
The role of the Mpv17 protein mutations of which cause mitochondrial DNA depletion syndrome (MDDS): lessons from homologs in different species
-
Stefanie Löllgen
Stefanie Löllgen studied Applied Biology (Bsc) and Biomedical Sciences (Msc) at the Hochschule Bonn-Rhein-Sieg. Her Master’s thesis work is in part represented in this publication.
Hans Weiher graduated from the Universität Heidelberg, did postdoctoral work at UC Berkeley, USA, Heinrich Pette Institut, Hamburg, Germany, and the Whitehead Institute at MIT, Cambridge, USA. He held leading positions at various research institutions within Germany (Deutsches Krebsforschungszentrum, Heidelberg (DKFZ), Forschungszentrum Karlsruhe, and Institut für Diabetesforschung, München), and is now in charge of Human- and Molecular Genetics at Hochschule Bonn-Rhein-Sieg in Rheinbach, Germany.


Abstract
Mitochondrial DNA depletion syndromes (MDDS) are severe pediatric diseases with diverse clinical manifestations. Gene mutations that underlie MDDS have been associated with alterations in the mitochondrial DNA (mtDNA) replication machinery or in mitochondrial deoxyribonucleoside triphosphate pools. However, the nuclear gene MPV17, whose mutated forms are associated with hepatocerebral MDDS in humans, plays a so-far unknown role in mtDNA maintenance. A high degree of conservation has been determined between MPV17 and its mouse (Mpv17), zebrafish (tra) and yeast (SYM1) homologs, respectively, whereby mutants in these cause very different phenotypes. While dysfunction in this gene in humans causes fatal liver disease, kidney pathology is induced in mice. Moreover, in zebrafish inactivation of the Mpv17 homolog was detected as a viable dyscolouration mutant. Knock out of the yeast ortholog results in a temperature-sensitive metabolic growth phenotype. Detailed analyses on common denominators between these different phenotypes strengthen the hypothesis that the Mpv17 protein forms a channel in the inner mitochondrial membrane, allowing small molecules – in vertebrates probably nucleotides, and in yeast probably intermediates of the tricarboxylic acid cycle – to pass. Moreover, a function modifying the pathologic manifestations of MPV17-related disease in mice has been identified. This signaling pathway remarkably involves the non-mitochondrial catalytic subunit of DNA-dependent protein kinase (PRKDC), important in double-strand break repair resistance against reactive oxygen-induced genotoxic stress.
Introduction
Mitochondria are indispensable organelles, in that they generate chemical energy via oxidative phosphorylation (OXPHOS) to fuel cellular activities (Wallace, 1999). These organelles contain their own genome distinct from that of the nucleus (Anderson et al., 1981). At least 1000 mitochondrial genome copies may be found per cell (Spinazzola and Zeviani, 2007). Human mitochondrial DNA (mtDNA) represents a double-stranded circular molecule that constitutes about 16.6 kb. It encodes 13 proteins, and 22 transfer RNAs and two ribosomal RNAs for protein synthesis (Anderson et al., 1981). The 13 proteins encoded in mtDNA represent subunits of the ATP synthase and components of the mitochondrial respiratory chain complexes. As the remaining majority of its subunit components are encoded in nuclear DNA, the respiratory chain is orchestrated by both nuclear and mitochondrial genome (Spinazzola and Zeviani, 2007). Replication of mtDNA occurs autonomously (Anderson et al., 1981) but depends entirely on proteins encoded in nuclear DNA (Spinazzola and Zeviani, 2007). Moreover, proteins required for mtDNA gene expression are contributed by the nucleus (Spinazzola and Zeviani, 2007). On account of the complex interplay of both genomes, the underlying causes of mitochondrial disease appear to be manifold (Copeland, 2012). Mitochondrial DNA depletion syndromes (MDDS) represent a remarkable group of mitochondrial diseases that is caused by mutation of nuclear genes (Copeland, 2012). These severe pediatric diseases are distinct from mtDNA deletion syndromes in that the mitochondrial genome remains intact while the copy number of mtDNA is low within cells (Copeland, 2012). As the catalytic subunits of respiratory chain complexes are encoded in the mitochondrial genome, deprivation of mtDNA results in compromised oxidative phosphorylation (OXPHOS) and thus, energy metabolism (Suomalainen and Isohanni, 2010).
MDDS are rare disorders of mitochondria. According to the observations of the Necker Hospital in Paris, the prevalence of MDDS appears to be about 8% of pediatric patients, who suffer from deficiencies of the respiratory chain (Rötig and Poulton, 2009). As the developing fetus uses glycolysis as the primary source of energy, the detrimental effects of mitochondrial dysfunction become evident after birth (Spinazzola, 2011). Then, the organ system requires OXPHOS for energy metabolism so that patients develop severe clinical manifestations and die at a young age (Moraes et al., 1991). Depending on the gene mutation underlying MDDS, distinct organs may be affected by mtDNA depletion (Suomalainen and Isohanni, 2010). MDDS appear as myopathic, encephalomyopathic or hepatocerebral forms with overlapping clinical presentations (Alberio et al., 2007). Consistently, depletion of mtDNA is usually most pronounced in liver and muscle tissue of patients (Dimmock et al., 2010) but little is known about how tissue specificity of MDDS is generated (Suomalainen and Isohanni, 2010). Mutations within the mtDNA appear not to be responsible in the depletion mechanism as mitochondria from MDDS patients restore normal mtDNA amounts when introduced into an intact nuclear environment (Bodnar et al., 1993; Taanman et al., 1997). Thus, MDDS is caused by mutations in nuclear genes. So far, mutations in at least 13 nuclear genes have been demonstrated to be associated with the instability of mtDNA (see Copeland, 2012; Kornblum et al., 2013). These includeTK2, TYMP, DGUOK, RRM2B, SUCLA2, SUCLG1, SLC25A4, OPA1, POLG, POLG2, PEO1 genes (Copeland, 2012). The corresponding gene products either regulate the deoxyribonucleosidetriphosphate (dNTP) pools or are directly involved in replication of mtDNA (Copeland, 2012). In addition, the mitochondrial genome maintenance exonuclease 1 (MGME1) gene was identified as new MDDS candidate gene (Kornblum et al., 2013). MGME1 cleaves 7S DNA, which constitutes the single-stranded displacement loop segment of premature mtDNA. Without MGME1 function, mtDNA replication is defective and accumulates stalled intermediates rather than mature mitochondrial genomes (Kornblum et al., 2013). Besides mutations in these well-characterized genes, mutation in the MPV17 gene has been found to be associated with MDDS, causing the hepatocerebral form of the disorder (Spinazzola et al., 2006). MPV17 is conserved in mammals (Karasawa et al., 1993), fish (Krauss et al., 2013), and yeast (Trott and Morano, 2004). However, the molecular function of the MPV17 protein has remained unclear (Spinazzola et al., 2006). Thus, we here review findings on MPV17 from different organisms to combine the current state of knowledge. From these data, the hypothesis that MPV17 protein represents a channel in the inner mitochondrial membrane conducting nucleotides for mtDNA synthesis is supported.
Human MPV17-related disease
The human MPV17 gene is located on chromosome 2p23-21 and encodes a protein composed of 176 amino acids (Karasawa et al., 1993), which was localized in the inner mitochondrial membrane (Spinazzola et al., 2006). Mutations of this gene were first described as causing hepatocerebral MDDS (Spinazzola et al., 2006) and Navajo neurohepathopathy (Karadimas et al., 2006). Since then, about 20 different Mpv17 mutations have been associated with disease. Patients are either homozygous or compound heterozygous for Mpv17 mutations, providing genetic evidence for them to be loss of function. Nonsense, missense, deletion and insertion mutations occur spread over the coding region of the gene and splicing mutations have also been found (El-Hattab et al., 2010). Figure 1 shows a model of the protein on which missense mutations and in frame deletions are indicated (adopted from El-Hattab et al., 2010).
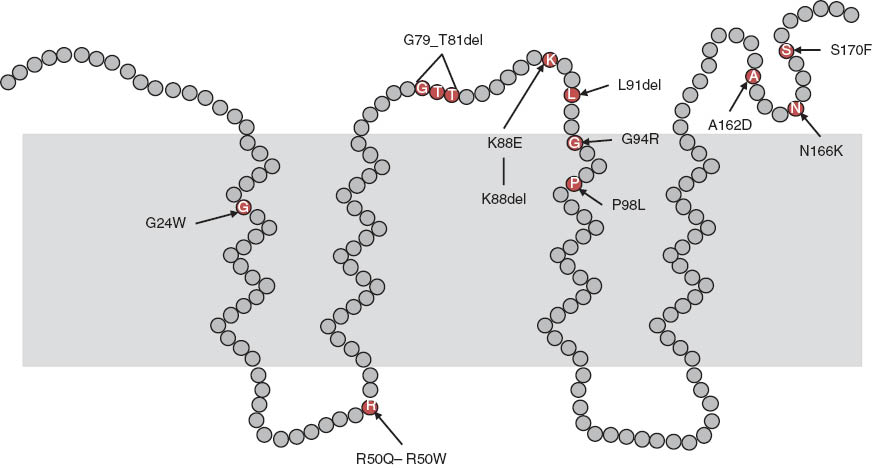
Model of the human MPV17 protein with point mutations and deletions leading to MDDS (data taken from El-Hattab et al., 2010).
In analogy to the SYM 1 protein in Sacharomyces cerevisiae (Reinhold et al. 2012) the N-and C-terminus reach into the intermembrane space. The inner membrane is indicated by shading. A theoretical PKC phosphorylation site is on position 80.
The spectrum of clinical presentations associated with Mpv17 mutations discovered since then is rather broad, whereby, generally, disease-onset is between neonatality and childhood (El-Hattab et al., 2010) although several patients with adult-onset symptoms have been described (Blakely et al., 2012; Garone et al., 2012). Hepatic and neurologic manifestations appear to be the most characteristic findings. All early-onset patients studied presented with liver dysfunction, comprising cholestasis, jaundice and coagulopathy (see Karadimas et al., 2006; Spinazzola et al., 2006, 2008; Wong et al., 2007; Navarro-Sastre et al., 2008; Kaji et al., 2009; Parini et al., 2009; El-Hattab et al., 2010; Merkle et al., 2012; Uusimaa et al., 2013). Patients developing cirrhosis or hepatocellular carcinoma were described less frequently (see Karadimas et al., 2006; El-Hattab et al., 2010). Most cases deteriorate to liver failure, being the main cause of death regarding this disorder. Most young patients present with neurologic manifestations, mainly involving developmental delay and hypotonia. Motor- and sensory peripheral neuropathy and leukoencephalopathy were also described. Besides hepatic and neurologic indications, hepatocerebral MDDS can result in metabolic manifestations, such as lactic acidosis and hypoglycemia, and it has frequently been observed that patients fail to thrive. Additional, less commonly noticed features are described in detail elsewhere (Karadimas et al., 2006; Spinazzola et al., 2006, 2008; Wong et al., 2007; Navarro-Sastre et al., 2008; Kaji et al., 2009; Parini et al., 2009; El-Hattab et al., 2010; Merkle et al., 2012; Uusimaa et al., 2013). Remarkably, the identical mutation might cause different severity, onset and pathology in different patients, as Blakely et al. describe a patient with adult-onset of neuropathy and leukoencephalopathy, carrying a homozygous mutation (P98L) described in infantile MDDS syndrome previously (El-Hattab et al., 2010; Blakely et al., 2012). It is interesting in this context, that a 65-year-old patient with an adult-onset multisystemic disorder was shown to carry compound heterozygous Mpv17 mutations (K-M 88-89 M-L, L143*; Garone et al., 2012), whereby mutations in pos. 88 have been described in infantile MDDS before (El-Hattab et al., 2010).
The cellular content of mtDNA has been found to be highly variable between patients, settling between 3 and 40% in liver in relation to that of age-matched controls. Milder depletion of mtDNA was reported for muscle, with mtDNA content between 8 and 100% of controls (see Karadimas et al., 2006; Spinazzola et al., 2006, 2008; Wong et al., 2007; Navarro-Sastre et al., 2008; Kaji et al., 2009; Parini et al., 2009; El-Hattab et al., 2010; Merkle et al., 2012; Uusimaa et al., 2013). In the adult-onset patients, multiple mitochondrial DNA deletions rather than depletion were found (Blakely et al., 2012; Garone et al., 2012).
The treatment of MDDS, whether related to mutation in MPV17 or other genes, is still insufficient comprising supplementation of vitamins, cofactors, substrates used in mitochondrial respiration, and glucose to support the metabolism. Acute cases of lactic acidosis may be rescued with sodium bicarbonate infusions (Spinazzola et al., 2008). Patients with progressed liver failure may receive liver transplantation, which is associated with poor prognosis, however. Regarding the experience with liver transplantation in such cases, one can estimate that less than half of the patients survive the procedure (Lee and Sokol, 2007). Recently, an MPV17 gene therapeutic approach was tested in a mouse model. Expression of Mpv17 was thereby induced in Mpv17 mutant mice through gene replacement via an adeno-associated virus carrying human MPV17. As a consequence, mtDNA copy number was enhanced in liver restoring oxidative phosphorylation activity (Bottani et al., 2014).
The Mpv17 mutant mouse model
Clinical manifestations and molecular biology
Before the role of the MPV17 gene in MDDS was established, a mouse strain, in which the Mpv17 gene was knocked out by insertional inactivation with a recombinant retrovirus had been generated and studied (Weiher et al., 1990). The mutagenic provirus was replication defective but its insertion produced loss of function mutations. In the Mpv17 mouse strain the expression of Mpv17 RNA was inactivated, leading to a phenotype in homozygous mutant animals. These developed renal disease characterized by focal segmental glomerulosclerosis (FSGS) and consequential nephrotic syndrome (Weiher et al., 1990). Specifically, homozygous Mpv17 mutant mice developed disease in early adulthood, presenting, at 6–8 weeks of age, with abnormal serum, blood, and urine parameters representative of renal failure followed by nephrotic syndrome. Abnormally high cholesterol, blood urea nitrogen, and creatinine levels were accompanied by proteinuria, and normocytic and normochromic anemia in progressed stages. Hence, beginning at 2–3 months of age, numerous animals lost weight, were inactive and developed pallor. Most Mpv17 transgenic mice died of renal failure after 18 weeks of life. Histologic analyses revealed that glomeruli were enlarged and that glomerular capillaries were degenerated and blocked by hyaline material containing plasma proteins and basement membrane residues. The resultant destruction of glomerular segments was classified as FSGS. The Mpv17 gene was found to be expressed in various tissues, including kidney, where the mutant phenotype was observed (Weiher et al., 1990). Human and murine coding regions of the Mpv17 gene were found to be conserved more than 90%, which indicated evolutionary significance (Karasawa et al., 1993). In mice, development of the mutant phenotype was prevented through transgenesis with human MPV17, indicating functional equivalence between the homologs (Schenkel et al., 1995). Further investigation of the Mpv17 mutant murine phenotype revealed that inner ear structures of Mpv17 mice were degenerated, involving deterioration of the organ of Corti, loss of inner and outer hair cells as well as spiral ganglion cells. In addition, the changes were obvious in the stria vascularis, and particularly the vascular basement membrane was dramatically thickened (Meyer zumGottesberge et al., 1996). The mice showed sensineural hearing loss progressing to deafness by 2 months of age (Müller et al., 1997). Remarkably, functional and structural glomerular and sensineural deterioration resembled the pathology of the inherited human disorder Alport’s syndrome, particularly with respect to basement membrane changes in the glomerula and the inner ear (Reuter et al., 1998). However, unlike in Alport’s, the basement membrane changes in kidney and inner ear were not caused by mutations in collagen genes (Lemmink et al., 1997). Instead, matrix metalloproteinase 2 (MMP-2), which has been shown to catalyze the breakdown of glomerular basement membranes (Johnson et al., 1992; Knowlden et al., 1995), was found to be over expressed in the mutant mice (Reuter et al., 1998). The kidney pathology was caused or at least mediated by excess of reactive oxygen (ROS), as it could be prevented by local administration of the scavenger dimethylthiourea (DMTU). Systemically, dietary supplementation with the lipid peroxidation scavenger probucol prevented glomerular disease in Mpv17 mutants (Binder et al., 1999). Detailed analysis showed that glomeruli of Mpv17-negative mice produced enhanced extracellular ROS production levels accompanied by the increased formation of intraglomerular lipid peroxidation adducts (Binder et al., 1999), products generated by the reaction of ROS with polyunsaturated fatty acids (Girotti, 1985). Thus, it appeared that in glomeruli, Mpv17 deficiency led to extracellular buildup of ROS, which resulted directly in noxic modifications of cellular components (Binder et al., 1999). An additional, indirect effect of ROS on basement membrane structure may be exerted via up regulation of MMP-2 (Reuter et al., 1998; Spallarossa et al., 2006). Of note, an early or primary liver phenotype was not detected. The left of Figure 2 depicts a scheme of the organismal pathology and its immediate causes in Mpv17 mutant mice.

Disturbed functions in the Mpv17 mouse model.
The tissue damage is indicated on the left, the cellular dysfunctions on the right. Prkdc stands for the modulating influence (mechanism unknown) of this kinase on mtDNA depletion and kidney phenotype (see text). Tissue damage might be through mtDNA depletion or caused by lack of Mpv17 function by a different pathway.
When mutant Mpv17 protein was found associated with human MDDS syndrome, the Mpv17 mutant mice were also investigated for mtDNA depletion. It revealed that the mutant mice displayed mtDNA depletion as well, which was highly variable among tissue types. While kidney and brain each contained 60% mtDNA of wt controls values, muscle exhibited 20%, and liver only 4% of the mtDNA content measured in wt animals (Spinazzola et al., 2006). It was furthermore shown, that in liver and skeletal muscle, around birth, mtDNA content of Mpv17 mutant and wild type cells were similar. Thereafter, the mtDNA copy number increased in wild type, while in Mpv17 mutant mice mtDNA levels stayed continuously low (Viscomi et al., 2009). This corresponded to the view that the developing fetus relies on glycolysis for energy production and that mitochondrial energy supply becomes indispensable only after birth (Spinazzola, 2011). In Figure 2, the causal relationship between the Mpv17 and mtDNA depletion and its potential action on the tissue pathology in Mpv17 mutant mice are schematically indicated. Despite severe mtDNA depletion in liver, however, a significant liver phenotype, characteristic for the human MDDS condition, was still not found. This might be explained by an upregulation of mitochondrial DNA gene expression under conditions of mtDNA depletion in these animals (Viscomi et al., 2009). As far as the kidney is concerned, glomerular podocytes were found to be depleted of mtDNA while renal tubules showed normal mtDNA levels, in line with the original concept, that the glomeruloscerosis phenotype in these animals is caused by a deficiency in glomerular podocytes (Weiher et al., 1990, Binder et al., 1999).
Thus, although the mtDNA depletion phenotype in the Mpv17 mutant mice is similar to the corresponding human phenotype, particularly being most prominent in liver, the clinical manifestations in both species appear to be very different. This may be because of, as mentioned above, compensatory mechanisms of mitochondrial gene expression in mouse liver or just reflect species-specific differences in tolerance to mtDNA depletion in different tissues.
The causal relationship between MPV17 mutations and mtDNA depletion in mice and humans on the one hand, and between Mpv17 knockout and ROS induced glomerular injury in mice on the other, raises the question of whether increased reactive oxygen (ROS) production – for instance within glomeruri – was actually the consequence of defective OXPHOS (Viscomi et al., 2009), or if ROS might constitute the driving force for mtDNA depletion through oxidative reactions with mtDNA. In fact, on the one hand, excess ROS can be generated at the mitochondrial electron transport chain (Shigenaga et al., 1994), and on the other, ROS are known to mediate genomic instability through the introduction of DNA single-strand or double-strand breaks (Woodbine et al., 2011).
An intriguing similarity to the phenotype expression in humans and mice in Mpv17 mutations is found with respect to mutations in the RRM2B gene. This gene encodes the p53-inducible small subunit (p53R2) of ribonucleotide reductase (RR) and is required for the maintenance of mtDNA copy number by contributing to de novo synthesis of desoxynucleotide triphosphate (dNTP) by way of conversion of ribonucleoside 5′-diphosphates into deoxyribonucleoside 5′-diphosphates. Consequently, mutations in this gene cause MDDS in humans (Bourdon et al., 2007). Rrm2b null mutants in mice, however, exhibit severe renal failure while the dNTP pools are attenuated (Kimura et al., 2003).
The molecular nature of the Mpv17 protein has been first deduced from its amino acid sequence. Bioinformatic analysis of the Mpv17 protein and its closest relatives – namely the mammalian peroxisomal proteins pxmp 22 (Kaldi et al., 1993) as well as the yeast homolog Sym 1 (Trott and Morano, 2004) – strongly suggested Mpv17 family proteins to be membrane proteins with four hydrophobic regions, likely to represent passages through a lipid membrane leaving the C- and N- terminus at the same side of the membrane (Figure 1). Over the years, a number of closely related proteins have been discovered in different species called the Mpv17 family of proteins. These share the general structure prediction, however, they appear to have different intracellular localizations. While pxmp 22, the closest homolog to Mpv17 (Kaldi et al., 1993), and Mpv17 like protein1 (Iida et al., 2006) are peroxisomal proteins, other family members, such as Mpv17 L2 in mammals (Dalla Rosa et al., 2014) and Sym 1 in yeast (Trott and Morano, 2004) localize to mitochondrial membranes. The mammalian Mpv17 protein itself had first been found in peroxisomes (Zwacka et al., 1994) but was later allocated to mitochondria (Spinazzola et al., 2006). This protein does not contain any known signal sequences and is not processed upon membrane transport or incorporation. It does contain a potential protein kinase C phosphopylation site at T80, which when deleted in patients, causes disease (El-Hattab et al., 2010; see Figure 1). However, Mpv17 has so-far not been demonstrated to be a phosphoprotein experimentally. From analogy with detailed in vitro data derived from the peroxisomal membrane protein pxmp22 (Rokka et al., 2009), the closest relative to Mpv17, one might expect the Mpv17 protein to form a channel through a lipid membrane. Pxmp 22 forms such a channel in vitro, with a diameter of 1.4 nm allowing the passage of molecules of about 500 Daltons. Analogous in vitro experiments will have to be engaged to clarify, if Mpv17 exhibits similar properties. It would be of interest to see which, if any, molecules can pass the proposed Mpv17 channel and how such channel passage would be regulated. The latter would be very important to keep the mitochondrial membrane potential intact. Mpv17 homo- or heteropolymeric complex formation has not been described in animal systems. However, the yeast homolog Sym 1 has been described to form large complexes of unknown composition (Reinhold et al., 2012). In this model a possible passage of metabolic intermediates through Sym 1 within the mitochondrial membrane has been demonstrated experimentally (Reinhold et al., 2012, see below).
The Mpv17 modulator Prkdc
Reviewing the pathology in the Mpv17 mouse model analyzed in various publications, it appeared that the glomerulosclerosis phenotype changed over time with respect to onset, severity, and lifespan of animals. In the initial study of Weiher et al. (1990), proteinuria was diagnosed in mice between 6 and 8 weeks of age. Subsequent studies demonstrated animals spared of proteinuria until 40 weeks of age (O’Bryan et al., 2000) or 18 months of age (Viscomi et al., 2009). Accordingly, Weiher et al. reported that most animals had died 18 weeks after birth owing to renal failure, whereas in other reports, mice survived for 1 year (O’Bryan et al., 2000) or for about 2 years (Viscomi et al., 2009). Thus, a function modifying the phenotypic severity, which was selected for by the breeding of the homozygous Mpv17 mutant mouse strain for survival seemed likely. Such a modifying gene activity was identified by Papeta et al., in 2010, when studying the Adriamycin (ADR) nephopathy mouse model (Papeta et al., 2010). This established model of renal disease involves diminution of glomerular podocytes upon ADR treatment with consequential development of glomerulosclerosis similar to the Mpv17 mutant phenotype (Papeta et al., 2010). Besides this, mtDNA mutation and depletion occur and are assumed to account for other cytotoxic effects of ADR, such as, for instance, ADR induced cardiomyopathy (Suliman et al., 2007; Papeta et al., 2010). When mice were ADR treated, it revealed, that some mouse strains develop kidney pathology upon treatment and some do not. Papeta et al. found that sensitivity or resistance to ADR depended on the activity and the genetics of the so-called Prkdc gene, which codes for the catalytic subunit of the DNA-dependent protein kinase. This kinase activity possesses a DNA-binding subunit as well (Davis et al., 2013) and repairs nuclear DNA double-strand breaks by means of non-homologous DNA end joining (Lieber, 2010). In short, it turned out that different mouse stains carried different polymorphisms, leading to different enzyme activity. In addition, loss of function mutants existed and were tested alongside. It revealed that PRKDC function inversely correlated with the ADR effect on mtDNA depletion and kidney pathology. Given the phenotypic similarity between the ADR model and the Mpv17 homozygous mutant with respect to kidney pathology and mtDNA depletion, a genetic interaction between Prkdc and Mpv17 was suspected and tested by interbreeding mutants in both genes. Indeed, mice homozygous for both, mutant Prkdc and mutant Mpv17 were shown to rapidly develop kidney disease, as well as liver and heart pathology in adulthood, even without ADR treatment, shortening the lifespan of these animals to about 16 weeks. By contrast, homozygous knock out animals for either Prkdc or Mpv17 survived up to 12 months. With respect to mtDNA content it was found, that loss of PRKDC function aggravated mtDNA depletion except in liver tissue, in which Mpv17 homozygous mice showed similarly low levels of mtDNA regardless of Prkdc gene dosage whereby in this tissue PRKDC is normally expressed only at low levels. Of note, despite of low PRKDC expression a liver phenotype similar to the human MDDS pathology did not develop in the animals with accelerated kidney pathology. This underlines the notion, that apparently mt DNA depletion has very different consequences in humans and mice on the organismal level. The modifying influence of the PRKDC function on Mpv17 mutant pathology is schematically illustrated in Figures 2 and 3.

The MDDS causing functions of Mpv17 and ribonucleotide reductase (RR) and their modifier repair kinases in mammals: analogous pathologies of RR- and Mpv17- organisms.
Humans develop MDDS and mice develop ROS mediated kidney disease in both cases. Both pathomechanisms are modified by kinases (ATM normally phosphorylates RR as indicated; relevant PRKDC target unknown) involved in double-strand break DNA repair.
ADR is an agent used in chemotherapy of hematologic malignancies as well as solid tumors. It intercalates into the DNA thereby inhibiting DNA replication, causing mtDNA depletion and also mutations in nuclear and mitochondrial DNA. The PRKDC protein, as stated above, constitutes the catalytic subunit of the DNA-dependent protein kinase (Papeta et al., 2010), which repairs nuclear DNA double-strand breaks by means of non-homologous DNA end joining (Lieber, 2010). However, the role of PRKDC in mtDNA maintenance remains a challenging subject.
Immunohistochemistry experiments revealed that PRKDC protein resides in the nucleus and not in mitochondria (Papeta et al., 2010) which makes it unlikely, that the PRKDC acts directly on the mitochondrial genome. Instead, indirect effects seem more likely, as it has been shown to be involved in a multitude of cellular functions. For example, it has been associated with regulation of nuclear respiratory factor 1, which plays an important role in the transcriptional activation of genes that guide function and biogenesis of mitochondria (Hossain et al., 2009). Targets for PRKDC phosphorylation might be the MPV17 protein itself or other proteins involved in mitochondrial genome maintenance (Papeta et al., 2010). The Mpv17 protein contains several theoretically possible phosphorylation targets but in fact, has never been shown to be a phosphoprotein. It remains to be determined whether functional PRKDC deficits might play a role not only in the murine Mpv17 phenotype, but may also contribute to MDDS in humans.
A similar relationship to the one between PRKDC and MPV17 in mice has been established between the ataxia-telangiectasia mutated (ATM) kinase, which is involved in the repair of DNA double-strand breaks, and RRM2B the small subunit (p53R2) of ribonucleotide reductase (RR) (Figure 3). RR contributes to desoxynucleotide triphosphate (dNTP) de novo synthesis by converting ribonucleoside 5′-diphosphates into deoxyribonucleoside 5′-diphosphates. As mentioned above, similar to mutations in the MPV17 gene, mutations in the RRM2B gene cause MDDS in humans and kidney disease in mice (Bourdon et al., 2007). It was reported that ATM positively regulates RR regarding expression and stability of its subunits (Eaton et al., 2007). Other work indicated that in response to DNA damage, ATM regulates p53R2 through the phosphorylation of serine 72, which supports protein stability (Chang et al., 2008) (Figure 3). Last but not least, loss of ATM function has been related to oxidative damage-mediated organ pathology in mice (Barlow et al., 1999; Reliene et al., 2004). Thus, as in the ADR and Mpv17 mutant models, imbalanced ROS production appears to mediate pathophysiology in the ATM and RRM2B model.
The significance of ROS in the ADR and Mpv17 models for the development of the organismal phenotype has been established. However, the underlying mechanisms are still unclear. ROS might be a byproduct or a mediator of mtDNA depletion, or both, whereby PRKDC might confer the cellular ability to cope with genotoxic stress conditions. However, other explanations might also be valid.
The yeast MPV17 ortholog SYM1
The Mpv17 gene was initially shown to be strongly conserved within the animal kingdom (Weiher et al., 1990; Karasawa et al., 1993). A functional ortholog of MPV17 in the yeast Saccharomyces cerevisiae, however, was identified by Trott and Moreno in 2004. As the gene is induced by heat stress, the open reading frame YLR251W was termed SYM1 (stress-inducible yeast Mpv17) (Trott and Morano, 2004). MPV17 and SYM1 are regarded to be orthologs based on the degree of sequence homology, which was reported with 48% similarity and 32% identity, as well as the similar design of transmembrane domains. Functional equivalence derived from the finding, that expression of human MPV17 in Sym-1 negative S. cerevisiae complemented the mutant yeast phenotype (Trott and Morano, 2004) while mutated human MPV17 variants did not (Spinazzola et al., 2006). Sym1 protein was located in the inner mitochondrial membrane and represents a heat shock protein associated with oxidative growth at elevated temperature (Trott and Morano, 2004; Spinazzola et al., 2006; Dallabona et al., 2010). In particular, while both, wild type and SYM1 mutant yeast strains grew on ethanol at 30°C, SYM1 mutants were not able to use ethanol (Trott and Morano, 2004) as well as other aerobic carbon sources at 37°C and thus failed to grow (Dallabona et al., 2010). Apparently, SYM1 mutants were defective of OXPHOS and supplementation with several non-essential amino acids corrected the defective OXPHOS phenotype. Therefore, Dallabona and coworkers reasoned that the Sym1 protein might be involved in anaplerotic reactions, which fuel the tricarboxylic acid (TCA) cycle with amino acid derivatives, such as oxalacetate and alpha-ketoglutarate (Dallabona et al., 2010). Correspondingly, the ability to store glycogen was found diminished SYM1 mutant, pointing to a compromised gluconeogenesis, which in turn depends on the flux of TCA cycle intermediates to the cytosol (Dallabona et al., 2010). Given the defective exchange of TCA cycle intermediates between the cytosol and mitochondria, central metabolic reactions would no longer be fueled, causing a disabled growth phenotype (Dallabona et al., 2010). Figure 4 depicts a schematical illustration on the role of SYM1 in S. cerevisiae.
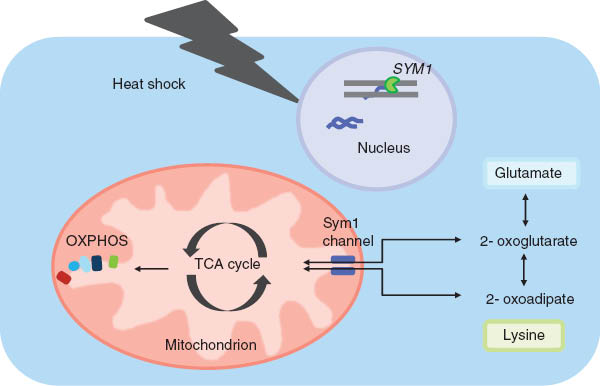
Stress-induced Sym1 channel permits bidirectional transport of metabolic intermediates across the inner mitochondrial membrane in Sacharomyces cerevisiae.
The mechanism was proposed by Dallabona et al. (2010): Heat shock (arrow) induces Sym 1 expression. The Sym 1 channel is essential for transport of TCA cycle intermediates in and out of mitochondria at non permissive temperature. Sym 1 function can be bypassed by delivery of intermediates through other pathways (here oxoadipate and oxoglutarate can be transported by Odc 1, which when overexpressed, complements a missing Sym 1 function).
Besides the growth arrest on aerobic carbon sources, the SYM1 mutant phenotype is characterized by the formation of small colonies. These respiratory-deficient clones were observed when SYM1 mutant yeast was grown in the presence of glucose in addition to ethanol as carbon source at elevated temperature (Spinazzola et al., 2006). Such cells were reminiscent of respiratory-defective yeast clones commonly referred to as ‘petite’ cells, caused by mutations in mtDNA (Clark-Walker and Miklos, 1975). In fact, SYM1 mutants showed rearrangements of mtDNA. Hence, similar to its mammalian ortholog MPV17, SYM1 might be involved in maintenance of mtDNA (Spinazzola et al., 2006). However, in yeast – unlike in mammals – genetic rescue experiments showed that the metabolic phenotype and mtDNA instability of Sym 1 deficient Sacharomyces cerevisiae can be separated from each other (Dallabona et al., 2010).
The phenotype of Sym1 negative yeast can be complemented to some extent by over expression of Ymc1 and Odc1. These two functions are involved in anaplerotic reactions in that they permit the passage of TCA cycle intermediates from the cytosol into mitochondria and vice versa (Dallabona et al., 2010). In particular, Odc1 has been shown to control the flux of 2-oxoadipate and 2-oxoglutarate from matrix to cytoplasm for lysine and glutamate synthesis (Palmieri et al., 2001). This suggests a possibly similar role of Sym1 during heat shock response (Dallabona et al., 2010; Figure 4).
Regarding the molecular function of Sym1, the genetic data suggested that Sym1 exhibited channel-like properties or acted like a pore to permit bidirectional transport of small molecules across the inner mitochondrial membrane under heat stress conditions. In line with these considerations, Sym1 has recently been shown to form a voltage-dependent channel in the inner mitochondrial membrane conducting cations. At physiological membrane potential, the channel closed, probably in order to prevent leakage of ions (Reinhold et al., 2012). The putative pore size of 1.6 nm would permit the passage of molecules as large as metabolites, and thus metabolic intermediates. For the transport of Sym1 into mitochondria, the translocase of the outer membrane was required and insertion of the protein into the inner mitochondrial membrane depended on the presequence translocase of the inner membrane (TIM23) (Reinhold et al., 2012). However, while the TIM23 complex normally transports proteins presenting an N-terminal presequence for recognition, which is cleaved later on (Geissler et al., 2002), Sym1 or its mammalian ortholog Mpv17 are not processed and thus, appear not to depend on this type of targeting signal. The organization of the Sym1 channel in the inner mitochondrial membrane is unknown, however. The model in Figure 1 constitutes only a very simplified representation with respect to Sym 1. Two larger Sym1 containing complexes of different molecular masses were identified within mitochondria and it might be possible that mature Sym1 complexes contain other components as well (Reinhold et al., 2012).
Taken together, Sym1 one represents a voltage dependent channel within the inner mitochondrial membrane, which is closed at physiological membrane potential and allows transport of metabolic intermediates, when opened.
The zebrafish Dario rerio MPV17 homolog tra
In the zebrafish Dario rerio, the transparent (tra) gene has been found to encode the homolog of mammalian Mpv17, which is localized to mitochondria as well. Human and fish Mpv17 proteins share 69% sequence identity and have conserved transmembrane domains (Krauss et al., 2013). Similar to the situation in mammals, tra is expressed ubiquitously and throughout development (Krauss et al., 2013). Meanwhile, deprivation of the functional gene product causes a tissue specific phenotype. However, unlike in mammals, the phenotype is not lethal, and the levels of mtDNA were found normal in essential tissues such as brain, liver and muscle (Krauss et al., 2013). Absence of functional Mpv17 results in severe diminution of pigment cells (particularly iridophores and melanophores) responsible for the characteristic stripe formation of zebrafish (Krauss et al., 2013). In unmutated fish, iridophores are responsible for the silvery to golden reflections, while the melanophores contribute the dark coloration. A regulatory hierarchy determines that in the absence of iridophores the melanophores cannot survive. Within iridophores there are cytoplasmic organelles, known as iridosomes, in which guanine is arranged into crystal-like reflective stacks (Figure 5), accounting for silvery to golden reflections. In the absence of these pigments, the iridosomes go into apoptosis leading to death of the melanosomes as well finally resulting in transparent fish. Krauss et al. explain the iridophore-restricted tra mutant phenotype by a special cellular requirement of guanine specifically in these cells (Figure 5): Because of the deposition of this molecule in large amounts in iridomes, guanosine, guanosine triphosphate (GTP), or deoxyguanosine triphosphate might be transported through the proposed tra channel into the mitochondria. However, in tra mutants, the absence of tra function might diminish the transport of nucleotides and precursors into mitochondria of iridophores, thereby impeding mitochondrial functions with consequential apoptosis (Krauss et al., 2013). In support of this hypothesis, the tra mutant phenotype resembles that of two further zebrafish mutants, gart and paics, in which the lack of the GTP de novo synthesis results in the absence of pigmentation patterns (Ng et al., 2009). In addition, experimental support for the idea that regulated cell death is involved in the pathomechanism comes from finding vesicles resembling autophagosomes within tra mutant iridophores but not in wild type pigment cells (Krauss et al., 2013).

Iridophore mitochondrial functions depend on intact guanosine metabolism in Dario rerio.
Pathomechanism as proposed by Krauss et al. (2013): the iridosome organelle within the iridosome cell requires large amounts of guanine, which is the main component of the silvery crystalline reflective stacks. In the absence of the Mpv17 homolog tra, guanosine or its phosphate derivatives are not taken up by the mitochondria, leading to mtDNA depletion. Lack of OXPHOS activity leads then to apoptosis of the iridophore cell.
Of note, one of the human genes causing – when mutated – an MDDS phenotype similar to that of Mpv17-related MDDS is the deoxyguanosine kinase (DGUOK) gene (Salviati et al., 2002; El-Hattab et al., 2010). This gene codes for one of several mitochondrial enzymes involved in the salvage pathway of purine nucleotides in that it delivers deoxyguanosine (Suomalainen and Isohanni, 2010).
Concluding remarks
MDDS are rare diseases, which are heterogeneous in their clinical manifestations and overlapping with other disorders. MPV17-related hepatocerebral MDDS is a remarkable subtype of this group of disorders, in which the function of the affected gene has remained unknown.
Loss of function mutations in the MPV17 gene and its homologs in different organisms cause a variety of different phenotypes, despite of more-or-less ubiquitous expression. While in humans this leads to the lethal MDDS disease, in mice glomerulosclerosis and inner ear dysfunction result from Mpv17 deficiency. A Dario rerio tra mutation leads to loss of color-causing guanine deposition and loss of the chromophoric iridophore cells. In yeast, temperature sensitivity of growth in ethanol results from inactivation of the Mpv17 homolog Sym1.
From their amino acid sequence, these proteins represent membrane proteins with four lipid layer passages. In yeast and human cells they have been localized to mitochondrial inner membranes. In mammals, they share remarkable homology to peroxisomal protein family members. The yeast channel appears to allow for influx of TCA cycle intermediates into the mitochondria, while in animals the most likely role in may be nucleotide transport. In zebrafish this must include guanine, guanosine or guanosine phosphate. Although in mammals this was originally subject to dispute (Zwacka et al., 1994; Spinazzola et al., 2006), in zebrafish and yeast the Mpv17 homoloques tra and Sym 1 localize to the mitochondria, the latter having been demonstrated to be a channel in the inner mitochondrial membrane (Reinhold et al., 2012). Given the functional conservation between the different MPV17 homologs it seems reasonable to hypothesize that vertebrate MPV17 forms a channel in the inner mitochondrial membrane, supplying the matrix with desoxynucleotide phosphates and/or nucleotide precursors.
Future work might focus first on the molecular function of the mammalian Mpv17 protein. This could include electrophysiological studies in vitro, analogous to the studies on pxmp22 (Rokka et al., 2009), aiming towards an understanding of potential channel properties of the molecule. Along these lines the potential to form a nucleotide conducting channel molecule could be addressed. This might also be explored in vivo by monitoring, if possible, the intracellular nucleotide levels in Mpv17 mutant and wild type cells. Further studies might address the question of potential formation of either homopolymers or heteropolymers with other proteins. Also, binding to potential ligands and identification of presumed other channel passenger molecules are desirable. Moreover, the question of selectivity and molecular regulation of this channel are interesting subjects for future research. As far as intracellular regulation is concerned, the finding of genetic interaction between the non-mitochondrial kinase PRKCD and a regulator of mitochondrial replication, MPV17, has an interesting parallel in the interaction between the Ataxia Teleangiectasia Mutant Kinase ATM and the MDDS gene RRMB2, both demonstrating nuclear-cytoplasmic-mitochondrial communication. These intriguing interactions also deserve further research effort.
To address these questions, biochemical tools are essential. For instance, molecular interactions might be studied with crosslinking approaches, but, more importantly, new and better anti-Mpv17 antibodies would be prerequisite. In this context, the different intracellular localizations of the different Mpv17 protein family members, and hence their different functions, remain another challenging question and lastly the distinct phenotypes produced by Mpv17 family member mutations in different species calls for the use of human primary cells from patients to uncover the pathomechanisms acting in the human disease.
Finally, it appears that the simple looking Mpv17 molecule cannot only cause devastating disease when dysfunctional in humans, but also very unexpected phenotypes in other biological systems, making it a fascinating, again and again surprising study subject, for so-far more than two decades.
About the authors

Stefanie Löllgen studied Applied Biology (Bsc) and Biomedical Sciences (Msc) at the Hochschule Bonn-Rhein-Sieg. Her Master’s thesis work is in part represented in this publication.

Hans Weiher graduated from the Universität Heidelberg, did postdoctoral work at UC Berkeley, USA, Heinrich Pette Institut, Hamburg, Germany, and the Whitehead Institute at MIT, Cambridge, USA. He held leading positions at various research institutions within Germany (Deutsches Krebsforschungszentrum, Heidelberg (DKFZ), Forschungszentrum Karlsruhe, and Institut für Diabetesforschung, München), and is now in charge of Human- and Molecular Genetics at Hochschule Bonn-Rhein-Sieg in Rheinbach, Germany.
Acknowledgments
We thank Heinz-Joachim Häbler (Hochschule Bonn-Rhein-Sieg) for expert supportive criticism during the generation of this work.
References
Alberio, S., Mineri, R., Tiranti, V., and Zeviani, M. (2007). Depletion of mtDNA: Syndromes and genes. Mitochondrion 7, 6–12.10.1016/j.mito.2006.11.010Suche in Google Scholar
Anderson, S., Bankier, A.T., Barrell, B.G., de Bruijn, M.H.L., Coulson, A.R., Drouin, J., Eperon, I.C., Nierlich, D.P., Roe, B.A., Sanger, F., et al. (1981). Sequence and organization of the human mitochondrial genome. Nature 290, 457–465.10.1038/290457a0Suche in Google Scholar
Barlow, C., Dennery, P.A., Shigenaga, M.K., Smith, M.A., Morrow, J.D., Roberts, L.J., Wynshaw-Boris, A., and Levine, R.L. (1999). Loss of the ataxia-telangiectasia gene product causes oxidative damage in target organs. Proc. Natl. Acad. Sci. USA 96, 9915–9919.10.1073/pnas.96.17.9915Suche in Google Scholar
Binder, C.J., Weiher, H., Exner, M., and Kerjaschki, D. (1999). Glomerular overproduction of oxygen radicals in Mpv17 gene-inactivated mice causes podocyte foot process flattening and proteinuria. Am. J. Pathol. 154, 1067–1075.10.1016/S0002-9440(10)65359-XSuche in Google Scholar
Blakely, E.L., Butterworth, A., Hadden R.D., Bodi I., He L., McFarland R., and Taylor, R.W. (2012). MPV17 mutation causes neuropathy and leukoencephalopathy with multiple mtDNA deletions in muscle. Neuromuscul. Disord. 22, 587–591.10.1016/j.nmd.2012.03.006Suche in Google Scholar PubMed PubMed Central
Bodnar, A.G., Cooper, J.M., Holt, I.J., Leonard, J.V., and Schapira, A.H.V. (1993). Nuclear complementation restores mtDNA levels in cultured cells from a patient with mtDNA depletion. Am. J. Hum. Genet. 53, 663–669.Suche in Google Scholar
Bottani, E., Giordano, C., Civiletto, G., Di Meo, I., Auricchio, A., Ciusani, E., Marchet, S., Lamperti, C., d’Amati, G., Viscomi, C., et al. (2014). AAV-mediated liver-specific MPV17 expression restores mtDNA levels and prevents diet-induced liver failure. Mol. Ther. 22, 10–17.10.1038/mt.2013.230Suche in Google Scholar PubMed PubMed Central
Bourdon, A., Minai, L., Serre, V., Jais, J.-P., Sarzi, E., Aubert, S., Chrétien, D., de Lonlay, P., Paquis-Flucklinger, V., Arakawa, H., et al. (2007). Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat. Genet. 39, 776–780.10.1038/ng2040Suche in Google Scholar PubMed
Chang, L., Zhou, B., Hu, S., Guo, R., Liu, X., Jones, S.N., and Yen, Y. (2008). ATM-mediated serine 72 phosphorylation stabilizes ribonucleotide reductase small subunit p53R2 protein against MDM2 to DNA damage. Proc. Natl. Acad. Sci. USA 105, 18519–18524.10.1073/pnas.0803313105Suche in Google Scholar PubMed PubMed Central
Clark-Walker, G.D. and Miklos, G.L.G. (1975). Complementation in cytoplasmic petite mutants of yeast to form respiratory competent cells. Proc. Natl. Acad. Sci. USA, 72, 372–375.10.1073/pnas.72.1.372Suche in Google Scholar PubMed PubMed Central
Copeland, W.C. (2012). Defects in mitochondrial DNA replication and human disease. Crit. Rev.Biochem. Mol. Biol. 47, 64–74.10.3109/10409238.2011.632763Suche in Google Scholar PubMed PubMed Central
Dalla Rosa, I., Durigon, R., Pearce, S.F., Rorbach, J., Hirst, E.M., Vidoni, S., Reyes, A., Brea-Calvo, G., Minczuk, M., Woellhaf, M.W., et al. (2014). MPV17L2 is required for ribosome assembly in mitochondria. Nucleic Acids Res. 2014 Jun 19. pii: gku513. [Epub ahead of print].Suche in Google Scholar
Dallabona, C., Marsano, R.M., Arzuffi, P., Ghezzi, D., Mancini, P., Zeviani, M., Ferrero, I., and Donnini, C. (2010). Sym1, the yeast ortholog of the MPV17 human disease protein, is a stress-induced bioenergetic and morphogenetic mitochondrial modulator. Hum. Mol. Genet. 19, 1098–1107.10.1093/hmg/ddp581Suche in Google Scholar
Davis, A.J., Lee, K.-J., and Chen, D.J. (2013). The N-terminal region of the DNA-dependent protein kinase catalytic subunit is required for its DNA double-stranded break-mediated activation. J. Biol. Chem. 288, 7037–7046.10.1074/jbc.M112.434498Suche in Google Scholar
Dimmock, D., Tang, L.-Y., Schmitt, E.S., and Wong, L.-J.C. (2010). Quantitative evaluation of the mitochondrial DNA depletion syndrome. Clin. Chem. 56, 1119–1127.10.1373/clinchem.2009.141549Suche in Google Scholar
Eaton, J.S., Lin, Z.P., Sartorelli, A.C., Bonawitz, N.D., and Shadel, G.S. (2007). Ataxia-telangiectasia mutated kinase regulates ribonucleotide reductase and mitochondrial homeostasis. J. Clin. Invest. 117, 2723–2734.10.1172/JCI31604Suche in Google Scholar
El-Hattab, A.W., Li, F.-Y., Schmitt, E., Zhang, S., Craigen, W.J., and Wong, L.-J.C. (2010). MPV17-associated hepatocerebral mitochondrial DNA depletion syndrome: New patients and novel mutations. Mol. Genet. Metab. 99, 300–308.10.1016/j.ymgme.2009.10.003Suche in Google Scholar
Garone, C., Rubio, J.C., Calvo, S.E., Naini. A., Tanji, K., Dimauro, S., Mootha, V.K., and Hirano, M. (2012). MPV17 mutations causing adult-onset multisystemic disorder with multiple mitochondrial DNA deletions. Arch. Neurol. 69, 1648–51.10.1001/archneurol.2012.405Suche in Google Scholar
Geissler, A., Chacinska, A., Truscott, K.N., Wiedemann, N., Brandner, K., Sickmann, A., Meyer, H.E., Meisinger, C., Pfanner, N., and Rehling, P. (2002). The mitochondrial presequencetranslocase: an essential role of Tim50 in directing preproteins to the import channel. Cell, 111, 507–518.10.1016/S0092-8674(02)01073-5Suche in Google Scholar
Girotti, A.W. (1985). Mechanisms of lipid peroxidation. Free Radic. Biol. Med. 1, 87–95.10.1016/0748-5514(85)90011-XSuche in Google Scholar
Hossain, M.B., Ji, P., Anish, R., Jacobson, R.H., and Takada, S. (2009). Poly(ADP-ribose). polymerase 1 interacts with nuclear respiratory factor 1 (NRF-1) and plays a role in NRF-1 transcriptional regulation. J. Biol. Chem., 284, 8621–8632.10.1074/jbc.M807198200Suche in Google Scholar PubMed PubMed Central
Iida, R., Yasuda, T., Tsubota, E., Takatsuka, H., Matsuki, T., and Kishi, K. (2006). Human Mpv17-like protein is localized in peroxisomes and regulates expression of antioxidant enzymes. Biochem. Biophys. Res. Commun. 344, 948–954.10.1016/j.bbrc.2006.04.008Suche in Google Scholar PubMed
Johnson, R., Yamabe, H., Chen, Y.P., Campbell, C., Gordon, K., Baker, P., Lovett, D., and Couser, W.G. (1992). Glomerular epithelial cells secrete a glomerular basement membrane-degrading metalloproteinase. J. Am. Soc. Nephrol. 2, 1388–1397.10.1681/ASN.V291388Suche in Google Scholar
Kaji, S., Murayama, K., Nagata, I., Nagasaka, H., Takayanagi, M., Ohtake, A., Iwasa, H., Nishiyama, M., Okazaki, Y., Harashima, H., et al. (2009). Fluctuating liver functions in siblings with MPV17 mutations and possible improvement associated with dietary and pharmaceutical treatments targeting respiratory chain complex II. Mol. Genet. Metab. 97, 292–296.10.1016/j.ymgme.2009.04.014Suche in Google Scholar
Kaldi, K., Diestelkötter, P., Stenbeck, G., Auerbach,S., Jäkle, U., Mägert, H.J. Wieland, F.T., and Just, W.W. (1993). Membrane Topology of the 22 kDa integral peroxisomal membrane protein. FEBS Lett. 315, 217–222.10.1016/0014-5793(93)81167-XSuche in Google Scholar
Karadimas, C.L., Vu, T.H., Holve, S.A., Chronopoulou, P., Quinzii, C., Johnsen, S.D., Kurth, J., Eggers, E., Palenzuela, L., Tanji, K., et al. (2006). Navajo neurohepatopathy is caused by a mutation in the MPV17 gene. Am. J. Hum. Genet. 79, 544–548.10.1086/506913Suche in Google Scholar PubMed PubMed Central
Karasawa, M., Zwacka, R.M., Reuter, A., Fink, T., Hsieh, C.L., Lichter, P., Francke, U., and Weiher, H. (1993). The human homolog of the glomerulosclerosis gene Mpv17: structure and genomic organization. Hum. Mol. Genet. 2, 1829–1834.10.1093/hmg/2.11.1829Suche in Google Scholar PubMed
Kimura, T., Takeda, S., Sagiya, Y., Gotoh, M., Nakamura, Y., and Arakawa, H. (2003). Impaired function of p53R2 in Rrm2b-null mice causes severe renal failure through attenuation of dNTP pools. Nat. Genet. 34, 440–445.10.1038/ng1212Suche in Google Scholar PubMed
Knowlden, J., Martin, J., Davies, M., and Williams, J.D. (1995). Metalloproteinase generation by human glomerular epithelial cells. Kidney Int. 47, 1682–1689.10.1038/ki.1995.233Suche in Google Scholar PubMed
Kornblum, C., Nicholls, T.J., Haack, T.B., Schöler, S., Peeva, V., Danhauser, K., Hallmann, K., Zsurka, G., Rorbach, J., Iuso, A., et al. (2013). Loss-of-function mutations in MGME1 impair mtDNA replication and cause multisystemic mitochondrial disease. Nat. Genet. 45, 214–219.10.1038/ng.2501Suche in Google Scholar PubMed PubMed Central
Krauss, J., Astrinides, P., Frohnhöfer, H.G., Walderich, B., and Nüsslein-Volhard, C. (2013). Transparent, a gene affecting stripe formation in Zebrafish, encodes the mitochondrial protein Mpv17 that is required for iridophore survival. Biology Open 2, 703–710.10.1242/bio.20135132Suche in Google Scholar PubMed PubMed Central
Lee, W.S. and Sokol, R.J. (2007). Liver disease in mitochondrial disorders. Semin. Liver Dis. 27, 259–273.10.1055/s-2007-985071Suche in Google Scholar PubMed PubMed Central
Lemmink, H.H., Schröder, C.H., Monnens, L.A.H., and Smeets, H.J.M. (1997). The clinical spectrum of type IV collagen mutations. Hum. Mutat. 9, 477–499.10.1002/(SICI)1098-1004(1997)9:6<477::AID-HUMU1>3.0.CO;2-#Suche in Google Scholar
Lieber, M.R. (2010). The mechanism of double-stranded DNA break repair by the nonhomologous DNA end joining pathway. Annu. Rev. Biochem. 79, 181–211.10.1146/annurev.biochem.052308.093131Suche in Google Scholar
Merkle, A.N., Nascene, D.R., and McKinney, A.M. (2012). MR imaging findings in the reticular formation in siblings with MPV17-related mitochondrial depletion syndrome. Am. J. Neuroradiol. 33, E34–E35.Suche in Google Scholar
Meyer zumGottesberge, A.-M., Reuter, A., and Weiher, H. (1996). Inner ear defect similar to Alport’s syndrome in the glomerulosclerosis mouse model Mpv17. Eur. Arch. Otorhinolaryngol. 253, 470–474.10.1007/BF00179952Suche in Google Scholar
Moraes, C.T., Shanske, S., Tritschler, H.-J., Aprille, J.R., Andreetta, F., Bonilla, E., Schon, E.A., and DiMauro, S. (1991). mtDNA depletion with variable tissue expression: a novel genetic abnormality in mitochondrial diseases. Am. J. Hum. Genet. 48, 492–501.Suche in Google Scholar
Müller, M., Smolders, J.W.T., Meyer zumGottesberge, A., Reuter, A., Zwacka, R.M., Weiher, H., and Klinke, R. (1997). Loss of auditory function in transgenic Mpv17-deficient mice. Hearing Res. 114, 259–263.10.1016/S0378-5955(97)00175-5Suche in Google Scholar
Navarro-Sastre, A., Martín-Hernández, E., Campos, Y., Quintana, E., Medina, E., de lasHeras, R.S., Lluch, M., Muñoz, A., del Hoyo, P., Martín, R., et al. (2008). Lethal hepatopathy and leukodystrophy caused by a novel mutation in MPV17 gene: Description of an alternative MPV17 spliced form. Mol. Genet. Metab. 94, 234–239.10.1016/j.ymgme.2008.01.012Suche in Google Scholar
Ng, A., Uribe, R.A., Yieh, L., Nuckels, R., and Gross, J.M. (2009). Zebrafish mutations in gart and paics identify crucial roles for de novo purine synthesis in vertebrate pigmentation and ocular development. Development 136, 2601–2611.10.1242/dev.038315Suche in Google Scholar
O’Bryan, T., Weiher, H., Rennke, H.G., Kren, S., and Hostetter, T.H. (2000). Course of renal injury in the Mpv17-deficient transgenic mouse. J. Am. Soc.Nephrol. 11, 1067–1074.10.1681/ASN.V1161067Suche in Google Scholar
Palmieri, L., Agrimi, G., Runswick, M.J., Fearnley, I.M., Palmieri, F., and Walker, J.E. (2001). Identification in Saccharomyces cerevisiae of two isoforms of a novel mitochondrial transporter for 2-oxoadipate and 2-oxoglutarate. J. Biol. Chem. 276, 1916–1922.10.1074/jbc.M004332200Suche in Google Scholar
Papeta, N., Zheng, Z., Schon, E.A., Brosel, S., Altintas, M.M., Nasr, S.H., Reiser, J., D’Agati, V.D., and Gharavi, A.G. (2010). Prkdc participates in mitochondrial genome maintenance and prevents Adriamycin-induced nephropathy in mice. J. Clin. Invest. 120, 4055–4064.10.1172/JCI43721Suche in Google Scholar
Parini, R., Furlan, F., Notarangelo, L., Spinazzola, A., Uziel, G., Strisciuglio, P., Concolino, D., Corbetta, C., Nebbia, G., Menni, F., et al. (2009). Glucose metabolism and diet-based prevention of liver dysfunction in MPV17 mutant patients. J. Hepatol. 50, 215–221.10.1016/j.jhep.2008.08.019Suche in Google Scholar
Reinhold, R., Krüger, V., Meinecke, M., Schulz, C., Schmidt, B., Grunau, S.D., Guiard, B., Wiedemann, N., van der Laan, M., Wagner, R., et al. (2012). The channel-forming Sym1 protein is transported by the TIM23 complex in a presequence-independent manner. Mol. Cell. Biol. 32, 5009–5021.10.1128/MCB.00843-12Suche in Google Scholar PubMed PubMed Central
Reliene, R., Fischer, E., and Schiestl, R.H. (2004). Effect of N-acetyl cysteine on oxidative damage and the frequency of DNA deletions in Atm-deficient mice. Cancer Res., 64, 5148–5153.10.1158/0008-5472.CAN-04-0442Suche in Google Scholar PubMed
Reuter, A., Nestl, A., Zwacka, R.M., Tuckermann, J., Waldherr, R., Wagner, E.-M., Höyhtyä, M., Meyer zumGottesberge, A.M., Angel, P., and Weiher, H. (1998). Expression of the recessive glomerulosclerosis gene Mpv17 regulates MMP-2 expression in fibroblasts, the kidney, and the inner ear of mice. Mol. Biol. Cell 9, 1675–1682.10.1091/mbc.9.7.1675Suche in Google Scholar PubMed PubMed Central
Rokka, A., Antonenkov, V.D., Soininen, R., Immonen, H.L., Pirilä, P.L., Bergmann, U., Sormunen, R.T., Weckström, M., Benz, R., and Hiltunen, J.K. (2009). Pxmp2 is a channel-forming protein in mammalian peroxisomal membrane. PLoS ONE, 4, e5090.10.1371/journal.pone.0005090Suche in Google Scholar PubMed PubMed Central
Rötig, A. and Poulton, J. (2009). Genetic causes of mitochondrial DNA depletion in humans. Biochim. Biophys. Acta 1792, 1103–1108.10.1016/j.bbadis.2009.06.009Suche in Google Scholar PubMed
Salviati, L., Sacconi, S., Mancuso, M., Otaegui, D., Camaño, P., Marina, A., Rabinowitz, S., Shiffman, R., Thompson, K., Wilson, C.M., et al. (2002). Mitochondrial DNA depletion and dGK gene mutations. Ann. Neurol. 52, 311–316.10.1002/ana.10284Suche in Google Scholar PubMed
Schenkel, J., Zwacka, R.M., Rutenberg, C., Reuter, A., Waldherr, R. ansWeiher, H. (1995). Functional rescue of the glomerulosclerosis phenotype in Mpv17 mice by transgenesis with the human Mpv17 homologue. Kidney Int. 48, 80–84.10.1038/ki.1995.270Suche in Google Scholar PubMed
Shigenaga, M.K., Hagen, T.M., and Ames, B.N. (1994). Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. USA 91, 10771–10778.10.1073/pnas.91.23.10771Suche in Google Scholar PubMed PubMed Central
Spallarossa, P., Altieri, P., Garibaldi, S., Ghigliotti, G., Barisione, C., Manca, V., Fabbi, P., Ballestrero, A., Brunelli, C., and Barsotti, A. (2006). Matrixmetalloproteinase-2 and -9 are induced differently by doxorubicin in H9c2 cells: The role of MAP kinases and NAD(P)H oxidase. Cardiovasc. Res. 69, 736–745.10.1016/j.cardiores.2005.08.009Suche in Google Scholar PubMed
Spinazzola, A. (2011). Mitochondrial DNA mutations and depletion in pediatric medicine. Semin. Fetal Neonat. M. 16, 190–196.10.1016/j.siny.2011.04.011Suche in Google Scholar PubMed
Spinazzola, A. and Zeviani, M. (2007). Disorders of nuclear-mitochondrial intergenomic communication. Biosci. Rep. 27, 39–51.10.1007/s10540-007-9036-1Suche in Google Scholar
Spinazzola, A., Viscomi, C., Fernandez-Vizarra, E., Carrara, F., D’Adamo, P., Calvo, S., Marsano, R.M., Donnini, C., Weiher, H., Strisciuglio, P., et al. (2006). MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat. Genet. 38, 570–575.10.1038/ng1765Suche in Google Scholar
Spinazzola, A., Santer, R., Akman, O.H., Tsiakas, K., Schaefer, H., Ding, X., Karadimas, C.L., Shanske, S., Ganesh, J., Di Mauro, S. et al. (2008). Hepatocerebral form of mitochondrial DNA depletion syndrome: novel MPV17 mutations. Arch. Neurol. 65, 1108–1113.10.1001/archneur.65.8.1108Suche in Google Scholar
Suliman, H.B., Carraway, M.S., Ali, A.S., Reynolds, C.M., Welty-Wolf, K.E., and Piantadosi, C.A. (2007). The CO/HO system reverses inhibition of mitochondrial biogenesis and prevents murine doxorubicin cardiomyopathy. J. Clin. Invest. 117, 3730–3741.10.1172/JCI32967Suche in Google Scholar
Suomalainen, A. and Isohanni, P. (2010). Mitochondrial DNA depletion syndromes – Many genes, common mechanisms. Neuromuscul. Disord. 20, 429–437.10.1016/j.nmd.2010.03.017Suche in Google Scholar
Taanman, J.-W., Bodnar, A.G., Cooper, J.M., Morris, A.A.M., Clayton, P.T., Leonard, J.V., and Schapira, A.H.V. (1997). Molecular mechanisms in mitochondrial DNA depletion syndrome. Hum. Mol. Genet. 6, 935–942.10.1093/hmg/6.6.935Suche in Google Scholar
Trott, A. and Morano, K.A. (2004). SYM1 is the stress-induced Saccharomyces cerevisiaeortholog of the mammalian kidney disease gene MPV17 and is required for ethanol metabolism and tolerance during heat shock. Eukaryot. Cell 3, 620–631.10.1128/EC.3.3.620-631.2004Suche in Google Scholar
Uusimaa, J., Evans, J., Smith, C., Butterworth, A., Craig, K., Ashley, N., Liao, C., Carver, J., Diot, A., Macleod, L., et al. (2013). Clinical, biochemical, cellular and molecular characterization of mitochondrial DNA depletion syndrome due to novel mutations in the MPV17 gene. Eur. J. Hum. Genet. 1–8.Suche in Google Scholar
Viscomi, C., Spinazzola, A., Maggioni, M., Fernandez-Vizarra, E., Massa, V., Pagano, C., Vettor, R., Mora, M., and Zeviani, M. (2009). Early-onset liver mtDNA depletion and late-onset proteinuric nephropathy in Mpv17 knockout mice. Hum. Mol. Genet. 18, 12–26.10.1093/hmg/ddn309Suche in Google Scholar
Weiher, H., Noda, T., Gray, D.A., Sharpe, A.H., and Jaenisch, R. (1990). Transgenic mouse model of kidney disease: insertional inactivation of ubiquitously expressed gene leads to nephrotic syndrome. Cell 62, 425–434.10.1016/0092-8674(90)90008-3Suche in Google Scholar
Wallace, D.C. (1999). Mitochondrial diseases in man and mouse. Science, 283, 1482–1488.10.1126/science.283.5407.1482Suche in Google Scholar PubMed
Wong, L.-J.C., Brunetti-Pierri, N., Zhang, Q., Yazigi, N., Bove, K.E., Dahms, B.B., Puchowicz, M.A., Gonzalez-Gomez, I., Schmitt, E.S., Truong, C.K., et al. (2007). Mutations in the MPV17 gene are responsible for rapidly progressive liver failure in infancy. Hepatology 46, 1218–1227.10.1002/hep.21799Suche in Google Scholar PubMed
Woodbine, L., Brunton, H., Goodarzi, A.A., Shibata, A., and Jeggo, P.A. (2011). Endogenously induced DNA double strand breaks arise in heterochromatic DNA regions and require ataxia telangiectasia mutated and Artemis for their repair. Nucleic Acids Res. 39, 6986–6997.10.1093/nar/gkr331Suche in Google Scholar PubMed PubMed Central
Zwacka, R.M., Reuter, A., Pfaff, E., Moll, J., Gorgas, K., Karasawa, M., and Weiher, H. (1994). The glomerulosclerosis gene Mpv17 encodes a peroxisomal protein producing reactive oxygen species. EMBO J. 13, 5129–5134.10.1002/j.1460-2075.1994.tb06842.xSuche in Google Scholar PubMed PubMed Central
©2014 by De Gruyter
Artikel in diesem Heft
- Frontmatter
- Reviews
- Cholesterol lowering: role in cancer prevention and treatment
- The role of the Mpv17 protein mutations of which cause mitochondrial DNA depletion syndrome (MDDS): lessons from homologs in different species
- Research Articles/Short Communications
- Genes and Nucleic Acids
- Identification of a novel PHEX mutation in a Chinese family with X-linked hypophosphatemic rickets using exome sequencing
- Protein Structure and Function
- Complement activation by salivary agglutinin is secretor status dependent
- Synthesis, biological evaluation, and docking studies of PAR2-AP-derived pseudopeptides as inhibitors of kallikrein 5 and 6
- Membranes, Lipids, Glycobiology
- Recombinant HDL (Milano) protects endotoxin-challenged rats from multiple organ injury and dysfunction
- Cell Biology and Signaling
- miR-126 regulates platelet-derived growth factor receptor-α expression and migration of primary human osteoblasts
- Legumain expression, activity and secretion are increased during monocyte-to-macrophage differentiation and inhibited by atorvastatin
- Proteolysis
- Cell surface serine protease matriptase-2 suppresses fetuin-A/AHSG-mediated induction of hepcidin
Artikel in diesem Heft
- Frontmatter
- Reviews
- Cholesterol lowering: role in cancer prevention and treatment
- The role of the Mpv17 protein mutations of which cause mitochondrial DNA depletion syndrome (MDDS): lessons from homologs in different species
- Research Articles/Short Communications
- Genes and Nucleic Acids
- Identification of a novel PHEX mutation in a Chinese family with X-linked hypophosphatemic rickets using exome sequencing
- Protein Structure and Function
- Complement activation by salivary agglutinin is secretor status dependent
- Synthesis, biological evaluation, and docking studies of PAR2-AP-derived pseudopeptides as inhibitors of kallikrein 5 and 6
- Membranes, Lipids, Glycobiology
- Recombinant HDL (Milano) protects endotoxin-challenged rats from multiple organ injury and dysfunction
- Cell Biology and Signaling
- miR-126 regulates platelet-derived growth factor receptor-α expression and migration of primary human osteoblasts
- Legumain expression, activity and secretion are increased during monocyte-to-macrophage differentiation and inhibited by atorvastatin
- Proteolysis
- Cell surface serine protease matriptase-2 suppresses fetuin-A/AHSG-mediated induction of hepcidin