organelle in HeLa cells treated with inducers of chromosome missegregation, as visualized by electron microscopy. This physiological change accompanied the transcriptional activation of UPRER (Ohashi et al. 2015). Acutely aneuploid RPE1-hTERT cells also experience ER stress and induce the UPRER pathway (Ippolito et al. 2024). Transcriptome and proteome profiling of engineered polysomic human cells also shows an upregulation of ER-related pathways (Durrbaum et al. 2014; Stingele et al. 2012). Analyses of data for nearly 1,000 tumor samples from the cancer genome atlas (TCGA) database revealed increased expression of UPRER genes in samples with high aneuploidy, which correlated with dysregulated expression of genes involved in immune surveillance (Xian et al. 2021). Interestingly, UPRER activation is beneficial to cancer cells as it helps to restore proteostasis and suppress immune functions (Clarke et al. 2014; Cubillos-Ruiz et al. 2015). It is conceivable that ER expansion and the associated UPRER activation observed in aneuploid cells occur in response to proteostasis imbalance, presumably due to erroneous protein synthesis and folding. In line with this reasoning, a similar expansion of ER membrane and UPRER activation was reported in budding yeast acutely treated with DTT and tunicamycin, drugs that induce ER stress by interfering with oxidative protein folding and glycosylation, respectively (Schuck et al. 2009, 2021). Here, the ER expansion was driven by lipid biosynthesis, a process which is itself affected by aneuploidy (Hwang et al. 2017; Tang et al. 2017). Future research would provide more insights into this aspect.
Other cellular organelles affected by aneuploidy include the autolysosome, ribosomes, and nuclei. As demonstrated above, aneuploidy places severe burden on lysosomal function and spurs expression of autolysosomal proteins through the activity of the transcription factor TFEB (Joy et al. 2021; Krivega et al. 2021; Santaguida and Amon 2015a; Santaguida et al. 2015). In acutely aneuploid human cells, lysosomal stress occurs due to persistent protein misfolding, and aggregation caused by the proteome imbalance (Santaguida and Amon 2015a; Santaguida et al. 2015). Owing to its role in removing mainly protein aggregates and damaged organelles, it is conceivable that lysosomal stress results from the nature of cargo accumulating in the autolysosome. Observations in constitutively polysomic human cells suggest that this cargo include cytosolic ribosomes (Amponsah et al. 2024), implying that the need to remove defective, bulky cellular components contributes to the autolysosomal stress in aneuploidy.
Ribosomes and the protein translation mechanism participate in the early steps of protein quality control. Abnormal mRNA accumulation, increased mRNA turnover, and aberrant translation are intuitive effects of gene dosage imbalance. Accordingly, a recent transcriptomic analysis revealed increased mRNA turnover and dependency on mRNA decay pathways in acutely aneuploid human cells (Ippolito et al. 2024). Transcriptomic and proteomic profiling of constitutively polysomic human cells also demonstrated a reduction in the global levels of cytosolic ribosomal subunits and translation machinery components compared to isogenic diploids (Durrbaum et al. 2014; Stingele et al. 2012). Genetic screens in aneuploid fission and budding yeasts versus euploid strains uncovered increased dependence on the CCR4-NOT complex, an essential and evolutionarily conserved multi-subunit complex that regulates the process of mRNA translation (Chikashige et al. 2007; Tange et al. 2012). Ribosome loss also occurs due to aneuploidy-induced environmental stress response in budding yeast (Terhorst et al. 2020). Thus, translation defects and mRNA-ribonucleoprotein turnover in aneuploidy might be more prevalent than currently appreciated.
Finally, the nuclear homeostasis may be altered in response to aneuploidy. In acutely aneuploid human cells that contain micronuclei; a small, extranuclear vesicle containing a fragment of or a whole chromosome, stability of the micronuclear membrane is regulated through oxidation, aggregation, and degradation of important membrane repair proteins (Di Bona et al. 2024; Martin et al. 2024). The collapse of micronuclei could occur through the production and release of ROS from mitochondria. Indeed, human aneuploid cells, which often form micronuclei, also harbor perinuclearly clustered mitochondria, which are oxidant-rich sites that enable ROS accumulation in the nucleus (Al-Mehdi et al. 2012; Amponsah et al. 2024). Thus, proteostasis imbalance and resulting mechanisms, including altered mitochondrial homeostasis, may feedback to regulate genome stability.
6 Aneuploidy-induced acute and chronic proteotoxic stress and disease
Research from numerous labs has provided substantial evidence that aneuploidy imposes proteotoxic stress, affecting multiple layers of the proteostasis network. While the gain or loss of a single chromosome may not disturb the protein stoichiometry sufficiently enough to trigger a severe proteotoxic stress response, the cumulative disturbances across multiple pathways can trigger an avalanche, leading to dysregulation of protein homeostasis maintenance. This vicious cycle significantly impacts several diseases, including cancer, trisomy syndromes, and aging. However, the extent to which aneuploidy-related changes in protein homeostasis contribute to disease pathology remains to be fully evaluated.
The intersection of protein homeostasis, aneuploidy, and cancer raises several pressing questions for future research. Despite significant efforts, the exact mechanisms of proteotoxic stress in cells with imbalanced karyotypes remain to be discovered, in particular the key molecular pathways that allow aneuploid cancer cells to tolerate proteotoxic stress. The survival of most cancer cells, despite highly aberrant multi-chromosomal changes, clearly demonstrates the existence of adaptive mechanisms that minimize the impact of protein homeostasis disruptions. So, what are the metabolic trade-offs cancer cells make to cope with proteotoxic stress, and can we use them to target cancer cells? With this, an increasingly important aspect of the role of quality control pathways comes to the focus, in particular the role of mRNA surveillance and ribosome quality control. Finally, the future challenge remains to understand the role of aneuploidy-instigated proteotoxic stress in vivo, within an organism. Here, the key question will be whether the proteotoxic stress in aneuploid cancer cells affects their immunogenicity and whether interventions in protein homeostasis make aneuploid cancers more susceptible to immune-based therapies. Addressing these questions will enhance our understanding of aneuploidy’s role in cancer and potentially lead to not only better understanding of the mechanisms of protein homeostasis, but also contribute to novel therapeutic approaches targeting the vulnerabilities triggered by protein imbalance.
Acknowledgments
We are grateful to Olha Kurpa for reading and providing feedback on the final draft of the manuscript. This work was supported by grants from the Deutsche Forschungsgemeinschaft (GRK2737-STRESSistance to ZS, and Walter Benjamin Programme Award AM 703/1 to PSA, Project number 510268075). PSA acknowledges support from the TU Nachwuchsring (RPTU Kaiserslautern) and an Add-on fellowship from the Joachim Herz Stiftung.
-
Research ethics: Not applicable.
-
Informed consent: Not applicable.
-
Author contributions: All authors have accepted responsibility for the entire content of this manuscript and approved its submission.
-
Use of Large Language Models, AI and Machine Learning Tools: None declared.
-
Conflict of interest: The authors state no conflict of interest.
-
Research funding: Deutsche Forschungsgemeinschaft: GRK2737-STRESSistance to ZS, and Walter Benjamin Programme Award AM 703/1 to PSA; TU Nachwuchsring Kaiserslautern and an Add-on fellowship from the Joachim Herz Stiftung to PSA.
-
Data availability: Not applicable.
References
Aivazidis, S., Coughlan, C.M., Rauniyar, A.K., Jiang, H., Liggett, L.A., Maclean, K.N., and Roede, J.R. (2017). The burden of trisomy 21 disrupts the proteostasis network in Down syndrome. PLoS One 12: e0176307, https://doi.org/10.1371/journal.pone.0176307.Search in Google Scholar PubMed PubMed Central
Al-Mehdi, A.B., Pastukh, V.M., Swiger, B.M., Reed, D.J., Patel, M.R., Bardwell, G.C., Pastukh, V.V., Alexeyev, M.F., and Gillespie, M.N. (2012). Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci. Signal. 5: ra47, https://doi.org/10.1126/scisignal.2002712.Search in Google Scholar PubMed PubMed Central
Amponsah, P.S., Bökenkamp, J.-E., Lenhard, S., Behrends, C., Herrmann, J.M., Räschle, M., and Storchová, Z. (2024). Aneuploidy-induced proteostasis disruption impairs mitochondrial functions and mediates aggregation of mitochondrial precursor proteins through SQSTM1/p62. bioRxiv, https://doi.org/10.1101/2024.07.29.605607.Search in Google Scholar
Arkinson, C., Dong, K.C., Gee, C.L., and Martin, A. (2025). Mechanisms and regulation of substrate degradation by the 26S proteasome. Nat. Rev. Mol. Cell Biol. 26: 104–122, https://doi.org/10.1038/s41580-024-00778-0.Search in Google Scholar PubMed PubMed Central
Balch, W.E., Morimoto, R.I., Dillin, A., and Kelly, J.W. (2008). Adapting proteostasis for disease intervention. Science 319: 916–919, https://doi.org/10.1126/science.1141448.Search in Google Scholar PubMed
Beach, R.R., Ricci-Tam, C., Brennan, C.M., Moomau, C.A., Hsu, P.H., Hua, B., Silberman, R.E., Springer, M., and Amon, A. (2017). Aneuploidy causes non-genetic individuality. Cell 169: 229–242.e21, https://doi.org/10.1016/j.cell.2017.03.021.Search in Google Scholar PubMed PubMed Central
Berkamp, S., Mostafavi, S., and Sachse, C. (2021). Structure and function of p62/SQSTM1 in the emerging framework of phase separation. FEBS J. 288: 6927–6941, https://doi.org/10.1111/febs.15672.Search in Google Scholar PubMed
Bökenkamp, J.E., Keuper, K., Redel, S., Barthel, K., Johnson, L., Becker, A., Wieland, A., Räschle, M., and Storchová, Z. (2025). Proteogenomic analysis reveals adaptive strategies for alleviating the consequences of aneuploidy in cancer. EMBO J. 44: 1829–1865, https://doi.org/10.1038/s44318-025-00372-w.Search in Google Scholar PubMed PubMed Central
Bosl, B., Grimminger, V., and Walter, S. (2006). The molecular chaperone Hsp104–a molecular machine for protein disaggregation. J. Struct. Biol. 156: 139–148, https://doi.org/10.1016/j.jsb.2006.02.004.Search in Google Scholar PubMed
Bossy-Wetzel, E., Petrilli, A., and Knott, A.B. (2008). Mutant huntingtin and mitochondrial dysfunction. Trends Neurosci. 31: 609–616, https://doi.org/10.1016/j.tins.2008.09.004.Search in Google Scholar PubMed PubMed Central
Brennan, C.M., Vaites, L.P., Wells, J.N., Santaguida, S., Paulo, J.A., Storchova, Z., Harper, J.W., Marsh, J.A., and Amon, A. (2019). Protein aggregation mediates stoichiometry of protein complexes in aneuploid cells. Genes Dev. 33: 1031–1047, https://doi.org/10.1101/gad.327494.119.Search in Google Scholar PubMed PubMed Central
Busciglio, J. and Yankner, B.A. (1995). Apoptosis and increased generation of reactive oxygen species in Down’s syndrome neurons in vitro. Nature 378: 776–779, https://doi.org/10.1038/378776a0.Search in Google Scholar PubMed
Cenini, G., Rub, C., Bruderek, M., and Voos, W. (2016). Amyloid beta-peptides interfere with mitochondrial preprotein import competence by a coaggregation process. Mol. Biol. Cell 27: 3257–3272, https://doi.org/10.1091/mbc.e16-05-0313.Search in Google Scholar PubMed PubMed Central
Chikashige, Y., Tsutsumi, C., Okamasa, K., Yamane, M., Nakayama, J., Niwa, O., Haraguchi, T., and Hiraoka, Y. (2007). Gene expression and distribution of Swi6 in partial aneuploids of the fission yeast Schizosaccharomyces pombe. Cell Struct. Funct. 32: 149–161, https://doi.org/10.1247/csf.07036.Search in Google Scholar PubMed
Chunduri, N.K. and Storchová, Z. (2019). The diverse consequences of aneuploidy. Nat. Cell Biol. 21: 54–62, https://doi.org/10.1038/s41556-018-0243-8.Search in Google Scholar PubMed
Chunduri, N.K., Menges, P., Zhang, X.X., Wieland, A., Gotsmann, V.L., Mardin, B.R., Buccitelli, C., Korbel, J.O., Willmund, F., Kschischo, M., et al.. (2021). Systems approaches identify the consequences of monosomy in somatic human cells. Nat. Commun. 12, https://doi.org/10.1038/s41467-021-25288-x.Search in Google Scholar PubMed PubMed Central
Clarke, H.J., Chambers, J.E., Liniker, E., and Marciniak, S.J. (2014). Endoplasmic reticulum stress in malignancy. Cancer Cell 25: 563–573, https://doi.org/10.1016/j.ccr.2014.03.015.Search in Google Scholar PubMed
Cohen, E., Bieschke, J., Perciavalle, R.M., Kelly, J.W., and Dillin, A. (2006). Opposing activities protect against age-onset proteotoxicity. Science 313: 1604–1610, https://doi.org/10.1126/science.1124646.Search in Google Scholar PubMed
Cowley, P.M., Nair, D.R., Deruisseau, L.R., Keslacy, S., Atalay, M., and Deruisseau, K.C. (2017). Oxidant production and SOD1 protein expression in single skeletal myofibers from Down syndrome mice. Redox Biol. 13: 421–425, https://doi.org/10.1016/j.redox.2017.07.003.Search in Google Scholar PubMed PubMed Central
Cubillos-Ruiz, J.R., Silberman, P.C., Rutkowski, M.R., Chopra, S., Perales-Puchalt, A., Song, M., Zhang, S., Bettigole, S.E., Gupta, D., Holcomb, K., et al.. (2015). ER stress sensor XBP1 controls anti-tumor immunity by disrupting dendritic cell homeostasis. Cell 161: 1527–1538, https://doi.org/10.1016/j.cell.2015.05.025.Search in Google Scholar PubMed PubMed Central
D’Acunzo, P., Perez-Gonzalez, R., Kim, Y., Hargash, T., Miller, C., Alldred, M.J., Erdjument-Bromage, H., Penikalapati, S.C., Pawlik, M., Saito, M., et al.. (2021). Mitovesicles are a novel population of extracellular vesicles of mitochondrial origin altered in Down syndrome. Sci. Adv. 7, https://doi.org/10.1126/sciadv.abe5085.Search in Google Scholar PubMed PubMed Central
Dekanty, A., Barrio, L., Muzzopappa, M., Auer, H., and Milan, M. (2012). Aneuploidy-induced delaminating cells drive tumorigenesis in Drosophila epithelia. Proc. Natl. Acad. Sci. U. S. A. 109: 20549–20554, https://doi.org/10.1073/pnas.1206675109.Search in Google Scholar PubMed PubMed Central
Dephoure, N., Hwang, S., O’sullivan, C., Dodgson, S.E., Gygi, S.P., Amon, A., and Torres, E.M. (2014). Quantitative proteomic analysis reveals posttranslational responses to aneuploidy in yeast. eLife 3: e03023, https://doi.org/10.7554/elife.03023.Search in Google Scholar
Deshaies, R.J. (2014). Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol. 12: 94, https://doi.org/10.1186/s12915-014-0094-0.Search in Google Scholar PubMed PubMed Central
Di Bona, M., Chen, Y., Agustinus, A.S., Mazzagatti, A., Duran, M.A., Deyell, M., Bronder, D., Hickling, J., Hong, C., Scipioni, L., et al.. (2024). Micronuclear collapse from oxidative damage. Science 385: eadj8691, https://doi.org/10.1126/science.adj8691.Search in Google Scholar PubMed PubMed Central
Di Domenico, F., Coccia, R., Cocciolo, A., Murphy, M.P., Cenini, G., Head, E., Butterfield, D.A., Giorgi, A., Schinina, M.E., Mancuso, C., et al.. (2013). Impairment of proteostasis network in Down syndrome prior to the development of Alzheimer’s disease neuropathology: redox proteomics analysis of human brain. Biochim. Biophys. Acta 1832: 1249–1259, https://doi.org/10.1016/j.bbadis.2013.04.013.Search in Google Scholar PubMed PubMed Central
Dodgson, S.E., Santaguida, S., Kim, S., Sheltzer, J., and Amon, A. (2016). The pleiotropic deubiquitinase Ubp3 confers aneuploidy tolerance. Genes Dev. 30: 2259–2271, https://doi.org/10.1101/gad.287474.116.Search in Google Scholar PubMed PubMed Central
Donnelly, N. and Storchová, Z. (2014). Dynamic karyotype, dynamic proteome: buffering the effects of aneuploidy. Biochim. Biophys. Acta Mol. Cell Res. 1843: 473–481, https://doi.org/10.1016/j.bbamcr.2013.11.017.Search in Google Scholar PubMed
Donnelly, N., Passerini, V., Durrbaum, M., Stingele, S., and Storchova, Z. (2014). HSF1 deficiency and impaired HSP90-dependent protein folding are hallmarks of aneuploid human cells. EMBO J. 33: 2374–2387, https://doi.org/10.15252/embj.201488648.Search in Google Scholar PubMed PubMed Central
Dowjat, W.K., Adayev, T., Kuchna, I., Nowicki, K., Palminiello, S., Hwang, Y.W., and Wegiel, J. (2007). Trisomy-driven overexpression of DYRK1A kinase in the brain of subjects with Down syndrome. Neurosci. Lett. 413: 77–81, https://doi.org/10.1016/j.neulet.2006.11.026.Search in Google Scholar PubMed PubMed Central
Durrbaum, M., Kuznetsova, A.Y., Passerini, V., Stingele, S., Stoehr, G., and Storchova, Z. (2014). Unique features of the transcriptional response to model aneuploidy in human cells. BMC Genom. 15: 139, https://doi.org/10.1186/1471-2164-15-139.Search in Google Scholar PubMed PubMed Central
Fassler, J.S., Skuodas, S., Weeks, D.L., and Phillips, B.T. (2021). Protein aggregation and disaggregation in cells and development. J. Mol. Biol. 433: 167215, https://doi.org/10.1016/j.jmb.2021.167215.Search in Google Scholar PubMed PubMed Central
Frieden, M., James, D., Castelbou, C., Danckaert, A., Martinou, J.C., and Demaurex, N. (2004). Ca(2+) homeostasis during mitochondrial fragmentation and perinuclear clustering induced by hFis1. J. Biol. Chem. 279: 22704–22714, https://doi.org/10.1074/jbc.m312366200.Search in Google Scholar
Geisler, S., Holmstrom, K.M., Skujat, D., Fiesel, F.C., Rothfuss, O.C., Kahle, P.J., and Springer, W. (2010). PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 12: 119–131, https://doi.org/10.1038/ncb2012.Search in Google Scholar PubMed
Glover, J.R. and Lindquist, S. (1998). Hsp104, Hsp70, and Hsp40: a novel chaperone system that rescues previously aggregated proteins. Cell 94: 73–82, https://doi.org/10.1016/s0092-8674(00)81223-4.Search in Google Scholar PubMed
Goetzl, E.J., Schwartz, J.B., Abner, E.L., Jicha, G.A., and Kapogiannis, D. (2018). High complement levels in astrocyte-derived exosomes of Alzheimer disease. Ann. Neurol. 83: 544–552, https://doi.org/10.1002/ana.25172.Search in Google Scholar PubMed PubMed Central
Halfmann, R., Jarosz, D.F., Jones, S.K., Chang, A., Lancaster, A.K., and Lindquist, S. (2012). Prions are a common mechanism for phenotypic inheritance in wild yeasts. Nature 482: 363–368, https://doi.org/10.1038/nature10875.Search in Google Scholar PubMed PubMed Central
Hipp, M.S. and Hartl, F.U. (2024). Interplay of proteostasis capacity and protein aggregation: implications for cellular function and disease. J. Mol. Biol. 436: 168615, https://doi.org/10.1016/j.jmb.2024.168615.Search in Google Scholar PubMed
Hipp, M.S., Kasturi, P., and Hartl, F.U. (2019). The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 20: 421–435, https://doi.org/10.1038/s41580-019-0101-y.Search in Google Scholar PubMed
Hose, J., Escalante, L.E., Clowers, K.J., Dutcher, H.A., Robinson, D., Bouriakov, V., Coon, J.J., Shishkova, E., and Gasch, A.P. (2020). The genetic basis of aneuploidy tolerance in wild yeast. eLife 9, https://doi.org/10.7554/elife.52063.Search in Google Scholar
Huettel, B., Kreil, D.P., Matzke, M., and Matzke, A.J. (2008). Effects of aneuploidy on genome structure, expression, and interphase organization in Arabidopsis thaliana. PLoS Genet. 4: e1000226, https://doi.org/10.1371/journal.pgen.1000226.Search in Google Scholar PubMed PubMed Central
Huiting, W. and Bergink, S. (2021). Locked in a vicious cycle: the connection between genomic instability and a loss of protein homeostasis. Genome Instabil. Dis. 2: 1–23, https://doi.org/10.1007/s42764-020-00027-6.Search in Google Scholar
Hwang, S., Gustafsson, H.T., O’sullivan, C., Bisceglia, G., Huang, X.H., Klose, C., Schevchenko, A., Dickson, R.C., Cavaliere, P., Dephoure, N., et al.. (2017). Serine-dependent sphingolipid synthesis is a metabolic liability of aneuploid cells. Cell Rep. 21: 3807–3818, https://doi.org/10.1016/j.celrep.2017.11.103.Search in Google Scholar PubMed PubMed Central
Hwang, S., Cavaliere, P., Li, R., Zhu, L.J., Dephoure, N., and Torres, E.M. (2021). Consequences of aneuploidy in human fibroblasts with trisomy 21. Proc. Natl. Acad. Sci. U. S. A. 118, https://doi.org/10.1073/pnas.2014723118.Search in Google Scholar PubMed PubMed Central
Ippolito, M.R., Zerbib, J., Eliezer, Y., Reuveni, E., Vigano, S., De Feudis, G., Shulman, E.D., Savir Kadmon, A., Slutsky, R., Chang, T., et al. (2024). Increased RNA and protein degradation is required for counteracting transcriptional burden and proteotoxic stress in human aneuploid cells. Cancer Discov. 14: 2532–2553, https://doi.org/10.1158/2159-8290.cd-23-0309.Search in Google Scholar
Isono, E., Saito, N., Kamata, N., Saeki, Y., and Toh, E.A. (2005). Functional analysis of Rpn6p, a lid component of the 26 S proteasome, using temperature-sensitive rpn6 mutants of the yeast Saccharomyces cerevisiae. J. Biol. Chem. 280: 6537–6547, https://doi.org/10.1074/jbc.m409364200.Search in Google Scholar
Izzo, A., Nitti, M., Mollo, N., Paladino, S., Procaccini, C., Faicchia, D., Cali, G., Genesio, R., Bonfiglio, F., Cicatiello, R., et al.. (2017). Metformin restores the mitochondrial network and reverses mitochondrial dysfunction in Down syndrome cells. Hum. Mol. Genet. 26: 1056–1069, https://doi.org/10.1093/hmg/ddx016.Search in Google Scholar PubMed
Izzo, A., Mollo, N., Nitti, M., Paladino, S., Cali, G., Genesio, R., Bonfiglio, F., Cicatiello, R., Barbato, M., Sarnataro, V., et al.. (2018). Mitochondrial dysfunction in down syndrome: molecular mechanisms and therapeutic targets. Mol. Med. 24: 2, https://doi.org/10.1186/s10020-018-0004-y.Search in Google Scholar PubMed PubMed Central
Jansen, J.M., Wanless, A.G., Seidel, C.W., and Weiss, E.L. (2009). Cbk1 regulation of the RNA-binding protein Ssd1 integrates cell fate with translational control. Curr. Biol. 19: 2114–2120, https://doi.org/10.1016/j.cub.2009.10.071.Search in Google Scholar PubMed PubMed Central
Joy, J., Barrio, L., Santos-Tapia, C., Romao, D., Giakoumakis, N.N., Clemente-Ruiz, M., and Milan, M. (2021). Proteostasis failure and mitochondrial dysfunction leads to aneuploidy-induced senescence. Dev. Cell 56: 2043–2058.e7, https://doi.org/10.1016/j.devcel.2021.06.009.Search in Google Scholar PubMed
Kaganovich, D., Kopito, R., and Frydman, J. (2008). Misfolded proteins partition between two distinct quality control compartments. Nature 454: 1088–1095, https://doi.org/10.1038/nature07195.Search in Google Scholar PubMed PubMed Central
Klaips, C.L., Jayaraj, G.G., and Hartl, F.U. (2018). Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 217: 51–63, https://doi.org/10.1083/jcb.201709072.Search in Google Scholar PubMed PubMed Central
Kramer, L., Dalheimer, N., Raschle, M., Storchova, Z., Pielage, J., Boos, F., and Herrmann, J.M. (2023). MitoStores: chaperone-controlled protein granules store mitochondrial precursors in the cytosol. EMBO J. 42: e112309, https://doi.org/10.15252/embj.2022112309.Search in Google Scholar PubMed PubMed Central
Krivega, M., Stiefel, C.M., Karbassi, S., Andersen, L.L., Chunduri, N.K., Donnelly, N., Pichlmair, A., and Storchova, Z. (2021). Genotoxic stress in constitutive trisomies induces autophagy and the innate immune response via the cGAS-STING pathway. Commun. Biol. 4: 831, https://doi.org/10.1038/s42003-021-02278-9.Search in Google Scholar PubMed PubMed Central
Kumar, A.V., Mills, J., and Lapierre, L.R. (2022). Selective autophagy receptor p62/SQSTM1, a pivotal player in stress and aging. Front. Cell Dev. Biol. 10: 793328, https://doi.org/10.3389/fcell.2022.793328.Search in Google Scholar PubMed PubMed Central
Kurischko, C., Kim, H.K., Kuravi, V.K., Pratzka, J., and Luca, F.C. (2011). The yeast Cbk1 kinase regulates mRNA localization via the mRNA-binding protein Ssd1. J. Cell Biol. 192: 583–598, https://doi.org/10.1083/jcb.201011061.Search in Google Scholar PubMed PubMed Central
Lakhani, A.A., Thompson, S.L., and Sheltzer, J.M. (2023). Aneuploidy in human cancer: new tools and perspectives. Trends Genet. 39: 968–980, https://doi.org/10.1016/j.tig.2023.09.002.Search in Google Scholar PubMed PubMed Central
Liu, Y., Borel, C., Li, L., Muller, T., Williams, E.G., Germain, P.L., Buljan, M., Sajic, T., Boersema, P.J., Shao, W., et al.. (2017). Systematic proteome and proteostasis profiling in human Trisomy 21 fibroblast cells. Nat. Commun. 8: 1212, https://doi.org/10.1038/s41467-017-01422-6.Search in Google Scholar PubMed PubMed Central
Ludtmann, M.H.R., Angelova, P.R., Horrocks, M.H., Choi, M.L., Rodrigues, M., Baev, A.Y., Berezhnov, A.V., Yao, Z., Little, D., Banushi, B., et al.. (2018). Alpha-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 9: 2293, https://doi.org/10.1038/s41467-018-04422-2.Search in Google Scholar PubMed PubMed Central
Marar, C., Starich, B., and Wirtz, D. (2021). Extracellular vesicles in immunomodulation and tumor progression. Nat. Immunol. 22: 560–570, https://doi.org/10.1038/s41590-021-00899-0.Search in Google Scholar PubMed PubMed Central
March, Z.M., Mack, K.L., and Shorter, J. (2019). AAA plus protein-based technologies to counter neurodegenerative disease. Biophys. J. 116: 1380–1385, https://doi.org/10.1016/j.bpj.2019.03.007.Search in Google Scholar PubMed PubMed Central
Martin, S., Scorzoni, S., Cordone, S., Mazzagatti, A., Beznoussenko, G.V., Gunn, A.L., Di Bona, M., Eliezer, Y., Leor, G., Ben-Yishay, T., et al.. (2024). A p62-dependent rheostat dictates micronuclei catastrophe and chromosome rearrangements. Science 385: eadj7446, https://doi.org/10.1126/science.adj7446.Search in Google Scholar PubMed PubMed Central
Moreira, P.I., Carvalho, C., Zhu, X., Smith, M.A., and Perry, G. (2010). Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochim. Biophys. Acta 1802: 2–10, https://doi.org/10.1016/j.bbadis.2009.10.006.Search in Google Scholar PubMed
Muenzner, J., Trebulle, P., Agostini, F., Zauber, H., Messner, C.B., Steger, M., Kilian, C., Lau, K., Barthel, N., Lehmann, A., et al.. (2024). Natural proteome diversity links aneuploidy tolerance to protein turnover. Nature 630: 149–157, https://doi.org/10.1038/s41586-024-07442-9.Search in Google Scholar PubMed PubMed Central
Nagaoka, S.I., Hassold, T.J., and Hunt, P.A. (2012). Human aneuploidy: mechanisms and new insights into an age-old problem. Nat. Rev. Genet. 13: 493–504, https://doi.org/10.1038/nrg3245.Search in Google Scholar PubMed PubMed Central
Nawa, N., Hirata, K., Kawatani, K., Nambara, T., Omori, S., Banno, K., Kokubu, C., Takeda, J., Nishimura, K., Ohtaka, M., et al.. (2019). Elimination of protein aggregates prevents premature senescence in human trisomy 21 fibroblasts. PLoS One 14: e0219592, https://doi.org/10.1371/journal.pone.0219592.Search in Google Scholar PubMed PubMed Central
Naylor, R.M. and Van Deursen, J.M. (2016). Aneuploidy in cancer and aging. Annu. Rev. Genet. 50: 45–66, https://doi.org/10.1146/annurev-genet-120215-035303.Search in Google Scholar PubMed PubMed Central
Newby, G.A. and Lindquist, S. (2013). Blessings in disguise: biological benefits of prion-like mechanisms. Trends Cell Biol. 23: 251–259, https://doi.org/10.1016/j.tcb.2013.01.007.Search in Google Scholar PubMed
Ohashi, A., Ohori, M., Iwai, K., Nakayama, Y., Nambu, T., Morishita, D., Kawamoto, T., Miyamoto, M., Hirayama, T., Okaniwa, M., et al.. (2015). Aneuploidy generates proteotoxic stress and DNA damage concurrently with p53-mediated post-mitotic apoptosis in SAC-impaired cells. Nat. Commun. 6: 7668, https://doi.org/10.1038/ncomms8668.Search in Google Scholar PubMed PubMed Central
Oromendia, A.B. and Amon, A. (2014). Aneuploidy: implications for protein homeostasis and disease. Dis. Model. Mech. 7: 15–20, https://doi.org/10.1242/dmm.013391.Search in Google Scholar PubMed PubMed Central
Oromendia, A.B., Dodgson, S.E., and Amon, A. (2012). Aneuploidy causes proteotoxic stress in yeast. Genes Dev. 26: 2696–2708, https://doi.org/10.1101/gad.207407.112.Search in Google Scholar PubMed PubMed Central
Ortega, M., De Toma, I., Fernández-Blanco, A., Calderón, A., Barahona, L., Trullàs, R., Sabidó, E., and Dierssen, M. (2022). Proteomic profiling reveals mitochondrial dysfunction in the cerebellum of transgenic mice overexpressing DYRK1A, a Down syndrome candidate gene. Front. Mol. Neurosci. 15, https://doi.org/10.3389/fnmol.2022.1015220.Search in Google Scholar PubMed PubMed Central
Pavelka, N., Rancati, G., Zhu, J., Bradford, W.D., Saraf, A., Florens, L., Sanderson, B.W., Hattem, G.L., and Li, R. (2010). Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature 468: 321–325, https://doi.org/10.1038/nature09529.Search in Google Scholar PubMed PubMed Central
Piccoli, C., Izzo, A., Scrima, R., Bonfiglio, F., Manco, R., Negri, R., Quarato, G., Cela, O., Ripoli, M., Prisco, M., et al.. (2013). Chronic pro-oxidative state and mitochondrial dysfunctions are more pronounced in fibroblasts from Down syndrome foeti with congenital heart defects. Hum. Mol. Genet. 22: 1218–1232, https://doi.org/10.1093/hmg/dds529.Search in Google Scholar PubMed
Rabanal-Ruiz, Y., Otten, E.G., and Korolchuk, V.I. (2017). mTORC1 as the main gateway to autophagy. Signal Mech. Autophagy 61: 565–584, https://doi.org/10.1042/ebc20170027.Search in Google Scholar PubMed PubMed Central
Regin, M., Lei, Y., De Deckersberg, E.C., Guns, Y., Verdyck, P., Verheyen, G., Van De Velde, H., Sermon, K., and Spits, C. (2023). Complex aneuploidy triggers autophagy and p53-mediated apoptosis and impairs the second lineage segregation in human preimplantation embryos. eLife.10.7554/eLife.88916.1Search in Google Scholar
Rinauro, D.J., Chiti, F., Vendruscolo, M., and Limbocker, R. (2024). Misfolded protein oligomers: mechanisms of formation, cytotoxic effects, and pharmacological approaches against protein misfolding diseases. Mol. Neurodegener. 19: 20, https://doi.org/10.1186/s13024-023-00651-2.Search in Google Scholar PubMed PubMed Central
Rodriguez-Sureda, V., Vilches, A., Sanchez, O., Audi, L., and Dominguez, C. (2015). Intracellular oxidant activity, antioxidant enzyme defense system, and cell senescence in fibroblasts with trisomy 21. Oxid. Med. Cell. Longev. 2015: 509241, https://doi.org/10.1155/2015/509241.Search in Google Scholar PubMed PubMed Central
Santaguida, S. and Amon, A. (2015a). Aneuploidy triggers a TFEB-mediated lysosomal stress response. Autophagy 11: 2383–2384, https://doi.org/10.1080/15548627.2015.1110670.Search in Google Scholar PubMed PubMed Central
Santaguida, S. and Amon, A. (2015b). Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat. Rev. Mol. Cell Biol. 16: 473–485, https://doi.org/10.1038/nrm4025.Search in Google Scholar PubMed
Santaguida, S., Vasile, E., White, E., and Amon, A. (2015). Aneuploidy-induced cellular stresses limit autophagic degradation. Genes Dev. 29: 2010–2021, https://doi.org/10.1101/gad.269118.115.Search in Google Scholar PubMed PubMed Central
Schuck, S., Prinz, W.A., Thorn, K.S., Voss, C., and Walter, P. (2009). Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J. Cell Biol. 187: 525–536, https://doi.org/10.1083/jcb.200907074.Search in Google Scholar PubMed PubMed Central
Schuck, S., Prinz, W.A., Thorn, K.S., Voss, C., and Walter, P. (2021). Correction: membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J. Cell Biol. 220, https://doi.org/10.1083/jcb.20090707402092021c.Search in Google Scholar PubMed PubMed Central
Schukken, K.M. and Sheltzer, J.M. (2022). Extensive protein dosage compensation in aneuploid human cancers. Genome Res. 32: 1254–1270, https://doi.org/10.1101/gr.276378.121.Search in Google Scholar PubMed PubMed Central
Sdeor, E., Okada, H., Saad, R., Ben-Yishay, T., and Ben-David, U. (2024). Aneuploidy as a driver of human cancer. Nat. Genet. 56: 2014–2026, https://doi.org/10.1038/s41588-024-01916-2.Search in Google Scholar PubMed
Shaid, S., Brandts, C.H., Serve, H., and Dikic, I. (2013). Ubiquitination and selective autophagy. Cell Death Differ. 20: 21–30, https://doi.org/10.1038/cdd.2012.72.Search in Google Scholar PubMed PubMed Central
Sheltzer, J.M. (2013). A transcriptional and metabolic signature of primary aneuploidy is present in chromosomally unstable cancer cells and informs clinical prognosis. Cancer Res. 73: 6401–6412, https://doi.org/10.1158/0008-5472.can-13-0749.Search in Google Scholar
Sheltzer, J.M., Torres, E.M., Dunham, M.J., and Amon, A. (2012). Transcriptional consequences of aneuploidy. Proc. Natl. Acad. Sci. U. S. A. 109: 12644–12649, https://doi.org/10.1073/pnas.1209227109.Search in Google Scholar PubMed PubMed Central
Shorter, J. (2017). Designer protein disaggregases to counter neurodegenerative disease. Curr. Opin. Genet. Dev. 44: 1–8, https://doi.org/10.1016/j.gde.2017.01.008.Search in Google Scholar PubMed PubMed Central
Singla, S., Iwamoto-Stohl, L.K., Zhu, M., and Zernicka-Goetz, M. (2020a). Autophagy-mediated apoptosis eliminates aneuploid cells in a mouse model of chromosome mosaicism. Nat. Commun. 11: 2958, https://doi.org/10.1038/s41467-020-16796-3.Search in Google Scholar PubMed PubMed Central
Singla, S., Iwamoto-Stohl, L.K., Zhu, M., and Zernicka-Goetz, M. (2020b). Autophagy-mediated apoptosis eliminates aneuploid cells in a mouse model of chromosome mosaicism. Nat. Commun. 11, https://doi.org/10.1038/s41467-020-16796-3.Search in Google Scholar PubMed PubMed Central
Skuodas, S., Clemons, A., Hayes, M., Goll, A., Zora, B., Weeks, D.L., Phillips, B.T., and Fassler, J.S. (2020). The ABCF gene family facilitates disaggregation during animal development. Mol. Biol. Cell 31: 1324–1345, https://doi.org/10.1091/mbc.e19-08-0443.Search in Google Scholar
Stefani, M. (2004). Protein misfolding and aggregation: new examples in medicine and biology of the dark side of the protein world. Biochim. Biophys. Acta 1739: 5–25, https://doi.org/10.1016/j.bbadis.2004.08.004.Search in Google Scholar PubMed
Stingele, S., Stoehr, G., Peplowska, K., Cox, J., Mann, M., and Storchova, Z. (2012). Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol. Syst. Biol. 8: 608, https://doi.org/10.1038/msb.2012.40.Search in Google Scholar PubMed PubMed Central
Sweeny, E.A. and Shorter, J. (2016). Mechanistic and structural insights into the prion-disaggregase activity of Hsp104. J. Mol. Biol. 428: 1870–1885, https://doi.org/10.1016/j.jmb.2015.11.016.Search in Google Scholar PubMed PubMed Central
Sweeny, E.A., Jackrel, M.E., Go, M.S., Sochor, M.A., Razzo, B.M., Desantis, M.E., Gupta, K., and Shorter, J. (2015). The Hsp104 N-terminal domain enables disaggregase plasticity and potentiation. Mol. Cell 57: 836–849, https://doi.org/10.1016/j.molcel.2014.12.021.Search in Google Scholar PubMed PubMed Central
Takahashi, A., Okada, R., Nagao, K., Kawamata, Y., Hanyu, A., Yoshimoto, S., Takasugi, M., Watanabe, S., Kanemaki, M.T., Obuse, C., et al.. (2017). Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat. Commun. 8: 15287, https://doi.org/10.1038/ncomms15287.Search in Google Scholar PubMed PubMed Central
Tan, K.L., Lee, H.C., Cheah, P.S., and Ling, K.H. (2023). Mitochondrial dysfunction in down syndrome: from pathology to therapy. Neuroscience 511: 1–12, https://doi.org/10.1016/j.neuroscience.2022.12.003.Search in Google Scholar PubMed
Tang, Y.C., Williams, B.R., Siegel, J.J., and Amon, A. (2011). Identification of aneuploidy-selective antiproliferation compounds. Cell 144: 499–512, https://doi.org/10.1016/j.cell.2011.01.017.Search in Google Scholar PubMed PubMed Central
Tang, Y.C., Hui, Y.W., Wang, K.Y., Bruno, P.M., Bullock, K., Deik, A., Santaguida, S., Trakala, M., Pfau, S.J., Zhong, N., et al.. (2017). Aneuploid cell survival relies upon sphingolipid homeostasis. Cancer Res. 77: 5272–5286, https://doi.org/10.1158/0008-5472.CAN-17-0049.Search in Google Scholar PubMed PubMed Central
Tange, Y., Kurabayashi, A., Goto, B., Hoe, K.L., Kim, D.U., Park, H.O., Hayles, J., Chikashige, Y., Tsutumi, C., Hiraoka, Y., et al.. (2012). The CCR4-NOT complex is implicated in the viability of aneuploid yeasts. PLoS Genet. 8, https://doi.org/10.1371/journal.pgen.1002776.Search in Google Scholar PubMed PubMed Central
Terhorst, A., Sandikci, A., Keller, A., Whittaker, C.A., Dunham, M.J., and Amon, A. (2020). The environmental stress response causes ribosome loss in aneuploid yeast cells. Proc. Natl. Acad. Sci. U. S. A. 117: 17031–17040, https://doi.org/10.1073/pnas.2005648117.Search in Google Scholar PubMed PubMed Central
Torres, E.M., Sokolsky, T., Tucker, C.M., Chan, L.Y., Boselli, M., Dunham, M.J., and Amon, A. (2007). Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317: 916–924, https://doi.org/10.1126/science.1142210.Search in Google Scholar PubMed
Torres, E.M., Dephoure, N., Panneerselvam, A., Tucker, C.M., Whittaker, C.A., Gygi, S.P., Dunham, M.J., and Amon, A. (2010). Identification of aneuploidy-tolerating mutations. Cell 143: 71–83, https://doi.org/10.1016/j.cell.2010.08.038.Search in Google Scholar PubMed PubMed Central
Vehvilainen, P., Koistinaho, J., and Gundars, G. (2014). Mechanisms of mutant SOD1 induced mitochondrial toxicity in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 8: 126, https://doi.org/10.3389/fncel.2014.00126.Search in Google Scholar PubMed PubMed Central
Venkatraman, P., Wetzel, R., Tanaka, M., Nukina, N., and Goldberg, A.L. (2004). Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol. Cell 14: 95–104, https://doi.org/10.1016/s1097-2765(04)00151-0.Search in Google Scholar PubMed
Vittoria, M.A., Quinton, R.J., and Ganem, N.J. (2023). Whole-genome doubling in tissues and tumors. Trends Genet. 39: 954–967, https://doi.org/10.1016/j.tig.2023.08.004.Search in Google Scholar PubMed PubMed Central
Wallace, E.W.J., Kear-Scott, J.L., Pilipenko, E.V., Schwartz, M.H., Laskowski, P.R., Rojek, A.E., Katanski, C.D., Riback, J.A., Dion, M.F., Franks, A.M., et al.. (2015). Reversible, specific, active aggregates of endogenous proteins assemble upon heat stress. Cell 162: 1286–1298, https://doi.org/10.1016/j.cell.2015.08.041.Search in Google Scholar PubMed PubMed Central
Weaver, B.A., Silk, A.D., Montagna, C., Verdier-Pinard, P., and Cleveland, D.W. (2007). Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell 11: 25–36, https://doi.org/10.1016/j.ccr.2006.12.003.Search in Google Scholar PubMed
Xian, S., Dosset, M., Almanza, G., Searles, S., Sahani, P., Waller, T.C., Jepsen, K., Carter, H., and Zanetti, M. (2021). The unfolded protein response links tumor aneuploidy to local immune dysregulation. EMBO Rep. 22: e52509, https://doi.org/10.15252/embr.202152509.Search in Google Scholar PubMed PubMed Central
Yamada, T., Murata, D., Adachi, Y., Itoh, K., Kameoka, S., Igarashi, A., Kato, T., Araki, Y., Huganir, R.L., Dawson, T.M., et al.. (2018). Mitochondrial stasis reveals p62-mediated ubiquitination in parkin-independent mitophagy and mitigates nonalcoholic fatty liver disease. Cell Metab. 28: 588–604.e5, https://doi.org/10.1016/j.cmet.2018.06.014.Search in Google Scholar PubMed PubMed Central
Yamada, T., Dawson, T.M., Yanagawa, T., Iijima, M., and Sesaki, H. (2019). SQSTM1/p62 promotes mitochondrial ubiquitination independently of PINK1 and PRKN/parkin in mitophagy. Autophagy 15: 2012–2018, https://doi.org/10.1080/15548627.2019.1643185.Search in Google Scholar PubMed PubMed Central
Yano, H., Baranov, S.V., Baranova, O.V., Kim, J., Pan, Y., Yablonska, S., Carlisle, D.L., Ferrante, R.J., Kim, A.H., and Friedlander, R.M. (2014). Inhibition of mitochondrial protein import by mutant huntingtin. Nat. Neurosci. 17: 822–831, https://doi.org/10.1038/nn.3721.Search in Google Scholar PubMed PubMed Central
Yim, A., Koti, P., Bonnard, A., Marchiano, F., Dürrbaum, M., Garcia-Perez, C., Villaveces, J., Gamal, S., Cardone, G., Perocchi, F., et al.. (2020). mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dynamics and mutations. Nucleic Acids Res. 48: 605–632, https://doi.org/10.1093/nar/gkz1128.Search in Google Scholar PubMed PubMed Central
Zhu, J., Tsai, H.J., Gordon, M.R., and Li, R. (2018). Cellular stress associated with aneuploidy. Dev. Cell 44: 420–431, https://doi.org/10.1016/j.devcel.2018.02.002.Search in Google Scholar PubMed PubMed Central
Zhu, P.J., Khatiwada, S., Cui, Y., Reineke, L.C., Dooling, S.W., Kim, J.J., Li, W., Walter, P., and Costa-Mattioli, M. (2019). Activation of the ISR mediates the behavioral and neurophysiological abnormalities in Down syndrome. Science 366: 843–849, https://doi.org/10.1126/science.aaw5185.Search in Google Scholar PubMed PubMed Central
© 2025 the author(s), published by De Gruyter, Berlin/Boston
This work is licensed under the Creative Commons Attribution 4.0 International License.
Articles in the same Issue
- Frontmatter
- Stress response pathways: machineries and mechanisms
- Computational strategies in systems-level stress response data analysis
- Back to the basics: the molecular blueprint of plant heat stress transcription factors
- Unfolded protein responses in Chlamydomonas reinhardtii
- Diversification of glutathione transferases in plants and their role in oxidative stress defense
- How neurons cope with oxidative stress
- The mitochondrial unfolded protein response: acting near and far
- MitoStores: stress-induced aggregation of mitochondrial proteins
- Unclogging of the TOM complex under import stress
- The mitochondrial intermembrane space – a permanently proteostasis-challenged compartment
- The nascent polypeptide-associated complex (NAC) as regulatory hub on ribosomes
- The evolution and diversification of the Hsp90 co-chaperone system
- The proteostasis burden of aneuploidy
Articles in the same Issue
- Frontmatter
- Stress response pathways: machineries and mechanisms
- Computational strategies in systems-level stress response data analysis
- Back to the basics: the molecular blueprint of plant heat stress transcription factors
- Unfolded protein responses in Chlamydomonas reinhardtii
- Diversification of glutathione transferases in plants and their role in oxidative stress defense
- How neurons cope with oxidative stress
- The mitochondrial unfolded protein response: acting near and far
- MitoStores: stress-induced aggregation of mitochondrial proteins
- Unclogging of the TOM complex under import stress
- The mitochondrial intermembrane space – a permanently proteostasis-challenged compartment
- The nascent polypeptide-associated complex (NAC) as regulatory hub on ribosomes
- The evolution and diversification of the Hsp90 co-chaperone system
- The proteostasis burden of aneuploidy