An improved and sustainable approach for the synthesis of α,β-dibromo ketones using ceric ammonium nitrate and ammonium bromide
-
Balaji B. Totawar
Balaji B. Totawar is a research scholar working for his doctoral degree at the School of Chemical Sciences, SRTM University, Nanded. He has been the recipient of a Rajiv Gandhi UGC fellowship, and his research interests include the development of CAN-mediated synthetic transformations.
Pramod S. Kulkarni completed his PhD at Swami Ramanand Teerth Marathwada University, Nanded. He published five research papers in his PhD work. He is working as an Assistant Professor of Chemistry in Hutatma Rajguru Mahavidyalaya, Rajgurunagar Pune, India. He completed two minor research projects.
Zubaidha K. Pudukulathan is a Professor at the School of Chemical Sciences, SRTM University, Nanded. She has been working on the development of synthetic methodologies and design of enzyme inhibitors.


Abstract
A simple and environmentally benign procedure for the bromination of substituted α,β-unsaturated ketones in good yield has been described using ammonium bromide as a brominating agent and ceric ammonium nitrate (CAN) as a single-electron oxidant to afford α,β-dibromoketones. The reaction involves C-Br bond formation by radical method generated by CAN. The reaction can be carried out by either at room temperature, stirring in solvent CH3CN or under aqueous moist condition with few drops of water by manual grinding in a mortar and pestle.
1 Introduction
The central theme of organic synthesis is carbon-carbon and carbon-heteroatom bond-forming reactions which comprise a major progress in modern synthesis, and this progress in turn results in development of novel methodologies. The application of carbon-centered radicals resulting from redox processes mediated by high-valent metal salts such as Mn (III), Co (III), V (V) and Ce (IV) has emerged as a powerful tool for carbon-carbon bond formation in recent years [1, 2]. The use of Ce (IV) reagents as convenient oxidants for a variety of substrates is well established [3–5]. Recently, C-C [6–9] and C-heteroatom [10–13] bond-forming reactions mediated by ceric ammonium nitrate (CAN) have been the subject of considerable interest [8–13]. One-electron oxidant CAN is one of the most important reagents, which appeals more and more with explored findings, as well as due to its use as Lewis acid catalyst from the group of lanthanide (IV) complexes in organic synthesis [8]. For the generation of radicals, CAN has been found to have the chemically upper hand in many respects to widely employed manganese triacetate [14]. CAN has some auxiliary advantages in terms of low toxicity, cost, solubility in many organic solvents and stability in air.
Organic bromine compounds have traditionally played an important role as intermediates in the production of agrochemicals, pharmaceuticals and dyes and others [15, 16]; some bromine compounds have been used as flame retardant and disinfectant. Bromination [17, 18] is termed as an important organic transformation, and bromination-debromination strategy for the protection-deprotection of double bonds has a substantial role in organic synthesis. Also, dibromoalkanes are important compounds widely used as versatile synthetic intermediates in a considerable number of useful transformations and for the protection-deprotection of unsaturated hydrocarbons [19–23]. The conventional method for bromination involves the use of bromine in chlorinated solvents [24], and unfortunately, a vast number of reported procedures employ highly polluting reaction conditions. In this context, several alternate green brominating agents have been reported to avoid the use of hazardous molecular bromine. Some of the methods worth mentioning are I2O5-KBr [25], potassium and ammonium halides [26] as the bromine source, oxybromination of activated aromatic compounds catalyzed by ammonium molybdate [27], environmentally benign electrophilic and radical bromination “in water”: H2 O2 -HBr system [28], NH4Br-oxone [29, 30], solvent-free bromination reactions with sodium bromide and oxone [31], bromination of chalcones using grinding technique [32], action of tetrabutylammonium tribromide on para-substituted chalcones in protic and aprotic media [33], in situ generated bromine under oxidative conditions [34], pyridiniumbromide-perbromide, quaternary ammonium tribromide or tribromide perbromide [35]. Though the search for sustainable protocol has led to tremendous progress in this area, most of the methods suffer from use of acid or organic solvents and are time consuming. Herein we present two approaches for bromination of enones under mild conditions and hence a step forward towards sustainable method development in tune with the current research scenario.
2 Materials and methods
Melting points were recorded by open capillary method on an electrothermal melting point apparatus and are uncorrected. The NMR spectra were recorded in CDCl3 with tetramethylsilane as an internal standard on Agilent VnmrJ 3.2 Spectroscopy 400/54 ASP, USA; IR spectra were recorded on a FT-IR 3.1 Win-BOMEM, Italy, apparatus. Flash chromatography was performed on thin layer chromatography (TLC) grade silica gel, and analytical TLC was performed on Merck silica gel 60 F254 pre-coated plates. The spots were visualized under UV light or by iodine vapors. Solvents were distilled before use. Ammonium bromide and ceric ammonium nitrate were procured from SD Fine chemicals, Mumbai, India, and in the preparation of chalcones we used benzaldehyde produced by Rankem, New Delhi, acetophenone from Hi-media, Mumbai, and NaOH from SD Fine Chemicals. All other commercial substrates were purchased from standard sources, and most of them were from SD Fine chemicals and were used as such.
2.1 Experimental procedure
2.1.1 Method A:
To a mixture of α,β-unsaturated carbonyl compound (1 mmol) and NH4Br (2 mmol) in acetonitrle (3–5 ml), CAN (2 mmol) was added at room temperature with stirring. After completion of the reaction (monitored by TLC, in 30–35 min), red orange color of reaction mixture decolorizes with formation of white solid of ceric (III) ammonium nitrate. The solid was filtered off and washed with chloroform. The combined organic layer was rotaevaporated under reduced pressure, and the residue was crystallized from ethanol.
2.1.2 Method B:
A mixture of NH4Br (2 mmol) and α,β-unsaturated carbonyl compound (1 mmol) was powdered in a mortar with pestle, and CAN (2 mmol) was added with grinding. The resulting mixture was ground for 5 min till it became sticky. Then the sticky mass was transformed to gel by addition of a few drops of water and left for 5 min. With one final grinding, the mixture was extracted with chloroform, and the combined organic layer was dried over sodium sulfate and concentrated under a vacuum. The residue obtained was crystallized from ethanol, and the product thus obtained was reasonably pure for all practical purposes.
2.2 Spectral analysis of synthesized compounds
2,3-Dichloro-1,3-diphenylpropan-1-one (1a): Yield: By method A: 52% and method B: 00%; IR: 1690, 796, 780; 1H NMR (CDCl3): δ 5.78 (d, J=12.4 Hz, 1Hβ), 5.84 (d, J=12.4 Hz, 1Hα), 7.41–7.53(m, 8H), 8.01 (d, J=8 Hz, 2H); 13C NMR (CDCl3): δ 52.8 (Cβ), 72.1 (Cα), 128.2, 128.9, 129.3, 129.5, 134.0, 135.0, 138.3, 191.9 (C=O).
3,4-Dibromobutan-2-one (2a): Yield: By method A: 67% and method B: 65%; IR: 1678, 710, 675, cm-1; 1H NMR (CDCl3): δ 2.36 (s, 3H, 1H), 3.64 (dd, J=8, 12 Hz, 1H), 3.90 (dd, J=8, 12 Hz, 1H), 4.51 (dd, J=8, 12 Hz, 1H); 13C NMR (CDCl3): δ 25.8, 28.1, 61.9, 198.2 (C=O).
3,4-Dibromo-4-phenylbutan-2-one (2b): Yield: By method A: 93% and method B: 91%; IR: 1662, 720, 712; 1H NMR (CDCl3): δ 2.47 (s, 3H), 4.93 (d, J=12 Hz, 1Hβ), 5.31 (d, J=12 Hz, 1Hα), 7.41 (m, 5H); 13C NMR (CDCl3): δ 27.1, 49.5 (Cβ), 52.7 (Cα), 128.9, 129.3, 137.7, 191.4 (C=O).
2,3-Dibromo-1,3-diphenylpropan-1-one (2c): Yield: By method A: 91% and method B: 90%; IR: 1646, 725, 715; 1H NMR (CDCl3): δ 5.85 (d, J=12 Hz, 1Hβ), 5.66 (d, J=12 Hz, 1Hα), 7.26–7.67 (m, 8H), 8.09 (d, J=8 Hz, 2H); 13C NMR (CDCl3): δ 46.8 (Cβ), 49.7 (Cα), 128.3, 128.9, 129.0, 129.3, 134.2, 191.2 (C=O).
2,3-Dibromo-3-phenyl-1-p-tolylpropan-1-one (2d): Yield: By method A: 94% and method B: 90%; IR: 1638, 727, 715; 1H NMR (CDCl3): δ 2.41 (s, 3H), 5.49 (d, J=12 Hz, 1Hβ), 5.62 (d, J=12 Hz, 1Hα), 7.46 (m, 5H), 7.10 (d, J=8 Hz, 2H), 7.91 (d, J=8 Hz, 2H); 13C NMR (CDCl3): δ 21.32, 44.7 (Cβ), 54.1 (Cα), 127.1, 128.0, 128.3, 128.8, 129.2, 135.0, 135.6, 139.9 (C-CH3), 189.1 (C=O).
2,3-Dibromo-1-(4-methoxyphenyl)-3-phenylpropan-1-one (2e): Yield: By method A: 20% and method B: 75%; IR: 1635, 1112, 727, 718; 1H NMR (CDCl3): δ 3.89 (s, 3H), 5.64 (d, J=12 Hz, 1Hβ), 5.80 (d, J=12 Hz, 1Hα), 7.01 (dd, J=8, 8 Hz, 2H), 7.39–7.43 (m, 3H), 7.52 (d, J=8 Hz, 2H), 8.09 (dd, J=8, 8 Hz, 2H); 13C NMR (CDCl3): δ 46.7 (Cβ), 50.0 (Cα), 55.6 (O-CH3), 114.2, 127.1, 128.3, 128.8, 129.2, 131.3, 138.4, 164.4 (C4′), 189.1 (C=O).
2,3-Dibromo-1-(4-nitrophenyl)-3-phenylpropan-1-one (2f): Yield: By method A: 83% and method B: 79%; IR: 1640, 1180, 722, 716; 1H NMR (CDCl3): δ 5.59 (d, J=11.6 Hz, 1Hβ), 5.71 (d, J=11.6 Hz, 1Hα), 7.34–7.44 (m, 5H), 7.69 (d, J=8 Hz, 2H), 8.01 (d, J=8 Hz, 2H); 13C NMR (CDCl3): δ 46.0 (Cβ), 52.9 (Cα), 126.8, 128.4, 128.9, 129.2, 135.2, 136.9, 139.9, 153.2 (C4′), 191.1 (C=O).
2,3-Dibromo-1-(4-fluorophenyl)-3-phenylpropan-1-one (2g): Yield: By method A: 87% and method B: 82%; IR: 1632, 1235, 722, 717; 1H NMR (CDCl3): δ 5.66 (d, J=11.2 Hz, 1Hβ), 5.73 (d, J=11.2 Hz, 1Hα), 7.30 (m, 3H), 7.36 (d, J=8.4 Hz, 2H), 7.53 (m, 2H), 8.15 (d, J=8.4 Hz, 2H); 13C NMR (CDCl3): δ 46.1 (Cβ), 51.2 (Cα), 117.1, 128.7, 129.7, 129.9, 130.8, 134.8, 136.8, 165.7 (C4′), 189.7 (C=O).
2,3-Dibromo-1-(4-chlorophenyl)-3-phenylpropan-1-one (2h): Yield: By method A: 93% and method B: 90%; IR: 1634, 780, 725, 720; 1H NMR (CDCl3): δ 5.66 (d, J=12.4 Hz, 1Hβ), 5.78 (d, J=12.4 Hz, 1Hα), 7.34 (m, 5H), 7.59 (d, J=8 Hz, 2H), 7.87 (d, J=8 Hz, 2H); 13C NMR (CDCl3): δ 46.1 (Cβ), 59.2 (Cα), 128.1, 128.7, 129.2, 129.4, 131.7, 135.2, 137.9, 140.7 (C4′), 189.0 (C=O).
2,3-Dibromo-1-(4-bromophenyl)-3-phenylpropan-1-one (2i): Yield: By method A: 91% and method B: 89%; IR: 1633, 726, 720, 630; 1H NMR (CDCl3): δ 5.63 (d, J=12 Hz, 1Hβ), 5.77 (d, J=12 Hz, 1Hα), 7.52 (m, 5H), 7.70 (d, J=8 Hz, 2H), 7.97 (d, J=8 Hz, 2H); 13C NMR (CDCl3): δ 46.5 (Cβ), 49.6 (Cα), 128.3, 128.8, 129.3, 129.5, 130.3, 132.3, 132.9, 137.9 (C4′), 190.2 (C=O).
2,3-Dibromo-1-(3-chlorophenyl)-3-phenylpropan-1-one (2j): Yield: By method A: 87% and method B: 82%; IR: 1630, 770, 726, 720; 1H NMR (CDCl3): δ 5.62 (d, J=12 Hz, 1Hβ), 5.76 (d, J=12 Hz, 1Hα), 7.52 (m, 6H), 8.03 (d, J=8 Hz, 2H); 13C NMR (CDCl3): δ 46.7 (Cβ), 49.6 (Cα), 128.3, 128.9, 129.0, 129.1, 129.9, 130.2, 132.9, 138.0 (C4′), 191.1 (C=O).
2,3-Dibromo-1,3-bis(4-fluorophenyl)propan-1-one (2k): Yield: By method A: 92% and method B: 90%; IR: 1631, 1290, 729, 722; 1H NMR (CDCl3): δ 5.65 (d, J=12 Hz, 1Hβ), 5.72 (d, J=12 Hz, 1Hα), 7.31 (m, 4H), 7.40 (d, J=8.4 Hz, 2H), 7.89 (d, J=8.4 Hz, 2H); 13C NMR (CDCl3): δ 45.7 (Cβ), 50.6 (Cα), 116.9, 117.7, 130.1, 130.8, 131.2, 134.2, 164.1, 165.9, 190.1 (C=O).
2,3-Dibromo-3-(2-fluorophenyl)-1-(4-fluorophenyl)propan-1-one (2l): Yield: By method A: 87% and method B: 80%; IR: 1631, 1280, 720, 724; 1H NMR (CDCl3): δ 5.66 (d, J=12 Hz, 1Hβ), 5.77 (d, J=12 Hz, 1Hα), 7.31 (m, 6H), 7.89 (d, J=8 Hz, 2H); 13C NMR (CDCl3): δ 46.2 (Cβ), 52.1 (Cα), 116.2, 117.2, 126.4, 129.6, 129.9, 130.7, 133.3, 161.2, 165.2, 189.7 (C=O).
2,3-Dibromo-1-phenyl-3-p-tolylpropan-1-one (2m): Yield: By method A: 91% and method B: 88%; IR: 1629, 724, 718; 1H NMR (CDCl3): δ 5.66 (d, J=12 Hz, 1Hβ), 5.77 (d, J=12 Hz, 1Hα), 7.31 (m, 6H), 7.89 (d, J=8 Hz, 2H); 13C NMR (CDCl3): δ 21.5, 46.2 (Cβ), 49.6 (Cα), 127.9, 128.4, 128.9, 129.1, 129.9, 135.2, 136.8, 140.1, 190.8 (C=O).
2,3-Dibromo-3-(4-methoxyphenyl)-1-phenylpropan-1-one (2n): Yield: By method A: 20% and method B: 75%; IR: 1632, 1105, 728, 718; 1H NMR (CDCl3): δ 3.81 (s, 3H), 5.61 (d, J=12 Hz, 1Hβ), 5.73 (d, J=12 Hz, 1Hα), 6.95 (dd, J=8.8 Hz, 2H), 7.26 (dd, J=8,8 Hz, 2H), 7.63–7.51 (m, 3H), 7.87 (d, J=8 Hz, 2H) 13C NMR (CDCl3): δ 43.9 (Cβ), 49.2 (Cα), 55.1, 114.9, 128.2, 128.7, 131.0, 132.2, 134.1, 137.0, 160.1 (C4), 190.8 (C=O).
2,3-Dibromo-3-(4-nitrophenyl)-1-phenylpropan-1-one (2o): Yield: By method A: 83% and method B: 80%; IR: 1638, 1370, 722, 716; 1H NMR (CDCl3): δ 5.71 (d, J=12 Hz, 1Hβ), 5.63 (d, J=12 Hz, 1Hα), 7.71–7.31 (m, 5H), 7.24 (d, J=8.4 Hz, 2H), 8.10 (d, J=8.4 Hz, 2H); 13C NMR (CDCl3): δ 44.9 (Cβ), 50.9 (Cα), 124.7, 128.2, 128.6, 129.0, 129.5, 134.7, 136.3, 149.3 (C4), 190.8 (C=O).
3 Results and discussion
CAN in the presence of NH4Br brings about bromination of enones and chalcones to afford the corresponding dibromide in good to excellent yields (Scheme 1).

Reaction of chalcone with different ammonium halides.
As the nature of the solvent is expected to play a crucial role in bromination, the reaction has been studied in different solvents commonly employed for the same. Table 1 displays the different solvents employed and their effect on yield and time. The reaction proceeded in most of the solvents screened, and acetonitrile was found to be the solvent of choice to afford the dibromo compound in 90% yield within 30–35 min at room temperature. Also, the reaction proceeded in appreciable way in DMF/DMSO in terms of conversion and time required for completion. The best results were obtained in grinding method under moist conditions to the tune of 90% yield within 20–25 min.
Effect of solvent on time and yield in the bromination of chalcone with NH4Br and CAN.a
| Sr. No. | Solvent | Time | Temperature | % Yield |
|---|---|---|---|---|
| 1 | CH2Cl2 | 5–6 h | RT (25–27°C) | 45 |
| 2 | CHCl3 | 5–6 h | RT (25–27°C) | 52 |
| 3 | Ethyl acetate | 12 h | RT (25–27°C) | 68 |
| 4 | Methanol | 2.5 h | RT (25–27°C) | 72 |
| 5 | Ethanol | 2 h | RT (25–27°C) | 76 |
| 6 | CH3CN | 35–40 min | RT (25–27°C) | 91 |
| 7 | DMF | 50–55 min | RT (25–27°C) | 84 |
| 8 | DMSO | 50–55 min | RT (25–27°C) | 86 |
| 9 | Water | 12 h | Reflux | 55 |
| 10 | Grinding | 20–25 min | RT (25–27°C) | 90 |
aStudy of reaction between chalcone and ammonium bromide was carried out with almost all solvents reported in literature.
Further, the applicability of the present conditions with other ammonium halides have been explored, and the results are presented in Table 2. As is obvious from the table, the present reaction conditions did not work with ammonium iodide and fluoride for the respective halogen addition in both the methods studied, while chlorination with ammonium chloride proceeded in moderate yield in acetonitrile, and no reaction was observed in the grinding method.
Halogenations of chalcones with CAN using different ammonium halides.a
| Sr. No. | Substrate | Ammonium halide | Product | % Yield | |
|---|---|---|---|---|---|
| Stirring with solvent A | Grinding with few drops of water B | ||||
| 1. | 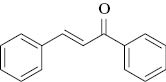 | NH4F | No reaction | No reaction | No reaction |
| 2. |  | NH4Cl |  | 52 | No reaction |
| 3 |  | NH4Br |  | 91 | 90 |
| 4 |  | NH4I | No reaction | No reaction | No reaction |
aHalogenations of chalcone in CH3CN with different ammonium halides were carried out under all possible conditions like mild heating and reflux to get good yields in minimum time, and the optimized results are presented here.
Having optimized the reaction conditions for bromination in solvent acetonitrile and by grinding method (Scheme 2), different substituted enones/chalcones were subjected to the reaction conditions, and the results are presented in Table 3. Methyl vinyl ketone and benzylideneacetone (Table 3, entries 1 and 2) required less time for conversion compared to other substituted chalcones. As expected, the reaction proceeded better with chalcones bearing electron-donating substituents as compared to the ones bearing electron-withdrawing groups. Substituents on the aromatic ring exert effect on the conversion and time. It is remarkable that bromination of methoxy-substituted chalcones (Table 3, entries 5 and 14) proceeded well under grinding method, and the same in acetonitrile gave low yield of 20% after 11–12 h of stirring at room temperature.
Bromination of different α,β unsaturated ketones with NH4Br and CAN.a
| Sr. No. | Substrate | Product | % Yieldb | Time | M. P. in °C | ||
|---|---|---|---|---|---|---|---|
| Stirred in solvent | Grinding in mortar and pestle | Stirred in solvent | Grinding in mortar and pestle | ||||
| 1 |  |  | 52 | – | 1 h | – | – |
| 2 |  |  | 67 | 65 | 10 min | 15–20 min | - |
| 3 |  |  | 93 | 91 | 25–30 min | 15–20 min | 123–125 |
| 4 |  |  | 91 | 90 | 35–40 min | 15–20 min | 156–158 |
| 5 |  |  | 94 | 90 | 35–40 min | 15–20 min | 147–149 |
| 6 |  |  | 20 | 75 | 4 h | 15–20 min | 146–148 |
| 7 |  |  | 83 | 79 | 35–40 min | 15–20 min | 188–190 |
| 8 |  |  | 87 | 82 | 35–40 min | 15–20 min | 156–158 |
| 9 |  | 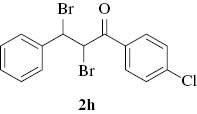 | 93 | 90 | 35–40 min | 15–20 min | 171–173 |
| 10 |  |  | 91 | 89 | 35–40 min | 15–20 min | 185–186 |
| 11 |  |  | 87 | 82 | 35–40 min | 15–20 min | 175–177 |
| 12 |  |  | 92 | 90 | 35–40 min | 15–20 min | 155–157 |
| 13 |  |  | 87 | 80 | 35–40 min | 15–20 min | 142–144 |
| 14 |  |  | 91 | 88 | 35–40 min | 15–20 min | 148–150 |
| 15 |  |  | 20 | 75 | 4 h | 15–20 min | 90–92 |
| 16 | 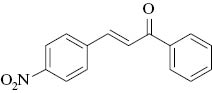 |  | 83 | 80 | 35–40 min | 15–20 min | 130–132 |
aMethod A: Reaction was performed at room temperature (28–30°C) with two equivalent of CAN in acetonitrile.
Method B: The reaction was carried out by grinding the reactants with mortar and pestle.
bYields refer to pure isolated products fully characterized by spectral and analytical methods.

Synthesis of dibromo chalcone using ammonium bromide and CAN.
Mechanistically, the bromination seems to proceed via addition of bromine radical formed by single-electron transfer to CAN (Figure 1).

Mechanism for synthesis of dibromo chalcone using ammonium bromide and CAN.
4 Conclusion
As dibromoalkanes are important compounds widely used as versatile synthetic intermediates in a considerable number of useful transformations, their easy access by the present method would find widespread use in organic synthesis. The present work highlights the usefulness of CAN and NH4Br as ecofriendly and mild reagent and salt, for economical, ecologically favorable bromination protocol with easy workup procedure coupled with short reaction times. The grinding method looks attractive in terms of conversion and reaction time, and the ease of handling cheaper ammonium bromide as a source of bromine makes the strategy an option of choice.
About the authors

Balaji B. Totawar is a research scholar working for his doctoral degree at the School of Chemical Sciences, SRTM University, Nanded. He has been the recipient of a Rajiv Gandhi UGC fellowship, and his research interests include the development of CAN-mediated synthetic transformations.

Pramod S. Kulkarni completed his PhD at Swami Ramanand Teerth Marathwada University, Nanded. He published five research papers in his PhD work. He is working as an Assistant Professor of Chemistry in Hutatma Rajguru Mahavidyalaya, Rajgurunagar Pune, India. He completed two minor research projects.

Zubaidha K. Pudukulathan is a Professor at the School of Chemical Sciences, SRTM University, Nanded. She has been working on the development of synthetic methodologies and design of enzyme inhibitors.
Acknowledgments
One of the authors, Balaji Totawar, would like to acknowledge UGC Scheme – Rajiv Gandhi National Fellowship support provided for SC/ST students.
Conflict of interest: The authors confirm that this article content has no conflicts of interest.
References
[1] De Klein WJ. In Organic Synthesis by Oxidation with Metal Compounds, Mijs, WJ, de Jonge, CRH, Eds., Plenum: New York, 1986, pp. 261–314.10.1007/978-1-4613-2109-5_4Search in Google Scholar
[2] Iqbal J, Bhatia B, Nayyar N. Chem. Rev. 1994, 94, 519–564.10.1021/cr00026a008Search in Google Scholar
[3] Molander GA. Chem. Rev. 1992, 92, 29–68.10.1021/cr00009a002Search in Google Scholar
[4] Ho T. Synthesis 1973, 6, 347–354.10.1055/s-1973-22210Search in Google Scholar
[5] Nair V, Panicker SB, Nair LG, George TG, Augustine A. Synlett 2003, 2, 156–165.Search in Google Scholar
[6] Baciocchi E, Ruzziconi R. J. Org. Chem. 1991, 56, 4772–4778.10.1021/jo00015a037Search in Google Scholar
[7] Citterio A, Sebastiano R, Carvaya M. J. Org.Chem. 1991, 56, 5335–5341.10.1021/jo00018a024Search in Google Scholar
[8] Nair V, Mathew J, Prabhakaran J. Chem. Soc.Rev. 1997, 26, 127–132.10.1039/CS9972600127Search in Google Scholar
[9] Linker T, Hartmann K, Sommermann T, Scheutzov D, Ruckdeschel E. Angew. Chem. Int. Ed. Engl. 1996, 35, 1730–1732.10.1002/anie.199617301Search in Google Scholar
[10] Bosman C, Annibale AD, Resta S, Trogolo C. Tetrahedron Lett. 1994, 35, 6525–6528.10.1016/S0040-4039(00)78263-3Search in Google Scholar
[11] Narasaka K, Mochizuk T, Hayakawa S. Chem. Lett. 1994, 23, 1705–1708.10.1246/cl.1994.1705Search in Google Scholar
[12] Nair V, George TG, Nair LG, Panicker S. Tetrahedron Lett. 1999, 40, 1195–1196.10.1016/S0040-4039(98)02563-5Search in Google Scholar
[13] Trahanovsky WS, Robbins M. J. Am. Chem. Soc. 1971, 93, 5256–5258.10.1021/ja00749a050Search in Google Scholar
[14] Holzweber M, Schnürch M, Stanetty P. Synlett 2007, 19, 3016–3018.10.1055/s-2007-992354Search in Google Scholar
[15] Gribble G. Chem. Soc. Rev. 1999, 28, 335–346.10.1039/a900201dSearch in Google Scholar
[16] Ioffe D, Kampf A. In Kirk-Othmer Encyclopedia of Chemical Technology, 5th ed., Kirk-Othmer, ed., John Wiley & Sons Inc: New York, 2004, Vol. 4, pp. 340.Search in Google Scholar
[17] Kutsumura N, Niwa K, Saito T. Org. Lett. 2010, 12, 3316–3319.10.1021/ol101110vSearch in Google Scholar
[18] Kuang C, Yang Q, Senboku H, Tokuda M. Tetrahedron 2005, 61, 4043–4052.10.1016/j.tet.2005.02.043Search in Google Scholar
[19] Maji T, Karmakar A, Reiser O. J. Org. Chem. 2011, 76, 736–739.10.1021/jo102239xSearch in Google Scholar
[20] Lee SH, Chang YM, Yoon C. Bull. Korean Chem. Soc. 2004, 25, 1723–1725.10.5012/bkcs.2004.25.11.1723Search in Google Scholar
[21] Totten LA, Jans U, Roberts A. Environ. Sci.Technol. 2001, 35, 2268–2274.10.1021/es0010195Search in Google Scholar
[22] Nair V, Panicker SB, Augusine A, George TG, Thomas S, Vairamani M. Tetrahedron 2001, 57, 7417–7422.10.1016/S0040-4020(01)00712-8Search in Google Scholar
[23] Ranu BC, Guchhait SK, Sarkar A. Chem. Commun. 1998, 19, 2113–2114.10.1039/a806530fSearch in Google Scholar
[24] Pavlinac J, Zupan M, Laali K, Stavber S. Tetrahedron 2009, 65, 5625–5665.10.1016/j.tet.2009.04.092Search in Google Scholar
[25] Hou J, Li Z, Jia XD, Liu Z. Synthetic Commun. 2014, 44, 181–187.10.1080/00397911.2013.796523Search in Google Scholar
[26] Kandepi VVKM, Narender N. Synthesis 2012, 44, 15–26.10.1055/s-0031-1289620Search in Google Scholar
[27] Choudary BM, Sudha Y, Reddy P. Synlett 1994, 6, 450.10.1055/s-1994-22886Search in Google Scholar
[28] Podgoršek A, Stavber S, Zupan M, Iskra J. Tetrahedron 2009, 65, 4429.10.1016/j.tet.2009.03.034Search in Google Scholar
[29] Kumar MA, Rohitha CN, Kulkarni SJ, Narender N. Synthesis 2010, 10, 1629.10.1055/s-0029-1218723Search in Google Scholar
[30] Naresh M, Kumar MA, Reddy MM, Swamy P, Nanubolu JB, Narender N. Synthesis 2013, 45, 1497.10.1055/s-0033-1338431Search in Google Scholar
[31] Wang GW, Gao J. Green Chem. 2012, 14, 1125.10.1039/c2gc16606bSearch in Google Scholar
[32] Jakhar K, Makrandi J. Indian J. Chem. 2013, 52B, 141–145.Search in Google Scholar
[33] Berthelot J, Benamma Y, Desmazieres B. Can. J. Chem. 1995, 73, 1526–1530.10.1139/v95-189Search in Google Scholar
[34] Eissen M, Lenoir D. Chem. Eur. J. 2008, 14, 9830–9841.10.1002/chem.200800462Search in Google Scholar
[35] Subbarayappa A, Gadde R, Bedekar AV, Ghosh S, Ranu BC, Ghosh P. Green Chem. 2006, 8, 916–922.10.1039/b606586dSearch in Google Scholar
©2016 by De Gruyter
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Articles in the same Issue
- Frontmatter
- In this issue
- Publisher’s note
- Happy Birthday Green Processing and Synthesis!
- Editorial
- Merging of the sciences and technologies: non-technological barriers
- Microwave energy treatment
- Microwave energy: a green synthesis and treatment solution
- Utilization of walnut shell as a feedstock for preparing high surface area activated carbon by microwave induced activation: effect of activation agents
- Optimization of drying ammonium tetramolybdate by microwave heating using response surface methodology
- Extraction of zinc from blast furnace dust in ammonia leaching system
- Kinetics of ultrasound-assisted silver leaching from sintering dust using thiourea
- Effects of roasting pretreatment on zinc leaching from complicated zinc ores
- Study on dechlorination kinetics from zinc oxide dust by clean metallurgy technology
- Microwave roasting of agglomerated flux for submerged-arc welding
- Glass-forming ability and mechanical properties of a Zr52.8Cu29.1Ni7.3Al9.8Y1 bulk metallic glass prepared by hereditary process
- Original articles
- An improved and sustainable approach for the synthesis of α,β-dibromo ketones using ceric ammonium nitrate and ammonium bromide
- Utilization of waste dried Mangifera indica leaves for extraction of mangiferin by conventional batch extraction and advance three-phase partitioning
- Research on the reduction of Guizhou oolitic hematite by hydrogen
- Sulfated metal oxides: eco-friendly green catalysts for esterification of nonanoic acid with methanol
- Application of Acacia modesta and Dalbergia sissoo gums as green matrix for silver nanoparticle binding
- Conference announcement
- Conferences 2016–2017
Articles in the same Issue
- Frontmatter
- In this issue
- Publisher’s note
- Happy Birthday Green Processing and Synthesis!
- Editorial
- Merging of the sciences and technologies: non-technological barriers
- Microwave energy treatment
- Microwave energy: a green synthesis and treatment solution
- Utilization of walnut shell as a feedstock for preparing high surface area activated carbon by microwave induced activation: effect of activation agents
- Optimization of drying ammonium tetramolybdate by microwave heating using response surface methodology
- Extraction of zinc from blast furnace dust in ammonia leaching system
- Kinetics of ultrasound-assisted silver leaching from sintering dust using thiourea
- Effects of roasting pretreatment on zinc leaching from complicated zinc ores
- Study on dechlorination kinetics from zinc oxide dust by clean metallurgy technology
- Microwave roasting of agglomerated flux for submerged-arc welding
- Glass-forming ability and mechanical properties of a Zr52.8Cu29.1Ni7.3Al9.8Y1 bulk metallic glass prepared by hereditary process
- Original articles
- An improved and sustainable approach for the synthesis of α,β-dibromo ketones using ceric ammonium nitrate and ammonium bromide
- Utilization of waste dried Mangifera indica leaves for extraction of mangiferin by conventional batch extraction and advance three-phase partitioning
- Research on the reduction of Guizhou oolitic hematite by hydrogen
- Sulfated metal oxides: eco-friendly green catalysts for esterification of nonanoic acid with methanol
- Application of Acacia modesta and Dalbergia sissoo gums as green matrix for silver nanoparticle binding
- Conference announcement
- Conferences 2016–2017